

Abstract
Protein-protein interfaces and architecture are critical to the function of multiprotein complexes. Mass spectrometry-based techniques have emerged as powerful strategies for characterization of protein complexes, particularly for heterogeneous mixtures of structures. In the present study, activation and dissociation of three tetrameric protein complexes (streptavidin, transthyretin, and hemoglobin) in the gas phase was undertaken by 193 nm ultraviolet photodissociation (UVPD) for the characterization of higher order structure. High pulse energy UVPD resulted in the production of dimers and low charged monomers exhibiting symmetrical charge partitioning among the sub-units (the so-called symmetrical dissociation pathways), consistent with the subunit organization of the complexes. In addition, UVPD promoted backbone cleavages of the monomeric subunits, the abundances of which corresponded to the more flexible loop regions of the proteins.
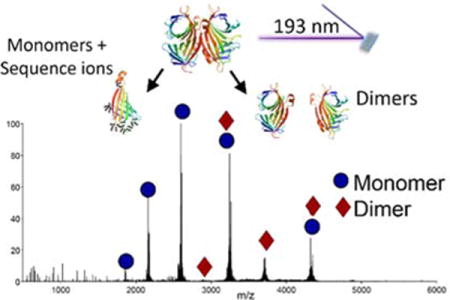
Introduction
Quaternary structures of proteins are mediated by non-covalent interactions that are responsible for modulating biological function and activity in cellular environments.1 Protein-protein complexes vary in size, complexity, and architecture, ranging from simple and symmetrical homodimers to single and double ring structures, to large heterocomplexes with little symmetry.2 The development of electrospray ionization and particularly native electrospray ionization has permitted transfer of multiprotein complexes into the gas phase and has consequently transformed the ability to examine protein complexes by mass spectrometry.3–6 Characterization of the assembly and architecture of multiprotein complexes via their solution and gas-phase disassembly pathways has gained momentum as a viable approach in structural biology.7–9 Dissociation of protein complexes in a manner that reflects the organization of the multi-protein complex, however, has proved challenging. Conventional collision induced dissociation (CID) typically results in the unfolding and subsequent ejection of a highly charged monomer, regardless of the structure of the multimer.7,10 This type of dissociation, termed asymmetric dissociation owing to the asymmetric charge partitioning between the ejected monomer and the rest of the complex, is a consequence of the numerous low energy collisions imparted by CID and is useful for determining stoichiometry of the complex but is generally uninformative regarding the quaternary structure.
In order to access higher energy dissociation pathways, the Wysocki group has developed surface induced dissociation, SID, as a promising alternative for characterization of multiprotein complexes.11,12 Using a variety of protein complexes of known subunit architecture, Wysocki and co-workers have successfully demonstrated that the higher energy, single collisions afforded by acceleration of a precursor into a target of infinite mass provides access to higher energy dissociation pathways.13,14 Higher, single-step energy deposition upon SID results in products that reflect both asymmetric and symmetric charge partitioning, the latter of which are consistent with the assembly of the protein complex.15–20 Reduction of the charge state of the precursor was shown to decrease unfolding of the proteins and resulted exclusively in observation of symmetric charge partitioning pathways consistent with the quaternary structures.17 Complexes in higher charge states (supercharged) were more likely to unfold and exhibited less preservation of higher order structure. This phenomenon was rationalized based on ions in higher charge states having lower stabilities and greater propensity to unravel in the gas phase compared to ions in lower charge states.17 More recently, Loo et al. examined CID and SID data and proposed that scrambling and heterolytic cleavage of interfacial salt bridges rather than subunit unfolding could account for asymmetric charge partitioning.21
Charge modulation has also emerged as a common strategy to alter collisional activated dissociation pathways of multiprotein complexes in the gas phase.17,22,23 Symmetric charge partitioning dissociation pathways of protein complexes have been observed in some cases following collisional activation and has motivated extensive study into the mechanisms that govern charge partitioning in protein complexes.22,24 The Williams and Robinson groups have explored charge partitioning as a function of charge state, using charge reducing and super-charging reagents to modulate the charge state envelope of the precursor multimer.23,25 Both groups have observed symmetric charge partitioning following CID of protein complexes in charge states greater than those found upon ESI from buffered solutions and asymmetric charge partitioning for proteins in normal and charged-reduced charge states, leading both groups to suggest that charge density dictates the observed dissociation routes to some extent.23,25 Upon examination of a series of complexes, it was found that symmetrical dissociation of supercharged complexes tended to occur for complexes having few interfacial salt bridges, small interfaces, and low subunit flexibility.25 Based on observed symmetric versus asymmetric dissociation in oxidized and reduced forms of α-lactalbumin and with supporting evidence derived from the fragmentation of cross-linked cytochrome C, subunit flexibility was also suggested to influence charge partitioning.23,26
Alternate methods for dissociation of protein complexes have recently received attention, including electron- and photon-based approaches. In contrast to collisional methods, electron capture dissociation does not cause disassembly of the non-covalently bound subunits and consequently the approach is more frequently used to examine the exposed and flexible regions of the protein complexes, from which cleavage of covalent bonds of the backbone is observed.27–29 Our group has recently shown that 193 nm ultraviolet photodissociation (UVPD) of protein-ligand complexes results in the production of both backbone and non-covalent fragment ions that reveal insight into the binding sites of the ligands and conformational changes as a function of ligand binding.30–32 In this study, we examine the 193 nm UVPD fragmentation pathways of two homotetrameric protein complexes, streptavidin and transthyretin, and a heterotetramer, hemoglobin, each having a dimer-of-dimers topology. We present evidence that UVPD provides a new means to characterize quaternary structures of protein complexes. In addition, sequence ions originating from backbone cleavages of the protein are observed, higher abundances of which correspond to the more flexible regions of the protein higher order structure.
Materials and Methods
Streptavidin (SA) from Streptomyces avidinii, recombinant human transthyretin (TTR), human hemoglobin (HG), ammonium acetate, triethylammonium acetate (TEAA), and m-nitrobenzyl alcohol (m-NBA) were obtained from Sigma-Aldrich (St. Louis, MO). Proteins were buffer exchanged into 100 mM ammonium acetate using BioRad micro P-6 spin columns (Hercules, CA) to a final tetramer concentration of approximately 10 uM prior to analysis by mass spectrometry. SA was sprayed from denaturing conditions and was found to be comprised of several proteoforms. Two of these are dominant and differ by the presence or absence of the N-terminal alanine residue (a nominal mass difference of 71 Da). Both proteoforms were terminated by Ser123/124. The relative abundances of the two proteoforms were observed to be follow a randomized probability distribution in the dimer products following dissociation of the tetrameric precursor; consequently, each proteoform was not treated separately, and the tabulated values (fractions of the an population in a particular charge state, described later in the results section) were calculated using the combined ion signal for both proteoforms.
