

Abstract
Introduction
Glioblastoma is the most common primary brain tumor in adults and prognosis remains poor with a median survival of approximately 15–17 months. This review provides an overview of recent advances in the field of glioblastoma immunotherapy.
Areas covered
Recent advances in dendritic cell vaccination immunotherapy are showing encouraging results in clinical trials and promise to extend patient survival. In this report we discuss current scientific knowledge regarding dendritic cell (DC) vaccines, including approaches to differentiating, priming, and injecting dendritic cells to achieve maximal anti-tumor efficacy in glioblastoma. These findings are compared to recently completed and currently ongoing glioblastoma clinical trials. Novel methods such as ‘fastDCs’ and vaccines targeting DCs in-vivo may offer more effective treatment when compared to traditional DC vaccines and have already entered the clinic.
Expert Commentary
Finally, we discuss the challenges of T-cell dysfunctions caused by glioblastoma immunosuppression and how they affect dendritic cell vaccinations approaches.
Keywords: Glioblastoma, Cancer Vaccine, Dendritic Cell, Immunotherapy, Clinical Trials, Immunosuppression, T-cell dysfunction, fastDCs, in-vivo DC vaccine, Recall antigens
1. Introduction to glioblastoma
Glioblastoma (GBM) represents approximately 50% of all malignant glioma and is the most common primary brain tumor in adults. At an incidence rate of 3.2 per 100,000 population, GBM patients have a median survival time of 15–17 months with median diagnosis occurring at age 64 [1]. In spite of the current gold-standard of care — maximal safe resection, radiotherapy, and temozolomide chemotherapy, GBM recurrence is almost always inevitable and the five-year survival rate is estimated at 5.1% [1, 2]. The severity and swift progression highlight the need for developing novel efficacious treatments for GBM patients.
1.1 CNS Immunocompetence
Literature has traditionally described the central nervous system as an immune-privileged site, marked by a stringent brain-blood barrier, the absence of a classical lymphatic drainage system, and low levels of T-cells, antigen-presenting cells (APCs), and major histocompatibility complexes (MHCs), aside from glial cells [2]. However, recent findings, including the discovery of MHC-1 expression by subsets of neurons, have challenged this viewpoint and credit the CNS with immunocompetence [3]. Of particular note is data by Louveau et al., which shows functional lymphatic vessels lining the dural sinuses in mice and draining cerebrospinal fluid, indicating the presence of a classical lymphatic system in the CNS [4]. In addition, angiogenesis around the growing brain tumor leads to the deterioration of microvasculature and relaxation of tight junction, thus increasing leaking.
The inherent brain immunocompetence, the ability of activated T-cells to enter the CNS [5–8], and the decreased barrier functions around the tumor ensure that the immune system has access to the malignant cells, making it feasible to develop immune-based treatment regimens that will improve the current standard of care.
2. Immunotherapy for GBM
Cancer growth is based on the principle that tumor cells are able to evade normal control of the immune system by using a number of complex mechanisms to escape immune detection, including low immunogenicity, antigenic modulation, and immune suppression [9–14]. Anti-cancer immunotherapies activate the immune system to specifically recognize and destroy malignant cells. Recent advances seen in immunotherapies targeting metastatic melanoma and other cancers show the extraordinary capacity of redirecting the immune system and warrant advancing immunotherapy research for treating glioblastoma [15–18].
2.1 Immunotherapy treatments pursued in GBM
A large portion of current GBM clinical trials are dedicated to testing the safety and efficacy of numerous immunotherapeutic approaches. Two main approaches are being pursued: passive immunotherapy and active immunotherapy. Passive immunotherapy is achieved by using therapeutic agents that activate the immune system and confer an antitumor response, often only for the duration of treatment. These include monoclonal antibodies (mAb), bispecific antibodies (bsAb), and immune checkpoint modulators [19]. Current clinical trials enrolling GBM patients are testing an anti-EGFR mAb (US clinical trial NCT02540161) and an anti-EGFR-CD3 bsAb (US clinical trial NCT02521090). Immune checkpoint modulators make up the majority of current passive immunotherapy trials for GBM (e.g. US clinical trial NCT02311920). Active immunotherapy, or tumor vaccination, is achieved by presenting tumor antigens which stimulate the body’s immune system to produce an endogenous anti-tumor response. The goal is to either amplify an existing tumor response or to elicit a de novo immune response against new antigens, thereby allowing long-term recognition and eradication of the tumor. Vaccinations being pursued for treating GBM include whole tumor lysate, peptides, viral vectors, and dendritic cells [19–21].
3. Dendritic cell vaccines
Dendritic cells (DC) play a crucial role in protecting the body from foreign antigens and form a link between the innate and adaptive immune system. Upon encountering foreign antigens, specifically pathogen-associated molecular patterns, DCs act as sentinels of the innate immune response by releasing activating cytokines. As orchestrators of the adaptive immune response, DCs take up, process, and present antigens on their cell surface to T-cells and B-cells, thereby activating naïve, effector, and memory immune cells or maintaining tolerance against self-antigens [22]. Ashley and colleagues describe DCs to be the most potent endogenous activators of de novo T-cell and B-cell responses, highlighting their vaccine potential in eliciting potent anti-tumor immune responses [23].
Spurred on by the invaluable efforts of Ralph Steinman, there is now a general consensus that DC vaccines can lead to long-lasting cancer immune responses by inducing tumor-specific T-cells and immunological memory [24]. Clinical trials testing DC vaccinations have, so far, shown encouraging, yet modest, results in patients with advanced cancers [25]. To date, sipuleucel-T is the only APC-based cancer vaccine approved in the United States and has shown an increase median survival of four months in prostate cancer patients [26]. However, several factors may be limiting the efficacy of current DC vaccines, including optimal maturation, tumor antigen loading, and the ability of DCs to migrate to vaccine-draining lymph nodes.
4. Dendritic Cell generation in-vitro
The most common approach to generating clinical-grade DCs in-vitro uses isolated CD14+ monocytes from patient PBMCs (Figure 1). Over a period of 5–7 days, monocytes are differentiated into immature DCs by culturing with GM-CSF and IL-4 [27, 28]. As originally developed and described by Helmut Jonuleit, DCs are subsequently matured in a cytokine cocktail consisting of GM-CSF, IL-4, TNF-α, IL-1β, IL-6, and, in some instances, prostaglandin E2 (PGE2) for 16 to 20 hours [29, 30]. However, PGE2 has been shown to skew T-cell differentiation to the Th2 phenotype by blocking the production of IL-12 p70 by DCs, the third signal needed for optimal T-cell expansion and acquisition of effector functions [22, 31, 32].
Figure 1. Dendritic cell vaccine production.
