

ABSTRACT
E-cadherin mediated cell-cell adhesion plays a critical role in epithelial cell polarization and morphogenesis. Our recent studies suggest that the desmosomal cadherin, desmoglein 3 (Dsg3) cross talks with E-cadherin and regulates its adhesive function in differentiating keratinocytes. However, the underlying mechanism remains not fully elucidated. Since E-cadherin trafficking has been recognized to be a central determinant in cell-cell adhesion and homeostasis we hypothesize that Dsg3 may play a role in regulating E-cadherin trafficking and hence the cell-cell adhesion. Here we investigated this hypothesis in cells with loss of Dsg3 function through RNAi mediated Dsg3 knockdown or the stable expression of the truncated mutant Dsg3ΔC. Our results showed that loss of Dsg3 resulted in compromised cell-cell adhesion and reduction of adherens junction and desmosome protein expression as well as the cortical F-actin formation. As a consequence, cells failed to polarize but instead displayed aberrant cell flattening. Furthermore, retardation of E-cadherin internalization and recycling was consistently observed in these cells during the process of calcium induced junction assembling. In contrast, enhanced cadherin endocytosis was detected in cells with overexpression of Dsg3 compared to control cells. Importantly, this altered cadherin trafficking was found to be coincided with the reduced expression and activity of Rab proteins, including Rab5, Rab7 and Rab11 which are known to be involved in E-cadherin trafficking. Taken together, our findings suggest that Dsg3 functions as a key in cell-cell adhesion through at least a mechanism of regulating E-cadherin membrane trafficking.
KEYWORDS: adherens junction, desmoglein 3, desmosome, E-cadherin trafficking, epithelial cells, intercellular junction
Introduction
The epithelial cells are attached to each other predominantly by adherens junctions and desmosomes that are essential in the maintenance of cell architecture and tissue integrity. These intercellular junctions are highly dynamic structures and coordinate with many physiological processes, including development, wound healing, cell polarization and tissue morphogenesis. Disruption of these junctions can have detrimental effect on these physiological processes resulting in a failure of tissue homeostasis leading to a variety of diseases.
The adhesion of both adherens junction and desmosome is mediated through the cadherin family proteins in a calcium-dependent manner. Thus, calcium ions are often used in vitro as a mediator to induce the junction formation in epithelial cultures.1,2 The adhesion receptors in adherens junctions belong to the classical cadherins and among them E-cadherin is the major molecule in most epithelial tissues. E-cadherin is crucial in many aspects of epithelial biogenesis and a key determinant for epithelial apical-basal polarity. The adhesion core proteins in desmosome, however, are the desmosomal cadherins, consisting of 2 subfamilies of desmoglein (Dsg1–4) and desmocollin (Dsc1-3). The cytoplasmic tails of desmosomal cadherins bind to plakoglobin, plakophilins and desmoplakin that in turn link to the intermediate filaments to form a network of desmosome-intermediate filament complex.3 Both classical cadherins (E-cadherin in epithelial and VE-cadherin in endothelial cells) and Dsgs (at least isoform 1/34) bind to p120 at the juxtamembrane domain and β-catenin/plakoglobin at the catenin-binding domain in the cytoplasmic tail. In contrast to desmosomal cadherins, the E-cadherin-catenin complex links to the actin cytoskeleton via proteins including α-catenin. There is accumulating evidence indicating that interaction of p120 and classical cadherins is critical in cadherin adhesion and stabilization, achieved through a mechanism of preventing cadherin endocytosis and degradation. Disruption of such an interaction causes the exposure of an endocytic signal motif within the juxtamembrane domain of cadherins that leads to junctional complex endocytosis.5,6
Dsg3 is a known major autoantigen in pemphigus vulgaris, an autoimmune disease with manifestation of blistering involving oral mucosa and skin. Despite many studies based on the pemphigus autoimmune antibodies, the molecular mechanism of blister formation remains not fully understood and is still under intensive research. Emerging evidence suggests a cross talk between Dsg3 and E-cadherin showing that Dsg3 regulates E-cadherin adhesion via signal pathways such as Src, Rho GTPases Rac1/cdc42 and Ezrin as well as transcription factor c-Jun/AP-1, all of which are involved in the organization of actin cytoskeleton associated with adherens junctions.7-10 This novel finding has recently been reported by independent studies in the literature that demonstrate existence of a complex formation containing non-junctional Dsg3, E-cadherin and Src in keratinocytes.7-10 Furthermore, it has been suggested that the stability of such a complex is Src dependent and the tyrosine phosphorylation of cadherins is required for recruiting Dsg3 to the cytoskeletal pool and for desmosome maturation.7 Moreover, it has been shown that overexpression of Dsg3 in cancer cell lines does not necessarily enhance cell-cell adhesion but rather causes a reduction of E-cadherin expression with concomitant accelerated cell migration and invasion.8,11 Knockdown of Dsg3, on the other hand, also showed a negative influence on desmosomes and cell cohesion with a consequence of failure in cell polarization.9,10 Furthermore, impaired E-cadherin coupled with enhanced phospho-Src expression was also detected in the oral mucosal membranes of pemphigus patients.9 However, the cross talk between Dsg3 and E-cadherin is still far from fully understood.
