

Abstract
Hepatic steatosis prevails each year. Autophagy is integral in mitochondrial quality control and lipid homeostasis in the liver. No pharmacological strategies are currently available to reduce hepatic steatosis, but exercise has been known to improve clinical outcomes of chronic liver disease, particularly non-alcoholic fatty liver disease (NAFLD). Recent studies suggest that exercise may improve NAFLD through enhancing autophagy.
Keywords: non-alcoholic fatty liver disease, liver, lipids, exercise, autophagy, mitochondria, mitophagy
Summary for TOC
We propose a hypothesis that activation of autophagy through exercise ameliorates cellular defects underlying the pathogenesis of nonalcoholic fatty liver disease
Introduction
The liver plays a central role in lipid homeostasis. This second largest organ in the body normally contains some fat. However, fatty liver whose fat content is greater than 5% – 10% percent of the liver’s weight, develops either by chronic and binge alcohol consumption or by excess fat intake. This pathological manifestation is becoming more common among nondrinkers, especially those who are overweight or have type 2 diabetes. Steatotic liver disorder independent of alcohol consumption is called nonalcoholic fatty liver disease (NAFLD). Despite few or no symptoms, 10–20% of the patients with NAFLD can develop more severe liver maladies accompanying hepatic inflammation, fibrosis and damage, a pathological condition termed nonalcoholic steatohepatitis (NASH) (48). Over a period of 10 years, 8 to 26% of NASH patients can develop liver cirrhosis (39).
NAFLD is recognized as one of the most common causes of chronic liver disease (CLD). Globally, CLD causes 1.2 million deaths annually and ranks as the 8th most common cause of death (36). This prevailing disease is also a significant health burden in the United States, and responsible for approximately 66,000 deaths annually (1). NAFLD and NASH are often observed in persons who are middle-aged, overweight, obese, diabetic or pre-diabetic. Notably, not every obese or diabetic person has NASH. Children even without any apparent risk factors can also develop NASH. Currently, no specific therapies are available to treat NAFLD or NASH except for the changes in life style. Treatments for very advanced conditions including cirrhosis and hepatocellular cancer are limited to surgical resection and liver transplantation. However, the patients with predisposed liver disease poorly tolerate surgical stresses during these operations.
Exercise or physical activity has shown promising results in clinical studies as well as in experimental models of obesity and NAFLD management (5, 8). The beneficial effects of exercise on fatty livers have been recently attributed to widespread stimulation of autophagy during physical activity (18). Autophagy is a catabolic process that eliminates surplus or abnormal cellular constituents and dysfunctional organelles. Mitophagy and lipophagy selectively clear dysfunctional mitochondria and excess lipids, respectively (14). As this selective autophagy plays an essential role in energy homeostasis and lipid metabolism, defective autophagy causes cells to accumulate abberrant mitochondria and surplus lipids, leading to bioenergetic failure and impaired lipid homeostasis. Here, we will review recent advances in the muscle-liver crosstalk after physical exercise. Additionally we propose that activation of autophagy through exercise ameliorates cellular defects underlying the pathogenesis of NAFLD.
Autophagy and NAFLD
Autophagy is a cellular degradation pathway for surplus or abnormal organelles, excess lipids, and protein aggregates. Importantly, this catabolic process is the only known pathway to selectively eliminate damaged or dysfunctional mitochondria. In general, autophagy protects cells against various stresses such as hypoxia, oxidative stress or invasion of intracellular pathogens by selectively or non-selectively sequestering cytotoxic contents (14). Besides its cytoprotective roles, autophagy is also directly linked to energy homeostasis in the cell because the autophagic degradation converts surplus cellular materials into energy substrates. Deficiencies in essential substrates for cell survival including amino acids and nitrogen, for example, are powerful stimuli for autophagy, whereas the cells with sufficient energy substrates have low basal autophagic activity.
Autophagy is divided into three categories: macroautophagy, microautophagy and chaperone-mediated autophagy. Macroautophagy represents the canonical autophagy where a nascent autophagic vesicle called the autophagosome is formed from an isolation membrane or phagophore, which subsequently recognizes and surrounds target constituents in the cytoplasm. After elongation of the phagophore, the double-membrane autophagosome fuses with the endosome or lysosome to remove their cargos by acidic hydrolases and proteases. Amino acids, lipids, and nucleotides are typical end products following autophagic degradation that are later recycled back to the cytoplasm for other purposes. The phagophore membrane can originate from several sources including the endoplasmic reticulum (ER), mitochondria, ER-mitochondria contact sites, and the ER-Golgi intermediate compartment as well as the plasma membrane (7, 14). Microautophagy and chaperone-mediated autophagy are two alternative routes to the lysosome. While the former directly engulfs cytosolic constituents by the lysosome, the latter selectively delivers cargo proteins containing the specific peptide sequence KFERQ to the lysosome, wherein heat shock cognate protein 70 (HSC70) plays a critical role in substrate translocation (14).