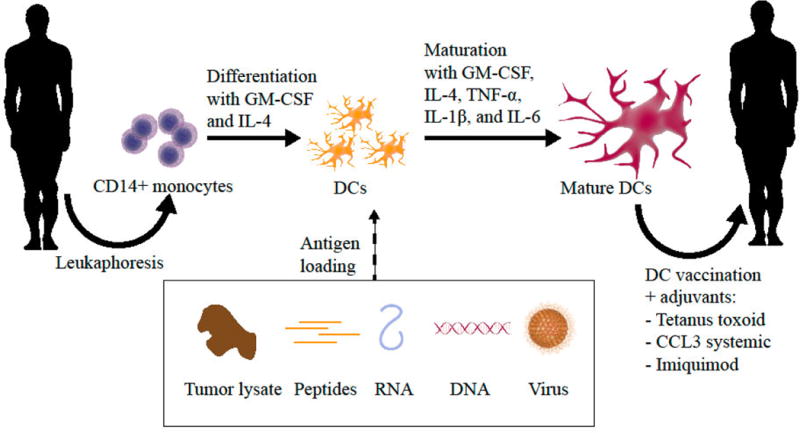
Dendritic cells (DC) vaccines for glioblastoma (GBM) immunotherapy are generated in-vitro using CD14+ monocytes isolated from patient PBMCs. Monocytes are typically differentiated into immature DCs by incubating with GM-CSF and IL-4 for a period of 5–7 days. Alternatively, monocytes can be differentiated in as little as 2 days using novel ‘fastDC protocols. DCs are subsequently matured in a cytokine cocktail for 16 to 20 hours and loaded with tumor antigen. DCs can be loaded with various formats of tumor antigen, including peptides, tumor lysate, DNA, and RNA. Finally, the DCs are injected back into the patient where they travel to vaccine-draining lymph nodes to elicit a tumor-specific immune response. Injection with adjuvants such as the tetanus toxoid can be used to increase DC migration to the lymph nodes and augment vaccine efficacy.
In order to improve the accessibility of DC vaccines to patients and to decrease cost and labor of DC generation, DC vaccine protocols have been developed that greatly reduce the ex-vivo culturing time required. ‘FastDCs’ have been obtained after mere 2-day culturing periods and retain the essential ability to induce antigen-specific T-cell responses [33, 34]. Important differences of fastDCs compared to standard DCs include higher yields in culture, lower release of IL-12 p70, higher CCL19-induced chemokinesis, better intracellular antigen processing, and more effective priming of tumor-specific cytotoxic T-cells [33, 35, 36]. A currently recruiting phase I/II clinical trial (NCT01734304), is using a 3-day protocol to produce fastDCs loaded with tumor antigens against acute myeloid leukemia and has so far concluded that vaccination with fastDCs is feasible and safe [37]. Furthermore, techniques that alleviate the need to differentiate DCs in-vitro from precursor cells via the direct isolation of myeloid or plasmacytoid DCs from PBMCs have recently proven safety and feasibility in clinical trials [38–40].
Cryopreservation is routinely used to store laboratory as well as clinical samples for extended time periods and has also been used for preserving mature dendritic cells, as first described by Feuerstein et al. [41]. Recent reports highlight that cryopreservation during DC vaccine preparation is feasible and allows patient leukaphoresis to be decoupled from the stringent DC generation process. In a small-scale clinical study, Nair and colleagues use cryopreserved PBMCs obtained from pediatric patients with medulloblastoma to generate functional DCs.[42] Alternatively, Fitzpatrick and colleagues showed that mature DCs can also be cryopreserved, alleviating stringent production timelines. The cryopreserved DCs showed no significant differences in viability and cytokine production when compared to freshly generated DCs [43]. Taken together, cryopreservation of DCs represents a practical approach for alleviating exhaustive production techniques and increasing accessibility of DC vaccines to sites without DC generation capabilities.
5. Tumor antigen targets in glioblastoma
DC vaccination is based on eliciting a potent immune response against malignant tumor cells. After DCs have been generated ex-vivo, the cells must be loaded with molecules that will allow the DCs to target harmful cells. So-called tumor antigens represent a set of molecular targets that can be used to direct DCs to initiate an immune response against the malignant cells. Broadly speaking, tumor antigens can be divided into tumor-associated antigens and tumor-specific antigens.
Tumor-associated antigens (TAAs) represent a class of molecules that are overexpressed on tumor cells due to changes in the genome that arise during the course of tumor progression. Based on their high expression level, TAAs can be used as targets for marking malignant cells. However, given the fact that TAAs are overexpressed yet normal proteins, they are also expressed in normal cells, which may lead to limited specificity for targeting tumor cells.
In GBM, studies have found numerous proteins, including survivin, HER2neu, EphA2, EGFR, and telomerase, which are over-expressed and could serve as immunological targets for potential immunotherapies [44–50]. For some of these antigens, clinical potential has been shown [49, 51].
Cancer/testis antigens (CTAs) represent a unique class of TAAs where normal expression is restricted to germ cells in the testis and expression is not found in healthy adult somatic cells. For example, the melanoma associated CTAs (MAGE, CAGE) are expressed across a wide range of cancers [52, 53]. In GBM, CTAs are variably expressed within the tumor and an extensive expression analysis by Freitas and colleagues of 153 CTAs identified 4 CTAs (ACTL8, CTCFL, OIP5, and XAGE3) uniquely expressed within GBM tumors when compared to normal brain.[54]
Tumor-specific antigens (TSAs) arise from mutations in the genome that lead to abnormal proteins found only on the tumor cells or a certain subset of tumor cells. Often, these abnormal proteins have a specific function and confer a survival advantage to the malignant cell. Targeting of TSAs makes it possible to use highly potent therapies without risking damage to normal cells. However, as TSAs may often be expressed in only a subset of tumor cells, treatment may prove to be inefficacious once the targeted cells have been removed, leaving the untargeted malignant cells behind.
In GBM patients there are currently two TSAs that are highly conserved among patients and have shown immunogenicity in multiple studies. 31 to 50% of patients with GBM express a conserved mutation of the epidermal growth factor receptor, called EGFRvIII [55–60]. In patients positive for EGFRvIII, the mutation is expressed in 37 to 87% of tumor cells [57].
The second TSA is a conserved mutation of isocitrate dehydrogenase type 1 (IDH1), which occurs at the critical arginine residue number 132 in the catalytic pocket and results in a neomorphic enzymatic function, genetic instability, and malignant transformation [61]. This mutation, termed IDH1(R132H), occurs in more than 70% of grade II and III gliomas and from a therapeutic viewpoint, represents a promising candidate for a tumor specific treatment of malignant glioma [62, 63].
Given the mutagenic nature of cancer cells, most mutations that arise spontaneously in GBM are patient-specific neoepitopes. Furthermore, since TSAs are only expressed in a subgroup of patients, researchers are pursuing the development of personalized cancer vaccines to develop treatments that benefit all patients.
In addition to mutations of native proteins, viral proteins that are specifically upregulated on malignant cells represent ideal TSAs as they have the unique advantage of being intrinsically foreign to the host. While viral expression is usually not restricted to malignant cells, expression of immunogenic epitopes is often undetectable on healthy tissue in patients with virus-associated cancers. We and others have shown that human cytomegalovirus (CMV) infection is associated with low-level viral gene expression in malignant glioma [64, 65]. Based on the success and safety of cellular immunotherapeutics targeting CMV in immunocompromised patients, immunodominant CMV antigens, such as immediate early 1 (IE1), phosphoprotein 65 (pp65), and glycoprotein B (gB), represent possible tumor specific targets for the development of immunotherapies targeting GBM [66–68].