A growing body of evidence suggests that the balance between assembly and disassembly of junctional complexes are the key determinant of cell-cell adhesion strength and stability. For instance, in the process of epithelial to mesenchymal transition (EMT) during tumor progression and embryonic development the junctional complexes are disassembled due to enhanced E-cadherin internalization and lysosomal degradation.12 On the other hand, in normal development of intestinal epithelium the assembly of adherens junction is enhanced by a mechanism of accelerated E-cadherin membrane trafficking, a process governed by the intestine-specific transcription factor Cdx2.13 It is known that E-cadherin endocytosis and recycling are regulated in part by Rab GTPases, the master regulators of membrane trafficking. Rab GTPases are molecular switches, cycling between GTP (active) and GDP (inactive) bound states and serving as scaffolds to integrate both membrane trafficking and intracellular signaling in a spatiotemporally sensitive manner.14 Rab5 and Rab7 which are involved in early endosome fusion and transport from early to late endosomes, respectively, are shown to be activated by Src signaling in MDCK cell line expressing a temperature-sensitive v-Src (MDCKpp60v-Src) and regulate E-cadherin shuttling from endosomes to lysosomes resulting in dissolution of adherens junctions and EMT.12,14 Rab11 which involves transport from Golgi to apical endocytic recycling endosomes is documented to regulate the sorting and basolateral targeting of E-cadherin during epithelial morphogenesis in MDCK cells.14-16
Since E-cadherin trafficking has emerged as an essential process in modulating cell-cell adhesion and dynamics, 16-18 we hypothesized that Dsg3 may play a role in the process of E-cadherin trafficking and thereby have an impact on cell-cell adhesion and epithelial morphogenesis. In the present study we investigated our hypothesis by using Dsg3 gain and loss-of-function approaches as well as dominant negative mutant of Dsg3, that were achieved by overexpression of full length Dsg3 and RNAi mediated transient knockdown of Dsg3 or stable expression of C-terminally truncated Dsg3, respectively. We demonstrate that disruption of normal Dsg3 activity affects the junction assembly of both adherens junction and desmosome proteins and also the E-cadherin membrane trafficking that were likely caused by, at least in part, altered expression of Rab proteins. As a result, cell-cell adhesion and epithelial polarity are largely affected, leading to abnormal cell flattening and spreading. These findings reinforce the concept that Dsg3 acts as a key regulator in epithelial junction formation, polarization and morphogenesis.