All mass spectrometry experiments were performed on a Thermo Scientific Orbitrap Elite mass spectrometer (San Jose, CA) coupled to a Coherent 193 nm Existar XS laser (Santa Clara, CA) as described previously.33 Proteins were introduced to the gas-phase by application of 0.9–1.5 kV to a platinum wire that was inserted into a glass capillary pulled to a tip of approximately 1 micron. The electrosprayed ions were transmitted through the source optics of the Orbitrap using as gentle conditions as possible and were accumulated in the high pressure region of the dual linear ion trap for a maximum of 200 ms prior to irradiation with the UV laser. Typical potential differences across the source region were 4 V or less between adjacent ion optics. More energetic source tuning and ion transfer conditions were observed to change the resulting fragmentation patterns of the tetramers such that more highly charged monomers were generated. We attributed this to protein unfolding/restructuring occurring in the source and during ion transfer akin to what has been shown with source activation for ECD and SID.34–36 Source parameters were consequently selected to minimize the possibility of unfolding. UVPD was performed using a single pulse of the laser, the energy of which was varied from 0.5 mJ to 3.0 mJ. As the laser energy increased, the probability of multiphoton excitation increased. Because the laser beam is unfocused, divergent, and passes through two apertures that skim the beam, the exact photon flux at each laser power is difficult to measure in the HCD cell but is estimated to be in the tens to hundreds of uJ depending on the selected pulse energy. HCD was performed using a normalized collision energy of 1 to 55 NCE, which corresponded to 5–80 eV (lab frame) depending on the charge state of the activated species. The lab frame is used throughout this study to facilitate comparisons. HCD collision energies were selected that resulted in product distributions that best mirrored the patterns observed by 1 mJ UVPD. MS1 and MS/MS spectra were acquired in the Orbitrap mass analyzer at a resolution of 240,000 or 480,000 for energy resolved experiments and backbone cleavage (sequence ion) analysis, respectively. Typically 15 uscans were collected per scan, and between 10 and 50 scans were averaged for each spectrum. Relative abundances of overlapping species were calculated for the energy-resolved experiments using the relative abundances of the overlapping isotope distributions, given that an ion with equal m/z but with twice or three times the charge has isotopes spaced more closely. The overlapping distribution was integrated and the contribution of the species calculated using the ratio of the five most abundant isotopes such that, for example, the isotope distribution of a singly charged monomer with twice the abundance of a doubly charged dimer represented a 50:50 mixture of the two species. Weighted average charge states were calculated for the observed charge states of the monomers and trimers (3+ to 13+) by: , where Ai is the abundance of a particular charge state of the trimer and Atrimer was the total abundance of the trimer population. Interfacial areas were calculated using the PISA tool in the European Protein Data Bank, and solvent accessible surface areas (SASA) were calculated using the POPs webserver.
Results and Discussion
Disruption of non-covalent interactions
Streptavidin (SA), transthyretin (TTR), and hemoglobin (HG) were selected as model tetrameric proteins to investigate the 193 nm UVPD dissociation pathways of the multiprotein complexes into their protein subunits. SA and TTR have been studied extensively by both SID and CID and feature a dimer-of-dimers topology from which it is possible to assess the degree to which the observed dissociation patterns reflect the assembly of the complexes.11,20,25,28 Hemoglobin is a heterotetramer comprised of two alpha and two beta subunits, the arrangement of which forms a dimer-of-dimer topology; however, HG features substantially different interfaces than do TTR and SA, having both lower interfacial areas and fewer intersubunit hydrogen bonds and salt bridges (corresponding to weaker binding energies). Mass spectra of SA, TTR, and HG in 150 mM ammonium acetate are shown in Figure S1, and the multimer distributions are consistent with those reported previously. The fragmentation patterns of the SA, TTR, and HG tetramers obtained by HCD and UVPD were examined in the present study as a function of charge state and energy deposition.
Streptavidin (SA)
In Figure 1, HCD spectra (left column) and UVPD spectra (1 mJ and 3 mJ, middle and right columns, respectively) are shown for two charge states (13+ and 15+) of apo-SA (net MW of tetramer is 52 kDa). Lower charge states were not evaluated due to the limitations in transmission of ions larger than m/z 4000. Activation of the 13+ charge state of SA by HCD and 1.0 mJ UVPD (Figure 1a and 1b) produced similar dissociation patterns, in which asymmetric charge-partitioning dissociation pathways were dominant and the most abundant products were the 6+ and 7+ monomers. This behavior is characteristic of unfolding and ejection of a highly charged monomer from the tetramer and yields little insight into sub-unit organization or higher order structure. In contrast, activation using a higher UVPD pulse energy of 3.0 mJ produced lower charge state monomers, dimers, a small population of trimers, and numerous fragment ions resulting from cleavage of the protein backbone (sequence type ions) (Figure 1c). This distribution of product ions is extremely similar to the SID spectrum reported for SA (11+ charge state).20 Comparison of the spectra collected for SA (13+) using 1.0 and 3.0 mJ UVPD pulse energies suggests that exposing the tetramers to a higher photon density, which consequently increases the probability of multiphoton absorption, may provide simultaneous access to symmetric charge partitioning pathways from disruption of the non-covalent interactions of the complexes and cleavage of the covalent bonds of the protein backbone to generate sequence ions. The symmetric charge partitioning pathways, and in particular the release of dimers, reveals substructure connectivity information. Activation of the 15+ charge state by 1.0 mJ and 3.0 mJ UVPD (Figure 1e,f) resulted in similar fragmentation patterns to that observed for the 13+ charge state, with the low energy UVPD favoring asymmetric charge partitioning of the protein subunits (via unfolding) similar to that observed upon CID, and high energy UVPD promoting symmetrical charge partitioning (reflecting higher order structural organization) in addition to cleavage of the protein backbone to yield diagnostic sequence ions. Use of higher laser energies thus favors the symmetric charge partitioning dissociation pathways, presumably due to the absorption of multiple photons.
Figure 1.

HCD and UVPD of 13+ and 15+ tetrameric streptavidin. In a) and d) HCD of the 13+ and 15+ charge states is shown, respectively. In b) and e) 1.0 mJ UVPD of the 13+ and 15+ charge states is shown and in c) and f) 3.0 mJ UVPD of the 13+ and 15+ charge states is shown. The bracket in c) denotes the region populated by fragments originating from cleavages of the protein backbone (i.e. sequence-type ions).
Figure 2a and 2b show energy-resolved mass spectrometry (ERMS) plots of the dissociation of the 13+ and 15+ charge states as a function of UVPD laser pulse energy, from which it is clear that the production of dimeric species is enhanced at the highest UVPD laser powers. Note that analogous HCD ERMS plots are shown in Figure S2. The low charged monomers (3+, 4+, and 5+) were found to track with the dimeric product ions, featuring a similar or identical onset energy and similar changes in relative abundance. The near identical onset energy for these two fragment types (low charge state monomers and dimers) suggests that the low charged monomers arise not from sequential dissociation of the dimers but from direct fragmentation of the tetrameric precursors (i.e. both the low charged monomers and the dimers arise from the same precursor population). For both the 13+ and 15+ charge states, the reduction in the relative abundances of highly charged monomers correlates with an increase in the fragments arising from backbone cleavages (sequence ions), implying that the latter products observed at high UVPD pulse energies originate from secondary dissociation of the highly charged monomers. This observation raises an interesting question as to whether the decrease in the average charge state of the monomer products as a function of UVPD energy is the result of the high laser energy providing access to symmetrical charge partitioning pathways or merely depletion of the highly charged monomer (via secondary dissociation to other products) and is discussed in more detail for TTR in the next section.
Figure 2.
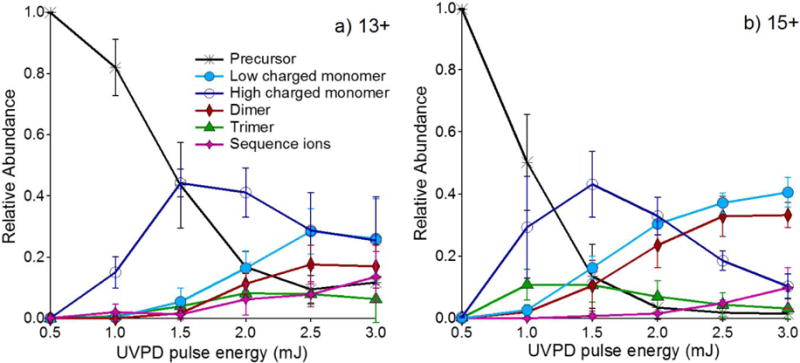
UVPD ERMS plots of the a) 13+ and b) 15+ charge states of tetrameric streptavidin. High charged monomers included charge states 6+ and higher, and low charged monomers included charge states 5+ and lower.