Macroautophagy, herein referred to as autophagy, is finely tuned by the energy status of the cell. The orchestrated interaction between the mechanistic target of rapamycin complex 1 (mTORC1), adenosine monophosphate activated protein kinase (AMPK), and UNC-51 like kinase 1 (ULK1) initiates autophagy (7). Under nutrient-rich conditions, ULK1 is phosphorylated at the residue of Ser757 by mTORC1 and subsequently dissociated from AMPK. This process promotes cell growth and proliferation by inducing the synthesis of proteins and lipids. In contrast, energy-deprived states activate AMPK, which in turn phosphorylates the residues of Ser317, 555, 777 of ULK1 and initiates autophagy. Phosphorylated ULK1 recruits and further phosphorylates both autophagy-related protein 13 (ATG13) and focal adhesion kinase family interacting protein of 200 kDa (FIP200). As a consequence, the ULK1-ATG13-FIP200 complex is formed on the surface of phagophores. Furthermore, ULK1 suppresses mTORC1 through its binding with raptor. Notably, exercise can promote AMPK and prevent mTORC1 in skeletal muscle (6). Following endurance physical activity, the rate of energy consumption outpaces the rate of production, resulting in a higher ratio of adenosine monophosphate (AMP) over adenosine triphosphate (ATP).
Beclin-1 (BECN1) is another important player in autophagy initiation. Initially identified as a B cell lymphoma-2 (BCL2) interacting protein, this autophagy-initiating protein has BCL2-homology-3 (BH3) domain that can bind to anti-apoptotic BCL2 family members (19). The interaction of ER-localized BCL2, but not mitochondrial-localized BCL2, with BECN1 governs autophagy onset. Under anti-autophagy conditions such as nutrient sufficiency, BCL2 firmly binds to BECN1. On the contrary, under pro-autophagy conditions like nutrient insufficiency, c-Jun N-terminal protein kinase 1 (JNK1) phosphorylates the residues Thr69, Ser70, and Ser97 of the non-structured loop of BCL2, leading to disruption of BCL2/BECN1 interaction. As a consequence, BECN1 becomes liberated from BCL2, which in turn forms the BECN1-ATG14-Vacuolar sorting protein 34 (VPS34)-VPS15 class III PI3K core, a molecular complex essential to autophagy initiation (14).
Mitochondria and NAFLD
The metabolic rate of the liver and brain is about 0.28 kcal/g/day, which is 20 times greater than that of resting skeletal muscles (33). Mitochondria supply hepatocytes with ATP through oxidative phosphorylation and β-oxidation. Therefore, mitochondrial dysfunction could adversely impact on liver function. The mitochondria in the fatty liver exhibit structural alterations such as matrix swelling, disrupted cristae, and changes in shape (21). Chronic high fat diet reduces mitochondrial electron transfer and impairs energy output in the liver (2) and skeletal muscle (45). In response to high concentrations of intracellular lipids, mitochondria initially burn more fatty acids to relieve the lipid burden. However, this metabolic adaptation could eventually injure mitochondria due to increased production of reactive oxygen species (ROS).
High fat or high glucose intake likely favors the onset of mitochondrial permeability transition (MPT) (50). Mitochondrial overloading of ROS culminates in MPT onset (25). The mitochondrial inner membrane is virtually impermeable to all solutes except for those having specific carriers or exchangers. However, the opening of MPT pores upon stresses allows an unregulated influx of solutes up to 1,500 Da into the mitochondrial matrix, leading to mitochondrial swelling, uncoupling of oxidative phosphorylation, depolarization of the mitochondrial membrane potential, ATP depletion, and ultimately necrotic cell death. MPT onset can also provoke apoptotic cell death. While cytochrome c is normally sequestered in the intermembrane space in functional mitochondria, mitochondrial swelling and subsequent rupture of the outer membrane following the MPT release cytochrome c to the cytosol. Released cytochrome c binds to the apoptosis-inducing factor-1 and pro-caspases to develop apoptosis (25).
Lipophagy and regulation of cellular lipid stores
While mitophagy prevents accumulation of dysfunctional or abnormal mitochondria, lipophagy specifically removes overloaded lipids (14). How lipophagy selectively targets and clears surplus lipids remains elusive, although the onset of lipophagy is likely to be modulated by the interaction between ancient ubiquitous protein 1 (AUP1) and E2 ubiquitin conjugase G2 (UBE2G2) at the surface of lipid droplets (14). During lipophagy, the autophagosome sequesters either small lipid droplets or portions of large lipid droplets in a microtubule-associated protein light chain 3 (LC3)- and small guanosine triphosphatase (GTPase) RAB7-dependent manner (14). Unlike nonselective canonical autophagy, chaperone-mediated autophagy appears to be involved in lipophagy activation (14). Mice deficient in chaperone-mediated autophagy fail to remove extra lipid droplets. Furthermore, these knockout animals rapidly accumulate lipid droplets in the liver without affecting macroautophagy.