6. Loading DCs with antigen
In practice, DCs can be loaded with a variety of antigen sources, including peptides, tumor lysate, DNA, or RNA (Figure 1). Antigen loading may occur after maturation or concurrent with cytokine maturation. The variety of antigen loading strategies in current clinical trials testing DC vaccinations in GBM patients reveals that the optimal antigen source for causing tumor regression remains a topic of much interest (see Table 1).
Table 1.
Recent clinical trials employing dendritic cell vaccination immunotherapy for glioblastoma
| NIH clinical trial | Type | Location / Sponsor | Phase | Eligibility Criteria | Primary Outcome | Dosing regimen | Treatment groups |
|---|---|---|---|---|---|---|---|
| NCT 02010606 | DC loaded with lysate | Cedars-Sinai Medical Center | 1 | Newly diagnosed or recurrent GBM | Safety, adverse events, treatment-related toxicities | Autologous DCs loaded with lysate from allogeneic GBM stem-like cell line given once every week for 4 weeks followed by once every 8 weeks | Cohort A: Newly diagnosed GBM Cohort B: Recurrent GBM |
| NCT 01808820 | DC loaded with lysate, Imiquimod, Tumor lysate | University of Miami | 1 | Recurrent GBM | Number of adverse events | DCs loaded with tumor lysate are injected into imiquimod-treated skin given once every week for 4 weeks followed by tumor lysate injections during weeks 8, 12, 16, and 28 | Single-arm |
| NCT 02049489 | DC loaded with peptide antigens | Multi-center / Immuno-Cellular Therapeutics | 1 | Recurrent GBM | Safety study | At least 4 doses of DCs loaded with CD133 peptides given (ICT-121), followed by additional maintenance vaccines | Single-arm |
| NCT 02649582 | DC loaded with RNA | Antwerp University Hospital | 1/2 | Newly diagnosed GBM | Overall survival | WT1 mRNA loaded DC vaccine given weekly for 3 weeks followed by maintenance vaccine on day 21 of every TMZ cycle | Single-arm |
| NCT 01567202 | DC loaded with lysate | Huashan University, Fudan University | 2 | Histologically confirmed GBM | Overall survival | 8–10E6 DCs loaded with autogeneic glioma stem-like cells associated antigens given once a week for 6 weeks | Triple-blind Arm I: DCs Arm II: Placebo |
| NCT 02366728 | DC loaded with RNA | Duke University Medical Center | 2 | Newly diagnosed GBM | Overall survival | CMV pp65-loaded DC vaccines #1–3 given every two weeks followed by vaccine #4 (only Arm I-II). Td given to all during vaccine #1. Arm III patients receive basilixumab 1 week before vaccine #1–2. Before vaccine #4, Arm I receives unloaded DCs, Arm II-III Td dose. | Arm I: Unloaded DCs + loaded DCs Arm II: Td + loaded DCs Arm III: Td +loaded DCs + basiliximab |
| NCT 02465268 | DC loaded with RNA | University of Florida | 2 | Newly diagnosed GBM | Overall survival | CMV pp65-loaded or influenza-loaded DCs matured with GM-CSF and injected ID into Td preconditioned patients at day 22–24 after first TMZ cycle then at 2 week intervals. Doses 4–10 given on day 22–24 of each TMZ cycle. | Arm I: pp65 DC + GM-CSF + Td Arm II: Influ. DC + GM-CSF + Td Arm III: PBMC + GM-CSF + Td Arm IV: pp65 DC + GM-CSF + Sal. Arm V: Influ. DC + GM-CSF + Sal. Arm VI: PBMC + GM-CSF + Sal |
| NCT 01204684 | DC loaded with lysate | University of Los Angeles / Jonsson Comprehensive Cancer Center | 2 | Newly diagnosed or recurrent grade 3 or 4 glioma | Most effective combination of DC vaccine components | DCs loaded with tumor lysate given alone or with adjuvants 0.2% resiquimod or polyICLC | Arm I: DCs Arm II: DCs + resiquimod Arm III: DCs + polyICLC |
| NCT 01759810 | DC loaded with protein, HSC, CTL | NeuroVita Clinic / Numerous sponsors | 2/3 | Histologically confirmed GBM | All-cause mortality | DCs loaded with recombinant proteins analogous to key tumor antigens, CTLs, and autologous proteome-modified HSCs or allogeneic HSCs given at different intervals to patients over the first couple of months. | Arm 1: allogeneic HSCs Arm II: autologous HSCs |
| NCT 02546102 | DC loaded with peptides | Multi-center / ImmunoCellular Therapeutics | 3 | Newly diagnosed GBM | Overall survival | DCs loaded with immunogenic peptides from 6 tumor-associated antigens (ICT-107) given once a week for four weeks ID followed by maintenance injections once a month for 11 months and once every 6 months therafter. | Arm I: ICT-107 Arm II: Placebo |
| NCT 00045968 | DC loaded with lysate | Multi-center / Northwest Biotherapeutics | 3 | Newly diagnosed GBM | PFS | Autologous DCs pulsed with tumor lysate antigen, called DCVax-L, or autologous PBMCs (placebo) given ID twice daily on days 0, 10, 20, and at weeks 8, 16, 32, 48, 72, 96, and 120 | Arm I: DCVax-L Arm II: Placebo |
CTL, cytotoxic T-lymphocyte; DC, dendritic cell; ID, intradermal; GBM, glioblastoma; GM-CSF, granulocyte macrophage colony-stimulating factor; HSC, hematopoietic stem cell; PBMC, peripheral blood mononuclear cells; PFS, progression-free survival; pp65, phosphoprotein 65; Sal., saline; Td, tetanus/diphtheria toxoid; TMZ, temozolomide
Peptide and tumor lysate loading is achieved by co-incubation, allowing the DCs to scavenge the antigens from their surroundings, process them internally, and present epitopes on their MHC molecules. For peptide loading, either a single peptide or a mixture of different peptides may be used for generating the DC vaccine. By giving conserved immunocompetent glioblastoma antigen peptides, vaccine preparation can be standardized across a patient subpopulation resulting in a focused immune response. CD133 is an antigen expressed on, albeit not all, GBM stem-like cells, whose expression levels correlate with patient survival in glioma [69, 70]. ICT-121, a DC vaccine trial targeting recurrent glioblastoma, uses a combination of purified peptides from the CD133 protein to load DCs which are injected into the patient over several months with the goal of eliciting an immune response against the tumor (Table 1, NCT02049489).
Alternatively, DCs can be pulsed with whole tumor lysate, which, when compared to peptide loading, has the advantage of containing the full tumor antigen repertoire of conserved and novel patient-specific neoepitopes, allowing DCs to naturally process the proteins and present the most immunogenic antigens. In a recent report, Garg et al. show that inducing immunogenic cell death of the tumor cells, instead of freeze/thawing-based necrosis, markedly improves DC vaccines by increasing the immunogenicity of the dying/dead cancer cells which they link to the exposure/release of potent danger signals [71]. Using autologous tumor cell lysate from each patient to load the DC’s could represent an important step towards personalized medicine in the treatment of GBM. The DCVax-L vaccine (autologous dendritic cells pulsed with autologous tumor cell lysate) showed a 3 year overall survival rate, 2.5 times the usual period of survival, in a phase 1/2 clinical trial in patients with newly diagnosed GBM, extended survival by 5 months or more in patients with recurrent GBM (Table 1, NCT00045968), and is currently being tested in a blinded randomized phase 3 trial [72–74].