Transthyretin (TTR)
Transthyretin (TTR) was examined by 193 nm UVPD to determine if the general UVPD fragmentation trends observed for streptavidin were reproduced for other complexes with dimer-of-dimer topologies. Akin to streptavidin, SID of TTR generates a mixture of oligomeric products, including abundant dimers.20 Spraying from 150 mM ammonium acetate predominantly generated the 14+ and 15+ charge states (Figure S1b). UVPD was performed on the 15+ charge state using a range of pulse energies, and low (1.0 mJ) and high (3.0 mJ) pulse energy UVPD mass spectra are shown in Figure 3. Like streptavidin, low pulse energy UVPD of the 15+ charge state resulted in CID-like dissociation patterns, with highly charged monomers and trimers being the only observed products. Higher pulse energy UVPD of the 15+ charge state resulted in the formation of dimers and an increase in the relative abundance of low charged monomers. In contrast to SA, for which trimers were observed in low abundance and typically at lower UVPD pulse energies (the exception being the 13+ charge state), trimers were observed for TTR upon both low and high energy UVPD. The higher abundances of the trimeric species at almost all UVPD pulse energies provides an opportunity to better understand the dissociation pathways of tetrameric complexes by UVPD and are discussed in more detail below.
Figure 3.
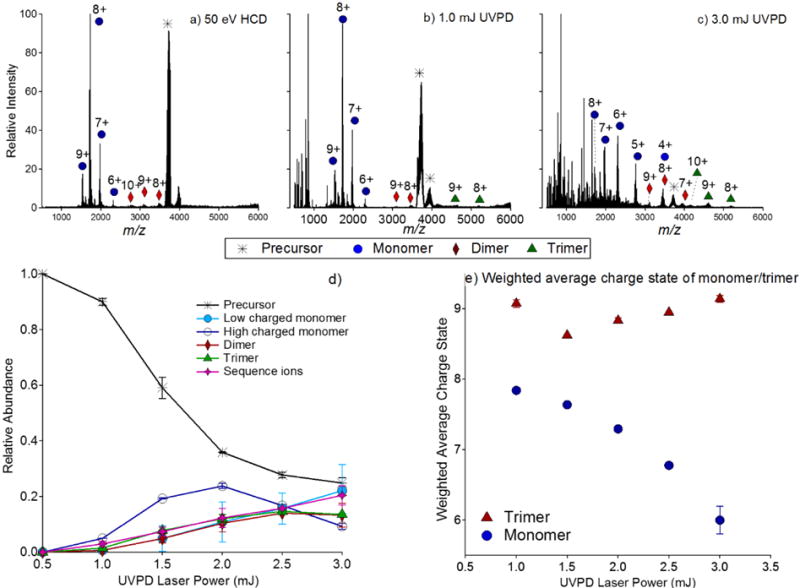
HCD and UVPD of tetrameric transthyretin (15+); in a) HCD (50 eV) is shown, in b) 1.0 mJ UVPD is shown, and in c) 3.0 mJ UVPD is shown. UVPD ERMS plot of tetrameric transthyretin (15+) is shown in d) and the weighted average charge states of the trimer and monomer products upon UVPD are shown in e). In d), high charged monomer includes charge states 7+ and higher, and low charged monomer includes charge states below 7+.
From the ERMS plots of SA it was surmised that ejection of low charged monomers (5+ charge state and lower) occurred concomitantly with ejection of dimers upon high pulse energy UVPD, and we postulated above that direct ejection of low charged, presumably folded, monomers and dimers occurred from the tetrameric complexes. This assertion was further evaluated by comparison of the fragmentation behavior of TTR. In Figure 3d, an ERMS plot as a function of UVPD laser energy is shown for the 15+ charge state of tetrameric TTR. Highly charged monomers and trimers exhibit low onset energies (≈0.5–1.0 mJ), and low charged monomers and dimers display slightly higher onset energies (≈0.5–1.5 mJ). The relative abundances of the low charged monomers and dimers are observed to track together when produced from the 15+ charge state. In contrast to the ERMS plots of SA, trimeric products are relatively abundant. This permits relatively accurate measurement of the weighted average charge state of the trimeric product ions for all but the lowest UVPD pulse energies.
From the analysis of SA described above, two possible explanations were proposed for the change in the average charge state of the monomers as a function of UVPD power. Depletion of the higher charged monomers by secondary backbone cleavages (creating sequence-type ions) may result in an observed increase in the relative abundance of the non-dissociating species, including low charged monomers. Alternatively, higher pulse energy (multiphoton) UVPD may allow access to higher energy pathways that result in the direct dissociation of compact monomers and dimers. Ejection of a low charged monomer directly from the tetramer, carrying approximately a quarter of the charge, is expected to produce a complementary trimer with three-quarters of the remaining charge. Thus, if monomer charge state varies as a function of accessible dissociation pathways rather than subsequent cleavage of the protein backbone of highly charged monomers, the average charge state of the trimers is also expected to vary as a function of laser power. In Figure 3e, the weighted average charge states of the monomers and trimers produced from UVPD of tetrameric TTR (15+) are plotted as a function of UVPD laser power, and it is clear that average trimer charge state increases with laser power. The decrease in monomer charge state is greater than the corresponding increase for the trimers, suggesting that monomer dissociation does contribute to the apparent reduction in monomer charge state; however, the increase in the average charge state confirms that the shift in product ion distribution observed as a function of laser power is the consequence of higher laser powers permitting access to higher energy dissociation pathways having symmetric charge partitioning.
Hemoglobin (HG)
Human hemoglobin is a heterotetramer that also features a dimer-of-dimer topology; however, the secondary structure and interfacial characteristics of human hemoglobin are different from those of TTR and SA. The native mass spectrum of HG is shown in Figure S1. The 16+ to 18+ charge states of the tetramer were observed upon electrospray from an ammonium acetate solution. In Figure 4a to 4c, HCD, 1 mJ UVPD, and 3 mJ UVPD spectra are shown for hemoglobin (17+). HCD of hemoglobin produced a mixture of monomers ranging from the 8+ to 10+ charge states and of which about half retained the heme group. A previous report of CID of tetrameric hemoglobin demonstrated preferential ejection of a holo or apo alpha subunit and a complementary αβ2 trimer containing two to four heme groups.37 In contrast to CID, 1 mJ and 3 mJ UVPD resulted in the production of monomers (in 4+ to 9+ charge states) with the relative abundance of apo and holo species varying as a function of laser energy. While some low abundance trimer products were observed and identified, many of the low abundance ions in the region between m/z 4000 and 5000 could not be unambiguously assigned due to overlap in the predicted m/z values of αβ2 and α2β trimers with variable numbers of heme groups. Dimer species were therefore not explicitly identified in this region; however, while it is possible that they were present in low abundance, the absence of distinguishable dimers in the 9+ charge state (m/z 3580) makes this rather unlikely. In Figure 4d, retention of heme is shown as a function of laser power, with the fraction of the holo form of the alpha and beta subunits plotted in red and blue, respectively. Retention of heme decreases linearly with increasing laser fluence and is generally higher for the beta subunit. Interestingly, the relative abundance of holo fragments was higher for the more highly charged monomers, which may result from charge reduction upon loss of a protonated heme group.
Figure 4.