The duration of fatty acid uptake distinctly influences lipophagy in the liver. While short-term treatment of free fatty acids stimulates hepatic lipophagy, prolonged exposure of fatty acids suppresses the fusion between autophagosomes and lysosomes, a final step for lipophagic clearance (14). In a genetic (ob/ob) model of murine obesity, hepatic levels of key autophagy components including LC3, BECN1, ATG5, and ATG7 have been known to decline (49).
Beneficial effects of exercise on NAFLD
Changes in life style such as weight loss and dietary modification have long been established as the first step in the management of NAFLD. Weight loss appears to independently improve hepatic function in NAFLD, although improved profile of intrahepatic lipids requires at least 3%–5% weight loss through physical activity and calorie control (5). Exercise alters various biochemical activities not only in the muscle, but also in the liver and adipose tissue. The muscle-liver crosstalk in energy and metabolic balance is also inferred from the observation that the patients with CLD manifest a higher incidence of sarcopenia, a loss of muscle mass (20). A recent study further suggests a strong association between sarcopenia and NAFLD in both non-obese and obese subjects (29). The importance of muscle-liver crosstalk is also implicated in a transgenic animal study where genetic ablation of myostatin, a TGF-beta superfamily member that regulates skeletal muscle mass (34), ameliorated high fat diet-induced elevation of liver weight (51). Considering multiple benefits on the metabolic syndrome by increasing physical activity, it has been proposed that the skeletal muscle could be a pharmacological target for treating metabolic disorders including NAFLD (35).
How exercise directly or indirectly diminishes intrahepatic lipids independently of diet modification, however, remains unclear. The simplest view could be mobilization of hepatic lipids to the muscle in order to fuel muscular energy deficit during physical activity. Exercise increases glucose uptake in muscles and concomitantly signals the liver to enhance glucose production to support continued energy expenditure. The demand for increased gluconeogenesis further stimulates the degradation of intracellular lipids to provide mitochondrial substrates for β-oxidation. Besides enhancing free fatty acid oxidation in mitochondria, physical activity, particularly chronic aerobic exercise, may also reduce hepatic lipogenesis. In a high fat-fed mouse model, treadmill exercise substantially decreases the expression of sterol regulatory element-binding protein-1c (SREBP1c), a transcription factor triggering triglyceride synthesis (8). Chronic consumption of high fat or high carbohydrate diet elevates levels of inflammatory cytokines such as tumor necrosis factor alpha (TNFα) and interleukin-1β (IL-1β) (38). We have previously shown that in livers of old rats there was a significantly increase in nuclear presence of nuclear factor κB (NF-κB) and several other proteins, demonstrating an increased pro-inflammatory response. The age-associated increase in the upregulation of pro-inflammatory proteins was substantially attenuated in the livers of old animals exposed to long-term voluntary exercise by wheel running (43). Although physical exercise may attenuate hepatic inflammation by reducing pro-inflammatory cytokines, the mechanisms of anti-inflammatory benefits by exercise remain to be elucidated.
Exercise and autophagy
Using a mouse treadmill model, He et al. recently showed that physical activity stimulates autophagy in a wide range of tissues, including skeletal muscle, heart, liver, pancreas and adipose tissue (18). The exercise-mediated autophagy induction is likely to occur at the initiation step of autophagy through the dissociation of BECN1 from BCL2. Mice lacking three conserved phosphorylation sites of Thr69, Ser70, and Ser97 in the non-structured loop of BCL2 failed to induce autophagy after exercise. Furthermore, these genetically modified animals were unable to run on a treadmill as long as their wild type counterparts, implying that BECN1-dependent autophagy may be uniquely launched by exercise. Intriguingly, analysis of p62 and LC3-II/LC3-I, markers of autophagy induction, revealed that exercise also markedly increases autophagy in extramuscular tissues like the liver and pancreas, though no apparent morphological or structural alterations were observed in hepatocytes and pancreatic β-cells from the mice expressing non-phosphoryltable BCL2. In particular, long-term exercise ameliorated high fat diet-induced glucose intolerance in wild-type mice, but not in BCL2 mutant mice, substantiating the importance of autophagy in exercise-mediated cytoprotection against metabolic disorder. Taken together, this study demonstrates that exercise is a natural stimulus of autophagy that can confer metabolic protection.