Antigen presentation on DCs can also be initiated by the transfer of nucleotides which subsequently drive protein expression and processing. DNA-based approaches use retroviral transduction to integrate antigen DNA into the DC genome using retrovirus transduction methods and therefore allow long-term antigen expression [75–77].
However, given the safety implications of genomic integration and the complication of driving efficient antigen expression in DCs, RNA-based approaches have come to be favored over DNA in the laboratory and the clinic. Our lab has shown indications of clinical efficacy in treating GBM patients with mRNA transfected DC’s [21]. RNA transfection of DC’s has the major advantage that this approach is applicable to a wide range of patients given that little tumor sample is needed to prepare the vaccine since RNA can be amplified from a small number of tumor cells. Furthermore, off-the-shelf RNA-DCs can be generated using conserved GBM tumor antigens such as the CMV antigen pp65. DC loading of whole tumor RNA or antigen-specific RNAs is achieved by electroporation followed by an overnight incubation. In terms of safety, stimulating DCs with RNA is a transient therapy that poses no risk of integration, unlike viral or DNA vectors [22]. In a recent phase 1/2 clinical trial, our group generated a dendritic cell vaccine using pp65 mRNA for treating glioblastoma (US clinical trial NCT00639639). Given the expression of pp65 in glioblastoma, as previously published [64, 78, 79], this viral antigen is ideal for eliciting a specific tumor response. Pre-conditioning patients with tetanus/diphtheria toxoid led to a significant increase in lymph node homing of pp65-specific DCs, anti-tumor immune responses, and patient survival [21]. Confirmatory randomized and double-blinded clinical trials are now testing the effects of Td preconditioning on survival in patients with newly-diagnosed GBM (Table 1, NCT02366728 and NCT02465268).
7. Overcoming limited DC migration
In addition to developing optimally matured and loaded DC vaccines, DCs must migrate from their injection site to the lymph node to induce immune responses. While previous DC vaccines in GBM have shown limited promise in patients, a recent finding by Mitchell et al. has shown that simply improving DC lymph node homing leads to significantly improved tumor responses in humans and mice [21].
Less than 5% of injected DCs reach the lymph nodes [80]. By preconditioning injection sites with inflammatory molecule TNF-α, researchers have previously shown improvements in DC lymph node homing in mice [81]. Mitchell and colleagues applied the idea of inflammatory preconditioning by injecting tetanus/diphtheria toxoid (Td), a standard vaccine used for the prevention of tetanus and diphtheria, into the vaccine site of GBM patients. Results showed that pp65 mRNA loaded DCs injected bilaterally into unilaterally TD preconditioned patients resulted in a doubling of DC migration bilaterally, indicating a systemic effect, and significantly improved survival. In both humans and mice, the Td preconditioning effects were dependent on chemokine (C-C motif) ligand 3 (CCL3) — knocking out CCL3 or depleting CD4+ T cells in mice abrogated improved survival and giving CCL3 or activated CD4+ T cells intravenously rescued DC migration to the lymph nodes [21]. These results expand our knowledge of the mechanisms that underlie successful DC vaccines by showing the relationship between systemic CCL3, CD4+ T cells, and DC cells. Future research will need to examine the downstream mechanisms of CCL3 to determine which CCL3 receptors are vital and whether other recall antigens or systemic administration of CCL3 will improve DC vaccines in GBM and other cancer patients.
8. Vaccines target DCs in-vivo
Given the labor and time intensive process of generating DC vaccines ex-vivo in addition to variable response rates, efforts are underway to develop vaccines that target DCs in-vivo. Targeting DCs in-vivo circumvents the issue of poor migration seen with ex-vivo generated DCs and allows the targeting of single or multiple DC subsets via their range of unique and shared pattern recognition receptors [82]. A finding by Allan and colleagues showed injected DCs may act indirectly through DCs already present in the lymph node, further supporting strategies that circumvent ex-vivo DC generation [83].
In-vivo DC vaccination is achieved by targeting mAbs to DC-specific cell surface receptors. CD205 is a novel endocytic receptor of the C-type lectin family. It mediates antigen uptake and cross-presentation to T cells and has been a major focus for the development of in-vivo DC vaccines. Delivery of tumor antigen bound to anti-CD205 antibodies stimulated CD4+ and CD8+ T cells in mice, resulting in tumor regression superior to in-vitro loaded DCs [84–86]. In a recent early-phase study using a vaccine composed of a human CD205 antibody fused to the full-length tumor antigen NY-ESO-1, 45 patients with tumors expressing NY-ESO-1 received various dose levels and adjuvant combinations (US clinical trial NCT00948961). 13 patients experienced stabilization of disease and two patients experienced tumor regression. This first-inhuman study of a protein vaccine targeting DCs demonstrated feasibility, safety, and biological activity, providing rationale for follow-up studies [87].
Given that DC cross-presentation can activate GBM-specific T cells in the periphery that travel to the brain, as seen in trials using in-vitro generated DCs, vaccines targeting DCs in-vivo present a promising alternative that use similar mechanisms while bypassing the need for DCs to travel from the injection site to the draining lymph nodes. Future experiments will show whether mAb-mediated delivery of GBM-specific antigens to DCs will lead to tumor regression and increase survival.
9. Remaining Questions
Recurrent tumors differ from primary tumors, since treatment with radiotherapy and chemotherapy promotes genetic changes in the primary tumor allowing it to overcome the treatment regimen. Patients with recurrent GBM have a poor overall performance status and their tumors are unresectable, requiring significant corticosteroid treatment to control cerebral edema. A common strategy in GBM — a strategy that needs to be re-visited — is to test new treatments in patients with recurrent disease and then advance those therapies with indications of efficacy into the newly diagnosed patient group. Testing of novel immunotherapies, such as DC vaccines, in patients with newly diagnosed GBM independently of patients with recurrent GBM, may help reveal novel efficacious immunotherapeutic treatments.
Patients with recurrent GBM have undergone multiple cycles of radiotherapy and cytotoxic chemotherapy resulting in a relative immunosuppression. Although TMZ chemotherapy induces lower CD4+ counts in patients and is linked to worse clinical outcomes, lymphopenia appears to play a complex role in studies testing vaccination strategies. A single-arm phase II trial of rindopepimut found lymphopenia enhances tumor-specific cellular and humoral immune responses [88], and in another phase II trial testing an autologous tumor vaccine, patients with grade 3 lymphopenia had improved outcomes when compared to grade 4 and grade 0–2 lymphopenia [89]. It remains to be seen whether lymphopenia can be leveraged to improve GBM vaccines.
10. Conclusion
GBM remains the most malignant tumor of the CNS and patient outlook is dismal. Novel therapies are needed for effective treatment, and the rise of immunotherapies in the field of cancer treatment is promising to revolutionize GBM treatment. Amongst the multitude of immunotherapeutic approaches, DC vaccines have in the past shown limited benefit in tumor therapy, though recent trials in GBM have shown anti-tumor effects for the first time. Combined with the misconceived dogma of an immune-incompetent CNS, DC vaccine research faces a number of hurdles ranging from optimal production, targeting, and antigen loading to overcoming limited migration.