Results
Expression of C-terminally truncated Dsg3 results in compromised cell-cell adhesion in epithelial cells
Our previous study showed that Dsg3 depletion in epithelial cells caused compromised cell cohesion resulting in enhanced fragmentation as assessed by the dispase dissociation assay.19 To evaluate whether cells expressing C-terminally truncated Dsg3ΔC proteins (Fig. 1A) also render similar effect, we, first of all, performed the dispase dissociation assay in SqCC/Y1 oral keratinocyte lines with stable expression of 3 different Dsg3 truncated proteins (Dsg3Δ238, Δ458 and Δ560) along with an empty vector control (Vect Ct) and full length hDsg3.myc (FL) cells.11 As indicated in Figure 1A, all Dsg3ΔC truncated proteins lack the transmembrane domain and cytoplasmic tail of Dsg3 and thus, it was predicted that the expression of these truncated proteins could result in a phenotype similar to that of Dsg3 depleted cells with compromised cell-cell adhesion. Western blotting was performed to confirm the truncated protein expression but with their molecular weights displaying slightly larger than predicted that was likely caused by protein glycosylation (Fig. 1B). The exogenous protein expression was also analyzed by immunostaining with rabbit anti-Myc tag that binds to the Myc epitope at C-terminus of exogenous proteins, including FL and 3 Dsg3ΔC proteins, and confirmed the cytoplasmic distribution of Dsg3ΔC proteins in contrast to FL cells that showed pronounced membrane binding of hDsg3.myc (data not shown). To assess cell-cell adhesion strength, the dispase dissociation assay was conducted in confluent cultures of various SqCC/Y1 cell lines grown in 6-well plates. Cells were treated with dispase (2.4 U/ml in PBS) to detach the entire epithelial sheets from the well, and then the epithelial sheets were subjected to mechanical stress to induce fragmentation. Five independent experiments were performed and Figure 1C shows the pooled data from 5 attempts that indicated a significant increase in the number of fragments in all Dsg3ΔC lines compared to Vect Ct or FL cells. Disruption of cell-cell adhesion was also observed in Dsg3ΔC culture, but not in control cells, during the dispase treatment (Fig. 1C arrows). Taken together, these results demonstrate a dominant negative function of the Dsg3ΔC truncated constructs on cell cohesion and epithelial integrity, and suggest that the expression of Dsg3ΔC proteins elicits compromised cell-cell adhesion with the result of a phenotype resemble that of Dsg3 depleted cells, as reported previously.19 This finding supports the general conclusion, based on ample data in pemphigus research,20 that Dsg3 plays a crucial role in cell-cell adhesion and the maintenance of epithelial homeostasis.
Figure 1.

The expression of Dsg3 C-terminally truncated mutant compromised epithelial cell cohesion. (A) Schematic of Dsg3 full length (FL) and its 3 C-terminally truncated proteins (lacking the transmembrane domain and cytoplasmic tail) denoted as Δ238, Δ458 and Δ560, respectively, all of which are tagged with a Myc epitope at C-terminus. The binding sites of 3 antibodies used in the study are indicated by arrow lines or in red. (B) Western blotting of exogenous Dsg3 FL and truncated proteins in SqCC/Y1 oral keratinocyte lines with rabbit anti-Myc tag. The molecular weights of truncated proteins (indicated by arrows) were slightly larger than expected, i.e. 35 kDa for Δ238, 67 kDa for Δ458 and 80 kDa for Δ560. The weak bands shown in the Vect Ct and other lanes are likely to be non-specific bands detected by the rabbit antibody. (C) Dispase dissociation assay in SqCC/Y1 lines with stable expression of empty vector control (Vect Ct), FL and 3 truncated proteins that showed significantly enhanced fragmentation of epithelial sheets, after subjected to mechanical stress, in all 3 truncated cell lines compared to Vect Ct or FL cells whose integrity was largely intact. Cells were seeded at confluent density in 6-well plates and grew for 2 d before treated with dispase (2.4units/ml) for ∼30 min till completely detached (Before). The fragment data from 5 independent experiments were pooled before statistical analysis (n = 13∼16, mean ± SD, *p < 0.05, ***p < 0.001). The phase contrast micrographs showed disruption of cell-cell adhesion in Dsg3Δ458 cells during the course of dispase treatment (arrows) as opposed to control cells. Scale bar, 20 µm.