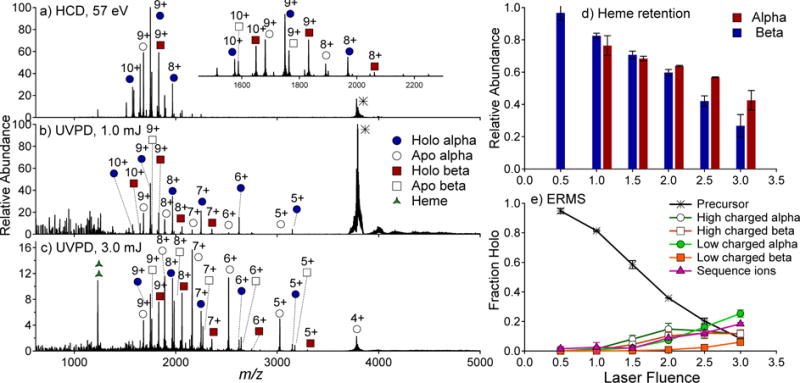
HCD (a), 1 mJ UVPD (b), and 3 mJ UVPD (c) mass spectra of the 17+ charge state of tetrameric HG. The relative abundance of holo monomers are shown in d) as a function of laser power. In e) a UVPD-ERMS plot is shown for the production of high charged (8+ to 10+) monomers, low charged monomers (4+ to 7+), and backbone cleavage products (sequence ions) of HG (17+) as a function of laser power. Trimer ions are not shown and (subunit) dimers were not observed as products.
The distributions of the charge states of the monomer products are examined as a function of laser power in Figure 4e for tetrameric HG, in which a UVPD-ERMS plot is shown for high charged monomers (8+ to 10+ charge states, as was observed by HCD) and low charged monomers (4+ to 7+) and backbone cleavage fragments (sequence ions). Depletion of the precursor featured a near linear trend with only mild sigmoidal character. This behavior contrasts the ERMS trends for TTR and SA, for which highly sigmoidal curves were observed. UVPD of peptides has been shown to result from single photon absorption, and linear depletion of the precursor as a function of laser fluence has been used to evaluate single versus multiphoton absorption.38 Inspection of the non-linear precursor depletion curves in Figures 2 and 3 implicates multiphoton absorption in UVPD of SA and TTR. In contrast, the linearity of the depletion of tetrameric HG (17+) may be a consequence of single-photon or nearly single-photon UVPD for this system. Sigmoidal character of precursor depletion curves may thus be related to the interfacial characteristics of multiprotein complexes.
Consideration of the structural differences between HG and SA/TTR is necessary to understand the differential UVPD dissociation observed. Although a previous study of holo and apo myoglobin using 193 nm UVPD showed some variations in fragmentation of holo and apo myoglobin along the backbone, global fragmentation yield was largely the same.32 It seems unlikely that the heme moieties solely account for the different UVPD dissociation pathways observed for HG versus SA and TTR tetramers. HG has a dimer-of-dimers topology with each monomer in contact with all three other subunits. In contrast to SA and TTR, the relative interfacial area of the stronger AB interface (877 Å2) is not considerably higher than the weaker BA or AA/BB interfaces (429 and 397/150 Å2, respectively). The Wysocki group has shown that interfacial area provides the best predictor of SID dissociation pathways and these same arguments can be invoked to some extent here.19,20 Ejection of an alpha or beta subunit from HG requires disrupting one AB interface (877 Å2), one BA (429 Å2) interface, and either an AA or BB interface (397 and 150 Å2, respectively), requiring cleavage of 1456 or 1703 Å2 interfacial area, two salt bridges, and ≈16 hydrogen bonds. In contrast, disruption of the dimer-dimer interface requires disruption of 1405 Å2 interfacial area and cleavage of two salt bridges and 13 hydrogen bonds. Given that the dimer abundance at its maximum only contributed 20–40% of the total product ion current for SA and TTR, which feature subunit interfaces with high and low interfacial areas differing by a much greater degree such that ejection of a monomer versus dimer requires cleavage of approximately 1.5–2 fold the interfacial area, it is likely that UVPD (through dimer production) cannot differentiate interfaces of the degree of similarity exhibited by hemoglobin.
In addition to interfacial strength, the more numerous hydrogen bonds that must be broken to produce monomers in SA and TTR (24 and 27, respectively) relative to HG (16) may explain why more photons appear to be needed for subunit dissociation in SA and TTR and fewer photons are seemingly needed for subunit dissociation of HG. The weakness of the HG interfaces may permit rapid ejection of subunits without preceding unfolding by asymmetric charge partitioning. This concept is illustrated by examination of the average charge state of monomer produced upon HCD and 1.0 mJ UVPD of hemoglobin (Figure 4). HCD of hemoglobin (17+) produces a distribution of alpha and beta monomers centered around the 9+ charge state. In contrast, 1.0 mJ UVPD produces a lower charged distribution in addition to one centered around the 9+ charge state, suggesting that symmetric dissociation pathways may be accessed at lower onset energies for those complexes having weaker interfaces. Thus, although the dimer-of-dimer topology was not represented in the UVPD dissociation of tetrameric hemoglobin through the production of dimeric fragments, the relationship between the number of absorbed photons and interfacial strength may provide an additional metric with which it is possible to extract biologically relevant information regarding multi-protein complex architecture from UVPD data. A systematic study of dimeric proteins is needed better to understand the relationship between precursor depletion and interfacial strength.
Backbone fragmentation of SA, TTR, and HG
Observation of products arising from both cleavage of backbone bonds (sequence ions) and disruption of non-covalent interactions of multiprotein complexes (production of monomers, dimers, trimers from tetramers) is a unique characteristic of UVPD, and results in simultaneous access to substructure connectivity and extensive sequence coverage that offers enormous potential for extracting structure information from protein complexes. Collisional activation approaches typically result in disruption of non-covalent interactions and when backbone cleavage products (sequence ions) are observed, the sequence coverage is limited.39 Electron-based approaches yield extensive backbone cleavage products (c and z ions) that characterize the protein sequence and have been shown to map onto the more surface-exposed regions of native proteins or complexes.27–29 Hence, the UVPD product ions from backbone cleavages of SA, TTR, and HG were examined in detail in order to better understand the origin of these fragments in the context of the subunit dissociation. A diverse array of product ions resulting from backbone cleavages is generated upon UVPD of tetrameric SA, and Figure S3 shows a UVPD mass spectrum collected using a single 3.0 mJ laser pulse to dissociate the 15+ charge state of SA. The inset displays the complexity of the fragmentation. Products arising from cleavage of the protein backbone can conceivably occur from one of three sources: directly from the tetrameric precursors, or via secondary dissociation of the folded dimeric or monomeric products, or via secondary dissociation of the unfolded monomers released by a typical CID-like pathway. We have previously demonstrated that it is possible to assign the charge sites of native-like protein ions by tabulating the relative abundances of the charge states of the a and x ion product ions.40 Using this strategy it was determined that 1) the charge state of the fragmenting protein can be deduced by identifying and tallying all charge sites, and 2) that charge site heterogeneity correlates with protein unfolding.40 Thus, we postulated that a similar charge site analysis of the products arising from backbone dissociation of tetrameric SA and TTR could be used to assess the fragmentation pathway(s) by which these products are generated from 193 nm UVPD of multi-protein complexes.