DCs are immune cells that form a bridge between the adaptive and innate immune system. Acting as sentinels, DCs scavenge for foreign material, which is taken up, processed, and presented on MHC molecules to T-cells and B-cells, thereby activating the adaptive immune system to mount an attack on the foreign antigens. DC vaccine approaches exploit this system by isolating monocytes from patients, maturing them into DCs and loading them with appropriate tumor antigens in-vitro before infusing them back into the patient where they will migrate to the lymph nodes and activate the immune system.
The ex-vivo maturation of monocytes to DCs is achieved by plating the cells with media containing various cytokines. Depending on the cytokines and the concentrations used, maturation normally takes between 5–7 days. Recently developed fastDC reduce the maturation time to about 2 days; the cells demonstrate comparable characteristics to traditionally matured DCs and are able to stimulate antigen-specific T-cell immune responses [33]. Furthermore, recent findings show that DC injection and subsequent migration to the draining lymph nodes is a critical step in obtaining therapeutic efficacy. By preconditioning the vaccine site with a recall antigen, researchers were able to significantly improve DC migration and show increased survival in patients and mice [21].
Taken together, advancements in DC vaccines and results from recent clinical trials substantiate the feasibility of using DC vaccines for treating GBM and provide support for the continued development of DC vaccines along all stages of vaccine development.
11. Expert Commentary
Optimal DC vaccines mirror the essential signals that dictate effective T-cell activation [90]. In-vitro generated DCs need to proficiently present antigens for a period long enough to engage the T-cell receptor (Signal 1) and provide T-cells with co-stimulation via cell surface receptors CD80/86 and CD70 (Signal 2). Finally, the cytokine IL-12p70 is thought to represent the third signal for optimal T-cell activation and proliferation provided by DCs. Given the vital interaction of DCs with T-cells to induce an antigen-specific immune response, T-cell dysfunction seen in GBM may play a major role in dictating efficacy of DC vaccine studies. An improved understanding of the various T-cell dysfunctions could lead to patient-specific combination therapies that will enhance DC-mediated immune activation.
During carcinogenesis, the tumor microenvironment induces T-cell tolerance, contributing to tumor growth. Although T-cell tolerance is an integral component of T-cell maturation that prevents autoimmunity, T-cell tolerance toward malignant cells is detrimental and leads to tumor immune escape [91]. Direct induction of T-cell tolerance occurs via down-regulation of MHC molecules and costimulatory molecules and the upregulation of inhibitory receptors such as PD-1 ligand, HLA-G, and HLA-E [92–95]. Blocking of PD-1 ligand in-vivo has been shown to enhance the host’s anti-tumor response, highlighting the potential of altering the tumor receptors [96]. In addition to altered surface reception molecules, tumor cells secrete various immunosuppressive factors, such as TGF-β, VEGF, IL-10, and CCL21, leading to an immunosuppressive microenvironment that results in the recruitment or promotion of immunosuppressive immune cells including regulatory T-cells (Tregs), immature DCs, and tumor-associated macrophages [97–103]. In GBM patients, immunosuppression is particularly severe, resulting in a shift toward Th2 cytokine production, increased Treg recruitment, and inhibiting T-cell proliferation [13, 104, 105]. Fecci and colleagues found that GBM patients had decreased CD4+ T-cell counts but that the fraction of Tregs in the remaining CD4+ T-cells was increased. Treg reduction removed T-cell proliferative defects and reversed Th2 cytokine shifts [13]. Breaking of T-cell tolerance in GBM patients may be required to obtain durable DC vaccine efficacy in patients. This could be done by changing the tumor microenvironment signals or decreasing Treg numbers.
In addition to tolerance, T-cell exhaustion in cancer patients is marked by the upregulation of negative regulators, a distinct regulation of transcription factors, and the hierarchical loss of effector functions on tumor-infiltrating lymphocytes [106]. T-cell inhibitory receptors prominently include LAG-3, PD-1, CTLA-4, and TIM-3, some of which are already being targeted in successful immunotherapies [15, 107]. Although trials testing checkpoint inhibitors in GBM have commenced, they may ultimately not prove advantageous when given as stand-alone therapy, but instead will prove successful as a way reverse immune suppression while treating with tumor-specific immunotherapies such as DC vaccines. Given the complexity of tumor-mediated effects on patient immune systems, researchers developing DC vaccines will need to consider tumor-mediated immunosuppression and how to intelligently design co-therapies to boost DC vaccine efficacy.
12. Five-year view
Dendritic cell vaccinations for glioblastoma have already shown promising anti-tumor effects in clinical trials. Determining whether DC vaccinations have the potential to become standard-of-care remains to be elucidated. Results from ongoing clinical trials, including the phase 3 DCVax-L and ICT-107 trial (Table 1), will reveal which DC vaccine protocols are efficacious. Based on recent findings that tetanus toxoid preconditioning acts as an adjuvant to boost systemic DC migration and increase survival, it is probable that over the next few years DC vaccination strategies for GBM will include novel recall antigens as adjuvants to increase efficacy and enhance immune responses. Furthermore, it is expected that DCs loaded with immunogenic neoepitopes will soon enter trials, mirroring current GBM peptide vaccinations strategies and bringing us closer to designing patient-specific tumor treatments. In parallel, immune checkpoint modulators are likely to be used in combination with DC vaccinations in an effort to ‘release the immunologic brakes’ and thereby enhance the immune response and magnitude of antitumor DC vaccines.
13. Key Issues.
Dendritic cell vaccinations have shown anti-tumor effects, although limited, in glioblastoma patients.
Efforts to improve DC vaccines focus on optimal maturation, tumor antigen loading, and the ability of DCs to migrate to vaccine-draining lymph nodes.
Fast-DC maturation protocols promise improvements in DC vaccine preparation, helping to reduce time and labor needed when compared to traditional DC vaccine protocols.
A variety of tumor antigen sources can be used to load DCs, including peptides, tumor lysate, DNA, and RNA. To date, no one antigen source has been found to be significantly superior, despite numerous ongoing GBM clinical trials testing antigen sources.
Tetanus toxoid has been used as an inflammatory adjuvant to boost DC migration and overall survival in patients and mice, hinting at a possible pervasive strategy to boost DC vaccine efficacy using recall antigens.
Vaccines targeting DCs in-vivo allow targeting of specific DC subsets and promise more effective immune responses.
Neoepitope-loaded DCs and combination approaches with immune checkpoint modulators are likely to be pivotal research areas in the next years.
Acknowledgments
Funding
JH Sampson’s research is funded by Celldex Therapeutics and also funding under the Duke University Faculty Plan from license fees paid to Duke University by Celldex Therapeutics.
