

ABSTRACT
We recently described the 4.5-year time course of the enteric bacterial microbiota and virome of a patient cured from recurrent Clostridium difficile infection (rCDI) by fecal microbiota transplantation (FMT). Here, we extended the virome analyses and found the patient's phage population to exhibit highly donor-similar characteristics following FMT, which remained stable for the whole period tested (up to 7 months). Moreover, the detected viral populations of donor and patient exhibited comparable diversity and richness. These findings were unexpected since enteric viromes are normally highly variable, assumed to influence the bacterial host community and change with environmental conditions. In contrast to the virome, the bacterial microbiota varied indeed for more than 7 months with ongoing dysbiosis before it reached donor similarity. Our findings that are based on sequence information and protein domain analysis seem to suggest that stable phage properties correlate with successful FMT better than the changing bacterial communities. We speculate that we here preferentially detected a stable core virome, which dominated over a variable flexible virome that may have been too heterogeneous for experimental detection or was underrepresented in the databases. It will be interesting to analyze whether the enteric virome allows predictions for the clinical outcome of FMT for rCDI and other diseases such as inflammatory bowel disease or obesity.
KEYWORDS: Clostridium difficile, sequencing, virome, microbiome, fecal microbiota transplantation, long-term analysis, core virome
Introduction
FMT is an effective treatment against recurrent C. difficile infection (rCDI) that may soon be applied to other dysbiotic gastrointestinal conditions including inflammatory bowel disease (IBD) and obesity.1,2 FMT-mediated cure of rCDI is associated with expanded beneficial Bacteroidetes and Firmicutes bacteria in the patient's intestine, whereas detrimental Proteobacteria populations contract.3,4 The 1014 gut bacteria per individual5 are conventionally considered the most relevant component of FMT. In contrast, the 10 times more numerous enteric viruses6,7 have remained largely unexplored in this context.4,8 The human gut virome is mainly composed of bacteriophages belonging to the Caudovirales order, which are tailed double-stranded (dsDNA) phages, and the ssDNA Microviridae family9-11 but also harbors a diverse community of eukaryotic viruses6 and probably giant viruses such as Chlorella viruses4 with a poorly understood impact on health and disease.
We have recently reported on a patient with antibiotic-refractory rCDI – termed the “Zurich patient” – who experienced immediate subjective improvement and clinical cure within 2 weeks following FMT after about 7 months of C. difficile-induced disease.12 To characterize enteric bacterial and viral communities over time, we have subjected total dsDNA isolated from patient and donor feces sampled up to 4.5 y post-FMT to 16S rRNA gene or metagenomic sequencing analyses, respectively (Fig. 1a). An overview of the respective sequencing yields is shown in Tables S1 and S2. Despite immediate clinical cure within days, the patient's bacterial microbiota remained dysbiotic during the time period tested up to 7 months post-FMT and were characterized by expanded Proteobacteria, a hallmark of gut inflammation13 (Fig. 1b). Prolonged dysbiosis was an unusual characteristic of this case report, as the majority of rCDI patients have been reported to attain highly donor-similar bacterial communities within days or weeks following successful FMT.4 Only after a longer period up to 4.5 years, bacterial communities have reached a highly donor-similar composition in the Zurich patient. We have previously characterized the fecal dsDNA viromes of donor and patient12 by querying open reading frames (ORFs) predicted from metagenomic contigs against the non-redundant protein (NRPROT) database14 using BLASTP.15 Thereby, we have identified about 20 Caudovirales phage species among all samples studied. This approach, however, only captured a very small fraction of the actual phage population, since ORFs of prophages (phages integrated within bacterial genomes) were frequently taxonomically assigned to the respective host bacterium (unpublished observation) and viral sequences are notoriously underrepresented in current sequence databases including NRPROT.11 Therefore, the results presented here are based on the rather small number of available phage sequences.
Figure 1.

(a) Timeline of fecal samples of donor (D0 and D4) and patient (P1 through P4) subjected to 16S rRNA gene sequencing (all samples) or metagenomic sequencing (samples D0 and P1-P3). (b) Relative abundances of bacterial phyla as inferred by 16S rRNA gene sequencing. (c) and (d) Relative proportions of bacterial phylum-specific (c) or phage-specific (d) Pfam domains identified from metagenomic ORFs. (e) Relative abundances of phage families identified by aligning phage-specific ORFs against tailed phage protein sequences of the NRPROT database. (f) Virus family-specific relative proportions of phage-specific Pfam domains identified from metagenomic ORFs.