The fraction of the an (or xn) population in a particular charge state is given by: , where , and p+, q+, and r+ are the observed charge states of a given a ion an and the abundance of a given charge state of that specific a ion is . In Figure S4, and values were calculated and are displayed as segmented histograms for the observed a/x ion fragments arising from 3.0 mJ UVPD of the 15+ charge state of SA. In brief, the different colors in the histograms indicate the charge states of the observed a and x ions, and bars that are segmented into two or more colors convey heterogeneous populations of charge states (with the sizes of the segments reflecting the portion in each charge state). Thus, mapping the charge states of the complementary a and x ions offers a convenient way to estimate the locations of charge sites of the protein.40
From the bisigmoidal transitions in fragment ion charge sites, evidenced by 10–20% of the a-ion population featuring an increase in charge state at a47 (Arg47), a72 (Arg72), a91 (Arg91), and a109 (Lys109), two charge site populations are evident. Approximately 80–90% of the population features charge state transitions for the fragments in the range a62 to a67 (Gly62-Lys67), a81 (G81), and a102 (Thr102). The smallest a and x ions detected are a5 (Ile5) and x3 (Lys122), respectively, suggesting protonation sites are present for both charge site populations at the N-terminus and the most C-terminal lysine residue, respectively. Similarly, a single transition occurs between the singly and doubly charged a ions at approximately residue 30, which suggests that both charge site populations have a charge site at this location. Note that the singly charged a ion series was difficult to discern in the range of a20 – a40 due to the coalescence of singly charged ions into single, broad peaks, even at the 480k resolving power used for the collection of the spectra. Thus, assignment of the specific location of the second charge site is tentative at best. Nonetheless, the presence of this charge is apparent by the clear emergence of the doubly charged a ion series and suggests, in conjunction with the assignment of the other charge sites, that the more abundant population, evidenced by fragment charge state transitions at Gly62-Lys67, G81, and Thr102, is best categorized as a fragmenting monomer (6+). In contrast, the minor population evidenced by fragment ion charge state transitions at Arg47, Arg72, Arg91, and Lys109 originates from a 7+ monomer. The sequence of SA is shown in Figure S4a with the proposed charge sites highlighted in red and blue corresponding to the sites associated with the fragmenting 6+ and 7+ monomers. The primary difference between the plots is the relative abundance of the fragmenting 6+ and 7+ populations, contributing roughly 80% and 20%, respectively. It is useful to consider subunit dissociation in terms of the charge sites and the three-dimensional crystal structure. The charge sites of the 6+ ion population are highlighted in red in Figure S4b. Four of the six charges of the fragmenting 6+ monomer were consistent with being located in solvent exposed surface sites of the folded protein and the remaining two charges were localized to the strong interface of the protein. Given that the tetrameric precursor had a 15+ charge state, these sites would be consistent with an even distribution of charges of the intact protein. Strepavidin has three salt bridges along the strong interface and one intra-subunit salt bridge, highlighted in green in Figure S4b. Although none of the basic residues in these salt bridges were found to be protonated, asymmetric partitioning of the salt bridges could in the formation of a 6+ monomer that undergoes charge relocation to the interfacial region in response to coulombic interactions.21
Having established that 193 nm UVPD accesses higher energy dissociation pathways and that the backbone cleavage products likely arise from secondary dissociation of the most highly charged monomers, a question is raised as to whether the fragmenting monomers are folded or unfolded and consequently whether the sequence ions from backbone cleavages reflect the native structure of the protein. By the monomer unfolding model of CID, collisional activation typically results in the unfolding and ejection of highly charged monomers; for the 15+ tetramer shown in Figure 1a, this results in the formation of 6+, 7+, and 8+ monomers. Given that these monomers are the same charge states from which backbone cleavage products are observed upon UVPD, it seems reasonable to conclude that the sequence ions arise from secondary dissociation of the highly charged, unfolded monomers. However, it bears noting that several of the charge sites of the fragmenting 6+ monomer observed in Figure S4 are found to be non-basic residues located on the surface of the protein in the crystal structure. We have previously shown that localization of charges to non-basic sites occurs with some regularity in native proteins and that localization of protons at such sites tends to correlate with more folded structures, for which charge density at the surface of the protein is modulated by secondary and tertiary structural characteristics.40 Moreover, the timescales associated with protein unfolding and backbone cleavage by photolytic pathways are different; backbone cleavages may occur on the picosecond timescale while unfolding following vibrational energy redistribution may require nanoseconds or longer.41 Thus, it is not unreasonable that high pulse energy UVPD results in the formation of a charge envelope of monomers that either remain folded and/or fragment via backbone cleavage prior to undergoing unfolding.
To explore this notion, the abundances of the a and x fragment ions from 3.0 mJ UVPD of the 15+ SA tetramer were studied. The maps of the summed relative abundances of the a and x fragment ions produced from the 15+ tetramer (black trace)) are plotted along the protein sequence in Figure 5. The B-factors from the crystallography data and solvent accessible surface areas (SASA) for SA are shown in Figure 5a as a function of sequence position. B-factor is a crystallographic packing parameter and loosely reflects protein flexibility. Incidentally, because of the B-barrel tertiary structure of the protein, SASA is found to correspond closely with the B-factor trend, and both roughly correlate with fragmentation yield of the 15+ tetramer upon UVPD as evidenced by more abundant fragmentation in several loop regions of the protein (Figure 5b). The average fragment abundances were calculated, and regions featuring fragments with above average abundance, typically the loop regions, are highlighted in red in the crystal structure of SA shown in Figure 5c. We have previously observed increased UVPD fragmentation in the loop regions of monomeric native proteins,41 and the fragmentation from tetrameric SA is consistent with these observations.
Figure 5.
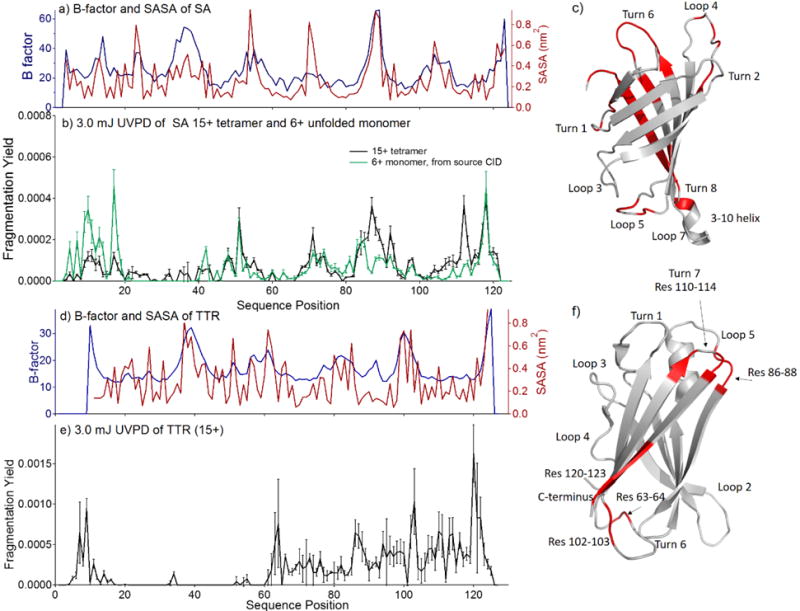
For streptavidin: a) B-factor values and solvent accessible surface areas (SASA), b) UVPD fragmentation yields (15+) and c) the crystal structure of SA (pdb 1SWB) is highlighted such that regions featuring enhanced UVPD fragmentation are shown in red. In b), the UVPD fragmentation yield of the 6+ monomer of SA, generated from source CID, is shown in green. For transthyretin: d) B-factor values and solvent accessible surface areas (SASA), e) UVPD fragmentation yields (15+), and f) the crystal structure of TTR (pdb 4TLT) is highlighted such that regions featuring enhanced UVPD fragmentation are shown in red.