How the muscle transduces autophagy initiation signal to the liver is currently unknown. The skeletal muscle is an endocrine organ secreting a plethora of cytokines, chemokines, growth factors, hormones and vasoactive factors, collectively termed myokines(42). Autophagy induction after physical training might involve these myokines. Myonectin or C1q/TNF-related protein 5 (CTRP5) is a nutrient-responsive myokine that enhances glucose uptake and stimulates fatty acid oxidation. This myokine is often released in response to feeding and insulin. Interestingly, exogenous administration of recombinant CTRP5 to the liver and cultured hepatocytes has been shown to prevent autophagy via activating mTOR pathway (42). Using a high fat diet rodent model, Lei and colleagues demonstrated that CTRP5-null animals exhibit reduced hepatic steatosis and improved insulin action, implying a negative correlation between CTRP5 and NAFLD onset (30). Intriguingly, aerorobic exercise has been reported to diminish levels of CTRP5 in humans (31). Although future studies are warrantied to determine how this muscle-derived cytokine is delivered to and triggers autophagic signals in the liver, these studies suggest that hepatic autophagy could be modulated by the changes in CTRP5 levels following physical activity (Fig. 1).
Figure. 1. Potential mechanisms underlying exercise-induced protection against fatty liver disease.
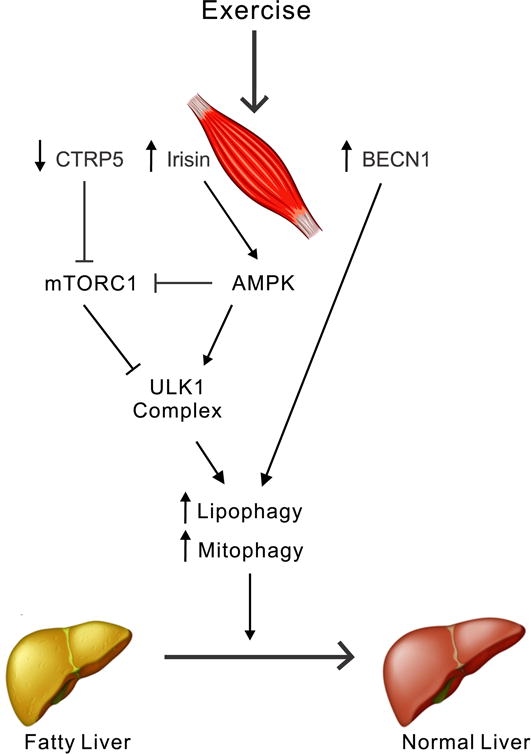
Physical activity may alter levels of muscle-derived myokines. A decrease in CTRP5 after exercise blocks mTORC1 activity, thereby inducing autophagy. At the same time, an increase in irisin promotes AMPK stimulation, which subsequently enhances autophagy in a ULK1-dependent fashion. Alternatively, exercise may directly initiate autophagy by releasing BECN1 from its complex with BCL2. Under these conditions, mitophagy and lipophagy in the liver also become activated to clear abnormal mitochondria and excess lipids, respectively. As a consequence, extra lipids in the fatty liver are more efficiently oxidized in mitochondria and selectively eliminated by lysosomes, leading to improvement of NAFLD.
Other myokines might also mediate autophagy stimulation in the liver. Irisin, an active form of the fibronectin type III domain containing 5 (FNDC5) protein, is a newly identified exercise-induced myokine (44). Irisin activates AMPK signaling in hepatocytes and reduces intracellular triglyceride accumulation. As AMPK is an essential player in autophagy initiation, irisin derived from the muscle might be an important signaling molecule that translocates to the liver and stimulates hepatic signaling of autophagy (Fig. 1). However, it is important to note that despite numerous studies, large controversy still exists as to how much irisin increases after exercise in humans. Tandem mass spectrometric analysis of 10 individuals found that high intensity aerobic exercise increases the level of circulating irisin by 19% (24). Although statistically significant, this elevation is rather small. Future studies are required with larger population of subjects to investigate how various exercise conditions affect irisin production in humans and how the liver interacts with this myokine released from the muscle.
Reduced calorie intake without malnutrition, or calorie restriction (CR) has long been shown to effectively expand life span in various species including primates (10). One potential mechanism of CR-mediated benefit may be its induction of autophagy. Several studies have demonstrated that chronic or long-term CR facilitates protein turnover by activating multiple regulatory pathways of autophagy (3). For instance, CR acts on the upstream events of autophagy initiation by suppressing mTORC1 and stimulating AMPK, which in turn leads to ULK1 activation (40). Furthermore, CR enhances sirtuin 1 activity (9), an enzyme that induces autophagy through deacetylating multiple cellular targets (4). Although autophagy is evidently launched by either CR or exercise, ongoing controversies exist as to whether the combination of dietary intervention with exercise could provide greater benefits than CR or exercise alone (11).