In an attempt to explore the possibility of Dsg3ΔC secretion into the culture media, we did the following with a focus on Vect Ct and Dsg3Δ458 cells: (1) ELISA assay with the conditioned media from 24 hrs culture of SqCC/Y1 cells. Comparing with the Vect Ct sample, there was a marginal but significant increase of ELISA signals in conditioned media of Dsg3Δ458 cells (Fig. S1A). (2) Co-IP analysis in conditioned media of 24 hrs cultures of MDCK-Vect Ct and Dsg3Δ458 cells with anti-Dsg3 5H10 that recognizes EC1-EC2 of the extracellular domain of Dsg38 (Fig. 1A). The purified immunoprecipitates were analyzed by Western blotting with an anti-Myc tag antibody, and a stronger band of ∼95 kDa was detected in the conditioned media of Dsg3Δ458 cells as compared to Vect Ct sample (Fig. S1B). Using mouse monoclonal antibody 5H10 for blotting, we detected a few other bands at molecular weights of approximately 120, 80, 35-40 kDa but these bands also appeared in Vect Ct sample albeit they were expressed at relatively lower levels (data not shown). (3) Treatment of HaCaTs with the conditioned media generated from Vect Ct and Dsg3Δ458 cells before immunofluorescence analysis. In this case, HaCaT cells grown on coverslips were fixed with formaldehyde or the ice cold methanol, respectively, for 10 min before immunostaining with anti-Myc tag and 5H10. Microscopic analysis revealed disruption of the Dsg3 staining in cells treated with the conditioned medium from Dsg3Δ458 cells with a concomitant decrease of surface Dsg3 (formaldehyde fixed sample, p < 0.05), but showed an increase in cells fixed with methanol, compared to the respective controls (p < 0.01, Fig. S1C). This result suggested a possibility of Dsg3Δ458 secretion that caused alteration of the Dsg3 surface expression as well as its junctional assembly. Nevertheless, no difference was detected for the surface Myc staining between control and test sample (formaldehyde fixed) but a significant increase of Myc signals in cells treated with Dsg3Δ458 conditioned medium and fixed with methanol (both surface and intracellular), indicating an enhanced internalization and accumulation of truncated proteins (p < 0.001, Fig. S1C). Confocal analysis of E-cadherin staining also revealed disruption and reduction of E-cadherin in cells treated with conditioned medium from Dsg3Δ458 in contrast to control cells that displayed strong E-cadherin staining at the junctions (Fig. S1D). Taken together, the above results suggest a possibility of Dsg3ΔC mutant secretion that might cause perturbation of Dsg3 action on cell surface which might attribute to the disrupted cell cohesion of epithelial cells.
Expression of Dsg3ΔC mutants caused altered protein distribution and cadherin assembly at the junctions
The disrupted cell-cell adhesion in Dsg3ΔC cell lines suggests a defect in junction protein assembly, as we observed in cells with Dsg3 depletion.9 Thus, we performed immunofluorescence for Dsg3 and E-cadherin in Dsg3Δ238 and Dsg3Δ458 lines alongside Vect Ct and FL cells. Cells were double labeled with mouse anti-Dsg3, 5H10 and rabbit anti-Myc tag and, as predicted, strong membrane staining was observed with mouse anti-Dsg3 in all cell lines (green in Fig. 2A) indicating the membrane localization of native and exogenous FL Dsg3. However, aberrant distribution of Dsg3 was observed at the cell borders of Dsg3ΔC cells that displayed a broad zone of punctate staining (Fig. 2A), with 19.2 ± 3.59% positive cells in Dsg3Δ238 population and 40.7 ± 3.75% positive cells in Dsg3Δ458. In contrast, a linear pattern was consistently observed in control cells and FL cells (with only 7.54 ± 2.81% showing punctate staining). Close inspection revealed a few co-localizations of both channels within the broad zone in Dsg3ΔC cells, suggesting a possibility of recruitment of truncated proteins to area adjacent to the plasma membrane (Fig. 2 arrows).
Figure 2.
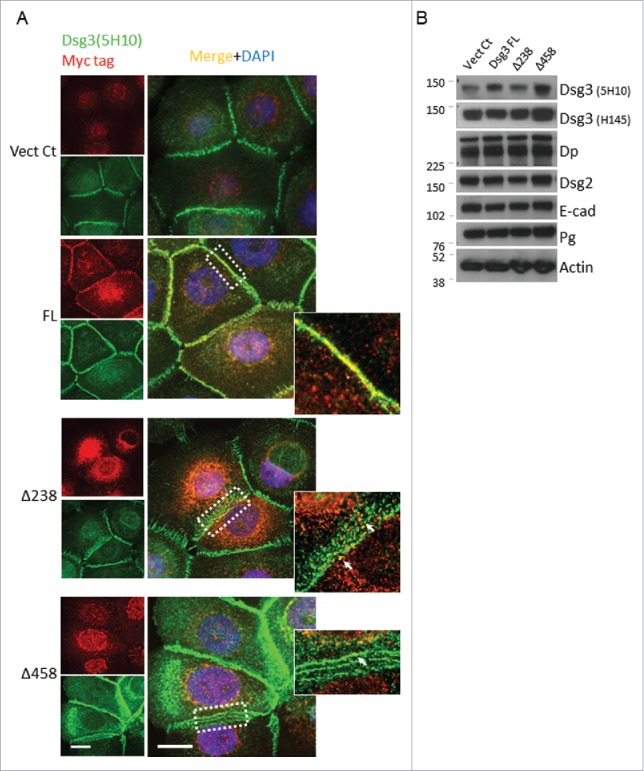
The expression of Dsg3ΔC proteins affects membrane distribution of native Dsg3. (A) SqCC/Y1 cells seeded on coverslips were double labeled with mouse anti-Dsg3 5H10 (green) and rabbit anti-Myc tag (red). The linear staining pattern of Dsg3 at the cell periphery was readily seen in both Vect Ct and FL cells (an enlarged image of dotted box is shown on the right). In contrast, a broad zone of abnormal, punctate staining was frequently observed in Dsg3ΔC cells with some dots displaying colocalisation of both channels (arrows). (B) Western blotting of total lysates of SqCC/Y1 cell lines with the indicated antibodies. No apparent reduction of Dsg3 and E-cadherin was observed in Dsg3ΔC cells as compared to control samples. Scale bar, 10 µm.