Results
To reveal the most abundant phage species within the fecal samples, we queried the metagenomic contigs against virus sequences deposited in the nucleotide collection of NCBI using BLASTN.15 About 0.04% of all contigs aligned to known viruses with sequence identity >80% (Table S2). We solely found Caudovirales phages stratifying into Myo-, Podo- and Siphoviridae families. The identification of strictly lytic viruses such as T4-like phages (Myoviridae), which do not form prophages,16 confirmed that the sequencing approach also captured free phages (Table S3). Not surprising, of the 26 phages identified among all samples, 22 had Enterobacteriaceae genera such Escherichia, Cronobacter, Citrobacter, Shigella and Salmonella as their hosts. Cronobacter phage vB CsaM_GAP161 was identified in both donor and patient (samples D0 and P2) and may have been transmitted during FMT. However, there were no overlapping contigs to confirm the sequence identity between samples of donor and patient.
For a more comprehensive characterization of enteric phages and to study their possible transmission during FMT, we re-analyzed the ORF sets of patient and donor described above using the Pfam protein families database of conserved protein domains17 and the UproC program.18 Of all ORFs, 64% contained recognizable Pfam motifs, 99.9% of which were taxonomically assigned to bacteria, a taxon that also includes phages in this database (Table S3). In accordance with the 16S data, Bacteroidetes-specific protein clusters were less abundant in the patient than in the donor, while those specific for Proteobacteria were expanded in the patient (Fig. 1c). About 2% of all Pfam domains were assigned to essential bacteriophage functions, as identified using a list of Pfam families described previously11 (Fig. 1d). The composition of phage-specific protein clusters was highly donor-similar in the patient and stable over time. In order to determine the taxonomic distribution of these phage-specific domains, the corresponding ORFs were aligned to tailed phages protein sequences of NRPROT using BLASTP (Fig. 1e). As expected, almost all of the phage-specific ORFs, 3,643 of 3,467, aligned to sequences of tailed phages (Table S4) that represent 96% of all known phages.19 For each ORF, the BLASTP hit with the lowest e-value was used for taxonomic assignment at the viral family level. In all samples of donor and patient, Siphoviridae were most abundant, followed by Myoviridae and Podoviridae, all 3 families belonging to the tailed Caudovirales order. Each identified viral family showed a distinct distribution of phage-specific Pfam domains (Fig. 1f).
To further compare phage populations among the different samples, we calculated their α diversity (Shannon's H’) and richness (Menhinick's D). The diversity and richness measures calculated from taxonomic assignment of NRPROT were highly similar among phage populations when all tailed phages were taken into account (Fig. 2a), whereby Shannon's H’ ranged between 4.83 (P1) and 5.06 (P3) and Menhinick's D between 9.11 (P3) and 9.32 (P1). The most diverse and rich subpopulations were the Siphoviridae, followed by Myoviridae, Podoviridae, unclassified Caudovirales and unclassified phages. All subpopulations exhibited comparable diversity and richness among samples. Similar trends were observed when diversity and richness were calculated from Pfam domains (Fig. 2b), whereby differences among samples in the least diverse subpopulations, unclassified Caudovirales and unclassified phages, were more pronounced. This was likely due to the relatively low numbers of Pfam identifiers (between 3 to 8 per sample, see Table S5) assigned to these subpopulations that resulted in less robust measures for H’ and D. The overall findings showed that, surprisingly, despite dysbiotic bacterial microbiota for at least 7 months post-FMT, phage populations in the same time period were highly donor-similar and stable in composition, diversity and richness.
Figure 2.