Streptavidin is a beta barrel protein featuring eight loops/turns, spanning the following regions; turn 1: Tyr10 – Ser15, turn 2: Ala21 – Gly29, loop 3: Glu32 – Ser40, loop 4: Tyr48 – Gly58, loop 5: Ala66 – His75, turn 6: Val85 – Arg91, loop 7: Ser100 – Glu104, and turn 8 (Trp108 – Thr111). These regions are labeled on the crystal structure of SA (1SWB) in Figure 5c. Enhanced fragmentation was observed for backbone cleavage C-terminal to residues 9–16, 47–54, 68–74, and 83–93 which correlates well with the residues constituting loops/turns 1, 4, 5, and 6. UVPD fragmentation was suppressed from residues 20– 40, likely due to limitations in the transfer and detection efficiency of singly charged fragments of high m/z (m/z 2000–4000). Hence, fragmentation in this region, which would be expected to be enhanced along loop 3, spanning residues 32 to 40, is not observed, and is attributed to the poor transfer efficiency of the fragments. Enhanced UVPD fragmentation corresponding to loop 7 is also not evident; instead an increase in UVPD efficiency is observed between residues 103 and 110, which corresponds to the 3–10 helix and turn 8 of SA. Substantial fragmentation was also observed along the β-strand closest to the C-terminus (residues 110–123), which is not entirely surprising given the elevated B-factor in this region. Thus, although enhanced UVPD fragmentation was not observed for every loop of streptavidin, the regions displaying the most dominant fragmentation were found to be exclusively loop/turn regions and the C-terminal strand of the β-barrel, which is also more flexible according to the B-factors of the protein. A reduction in fragmentation efficiency along the interfacial region of SA was not observed; the strong interface of SA comprises the bulk of the C-terminal half of the protein (excluding the loop regions), and from examination of Figure 5 it is clear that fragmentation was prominent in these regions. It is possible that some structural differences exist between the gas-phase protein and crystal structure, which could explain some of the aforementioned deviations.
The fragmentation yield of the SA tetramer (15+) by UVPD was subsequently compared to the UVPD fragmentation yield of the 6+ monomer produced by collisional activation of the SA tetramer in the source region. The 6+ monomer was selected for this comparison because the dominant fragmenting species upon 3.0 mJ UVPD of the 15+ tetramer was attributed to a 6+ monomer. Because the 6+ monomer arises from a monomer ejection/unfolding CID pathway, the UVPD fragments from this experiment are expected to pertain to the unfolded monomer. The UVPD fragmentation yield of the 6+ monomer is shown in green in Figure 5b. Comparison of the variations in the abundances of a and x ions suggest that UVPD of tetrameric SA is sensitive to the structure of the protein and that some structural differences exist between the presumably unfolded monomer and tetramer. Interestingly, the regions in which the greatest differences in UVPD fragmentation yield are observed between the folded 15+ tetramer and presumably unfolded 6+ monomer are turn 1 (Tyr10–Ser15), turn 6 (Val85–Arg91), and loop 7 (Ser100– Glu104). Relative to the fragmentation of the tetramer, the fragmentation of the 6+ monomer is significantly increased across the first 20 residues of the protein, suggesting that the first strand of the beta barrel unfolds or frays upon collisional activation. A difference plot is shown in Figure S5 that highlights these differences. On the C-terminal side of the protein, suppressed fragmentation of the 6+ monomer is observed relative to the tetramer, particularly for residues 85 to 93 and 103 to 115. This could indicate unraveling and restructuring in this region. Nearly identical UVPD fragmentation efficiency is observed in the center of the protein sequence, including loops 4 and 5 and the connecting beta strand (residues 59-65). These differences confirm the sensitivity of UVPD to secondary structure, demonstrate that UVPD of the 15+ tetramer reflects more folded conformations, and suggests that source-CID of SA results in the ejection of a partially unfolded monomer that predominately unravels from the termini while retaining some of the central structure.
Transthyretin is a beta sandwich protein featuring seven loops/turns, spanning the following regions; turn 1: Asp18 – Ser23, an unstructured region between Pro24 to Ala29, loop 2: Lys35 – Trp41, loop 3: Lys35 – Glu54, loop 4: His56 – Gly67, loop 5: Leu82 – His88, loop 6: Ala97 – Arg104, and turn 7: Ser112 –Ser115. These regions are labeled on the crystal structure of TTR (4TLT) in Figure 5f. The fragmentation yield of the TTR tetramer (15+) is shown in Figure 5e, with the corresponding B-factor and SASA analysis shown in Figure 5d, and a crystal structure highlighted in the same manner as SA shown in Figure 5f. Backbone (a/x) fragmentation was observed primarily along the C-terminal half of TTR and exhibits less satisfying agreement with the crystal structure of TTR because the fragmentation maxima were less enhanced relative to the basal level of fragmentation observed from residue 60 to 125. Nonetheless, enhanced fragmentation was observed at residues 63–64, 86–88, 102–103, 110–111, 113– 114, and 120–123, all of which correspond to loop regions or to the C-terminal strand of the β-sandwich. Like streptavidin, enhanced fragmentation was not observed in all loop regions, but all enhanced fragmentation was found to correspond to loops or regions with higher B-factors. B-factor and SASA trend together as well; consequently, it is not possible to discern whether fragmentation corresponds to more flexibility or to surface accessibility.
The alpha and beta subunits of hemoglobin feature similar structures having helical secondary structures and six to seven loops/turns, and having homologous loop/turns in the following regions; turn 1: residues 18–21 (α) and 17–20 (β), turn 2: residues 26–28 (α) and 34–36 (β), loop 3: residues 42–50 (α) and 41–50 (β), turn 4: residues 56–59 (β only), loop 5: residues 71–81 (α) and 76–85 (β), loop 6: residues 88–94 (α) and 93–99 (β), and loop 7: residues 112–119 (α) and 117–123 (β). These regions are labeled on the crystal structures of HG (1BBB) in Figure 6. Backbone fragmentation of hemoglobin is unique because mass analysis provides the ability to discern fragments from the alpha and beta subunits. In Figure 6b and 6e, UVPD fragmentation yield is shown for the alpha and beta subunits of hemoglobin. In considering the a/x ions of the two subunits, only a ions were observed, extending through residue 57 in alpha hemoglobin and through residue 63 in beta hemoglobin. SASA and B-factor values for each subunit are shown in Figure 6a and 6d. Several phenylalanine residues are present in both alpha and beta hemoglobin, and enhanced fragmentation is observed C-terminal to these residues, particularly those that precede a proline. Inspection of the fragmentation yield beyond these specific preferential backbone cleavages reveals several regions in both alpha and beta hemoglobin that feature enhanced fragmentation. Interestingly, these regions do not correspond well with the B-factor or SASA values predicted from the crystal structure but do agree well with one another. For example, residues 15 to 23 in both subunits feature enhanced fragmentation; this region corresponds to a turn 1 between α-helix 1 and 2 and is correspondingly highlighted in red in Figure 6c and 6f. A recent study reported the nearly exclusive fragmentation of hemoglobin along the first 23 residues of the protein upon ECD; application of collision induced unfolding prior to ECD resulted in more extensive fragmentation up to approximately the 50th residue, presumably due to unfolding of the N-terminus prior to ECD.42 Our results feature many of the same trends, with the most notable difference being the observation of fragments up to the 60th residue in the absence of preactivation. Similar enhancement to that observed for residues 15–23 is observed for a ions from UVPD covering residues 30–35 in both subunits and corresponds to the C-terminal end of helix 2 and a turn between helix 2 and 3 (turn 2–3). Enhancement of fragmentation of helix 2 may suggest that fraying of this region occurs before or during fragmentation. A third region exhibiting enhanced fragmentation occurs between residues 40 and 50 in both subunits. Although preferential Phe- and Pro-modulated cleavages dominate this stretch, it is evident from the beta subunit that fragmentation is indeed enhanced in this region. This region corresponds to loop 3 in the beta subunit and to the end of helix 3 and loop 3 in the alpha subunit. Fragmentation of both proteins greatly diminishes around residue 50 in both subunits, possibly owing to overlap of fragments with the monomers in the same m/z region of the spectrum and reduced transfer efficiency for large fragment ions in low charge states. Interestingly, neither SASA nor B-factor values appear to represent the structural features of the protein. We speculate that the alpha-helical nature of the subunits allows greater solvent exposure across the entire backbone. B factor also has only limited applicability as a metric of flexibility as it is derived from a packing parameter in crystallography (solid state).