While strength and resistance exercise increases the strength and cross-sectional area of muscle fibers (17), endurance exercise, also known as aerobic exercise, augments the mitochondrial function and content of the muscle (16). Though either type of exercise profoundly influences cellular protein turnover, accumulating evidence indicates differential effects on protein homeostasis between resistance and endurance exercise. Within 24 h after acute endurance exercise, mRNA of key autophagy factors, including LC3, ATG4B, ATG12, BNIP3, and cathepsin L, is upregulated (22). A study with a mouse model of 40 min-exercise duration showed that moderate-to-low intensity exercise rapidly promotes the phosphorylation of the residues of Ser317 and 555 of ULK1, while preventing mTORC1-dependent ULK1 phosphorylation, events indicative of autophagy initiation (37). It is, however, noteworthy that the increase in autophagy in response to a common endurance exercise is not always observed in other studies wherein animals are exposed to 50 to 90 min of tread mill exercise (27, 41). This could imply that exercise duration may be an important factor contributing to autophagy induction in the muscle. Another important factor in endurance exercise-mediated autophagy induction is a feeding status prior to exercise. When autophagy onset during endurance exercise is compared between fast and fed state, stimulation of autophagy becomes more robust in the fasted state, as evidenced by a higher increase in LC3, BNIP3 and Parkin (23).
In contrast to endurance exercise, a decrease in autophagy has been reported after resistance exercise (15). In humans, while resistance exercise reduces the lipidation of LC3-I, an integral event for autophagy induction, E3-ligase activity in the ubiquitin-proteasome pathway appears to be upregulated after this exercise regimen (15). Increased expression of class III PI3K has been reported after resistance exercise (32). However, since this kinase is also involved in multiple pathways other than autophagy, these studies do not necessarily reflect autophagy involvement in response to resistance exercise (47). Although current literatures favor non-essential roles of autophagy in strength and resistance exercise, future studies are needed to better clarify how autophagy is associated with this exercise regimen. It is also important to understand potential impacts of a combination of resistance and endurance exercise on muscle autophagy.
Effects of muscle autophagy on the liver
Growing evidence supports the presence of a remote communication between individual organs (12). Skeletal muscles are a major provider of gluconeogenic and ketogenic amino acids during prolonged starvation in mammals (13). As the liver is a primary tissue controlling both gluconeogenesis and ketogenesis and starvation is a powerful autophagy inducer in the liver and muscle as well, it is plausible to speculate that muscle autophagy could directly impact the liver or vice versa. Although defective autophagy in the muscle causes accumulation of intramyocellular triglycerides and enhanced autophagy facilitates removal of lipids from muscle cells (28), Takagi et al. recently demonstrated in tissue-specific ATG5 knockout mice that the mice lacking ATG5 in both the liver and muscle indeed exhibited the improvement of metabolic profile, as compared to liver-specific knockout counterparts, suggesting that autophagy in the skeletal muscle may be metabolically distinct from that in the liver (46). In an independent study, skeletal muscle–specific ATG7 knockout mice also showed lower lipid accumulation and higher expression of β-oxidation–related genes, as compared to control mice (26). Furthermore, when fed with high fat diet, these transgenic animals displayed lower expression of lipogenic genes in the liver, and were protected from diet-induced obesity and insulin resistance. As high fat diet markedly causes accumulation of lipid droplets in autophagy-deficient livers (48), it is likely that metabolic outcomes after the onset of autophagy or lipophagy in the muscle may be different from those in the liver.
Conclusions
Exercise is a natural intervention that amends liver function of the patients with fatty liver disease. Here, we propose a hypothesis that autophagy activation in the liver and muscle after exercise improves underlying defects in lipid metabolism in NAFLD patients (Fig. 1). There are, however, many questions to be answered. For instance, it is unknown how exercise directly or indirectly stimulates autophagy in the liver. Hepatic enhancement of autophagy following physical activity could be mediated either by muscle-derived myokines or by unidentified signaling molecules originated from extramuscular tissues. Another open question is to determine the most appropriate physical training regimen for NAFLD patients. It is also important to understand how the duration and intensity of exercise influences the cross talk between the muscle and liver.
Key Points.
Exercise has been showing promising outcomes both in clinical settings and in animal models of NAFLD. However, underlying mechanisms are unknown.
Recent studies suggest that exercise stimulates autophagy in both muscles and extramuscular tissues. Furthermore, muscle-derived myokines following exercise are likely to induce autophagy in liver cells.
As lipophagy and mitophagy selectively remove excess lipids and dysfunctional mitochondria, respectively, beneficial effects of exercise on NAFLD could be associated with autophagy induction after physical activity.
Acknowledgments
Funding: This work was supported in part by US National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases grant DK079879 and DK090115 (J-S Kim) and National Institute on Aging AG028740 (J-S Kim & C Leeuwenburgh).