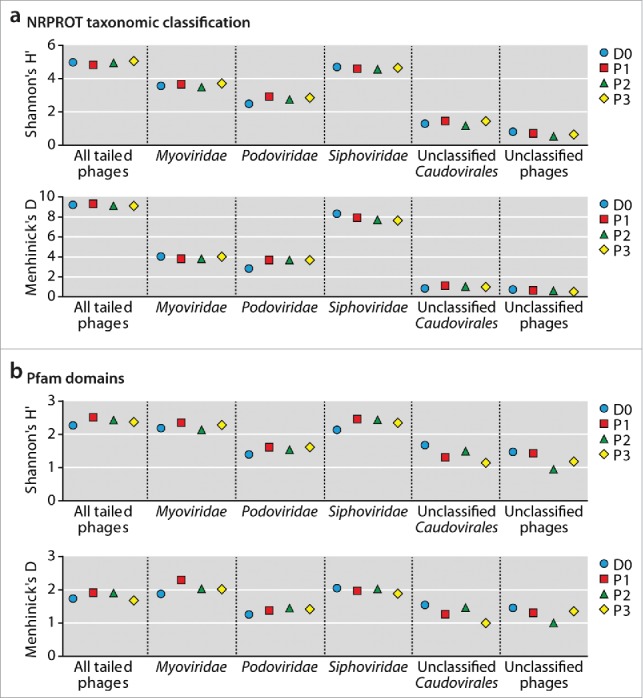
α diversity (Shannon's H’) and richness (Menhinick's D) measurements based on phage species taxonomically assigned by NRPROT (a) and on Pfam domain annotation (b). Sample identifiers D0 and P1-P3 are explained in Fig. 1a.
Discussion
Phage communities of the herein described patient were unexpectedly similar to the donor in composition (Fig. 1e, f), diversity and richness (Fig. 2) and stable even when bacterial microbiota were dysbiotic and variable. This is especially surprising since the enteric virome, as determined by sequencing of isolated virus-like particle (VLP) derived DNA, is known to be variable individually over time and, more substantially so, between individuals.11,20 Our findings suggest that a predominant fraction of enteric phages was transferred from donor to the patient during FMT and then remained donor-like and stable.
The approach described here sought to overcome several challenges encountered during virome analysis. Due to their low abundance in feces, viral genomes are usually enriched by removing bacteria through centrifugation and filtration followed by VLP isolation with cesium chloride density gradients.9,11,20 Efficient virome sequencing then requires promiscuous amplification of viral genomes, for instance, with the phi29 polymerase.9,11,20 Assignment of the generated reads to virus species remains incomplete since the majority of sequences have no homologues in the current viral nucleotide databases.21 Therefore, the reference databases need to be expanded to allow for more comprehensive virome analyses in the future.22 The extensive sequence diversity found in the virosphere requires innovative means of virus sequence discovery.21-23 Novel phages have recently been identified by assembling reads from different metagenomic samples (cross-assembly) that overcomes the low abundance of phage sequences within single metagenomes,21 or by searching metagenomes for virus-specific sequence signatures such as oligonucleotide repeats.24 The discovery of new viral species may also benefit from improvements of sequencing technology with respect to sequencing depth, read length and accuracy that facilitate the identification of low abundance microbial taxa such as viruses.25 These advancements may also improve the quality of viromes inferred from total metagenomes.
Here, we tackled the aforementioned experimental and technical difficulties by identifying viral protein domains from total dsDNA metagenomes. The metagenomes of the patient exhibited an expansion of Proteobacteria and a depletion of Bacteroidetes as observed by Pfam domain analysis (Fig. 1c). Thus, although taxa with larger genomes may be overrepresented in the dsDNA metagenomes,12 they at least partially reflected the dysbiosis of the patient observed by 16S rRNA gene sequencing (Fig. 1b). The analysis of viromes through querying ORFs predicted from metagenomic contigs in the NRPROT and Pfam databases allowed for a more comprehensive characterization of phage communities than direct contig alignment against the NCBI nucleotide database that only yielded 3–14 identified viral species per sample (Table S3). Since we used total dsDNA to identify viral sequences, the herein described phage populations represent both free phages and prophages that were indistinguishable by our approach. Also, the number and type of isolated phage genetic material depends on the isolation method, which was adjusted for bacterial DNA and might have excluded some phage populations. Another limitation of this study is that the patient sample before FMT was missing. However, it can be assumed that the patient's microbiota at that time was severely dysbiotic due to month-long antibiotic therapy prior to FMT.12 It has been shown previously that antibiotic treatment against rCDI leads to an expansion of Proteobacteria and a depletion of Bacteroidetes and Firmicutes,4 characteristics that were partially observed in the patient after FMT as well (Figs. 1b, c). To what extent the virome is altered during rCDI remains to be shown. Future studies with more FMT-treated patients are needed to confirm the transmissibility of phage populations observed in this case report.