Figure 6.
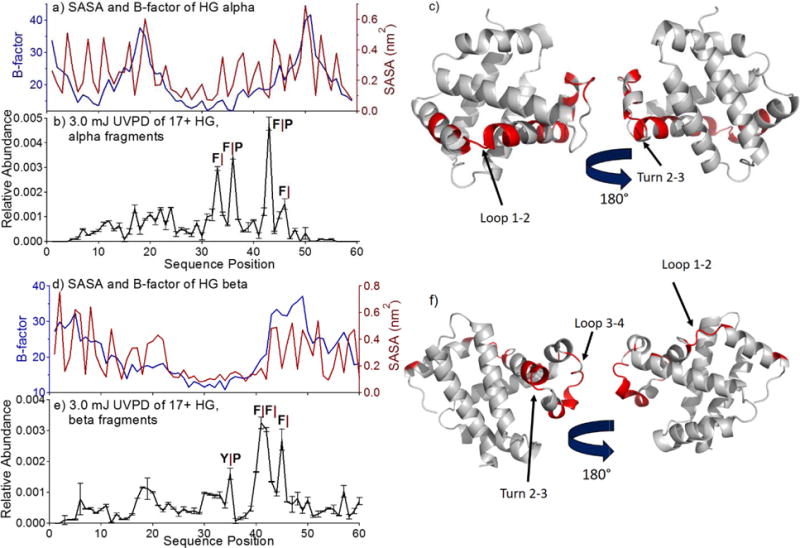
SASA values, B-factors, and UVPD fragmentation yields are shown for alpha (a–c) and beta (d–f) hemoglobin. B-factor is plotted in blue and SASA in red in a) and d). Fragmentation yield following 3.0 mJ UVPD of the 17+ charge state is shown in for alpha HG (b) and beta HG (e). In c) and f), the crystal structures of alpha and beta hemoglobin (pdb 1BBB) are highlighted such that regions featuring enhanced UVPD fragmentation are shown in red. Cleavages C-terminal to Phe or Tyr are denoted F| and Y|. Cleavages N-terminal to Pro are denoted |P.
Discussion
Aggregate consideration of a number of factors, including (i) the apparent charge state of the fragmenting monomers following UVPD of the tetrameric precursors, (ii) the relative abundances of the fragment ions, and (iii) the time-scales associated with backbone cleavage versus unfolding and ejection of monomers by CID-like pathways illuminates an integrated view of dissociation pathways of tetrameric protein complexes by UVPD. UVPD of tetrameric complexes possessing strong interfaces likely requires absorption and subsequent intramolecular vibrational energy redistribution (IVR) to induce dissociation of non-covalently bound subunits. In contrast, complexes featuring weaker interfaces may require fewer photons to achieve subunit dissociation. Multiphoton absorption by use of high pulse energies is proposed to provide access to the high energy deposition, fast dissociation pathways previously demonstrated with SID for tetramers having dimer-of-dimer topology and strong interfaces. As the size of the protein (or protein complex) increases, the number of chromophores also increases, thus increasing the probability for multiphoton absorption.
The specific mechanistic details of the UVPD process remain uncertain. There have been evidence and arguments for two general pathways: excitation to dissociative electronic states that results in fast dissociation directly from those states, and excitation followed by deactivation to ground electronic states from which the dissipated energy is sufficient to cleave the peptide backbone.43–49 Both pathways may be operative in the present work. It is also interesting to note that the dichotomy between the fragmentation yield reflecting secondary structure and the observed charge state of the fragmenting monomers of SA, which were consistent with an asymmetric charge partitioning pathway, can be rationalized together in terms of a recent model proposed by Loo et al. based on heterolytic cleavage of interfacial salt bridges instead of subunit unfolding to rationalize asymmetric charge partitioning.21
Conclusions
193 nm UVPD of three tetrameric proteins featuring a dimer-of-dimers topology provided access to symmetrical and asymmetrical charge partitioning pathways akin to what has been observed by SID and CID, respectively.20,25 Modulation of these pathways as a function of laser power suggests multi-photon absorption plays a prominent role in multiprotein complex dissociation by UVPD. Interestingly, the production of dimers upon UVPD of TTR and SA is consistent with the quaternary structure of the complexes. In contrast, dimers were not observed from tetrameric hemoglobin, which also features a dimer-of-dimers topology, and this result is consistent with HG having weaker interfaces than SA and TTR. Extensive studies of additional multimeric proteins, particularly those having other architectures, are needed to evaluate whether the production of dimers is unique to complexes with dimer-of-dimer topology and consequently whether UVPD is broadly suitable for characterization of protein complex architecture as already demonstrated for SID.
Charge state analysis of the backbone cleavage fragments produced upon UVPD of the tetrameric proteins reflects the highest charge states of monomeric sub-units observed in a given UVPD spectrum. Modulation of laser energy consequently influences the monomers that undergo secondary dissociation and generate backbone cleavage products (sequence ions). Enhancement of the abundances of the sequence ions from 3.0 mJ UVPD in the loop regions of the crystal structures of SA and TTR implies that 193 nm UVPD yields information about both quaternary and secondary structures of protein complexes.
Supplementary Material
Acknowledgments
Funding from the NSF (CHE-1402753), the Welch Foundation (F-1155) and NIH 1K12GM102745 (fellowship to LM) is acknowledged.