Footnotes
Conflicts of interest: None
References
- 1.Asrani SK, Larson JJ, Yawn B, Therneau TM, Kim WR. Underestimation of liver-related mortality in the United States. Gastroenterology. 2013;145(2):375–82. doi: 10.1053/j.gastro.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Begriche K, Massart J, Robin MA, Bonnet F, Fromenty B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology. 2013;58(4):1497–507. doi: 10.1002/hep.26226. [DOI] [PubMed] [Google Scholar]
- 3.Bergamini E, Cavallini G, Donati A, Gori Z. The role of autophagy in aging: its essential part in the anti-aging mechanism of caloric restriction. Ann N Y Acad Sci. 2007;1114:69–78. doi: 10.1196/annals.1396.020. [DOI] [PubMed] [Google Scholar]
- 4.Biel TG, Lee S, Flores-Toro JA, Dean JW, Go KL, Lee MH, Law BK, Law ME, Dunn WA, Jr, Zendejas I, et al. Sirtuin 1 suppresses mitochondrial dysfunction of ischemic mouse livers in a mitofusin 2-dependent manner. Cell Death Differ. 2016;23(2):279–90. doi: 10.1038/cdd.2015.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chalasani N, Younossi Z, Lavine JE, Diehl AM, Brunt EM, Cusi K, Charlton M, Sanyal AJ. The diagnosis and management of non-alcoholic fatty liver disease: practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology. 2012;55(6):2005–23. doi: 10.1002/hep.25762. [DOI] [PubMed] [Google Scholar]
- 6.Chen ZP, Stephens TJ, Murthy S, Canny BJ, Hargreaves M, Witters LA, Kemp BE, McConell GK. Effect of exercise intensity on skeletal muscle AMPK signaling in humans. Diabetes. 2003;52(9):2205–12. doi: 10.2337/diabetes.52.9.2205. [DOI] [PubMed] [Google Scholar]
- 7.Chun SK, Go K, Yang MJ, Zendejas I, Behrns KE, Kim J-S. Autophagy in Ischemic Livers: A Critical Role of Sirtuin 1/Mitofusin 2 Axis in Autophagy Induction. Toxicol Res. 2016;32(1):35–46. doi: 10.5487/TR.2016.32.1.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cintra DE, Ropelle ER, Vitto MF, Luciano TF, Souza DR, Engelmann J, Marques SO, Lira FS, de Pinho RA, Pauli JR, et al. Reversion of hepatic steatosis by exercise training in obese mice: the role of sterol regulatory element-binding protein-1c. Life Sci. 2012;91(11–12):395–401. doi: 10.1016/j.lfs.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 9.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305(5682):390–2. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 10.Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science. 2009;325(5937):201–4. doi: 10.1126/science.1173635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cox KL, Burke V, Morton AR, Beilin LJ, Puddey IB. The independent and combined effects of 16 weeks of vigorous exercise and energy restriction on body mass and composition in free-living overweight men–a randomized controlled trial. Metabolism. 2003;52(1):107–15. doi: 10.1053/meta.2003.50017. [DOI] [PubMed] [Google Scholar]
- 12.Droujinine IA, Perrimon N. Interorgan Communication Pathways in Physiology: Focus on Drosophila. Annu Rev Genet. 2016;50:539–570. doi: 10.1146/annurev-genet-121415-122024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Felig P, Marliss E, Pozefsky T, Cahill GF., Jr Amino acid metabolism in the regulation of gluconeogenesis in man. Am J Clin Nutr. 1970;23(7):986–92. doi: 10.1093/ajcn/23.7.986. [DOI] [PubMed] [Google Scholar]
- 14.Flores-Toro JA, Go KL, Leeuwenburgh C, Kim J-S. Autophagy in the liver: cell’s cannibalism and beyond. Arch Pharm Res. 2016;39(8):1050–61. doi: 10.1007/s12272-016-0807-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fry CS, Drummond MJ, Glynn EL, Dickinson JM, Gundermann DM, Timmerman KL, Walker DK, Volpi E, Rasmussen BB. Skeletal muscle autophagy and protein breakdown following resistance exercise are similar in younger and older adults. J Gerontol A Biol Sci Med Sci. 2013;68(5):599–607. doi: 10.1093/gerona/gls209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gollnick PD, Armstrong RB, Saubert CW, 4th, Piehl K, Saltin B. Enzyme activity and fiber composition in skeletal muscle of untrained and trained men. J Appl Physiol. 1972;33(3):312–9. doi: 10.1152/jappl.1972.33.3.312. [DOI] [PubMed] [Google Scholar]
- 17.Hasten DL, Pak-Loduca J, Obert KA, Yarasheski KE. Resistance exercise acutely increases MHC and mixed muscle protein synthesis rates in 78–84 and 23–32 yr olds. Am J Physiol Endocrinol Metab. 2000;278(4):E620–6. doi: 10.1152/ajpendo.2000.278.4.E620. [DOI] [PubMed] [Google Scholar]