Previously, we showed that Dsg3 cross talks with E-cadherin and regulates its junctional assembly.8,9 Next, the effect of Dsg3ΔC expression on E-cadherin junction assembly, as well as the native Dsg3 and F-actin, was examined by immunofluorescence in SqCC/Y1 cells that were subject to calcium switch9 for 1 hr and 6 hrs. Cells seeded on coverslips were grown in EpiLife (containing low calcium at 60 µM) to fresh confluence before calcium addition for different time periods prior to staining for E-cadherin, Dsg3 and F-actin, respectively. Fluorescent microscopy revealed that within 1 hr of calcium addition both cadherins were relocation at cell periphery with a well-organized distribution in both control and FL cells. In contrast, this process was severely perturbed in Dsg3ΔC cells with pronounced cytoplasmic accumulation, in particular for Dsg3 staining (Figs. 3A and S2). Enlarged cells with irregular morphology and diffuse E-cadherin staining were frequently observed in these populations (Fig. S3). Remarkably, disrupted E-cadherin was observed at the cell borders, especially at an early time point (1 hr) of calcium switching (Fig. 3A). At 6 hrs, more cadherins were detectable at the cell borders in Dsg3ΔC cells but remained poorly organized and fragmented, and cells appeared loosely attached with one another. Quantitation of junctional cadherins supports the visual inspection indicating a reduction of E-cadherin and Dsg3 at junctions in Dsg3ΔC cells, particularly after 1 hr of calcium switching. By contrast, a significant increase of cadherin junction assembly was detected in FL cells compared to control (Figs. 3A and S2). Furthermore, Dsg3 expression at the junctions was detectable in FL cells even without calcium switch (Fig. S2B). Similar expression profile was also seen for cortical F-actin staining at both 1 hr and 6 hrs of calcium switching (Fig. S2B). In support, close inspection of confocal stacks of SqCC/Y1 Vect Ct and Dsg3Δ458 cells revealed characteristic adhesion zippers of cadherin in the apical plane of control cells (Fig. 3B arrows) but this was missing in Dsg3Δ458 cells that, instead, showed abundant cytoplasmic accumulation. To examine any changes on total protein expression of endogenous Dsg3 and E-cadherin they were analyzed by Western blotting along with other junctional proteins and the results showed little or no reduction in the expression of junctional proteins analyzed, as shown in Fig. 3B. Taken together, these results agree with our conclusion that Dsg3ΔC truncated proteins exhibit a dominant negative function in junction assembly for E-cadherin and Dsg3, and these findings are also consistent with our previous report based on RNAi study that showed perturbed junctional assembly of E-cadherin and its associated adhesive function.9
Figure 3.