Footnotes
Declaration of Interests
JH Sampson has served as a consultant or in an advisory role for Celldex Therapeutics. Duke University has the potential to receive patent-related royalties. JH Sampson has an equity interest in Annias Therapeutics, which has optioned intellectual property from Duke related to the use of the pepCMV Vaccine in the treatment of glioblastoma multiforme. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Reference annotations
*Of interest
**Of considerable interest
- 1*.Ostrom QT, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro Oncol. 2015;17(Suppl 4):iv1–iv62. doi: 10.1093/neuonc/nov189. comprehensive summary of the current epidemiology of primary brain and CNS tumors in the USA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Preusser M, et al. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol. 2015;11(9):504–14. doi: 10.1038/nrneurol.2015.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cebrian C, et al. MHC-I expression renders catecholaminergic neurons susceptible to T-cell-mediated degeneration. Nat Commun. 2014;5:3633. doi: 10.1038/ncomms4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Louveau A, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–41. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ludowyk PA, Willenborg DO, Parish CR. Selective localisation of neuro-specific T lymphocytes in the central nervous system. J Neuroimmunol. 1992;37(3):237–50. doi: 10.1016/0165-5728(92)90008-9. [DOI] [PubMed] [Google Scholar]
- 6.Odoardi F, et al. T cells become licensed in the lung to enter the central nervous system. Nature. 2012;488(7413):675–9. doi: 10.1038/nature11337. [DOI] [PubMed] [Google Scholar]
- 7.Ransohoff RM, Kivisakk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol. 2003;3(7):569–81. doi: 10.1038/nri1130. [DOI] [PubMed] [Google Scholar]
- 8*.Prins RM, et al. Anti-tumor activity and trafficking of self, tumor-specific T cells against tumors located in the brain. Cancer Immunol Immunother. 2008;57(9):1279–89. doi: 10.1007/s00262-008-0461-1. Confirmation of effective penetration of tumor-specific T cells into the CNS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bhatia A, Kumar Y. Cancer-immune equilibrium: questions unanswered. Cancer Microenviron. 2011;4(2):209–17. doi: 10.1007/s12307-011-0065-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sampson JH, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–9. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21(4):687–92. doi: 10.1158/1078-0432.CCR-14-1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stevenson GT. Three major uncertainties in the antibody therapy of cancer. Haematologica. 2014;99(10):1538–46. doi: 10.3324/haematol.2013.084640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fecci PE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66(6):3294–302. doi: 10.1158/0008-5472.CAN-05-3773. [DOI] [PubMed] [Google Scholar]
- 14.Fecci PE, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13(7):2158–67. doi: 10.1158/1078-0432.CCR-06-2070. [DOI] [PubMed] [Google Scholar]
- 15.Kim T, et al. Combining targeted therapy and immune checkpoint inhibitors in the treatment of metastatic melanoma. Cancer Biol Med. 2014;11(4):237–46. doi: 10.7497/j.issn.2095-3941.2014.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Callahan MK. Immune Checkpoint Therapy in Melanoma. Cancer J. 2016;22(2):73–80. doi: 10.1097/PPO.0000000000000183. [DOI] [PubMed] [Google Scholar]
- 17.Topalian SL, et al. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer. 2016;16(5):275–87. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kazandjian D, et al. FDA Approval Summary: Nivolumab for the Treatment of Metastatic Non-Small Cell Lung Cancer With Progression On or After Platinum-Based Chemotherapy. Oncologist. 2016;21(5):634–42. doi: 10.1634/theoncologist.2015-0507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reardon DA, et al. Immunotherapy advances for glioblastoma. Neuro Oncol. 2014;16(11):1441–58. doi: 10.1093/neuonc/nou212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brown MC, et al. Oncolytic polio virotherapy of cancer. Cancer. 2014;120(21):3277–86. doi: 10.1002/cncr.28862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21**.Mitchell DA, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519(7543):366–9. doi: 10.1038/nature14320. Tetanus/diphtheria toxoid precondition significantly improves survival in patients and mice with glioblastoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Batich KA, Swartz AM, Sampson JH. Enhancing dendritic cell-based vaccination for highly aggressive glioblastoma. Expert Opin Biol Ther. 2015;15(1):79–94. doi: 10.1517/14712598.2015.972361. [DOI] [PubMed] [Google Scholar]
- 23.Ashley DM, et al. Bone marrow-generated dendritic cells pulsed with tumor extracts or tumor RNA induce antitumor immunity against central nervous system tumors. J Exp Med. 1997;186(7):1177–82. doi: 10.1084/jem.186.7.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steinman RM. Decisions about dendritic cells: past, present, and future. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 25.Palucka K, Banchereau J. Dendritic-cell-based therapeutic cancer vaccines. Immunity. 2013;39(1):38–48. doi: 10.1016/j.immuni.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kantoff PW, et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med. 2010;363(5):411–22. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 27.Romani N, et al. Proliferating dendritic cell progenitors in human blood. J Exp Med. 1994;180(1):83–93. doi: 10.1084/jem.180.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sallusto F, Lanzavecchia A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J Exp Med. 1994;179(4):1109–18. doi: 10.1084/jem.179.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nair S, Archer GE, Tedder TF. Isolation and generation of human dendritic cells. Curr Protoc Immunol. 2012 doi: 10.1002/0471142735.im0732s99. Chapter 7. p. Unit7 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jonuleit H, et al. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27(12):3135–42. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]
- 31.Curtsinger JM, et al. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162(6):3256–62. [PubMed] [Google Scholar]
- 32.Trinchieri G, Pflanz S, Kastelein RA. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity. 2003;19(5):641–4. doi: 10.1016/s1074-7613(03)00296-6. [DOI] [PubMed] [Google Scholar]
- 33.Dauer M, et al. Mature dendritic cells derived from human monocytes within 48 hours: a novel strategy for dendritic cell differentiation from blood precursors. J Immunol. 2003;170(8):4069–76. doi: 10.4049/jimmunol.170.8.4069. [DOI] [PubMed] [Google Scholar]
- 34.Ramadan G. Generation of functional monocyte-derived fast dendritic cells suitable for clinical application in the absence of interleukin-6. Cytotechnology. 2011;63(5):513–21. doi: 10.1007/s10616-011-9375-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burdek M, et al. Three-day dendritic cells for vaccine development: antigen uptake, processing and presentation. J Transl Med. 2010;8:90. doi: 10.1186/1479-5876-8-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kvistborg P, et al. Fast generation of dendritic cells. Cell Immunol. 2009;260(1):56–62. doi: 10.1016/j.cellimm.2009.09.003. ** Evaluation of fastDC generation, showing functionality that is comparable to the standard 8-day DC protocol. [DOI] [PubMed] [Google Scholar]