Compared to healthy adults, the enteric virome/microbiome of IBD patients has been described to harbor increased numbers of unintegrated phages and reduced bacterial diversity as disease indicators.9 In the case reported here, the patient, due to month-long repeated antibiotic treatment, was likely deprived from most of the endogenous gut microbiota that was replenished by the donor's.12 In a healthy microbiome/virome, phages are less abundant than during inflammatory diseases and predominantly exist as integrated prophages.4,9 Since our sequencing approach included both free phages and prophages that we could not distinguish, DNA sequence information specifically from isolated VLPs11,20 will be required to reveal whether free phage communities likewise exhibit donor similarity and stability following FMT as observed here.
It is known that enteric phages normally influence the complexity of the host community.4,6,7 The bacteria in the case described here indeed exhibited heterogeneity and variability, which stood in stark contrast to the phage community. We consider the possibility that a dominating subpopulation of phages and/or a selection of genetic information and protein domains with lower complexity was preferentially detected here.
The hypothesis of a dominating phage subpopulation is supported by studies of the ocean microbiome by the Tara Oceans expedition in which viral community patterns were analyzed with respect to protein clusters and the DNA sequence space that is unknown by up to 93%.26 Viral protein cluster analysis revealed the existence of a relatively small “core virome” shared among samples that is present in a global “pan virome” whose richness is several logs bigger. According to the recently described seed-bank hypothesis,27,28 the diversity of viral populations at different sites in the ocean is generated by incorporating viruses of the pan virome moving between environments through ocean currents. Local adaptation and diversification then occurs due to environmental constraints.26-28 Thus, we hypothesize that we here detected the transmission and acceptance of a predominant core fecal virome of limited complexity, as described for marine samples, in which highly diverse sequences of low abundancy seem to be undetectable.26 This phenomenon may also apply here for fecal samples. Furthermore, exchange of viruses between individuals by FMT may correspond to the described dispersal of marine viruses by ocean currents.26
Individual fecal virome diversity may result from local adaptation of the core virome to the host's intestinal environment. We speculate that in the case described here the core virome was detected, whereby more variable virus populations probably present in the feces were either too low in abundance or not distinguishable from the core with respect to protein domains. Detection of viral variants arising from recombination events, horizontal gene transfer or single nucleotide mutations would require more comprehensive sequencing of VLP-enriched DNA, as described.20 Thus, the gut ecosystem may resemble the marine ecosystem and the analysis of both systems may support each other.
In line with our findings, it was recently shown – at the DNA level – that phages are transmitted during FMT.8 Phages are a major vehicle for horizontal gene transfer in the human gut and thereby largely influence bacterial evolution, diversity and metabolism.6,7,20 Thus, the transfer of undesired phage-encoded genes, such as those conferring antibiotic resistance29 or bacterial virulence30 may be a consequence of FMT-transmitted phages and should be avoided by better understanding of the virome. The composition of phage populations may be more complex than just reflecting bacterial communities and therefore by themselves contribute to inflammatory diseases such as IBD9,10 and obesity31 in humans and mice, respectively. Conversely, transmitted phages may also exert beneficial functions, such as replenishing the recently described non-host-derived mucosal immunity to bacterial pathogens.32 These data suggest that, similar to bacterial microbiota, the virome can be pathologically altered in patients, and its return to a “healthy” state by FMT may result in clinical improvement. Screening FMT donors for “healthy” viromes, in addition to bacterial microbiota, may therefore become an important aim of clinical metagenomics.33