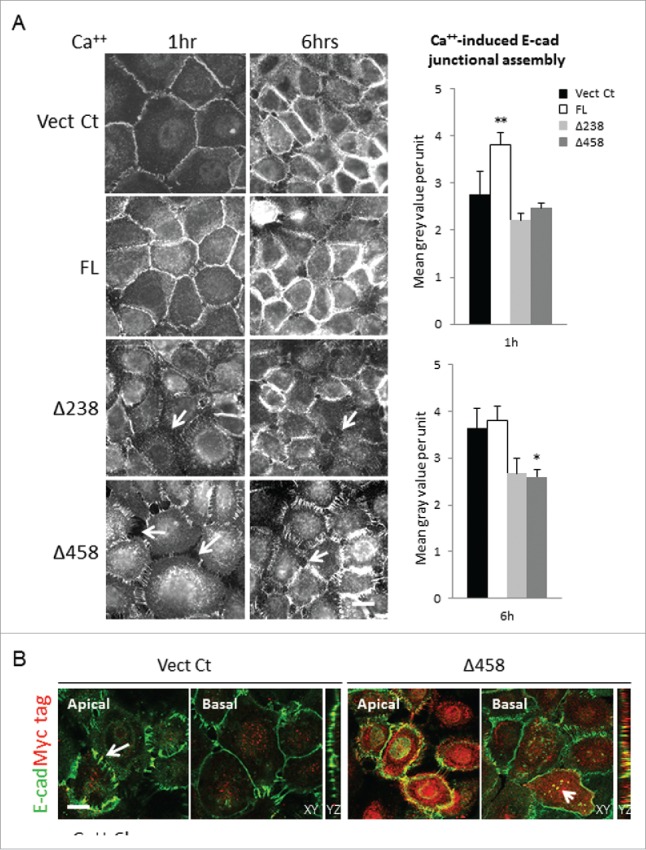
The E-cadherin junction assembly was perturbed in Dsg3ΔC cells. (A) SqCC/Y1 cell lines subjected to calcium switching (1.8 mM) for 1 hr and 6 hrs before immunostaining for E-cadherin with mAb HECD-1. Both Vect Ct and FL cells showed linear staining pattern at the cell borders at 1 hr of calcium switch and this was seen to be further enhanced at 6 hrs time point. However, in both Dsg3ΔC lines, the disrupted E-cadherin staining coupled with widened intercellular gaps was frequently seen in cell populations (arrows), indicating a retardation of E-cadherin junction assembly. Bar charts on the right were the quantitation of junctional cadherin that showed a significant increase in FL cells at 1 hr of calcium switch and a decrease in Dsg3Δ458 at 6 hrs, compared to Vect Ct (n > 50 cells, *p < 0.05, **p < 0.01). In addition, a trend of reduction of junctional cadherin was indicated in both Dsg3ΔC lines, compared to control or FL cells. (B) Confocal images of apical and basal frames as well as Z sections of Vect Ct and Δ458 cells with calcium switch for 6 hrs, and cells were double stained for E-cadherin (green) and Myc tag (red). The characteristic E-cadherin adhesion zippers were observed in the apical plane of control cells (arrows) but these were largely missing in Δ458 cells with abundant Myc positive, truncated protein expression. Colocalisation of both proteins was also observed in the cytoplasm of Δ458 cells (arrowhead in XY, and also YZ). Scale bar, 10 µm.
Expression of Dsg3ΔC protein caused a reduction of surface cadherins and defect in cadherin complex formation associated with Pg/p120-catenins
The observation of altered junctional cadherins without apparent changes in total proteins prompted us to speculate that junctional homeostasis might be affected in Dsg3ΔC cells. To gain insight of molecular changes at the junctions in mutant cells, we analyzed protein expression in subcellular fractions using various biochemical methods: 1) detergent soluble and insoluble fractions, 2) surface verses intracellular pool of proteins, and 3) protein expression in cadherin complexes. Firstly, Triton soluble and insoluble proteins in MDCK-Vect Ct and Dsg3Δ458 cell lines were extracted following the protocol as previously described.9 Prior to extraction, cells were subject to calcium depletion and then repletion for 4 hrs to restore cell-cell adhesion. Equal amounts of proteins in both fractions in control and mutant cells were analyzed by Western blotting and the results were shown in Figure 4A and B. In addition to Dsg3 that showed a reduction in Triton insoluble fraction in mutant cells, the expression of plakoglobin (Pg) and p120 also exhibited a significant or a trend of reduction when compared to control samples. Little or no difference was detected for β-catenin between control and mutant cells. This result suggested a defect in recruitment of Pg and p120-catenins to the cytoskeletal associated fraction that contains both adherens junction and desmosome proteins.
Figure 4.

The Dsg3ΔC protein expression caused a reduction of cytoskeletal or membrane association of cadherins, Pg and p120-catenins. (A) Western blotting analysis of Triton soluble and insoluble fractions of MDCK Vect Ct and Δ458 lines that were subjected to calcium switch for 3 hrs. Left panels showed detection of Dsg3ΔC protein (top blot, arrow) and a slight reduction of native Dsg3 in Δ458 cells (bottom blot) compared to control with the indicated Abs. Reduced expression of Dsg3, Pg and p120 was observed in Δ458 cells, in particular in insoluble fraction, but no change was seen for E-cadherin, compared to control. (B) Densitometry of Pg and p120 blots in Triton soluble and insoluble fractions that showed a significant or a trend of reduction in insoluble fraction of Δ458 cells compared to the respective controls (n = 4, *p < 0.05). (C) Western blotting of purified surface vs intracellular proteins in MDCK and SqCC/Y1 cells subjected to calcium switch for 3 hrs. Reduced expression of cadherins, Pg and p120 was detected in surface pool of Δ458 cells compared to the respective control samples. Altered expression of cadherins was also seen in FL cells in both surface and intracellular pools.