- 37.Lichtenegger FS, et al. Next-generation dendritic cell vaccination in postremission therapy of AML: Results of a clinical phase I trial. [abstract] In: Proceedings of the CRI-CIMT-EATI-AACR Inaugural International Cancer Immunotherapy Conference: Translating Science into Survival. Cancer Immunol Res. 2016;4(1 Suppl) p. Abstract nr B146. [Google Scholar]
- 38.Schreibelt G, et al. Effective Clinical Responses in Metastatic Melanoma Patients after Vaccination with Primary Myeloid Dendritic Cells. Clin Cancer Res. 2016;22(9):2155–66. doi: 10.1158/1078-0432.CCR-15-2205. [DOI] [PubMed] [Google Scholar]
- 39.Prue RL, et al. A phase I clinical trial of CD1c (BDCA-1)+ dendritic cells pulsed with HLA-A*0201 peptides for immunotherapy of metastatic hormone refractory prostate cancer. J Immunother. 2015;38(2):71–6. doi: 10.1097/CJI.0000000000000063. [DOI] [PubMed] [Google Scholar]
- 40.Bol KF, et al. Dendritic Cell-Based Immunotherapy: State of the Art and Beyond. Clin Cancer Res. 2016;22(8):1897–906. doi: 10.1158/1078-0432.CCR-15-1399. [DOI] [PubMed] [Google Scholar]
- 41.Feuerstein B, et al. A method for the production of cryopreserved aliquots of antigen-preloaded, mature dendritic cells ready for clinical use. J Immunol Methods. 2000;245:1–2. 15–29. doi: 10.1016/s0022-1759(00)00269-6. [DOI] [PubMed] [Google Scholar]
- 42.Nair SK, et al. Ex vivo generation of dendritic cells from cryopreserved, post-induction chemotherapy, mobilized leukapheresis from pediatric patients with medulloblastoma. J Neurooncol. 2015;125(1):65–74. doi: 10.1007/s11060-015-1890-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fitzpatrick E, et al. Cryopreservation of activated DC1 makes large scale dendritic cell vaccines feasible in cancer therapy. Cytotherapy. 2015;17(6):S22–S23. [Google Scholar]
- 44.Fenstermaker RA, Ciesielski MJ. Challenges in the development of a survivin vaccine (SurVaxM) for malignant glioma. Expert Rev Vaccines. 2014;13(3):377–85. doi: 10.1586/14760584.2014.881255. [DOI] [PubMed] [Google Scholar]
- 45.Liu R, Mitchell DA. Survivin as an immunotherapeutic target for adult and pediatric malignant brain tumors. Cancer Immunol Immunother. 2010;59(2):183–93. doi: 10.1007/s00262-009-0757-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Komata T, et al. Telomerase as a therapeutic target for malignant gliomas. Oncogene. 2002;21(4):656–63. doi: 10.1038/sj.onc.1205072. [DOI] [PubMed] [Google Scholar]
- 47.Akiyama Y, et al. Novel cancer-testis antigen expression on glioma cell lines derived from high-grade glioma patients. Oncol Rep. 2014;31(4):1683–90. doi: 10.3892/or.2014.3049. [DOI] [PubMed] [Google Scholar]
- 48.Ahmed N, et al. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474–85. doi: 10.1158/1078-0432.CCR-09-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okada H, et al. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J Neurooncol. 2008;88(3):245–50. doi: 10.1007/s11060-008-9566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pedersen MW, et al. Sym004: a novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res. 2010;70(2):588–97. doi: 10.1158/0008-5472.CAN-09-1417. [DOI] [PubMed] [Google Scholar]
- 51.Tandon M, Vemula SV, Mittal SK. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin Ther Targets. 2011;15(1):31–51. doi: 10.1517/14728222.2011.538682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zendman AJ, Ruiter DJ, Van Muijen GN. Cancer/testis-associated genes: identification, expression profile, and putative function. J Cell Physiol. 2003;194(3):272–88. doi: 10.1002/jcp.10215. [DOI] [PubMed] [Google Scholar]
- 53.Bolli M, et al. Tissue microarray evaluation of Melanoma antigen E (MAGE) tumor-associated antigen expression: potential indications for specific immunotherapy and prognostic relevance in squamous cell lung carcinoma. Ann Surg. 2002;236(6):785–93. doi: 10.1097/01.SLA.0000036266.09823.6C. discussion 793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Freitas M, et al. Expression of cancer/testis antigens is correlated with improved survival in glioblastoma. Oncotarget. 2013;4(4):636–46. doi: 10.18632/oncotarget.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aldape KD, et al. Immunohistochemical detection of EGFRvIII in high malignancy grade astrocytomas and evaluation of prognostic significance. J Neuropathol Exp Neurol. 2004;63(7):700–7. doi: 10.1093/jnen/63.7.700. [DOI] [PubMed] [Google Scholar]
- 56.Frederick L, et al. Diversity and frequency of epidermal growth factor receptor mutations in human glioblastomas. Cancer Res. 2000;60(5):1383–7. [PubMed] [Google Scholar]
- 57.Wikstrand CJ, et al. Cell surface localization and density of the tumor-associated variant of the epidermal growth factor receptor, EGFRvIII. Cancer Res. 1997;57(18):4130–40. [PubMed] [Google Scholar]
- 58.Wong AJ, et al. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci U S A. 1992;89(7):2965–9. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cunningham MP, et al. Coexpression, prognostic significance and predictive value of EGFR, EGFRvIII and phosphorylated EGFR in colorectal cancer. Int J Oncol. 2005;27(2):317–25. [PubMed] [Google Scholar]
- 60.Garcia de Palazzo IE, et al. Expression of mutated epidermal growth factor receptor by non-small cell lung carcinomas. Cancer Res. 1993;53(14):3217–20. [PubMed] [Google Scholar]
- 61.Schumacher T, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512(7514):324–7. doi: 10.1038/nature13387. [DOI] [PubMed] [Google Scholar]
- 62.Yan H, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360(8):765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Turkalp Z, Karamchandani J, Das S. IDH mutation in glioma: new insights and promises for the future. JAMA Neurol. 2014;71(10):1319–25. doi: 10.1001/jamaneurol.2014.1205. [DOI] [PubMed] [Google Scholar]
- 64.Mitchell DA, et al. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008;10(1):10–8. doi: 10.1215/15228517-2007-035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cobbs CS, et al. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002;62(12):3347–50. [PubMed] [Google Scholar]
- 66.Sampson JH, Mitchell DA. Vaccination strategies for neuro-oncology. Neuro Oncol. 2015;17(Suppl 7):vii15–vii25. doi: 10.1093/neuonc/nov159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fuji S, et al. Adoptive immunotherapy with virus-specific T cells. Best Pract Res Clin Haematol. 2011;24(3):413–9. doi: 10.1016/j.beha.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 68.Riddell SR, Greenberg PD. Cellular adoptive immunotherapy after bone marrow transplantation. Cancer Treat Res. 1995;76:337–69. doi: 10.1007/978-1-4615-2013-9_16. [DOI] [PubMed] [Google Scholar]
- 69.Zeppernick F, et al. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin Cancer Res. 2008;14(1):123–9. doi: 10.1158/1078-0432.CCR-07-0932. [DOI] [PubMed] [Google Scholar]
- 70.Gaedicke S, et al. Noninvasive positron emission tomography and fluorescence imaging of CD133+ tumor stem cells. Proc Natl Acad Sci U S A. 2014;111(6):E692–701. doi: 10.1073/pnas.1314189111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Garg AD, et al. Dendritic cell vaccines based on immunogenic cell death elicit danger signals and T cell-driven rejection of high-grade glioma. Sci Transl Med. 2016;8(328):328ra27. doi: 10.1126/scitranslmed.aae0105. [DOI] [PubMed] [Google Scholar]