In summary we showed, to our surprise, that phages assume highly donor-similar characteristics following FMT with little change over time, despite contemporaneous bacterial dysbiosis. Bacterial communities attained donor similarity only later and then remained stable for at least 4.5 y post-FMT. Thus, stable core phage populations may be an early indicator of the properties of the enteric community transferred during FMT. We speculate that the phage stability may be due to transmission of a core population within the gut virome, in analogy to population dynamics in the ocean.26 The core population is defined by a distinct composition of phage protein domains and presumably predominates a flexible virome. Variations derived from this core may have been too low in abundance to be detectable here. Further studies are required to better understand these results and compare donor and recipient viromes for a putative predictive value for the clinical success of FMT in rCDI and, more generally, in the increasing number of diseases associated with viral dysbiosis, including IBD and obesity.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Khoruts A, Sadowsky MJ. Understanding the mechanisms of faecal microbiota transplantation. Nat Rev Gastroenterol Hepatol 2016; 13:508-16; PMID:27329806; http://dx.doi.org/ 10.1038/nrgastro.2016.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Xu MQ, Cao HL, Wang WQ, Wang S, Cao XC, Yan F, Wang BM. Fecal microbiota transplantation broadening its application beyond intestinal disorders. World J Gastroenterol 2015; 21:102-11; PMID:25574083; http://dx.doi.org/ 10.3748/wjg.v21.i1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Seekatz AM, Young VB. Clostridium difficile and the microbiota. J Clin Invest 2014; 124:4182-9; PMID:25036699; http://dx.doi.org/ 10.1172/JCI72336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Broecker F, Klumpp J, Moelling K. Long-term microbiota and virome in a Zürich patient after fecal transplantation against Clostridium difficile infection. Ann N Y Acad Sci 2016; 1372:29-41; PMID:27286042; http://dx.doi.org/ 10.1111/nyas.13100 [DOI] [PubMed] [Google Scholar]
- [5].Savage DC. Microbial ecology of the gastrointestinal tract. Annu Rev Microbiol 1977; 31:107-33; PMID:334036. [DOI] [PubMed] [Google Scholar]
- [6].Scarpellini E, Ianiro G, Attili F, Bassanelli C, De Santis A, Gasbarrini A. The human gut microbiota and virome: Potential therapeutic implications. Dig Liver Dis 2015; 47:1007-12; PMID:26257129; http://dx.doi.org/ 10.1016/j.dld.2015.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Dalmasso M, Hill C, Ross RP. Exploiting gut bacteriophages for human health. Trends Microbiol 2014; 22:399-405; PMID:24656964; http://dx.doi.org/ 10.1016/j.tim.2014.02.010 [DOI] [PubMed] [Google Scholar]
- [8].Chehoud C, Dryga A, Hwang Y, Nagy-Szakal D, Hollister EB, Luna RA, Versalovic J, Kellermayer R, Bushman FD. Transfer of Viral Communities between Human Individuals during Fecal Microbiota Transplantation. MBio 2016; 7:e00322; PMID:27025251; http://dx.doi.org/ 10.1128/mBio.00322-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Norman JM, Handley SA, Baldridge MT, Droit L, Liu CY, Keller BC, Kambal A, Monaco CL, Zhao G, Fleshner P, et al.. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015; 160:447-60; PMID:25619688; http://dx.doi.org/ 10.1016/j.cell.2015.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lepage P, Colombet J, Marteau P, Sime-Ngando T, J Doré, Leclerc M. Dysbiosis in inflammatory bowel disease: a role for bacteriophages? Gut 2008; 57:424-5; PMID:18268057; http://dx.doi.org/ 10.1136/gut.2007.134668 [DOI] [PubMed] [Google Scholar]
- [11].Minot S, Sinha R, Chen J, Li H, Keilbaugh SA, Wu GD, Lewis JD, Bushman FD. The human gut virome: inter-individual variation and dynamic response to diet. Genome Res 2011; 21:1616-25; PMID:21880779; http://dx.doi.org/ 10.1101/gr.122705.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Broecker F, Kube M, Klumpp J, Schuppler M, Biedermann L, Hecht J, Hombach M, Keller PM, Rogler G, Moelling K. Analysis of the intestinal microbiome of a recovered Clostridium difficile patient after fecal transplantation. Digestion 2013; 88:243-51; PMID:24335204; http://dx.doi.org/ 10.1159/000355955 [DOI] [PubMed] [Google Scholar]
- [13].Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe 2007; 2:204; PMID:18030708. [DOI] [PubMed] [Google Scholar]
- [14].Benson DA, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW. GenBank. Nucleic Acids Res 2015; 43(Database issue):D30-5; PMID:25414350; http://dx.doi.org/ 10.1093/nar/gku1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403-10; PMID:2231712 [DOI] [PubMed] [Google Scholar]