To consolidate the above finding, we next analyzed surface proteins in both MDCK and SqCC/Y1 lines using a biotinylation reagent (Pierce Cell Surface Protein Isolation Kit). Again, the cell-cell contacts and junction formation were restored by calcium switching for 4 hrs. Cells were treated with a cell-impermeable, cleavable biotinylation reagent, Sulfo-NHS-SS-Biotin on ice before protein extraction with lysis buffer. Equal amount of labeled surface proteins was affinity-purified with NeutrAvidin Agarose and eluates were analyzed by Western blot alongside the intracellular non-biotinylated proteins. As shown in Figure 4C, in addition to Dsg2/3 and E-cadherin that exhibited a significant decrease a marked reduction was also detected for Pg and p120 in the surface pool of mutant cells in both epithelial cell types. Correspondingly, an increase of cadherins and Pg was observed in the intracellular pool of mutant cells as opposed to control samples. Again, little change was seen for β-catenin in both surface and intracellular fractions. Of note, a decrease in the expression of E-cadherin, Dsg2 and Pg was also found in the surface pool of FL cells compared to control (Fig. 4C), and this observation was consistent with our previous report that showed enhanced activation of Src signaling in the Dsg3 overexpressing cells that in turn caused tyrosine phosphorylation and protein degradation of junctional molecules including E-cadherin.8 Thus, it is likely that the observed reduction of junctional proteins is caused by elevated protein tyrosine phosphorylation or an enhanced protein membrane trafficking (discussed below). Furthermore, the effect of Dsg3 modulation on junctional Pg and p120 was also supported by immunofluorescence in Dsg3ΔC and cells with Dsg3 knockdown (Fig. S4 or not shown). Besides, the fluorescent intensities of Pg and p120 at the junctions were quantitatively analyzed and showed a significant reduction in mutant cells as well as cells with Dsg3 knockdown in all cell lines, such as MDCK, SqCC/Y1 and A431 (data not shown).
Next, protein expression in the cadherin complexes (E-cadherin and Dsg3) was analyzed by co-immunoprecipitation (IP) in order to verify that the integrity of junctional complexes was indeed affected by the expression of Dsg3ΔC mutant. In this case, SqCC/Y1 and A431 cells were examined and cells at ∼80% confluence were subjected to calcium switch for 4 hrs before protein extraction with RIPA buffer.8 Equal amounts of lysates of Vect Ct and Dsg3Δ458 cells were immune-purified with HECD-1 for E-cadherin or 5H10 for Dsg3, respectively, and the immunoprecipitates were analyzed by Western blotting for Pg and p120 expression as well as β-catenins (Fig. 5). For E-cadherin IP, a small amount of Pg was detected in control cells but this was significantly attenuated in mutant, especially in A431 cells. Little or no detection was seen for p120 in 3 attempts in each cell type albeit the abundant expression was found in the inputs of both cell types (Fig. 5A), indicating low affinity of the interaction between p120 and E-cadherin. No difference was observed for β-catenin in the E-cadherin complex between control and mutant lines, suggesting that the interaction between β-catenin and E-cadherin was not affected by mutant expression. Using the same protocol for Dsg3 IP, both Pg and p120 were found to bind to Dsg3 in control cells, especially for p120, which was not detectable in the E-cadherin complex with current regime, indicating a relatively higher affinity in binding to Dsg3 than to E-cadherin (Fig. 5B). In contrast, such an association was significantly attenuated in mutant cells.
Figure 5.