- 18.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al. Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature. 2012;481(7382):511–5. doi: 10.1038/nature10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22(2):140–9. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hong HC, Hwang SY, Choi HY, Yoo HJ, Seo JA, Kim SG, Kim NH, Baik SH, Choi DS, Choi KM. Relationship between sarcopenia and nonalcoholic fatty liver disease: the Korean Sarcopenic Obesity Study. Hepatology. 2014;59(5):1772–8. doi: 10.1002/hep.26716. [DOI] [PubMed] [Google Scholar]
- 21.Ibdah JA, Perlegas P, Zhao Y, Angdisen J, Borgerink H, Shadoan MK, Wagner JD, Matern D, Rinaldo P, Cline JM. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology. 2005;128(5):1381–90. doi: 10.1053/j.gastro.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 22.Jamart C, Benoit N, Raymackers JM, Kim HJ, Kim CK, Francaux M. Autophagy-related and autophagy-regulatory genes are induced in human muscle after ultraendurance exercise. Eur J Appl Physiol. 2012;112(8):3173–7. doi: 10.1007/s00421-011-2287-3. [DOI] [PubMed] [Google Scholar]
- 23.Jamart C, Naslain D, Gilson H, Francaux M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J Physiol Endocrinol Metab. 2013;305(8):E964–74. doi: 10.1152/ajpendo.00270.2013. [DOI] [PubMed] [Google Scholar]
- 24.Jedrychowski MP, Wrann CD, Paulo JA, Gerber KK, Szpyt J, Robinson MM, Nair KS, Gygi SP, Spiegelman BM. Detection and Quantitation of Circulating Human Irisin by Tandem Mass Spectrometry. Cell Metab. 2015;22(4):734–40. doi: 10.1016/j.cmet.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim J-S, Wang JH, Lemasters JJ. Mitochondrial permeability transition in rat hepatocytes after anoxia/reoxygenation: role of Ca2+-dependent mitochondrial formation of reactive oxygen species. Am J Physiol Gastrointest Liver Physiol. 2012;302(7):G723–31. doi: 10.1152/ajpgi.00082.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim KH, Jeong YT, Oh H, Kim SH, Cho JM, Kim YN, Kim SS, Kim DH, Hur KY, Kim HK, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19(1):83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- 27.Kim YA, Kim YS, Song W. Autophagic response to a single bout of moderate exercise in murine skeletal muscle. J Physiol Biochem. 2012;68(2):229–35. doi: 10.1007/s13105-011-0135-x. [DOI] [PubMed] [Google Scholar]
- 28.Lam T, Harmancey R, Vasquez H, Gilbert B, Patel N, Hariharan V, Lee A, Covey M, Taegtmeyer H. Reversal of intramyocellular lipid accumulation by lipophagy and a p62-mediated pathway. Death Discov. 2016;2:16061. doi: 10.1038/cddiscovery.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee YH, Jung KS, Kim SU, Yoon HJ, Yun YJ, Lee BW, Kang ES, Han KH, Lee HC, Cha BS. Sarcopaenia is associated with NAFLD independently of obesity and insulin resistance: Nationwide surveys (KNHANES 2008–2011) J Hepatol. 2015;63(2):486–93. doi: 10.1016/j.jhep.2015.02.051. [DOI] [PubMed] [Google Scholar]
- 30.Lei X, Rodriguez S, Petersen PS, Seldin MM, Bowman CE, Wolfgang MJ, Wong GW. Loss of CTRP5 improves insulin action and hepatic steatosis. Am J Physiol Endocrinol Metab. 2016;310(11):E1036–52. doi: 10.1152/ajpendo.00010.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lim S, Choi SH, Koo BK, Kang SM, Yoon JW, Jang HC, Choi SM, Lee MG, Lee W, Shin H, et al. Effects of aerobic exercise training on C1q tumor necrosis factor α-related protein isoform 5 (myonectin): association with insulin resistance and mitochondrial DNA density in women. J Clin Endocrinol Metab. 2012;97(1):E88–93. doi: 10.1210/jc.2011-1743. [DOI] [PubMed] [Google Scholar]
- 32.MacKenzie MG, Hamilton DL, Murray JT, Taylor PM, Baar K. mVps34 is activated following high-resistance contractions. J Physiol. 2009;587(1):253–60. doi: 10.1113/jphysiol.2008.159830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGarry JD, Foster DW. Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem. 1980;49:395–420. doi: 10.1146/annurev.bi.49.070180.002143. [DOI] [PubMed] [Google Scholar]
- 34.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387(6628):83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- 35.Merli M, Dasarathy S. Sarcopenia in non-alcoholic fatty liver disease: Targeting the real culprit? J Hepatol. 2015 Aug;63(2):309–11. doi: 10.1016/j.jhep.2015.05.014. [DOI] [PubMed] [Google Scholar]