Footnotes
Supplemental Information: Supplemental information includes ESI-mass spectra of streptavidin, transthyretin, and hemoglobin; energy-resolved MS curves obtained upon HCD of streptavidin; representative UVPD mass spectrum of streptavidin tetramers; charge site analysis of the a/x fragments of streptavidin (15+), and a difference plot for the fragmentation yield of tetrameric and monomeric streptavidin (15+) upon UVPD. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Marsh JA, Teichmann SA. Annu Rev Biochem. 2015;84:551. doi: 10.1146/annurev-biochem-060614-034142. [DOI] [PubMed] [Google Scholar]
- 2.Shen H-B, Chou K-C. J Proteome Res. 2009;8:1577. doi: 10.1021/pr800957q. [DOI] [PubMed] [Google Scholar]
- 3.Konijnenberg A, van Dyck JF, Kailing LL, Sobott F. Biol Chem. 2015;396:991. doi: 10.1515/hsz-2015-0136. [DOI] [PubMed] [Google Scholar]
- 4.Mehmood S, Allison TM, Robinson CV. Annu Rev Phys Chem. 2015;66:453. doi: 10.1146/annurev-physchem-040214-121732. [DOI] [PubMed] [Google Scholar]
- 5.Snijder J, Heck AJR. Annu Rev Anal Chem. 2014;7:43. doi: 10.1146/annurev-anchem-071213-020015. [DOI] [PubMed] [Google Scholar]
- 6.Chait BT, Cadene M, Olinares PD, Rout MP, Shi Y. J Am Soc Mass Spectrom. 2016;27:952. doi: 10.1007/s13361-016-1385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sciuto S, Liu J, Konermann L. J Am Soc Mass Spectrom. 2011;22:1679. doi: 10.1007/s13361-011-0205-x. [DOI] [PubMed] [Google Scholar]
- 8.Hall Z, Politis A, Robinson Carol V. Structure. 2012;20:1596. doi: 10.1016/j.str.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 9.Sharon M. J Am Soc Mass Spectrom. 2010;21:487. doi: 10.1016/j.jasms.2009.12.017. [DOI] [PubMed] [Google Scholar]
- 10.Benesch JL, Aquilina JA, Ruotolo BT, Sobott F, Robinson CV. Chem Biol. 2006;13:597. doi: 10.1016/j.chembiol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Zhou M, Huang C, Wysocki VH. Anal Chem. 2012;84:6016. doi: 10.1021/ac300810u. [DOI] [PubMed] [Google Scholar]
- 12.Galhena AS, Dagan S, Jones CM, Beardsley RL, Wysocki VH. Anal Chem. 2008;80:1425. doi: 10.1021/ac701782q. [DOI] [PubMed] [Google Scholar]
- 13.Blackwell AE, Dodds ED, Bandarian V, Wysocki VH. Anal Chem. 2011;83:2862. doi: 10.1021/ac200452b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wysocki VH, Jones CM, Galhena AS, Blackwell AE. J Am Soc Mass Spectrom. 2008;19:903. doi: 10.1016/j.jasms.2008.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou M, Dagan S, Wysocki VH. Angew Chem Int Ed. 2012;51:4336. doi: 10.1002/anie.201108700. [DOI] [PubMed] [Google Scholar]
- 16.Zhou M, Jones CM, Wysocki VH. Anal Chem. 2013;85:8262. doi: 10.1021/ac401497c. [DOI] [PubMed] [Google Scholar]
- 17.Zhou M, Dagan S, Wysocki VH. Analyst. 2013;138:1353. doi: 10.1039/c2an36525a. [DOI] [PubMed] [Google Scholar]
- 18.Zhou M, Wysocki VH. Acc Chem Res. 2014;47:1010. doi: 10.1021/ar400223t. [DOI] [PubMed] [Google Scholar]
- 19.Song Y, Nelp MT, Bandarian V, Wysocki VH. ACS Cent Sci. 2015;1:477. doi: 10.1021/acscentsci.5b00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quintyn Royston S, Yan J, Wysocki Vicki H. Chem Biol. 2015;22:583. doi: 10.1016/j.chembiol.2015.03.019. [DOI] [PubMed] [Google Scholar]
- 21.Loo RRO, Loo JA. J Am Soc Mass Spectrom. 2016;27:975. doi: 10.1007/s13361-016-1375-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boeri Erba E, Ruotolo BT, Barsky D, Robinson CV. Anal Chem. 2010;82:9702. doi: 10.1021/ac101778e. [DOI] [PubMed] [Google Scholar]
- 23.Jurchen JC, Williams ER. J Am Chem Soc. 2003;125:2817. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van den Heuvel RHH, van Duijn E, Mazon H, Synowsky SA, Lorenzen K, Versluis C, Brouns SJJ, Langridge D, van der Oost J, Hoyes J, Heck AJR. Anal Chem. 2006;78:7473. doi: 10.1021/ac061039a. [DOI] [PubMed] [Google Scholar]
- 25.Hall Z, Hernández H, Marsh Joseph A, Teichmann Sarah A, Robinson Carol V. Structure. 2013;21:1325. doi: 10.1016/j.str.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jurchen JC, Garcia DE, Williams ER. J Am Soc Mass Spectrom. 2004;15:1408. doi: 10.1016/j.jasms.2004.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. J Am Soc Mass Spectrom. 2010;21:1966. doi: 10.1016/j.jasms.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang H, Cui W, Wen J, Blankenship RE, Gross ML. Anal Chem. 2011;83:5598. doi: 10.1021/ac200695d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lermyte F, Konijnenberg A, Williams JP, Brown JM, Valkenborg D, Sobott F. J Am Soc Mass Spectrom. 2014;25:343. doi: 10.1007/s13361-013-0798-3. [DOI] [PubMed] [Google Scholar]
- 30.O’Brien JP, Li W, Zhang Y, Brodbelt JS. J Am Chem Soc. 2014;136:12920. doi: 10.1021/ja505217w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cammarata MB, Thyer R, Rosenberg J, Ellington A, Brodbelt JS. J Am Chem Soc. 2015;137:9128. doi: 10.1021/jacs.5b04628. [DOI] [PubMed] [Google Scholar]
- 32.Cammarata MB, Brodbelt JS. Chem Sci. 2015;6:1324. doi: 10.1039/c4sc03200d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaw JB, Li W, Holden DD, Zhang Y, Griep-Raming J, Fellers RT, Early BP, Thomas PM, Kelleher NL, Brodbelt JS. J Am Chem Soc. 2013;135:12646. doi: 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quintyn RS, Zhou M, Yan J, Wysocki VH. Anal Chem. 2015;87:11879. doi: 10.1021/acs.analchem.5b03441. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Malmirchegini GR, Clubb RT, Loo JA. Eur J Mass Spectrom. 2015;21:221. doi: 10.1255/ejms.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Breuker K, Brüschweiler S, Tollinger M. Angew Chem Int Ed. 2011;50:873. doi: 10.1002/anie.201005112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Versluis C, Heck AJR. Int J Mass Spectrom. 2001;210–211:637. [Google Scholar]
- 38.Gimon-Kinsel ME, Kinsel GR, Edmondson RD, Russell DH. J Am Soc Mass Spectrom. 1995;6:578. doi: 10.1016/1044-0305(95)00247-B. [DOI] [PubMed] [Google Scholar]
- 39.Pagel K, Hyung S-J, Ruotolo BT, Robinson CV. Anal Chem. 2010;82:5363. doi: 10.1021/ac101121r. [DOI] [PubMed] [Google Scholar]
- 40.Morrison LJ, Brodbelt JS. Analyst. 2016;141:166–176. doi: 10.1039/c5an01819f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aravind G, Klærke B, Rajput J, Toker Y, Andersen LH, Bochenkova AV, Antoine R, Lemoine J, Racaud A, Dugourd P. J Chem Phys. 2012;136:014307. doi: 10.1063/1.3671943. [DOI] [PubMed] [Google Scholar]
- 42.Cui W, Zhang H, Blankenship RE, Gross ML. Protein Sci. 2015;24:1325. doi: 10.1002/pro.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moon JH, Yoon SH, Kim MS. Bull Korean Chem Soc. 2005;26:763. [Google Scholar]
- 44.Moon JH, Yoon SH, Bae YJ, Kim MS. J Am Soc Mass Spectrom. 2010;21:1151. doi: 10.1016/j.jasms.2010.03.025. [DOI] [PubMed] [Google Scholar]
- 45.Yoon SH, Moon JH, Kim MS. J Mass Spectrom. 2010;45:806. doi: 10.1002/jms.1773. [DOI] [PubMed] [Google Scholar]
- 46.Cui W, Thompson MS, Reilly JP. J Am Soc Mass Spectrom. 2005;16:1384. doi: 10.1016/j.jasms.2005.03.050. [DOI] [PubMed] [Google Scholar]
- 47.Parthasarathi R, He Y, Reilly JP, Raghavachari K. J Am Chem Soc. 2010;132:1606. doi: 10.1021/ja907975v. [DOI] [PubMed] [Google Scholar]
- 48.Girod M, Sanader Z, Vojkovic M, Antoine R, MacAleese L, Lemoine J, Bonacic-Koutecky V, Dugourd P. J Am Soc Mass Spectrom. 2015;26:432. doi: 10.1007/s13361-014-1038-1. [DOI] [PubMed] [Google Scholar]
- 49.Halim MA, Girod M, MacAleese L, Lemoine J, Antoine R, Dugourd P. J Am Soc Mass Spectrom. 2015;27:474. doi: 10.1007/s13361-015-1297-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.