- 72*.Hdeib A, Sloan AE. Dendritic cell immunotherapy for solid tumors: evaluation of the DCVax(R) platform in the treatment of glioblastoma multiforme. CNS Oncol. 2015;4(2):63–9. doi: 10.2217/cns.14.54. Summary of the clinical experience with the DCVax platform with a focus on GBM treatment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Polyzoidis S, Ashkan K. DCVax(R)-L–developed by Northwest Biotherapeutics. Hum Vaccin Immunother. 2014;10(11):3139–45. doi: 10.4161/hv.29276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bosch M, Prins R, Liau L. Abstract 2491: Treatment with tumor lysate-pulsed autologous dendritic cells prolongs survival in patients with recurrent glioblastoma multiforme. Cancer Res. 2015;(75):2491. [Google Scholar]
- 75.Kang TH, et al. Enhancement of dendritic cell-based vaccine potency by targeting antigen to endosomal/lysosomal compartments. Immunol Lett. 2006;106(2):126–34. doi: 10.1016/j.imlet.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 76.Boudreau JE, et al. Engineering dendritic cells to enhance cancer immunotherapy. Mol Ther. 2011;19(5):841–53. doi: 10.1038/mt.2011.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shurin MR, et al. Genetically modified dendritic cells in cancer immunotherapy: a better tomorrow? Expert Opin Biol Ther. 2010;10(11):1539–53. doi: 10.1517/14712598.2010.526105. [DOI] [PubMed] [Google Scholar]
- 78.Wakefield A, et al. Is CMV a target in pediatric glioblastoma? Expression of CMV proteins, 65 and IE1 72 and CMV nucleic acids in a cohort of pediatric glioblastoma patients. J Neurooncol. 2015;125(2):307–15. doi: 10.1007/s11060-015-1905-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scheurer ME, et al. Detection of human cytomegalovirus in different histological types of gliomas. Acta Neuropathol. 2008;116(1):79–86. doi: 10.1007/s00401-008-0359-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.De Vries IJ, et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003;63(1):12–7. [PubMed] [Google Scholar]
- 81.MartIn-Fontecha A, et al. Regulation of dendritic cell migration to the draining lymph node: impact on T lymphocyte traffic and priming. J Exp Med. 2003;198(4):615–21. doi: 10.1084/jem.20030448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Radford KJ, Tullett KM, Lahoud MH. Dendritic cells and cancer immunotherapy. Curr Opin Immunol. 2014;27:26–32. doi: 10.1016/j.coi.2014.01.005. [DOI] [PubMed] [Google Scholar]
- 83.Allan RS, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25(1):153–62. doi: 10.1016/j.immuni.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 84.Bonifaz LC, et al. In vivo targeting of antigens to maturing dendritic cells via the DEC-205 receptor improves T cell vaccination. J Exp Med. 2004;199(6):815–24. doi: 10.1084/jem.20032220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mahnke K, et al. Targeting of antigens to activated dendritic cells in vivo cures metastatic melanoma in mice. Cancer Res. 2005;65(15):7007–12. doi: 10.1158/0008-5472.CAN-05-0938. [DOI] [PubMed] [Google Scholar]
- 86.Johnson TS, et al. Inhibition of melanoma growth by targeting of antigen to dendritic cells via an anti-DEC-205 single-chain fragment variable molecule. Clin Cancer Res. 2008;14(24):8169–77. doi: 10.1158/1078-0432.CCR-08-1474. [DOI] [PubMed] [Google Scholar]
- 87**.Dhodapkar MV, et al. Induction of antigen-specific immunity with a vaccine targeting NY-ESO-1 to the dendritic cell receptor DEC-205. Sci Transl Med. 2014;6(232):232ra51. doi: 10.1126/scitranslmed.3008068. First-in-human study of a protein vaccine targeting DCs in-vivo demonstrates safety and activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sampson JH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13(3):324–33. doi: 10.1093/neuonc/noq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ishikawa E, et al. Phase I/IIa trial of fractionated radiotherapy, temozolomide, and autologous formalin-fixed tumor vaccine for newly diagnosed glioblastoma. J Neurosurg. 2014;121(3):543–53. doi: 10.3171/2014.5.JNS132392. [DOI] [PubMed] [Google Scholar]
- 90.Arens R, Schoenberger SP. Plasticity in programming of effector and memory CD8 T-cell formation. Immunol Rev. 2010;235(1):190–205. doi: 10.1111/j.0105-2896.2010.00899.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nurieva R, Wang J, Sahoo A. T-cell tolerance in cancer. Immunotherapy. 2013;5(5):513–31. doi: 10.2217/imt.13.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yamamoto R, et al. PD-1-PD-1 ligand interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood. 2008;111(6):3220–4. doi: 10.1182/blood-2007-05-085159. [DOI] [PubMed] [Google Scholar]
- 93.Agaugue S, Carosella ED, Rouas-Freiss N. Role of HLA-G in tumor escape through expansion of myeloid-derived suppressor cells and cytokinic balance in favor of Th2 versus Th1/Th17. Blood. 2011;117(26):7021–31. doi: 10.1182/blood-2010-07-294389. [DOI] [PubMed] [Google Scholar]
- 94.Derre L, et al. Expression and release of HLA-E by melanoma cells and melanocytes: potential impact on the response of cytotoxic effector cells. J Immunol. 2006;177(5):3100–7. doi: 10.4049/jimmunol.177.5.3100. [DOI] [PubMed] [Google Scholar]
- 95.Marincola FM, et al. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol. 2000;74:181–273. doi: 10.1016/s0065-2776(08)60911-6. [DOI] [PubMed] [Google Scholar]
- 96.Okudaira K, et al. Blockade of B7-H1 or B7-DC induces an anti-tumor effect in a mouse pancreatic cancer model. Int J Oncol. 2009;35(4):741–9. doi: 10.3892/ijo_00000387. [DOI] [PubMed] [Google Scholar]
- 97.Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat Rev Immunol. 2004;4(12):941–52. doi: 10.1038/nri1498. [DOI] [PubMed] [Google Scholar]
- 98.Ruffell B, Affara I, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012;33(3):119–26. doi: 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–64. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li MO, et al. Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol. 2006;24:99–146. doi: 10.1146/annurev.immunol.24.021605.090737. [DOI] [PubMed] [Google Scholar]
- 101.Gerlini G, et al. Metastatic melanoma secreted IL-10 down-regulates CD1 molecules on dendritic cells in metastatic tumor lesions. Am J Pathol. 2004;165(6):1853–63. doi: 10.1016/S0002-9440(10)63238-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gabrilovich DI, et al. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat Med. 1996;2(10):1096–103. doi: 10.1038/nm1096-1096. [DOI] [PubMed] [Google Scholar]
- 103.Shields JD, et al. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science. 2010;328(5979):749–52. doi: 10.1126/science.1185837. [DOI] [PubMed] [Google Scholar]
- 104.Wei J, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010;9(1):67–78. doi: 10.1158/1535-7163.MCT-09-0734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wainwright DA, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18(22):6110–21. doi: 10.1158/1078-0432.CCR-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36(4):265–76. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nguyen LT, Ohashi PS. Clinical blockade of D1 and LAG3–potential mechanisms of action. Nat Rev Immunol. 2015;15(1):45–56. doi: 10.1038/nri3790. [DOI] [PubMed] [Google Scholar]