- [16].Miller ES, Kutter E, Mosig G, Arisaka F, Kunisawa T, Rüger W. Bacteriophage T4 genome. Microbiol Mol Biol Rev 2003; 67:86-156; PMID:12626685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL, Potter SC, Punta M, Qureshi M, Sangrador-Vegas A, et al.. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 2016; 44(D1):D279-85; PMID:26673716; http://dx.doi.org/ 10.1093/nar/gkv1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Meinicke P. UProC: tools for ultra-fast protein domain classification. Bioinformatics 2015; 31:1382-8; PMID:25540185; http://dx.doi.org/ 10.1093/bioinformatics/btu843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ackermann HW. 5500 Phages examined in the electron microscope. Arch Virol 2007; 152:227-243; PMID:17051420; http://dx.doi.org/ 10.1007/s00705-006-0849-1 [DOI] [PubMed] [Google Scholar]
- [20].Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A 2013; 110:12450-5; PMID:23836644; http://dx.doi.org/ 10.1073/pnas.1300833110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dutilh BE, Cassman N, McNair K, Sanchez SE, Silva GG, Boling L, Barr JJ, Speth DR, Seguritan V, Aziz RK, et al.. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat Commun 2014; 5:4498; PMID:25058116; http://dx.doi.org/ 10.1038/ncomms5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dutilh BE. Metagenomic ventures into outer sequence space. Bacteriophage 2014; 4:e979664; PMID:26458273; http://dx.doi.org/ 10.4161/21597081.2014.979664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Mokili JL, Rohwer F, Dutilh BE. Metagenomics and future perspectives in virus discovery. Curr Opin Virol 2012; 2:63-77; PMID:22440968; http://dx.doi.org/ 10.1016/j.coviro.2011.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ogilvie LA, Caplin J, Dedi C, Diston D, Cheek E, Bowler L, Taylor H, Ebdon J, Jones BV. Comparative (meta)genomic analysis and ecological profiling of human gut-specific bacteriophage φB124-14. PLoS One 2012; 7:e35053; PMID:22558115; http://dx.doi.org/ 10.1371/journal.pone.0035053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Smith DP, Peay KG. Sequence depth, not PCR replication, improves ecological inference from next generation DNA sequencing. PLoS One 2014; 9:e90234; PMID:24587293; http://dx.doi.org/ 10.1371/journal.pone.0090234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Brum JR, Ignacio-Espinoza JC, Roux S, Doulcier G, Acinas SG, Alberti A, Chaffron S, Cruaud C, de Vargas C, Gasol JM, et al.. Ocean plankton. Patterns and ecological drivers of ocean viral communities. Science 2015; 348:1261498; PMID:25999515; http://dx.doi.org/ 10.1126/science.1261498 [DOI] [PubMed] [Google Scholar]
- [27].Breitbart M, Rohwer F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol 2005; 13:278-84; PMID:15936660; http://dx.doi.org/ 10.1016/j.tim.2005.04.003 [DOI] [PubMed] [Google Scholar]
- [28].Ignacio-Espinoza JC, Solonenko SA, Sullivan MB. The global virome: not as big as we thought? Curr Opin Virol 2013; 3:566-71; PMID:23896279; http://dx.doi.org/ 10.1016/j.coviro.2013.07.004 [DOI] [PubMed] [Google Scholar]
- [29].Abeles SR, Ly M, Santiago-Rodriguez TM, Pride DT. Effects of Long Term Antibiotic Therapy on Human Oral and Fecal Viromes. PLoS One 2015; 10:e0134941; PMID:26309137; http://dx.doi.org/ 10.1371/journal.pone.0134941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Quesada-Gómez C, López-Ureña D, Acuña-Amador L, Villalobos-Zúñiga M, Du T, Freire R, Guzmán-Verri C, del Mar Gamboa-Coronado M, Lawley TD, Moreno E, et al.. Emergence of an outbreak-associated Clostridium difficile variant with increased virulence. J Clin Microbiol 2015; 53:1216-26; PMID:25653402; http://dx.doi.org/ 10.1128/JCM.03058-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Yadav H, Jain S, Nagpal R, Marotta F. Increased fecal viral content associated with obesity in mice. World J Diabetes 2016; 7:316-20; PMID:27555892; http://dx.doi.org/ 10.4239/wjd.v7.i15.316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Barr JJ, Auro R, Furlan M, Whiteson KL, Erb ML, Pogliano J, Stotland A, Wolkowicz R, Cutting AS, Doran KS, et al.. Bacteriophage adhering to mucus provide a non-host-derived immunity. Proc Natl Acad Sci U S A 2013; 110:10771-6; PMID:23690590; http://dx.doi.org/ 10.1073/pnas.1305923110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hall RJ, Draper JL, Nielsen FG, Dutilh BE. Beyond research: a primer for considerations on using viral metagenomics in the field and clinic. Front Microbiol 2015; 6:224; PMID:25859244; http://dx.doi.org/ 10.3389/fmicb.2015.00224 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.