- 36.Murray CJ, Barber RM, Foreman KJ, Abbasoglu Ozgoren A, Abd-Allah F, Abera SF, Aboyans V, Abraham JP, Abubakar I, Abu-Raddad LJ, et al. Global, regional, and national disability-adjusted life years (DALYs) for 306 diseases and injuries and healthy life expectancy (HALE) for 188 countries, 1990–2013: quantifying the epidemiological transition. Lancet. 2015;386(10009):2145–91. doi: 10.1016/S0140-6736(15)61340-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pagano AF, Py G, Bernardi H, Candau RB, Sanchez AM. Autophagy and protein turnover signaling in slow-twitch muscle during exercise. Med Sci Sports Exerc. 2014;46(7):1314–25. doi: 10.1249/MSS.0000000000000237. [DOI] [PubMed] [Google Scholar]
- 38.Polyzos SA, Kountouras J, Zavos C. Nonalcoholic fatty liver disease: the pathogenetic roles of insulin resistance and adipocytokines. Curr Mol Med. 2009;9(3):299–314. doi: 10.2174/156652409787847191. [DOI] [PubMed] [Google Scholar]
- 39.Powell EE, Cooksley WG, Hanson R, Searle J, Halliday JW, Powell LW. The natural history of nonalcoholic steatohepatitis: a follow-up study of forty-two patients for up to 21 years. Hepatology. 1990;11(1):74–80. doi: 10.1002/hep.1840110114. [DOI] [PubMed] [Google Scholar]
- 40.Rubinsztein DC, Mariño G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–95. doi: 10.1016/j.cell.2011.07.030. [DOI] [PubMed] [Google Scholar]
- 41.Saleem A, Carter HN, Hood DA. p53 is necessary for the adaptive changes in cellular milieu subsequent to an acute bout of endurance exercise. J Physiol Cell Physiol. 2014;306(3):C241–9. doi: 10.1152/ajpcell.00270.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seldin MM, Lei X, Tan SY, Stanson KP, Wei Z, Wong GW. Skeletal muscle-derived myonectin activates the mammalian target of rapamycin (mTOR) pathway to suppress autophagy in liver. J Biol Chem. 2013;288(50):36073–82. doi: 10.1074/jbc.M113.500736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Seo AY, Hofer T, Sung B, Judge S, Chung HY, Leeuwenburgh C. Hepatic oxidative stress during aging: effects of 8% long-term calorie restriction and lifelong exercise. Antioxid Redox Signal. 2006;8(3–4):529–38. doi: 10.1089/ars.2006.8.529. [DOI] [PubMed] [Google Scholar]
- 44.So WY, Leung PS. Irisin ameliorates hepatic glucose/lipid metabolism and enhances cell survival in insulin-resistant human HepG2 cells through adenosine monophosphate-activated protein kinase signaling. Int J Biochem Cell Biol. 2016;78:237–47. doi: 10.1016/j.biocel.2016.07.022. [DOI] [PubMed] [Google Scholar]
- 45.Sparks LM, Xie H, Koza RA, Mynatt R, Hulver MW, Bray GA, Smith SR. A high-fat diet coordinately downregulates genes required for mitochondrial oxidative phosphorylation in skeletal muscle. Diabetes. 2005;54(7):1926–33. doi: 10.2337/diabetes.54.7.1926. [DOI] [PubMed] [Google Scholar]
- 46.Takagi A, Kume S, Kondo M, Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Haneda M, Chano T, et al. Mammalian autophagy is essential for hepatic and renal ketogenesis during starvation. Sci Rep. 2016;6:18944. doi: 10.1038/srep18944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11(5):329–41. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 48.Williams CD, Stengel J, Asike MI, Torres DM, Shaw J, Contreras M, Landt CL, Harrison SA. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: a prospective study. Gastroenterology. 2011;140(1):124–31. doi: 10.1053/j.gastro.2010.09.038. [DOI] [PubMed] [Google Scholar]
- 49.Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467–78. doi: 10.1016/j.cmet.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yin X, Zheng F, Pan Q, Zhang S, Yu D, Xu Z, Li H. Glucose fluctuation increased hepatocyte apoptosis under lipotoxicity and the involvement of mitochondrial permeability transition opening. J Mol Endocrinol. 2015;55(3):169–81. doi: 10.1530/JME-15-0101. [DOI] [PubMed] [Google Scholar]
- 51.Zhang C, McFarlane C, Lokireddy S, Bonala S, Ge X, Masuda S, Gluckman PD, Sharma M, Kambadur R. Myostatin-deficient mice exhibit reduced insulin resistance through activating the AMP-activated protein kinase signalling pathway. Diabetologia. 2011;54(6):1491–501. doi: 10.1007/s00125-011-2079-7. [DOI] [PubMed] [Google Scholar]