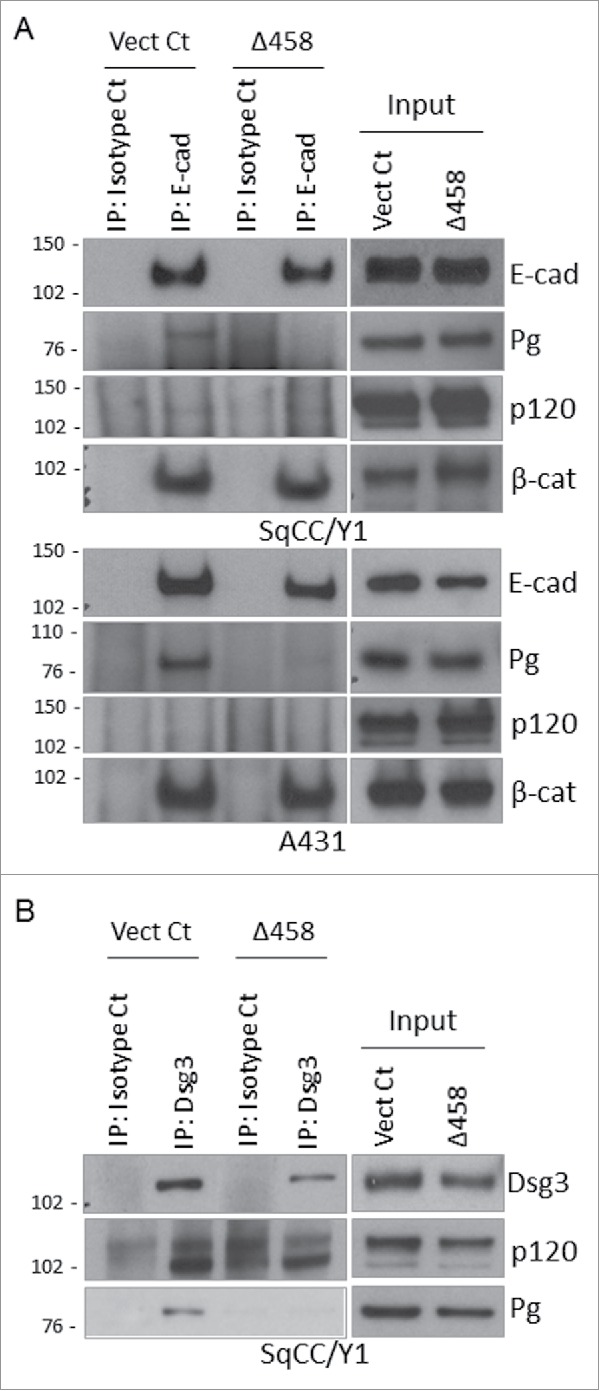
Co-immunoprecipitation analysis showed a reduction of Pg and p120 in E-cadherin complex of Dsg3ΔC cells. (A) The E-cadherin complex in Vect Ct and Δ458 lines in both SqCC/Y1 and A431 purified with mAb HECD-1 was analyzed by Western blotting for Pg, p120 and β-catenins in addition to E-cadherin. Although little expression was seen for p120, an evident reduction of Pg, but not β-catenin, was detected in Δ458 cells relative to Vect Ct. (B) Western blotting analysis of immune complex purified with mAb 5H10 for Dsg3 in SqCC/Y1. A lack of detection for Pg and a reduction of p120 expression were observed in purified Dsg3 complex of Δ458 cells compared to control.
Taken together, the above results suggest that the Dsg3ΔC mutant expression causes defects in complex formation of both classical and desmosomal cadherins, junction assembly and stabilization at the cell surface during the process of cell-cell adhesion assembly.
E-cadherin internalization and recycling were altered in Dsg3ΔC cells or cells with Dsg3 depletion that was coincided with Rab reduction
E-cadherin trafficking has been recognized to be a central determinant in cell-cell adhesion and maintenance. In addition, our observed reduction of surface cadherins may also suggest an abnormality in cadherin membrane trafficking. Thus, we hypothesized that Dsg3 may play a role in regulating E-cadherin trafficking and thereby cell-cell adhesions. To test this hypothesis, we analyzed cadherin trafficking in SqCC/Y1 cells with the loss of Dsg3 function, including Dsg3ΔC mutant expression and depletion, using the fluorescence antibody-based internalization and recycling assays.5,21 Dsg3ΔC mutant and its matched Vect Ct, as well as parental cells treated with either Dsg3 RNAi or scrambled siRNA control, were seeded on coverslips in 24-well plates. Live cells were surface labeled on ice for E-cadherin with HECD-1 followed by A488-IgG. The labeled antibodies were then allowed to be internalized at 18°C for 30 min17 followed by surface antibody quenching before fixation and nuclear counter stain with DAPI (see Materials and Methods). Multiple fluorescence images were acquired in arbitrary fields in each sample and the fluorescence intensities of both surface and internalized E-cadherin were measured with ImageJ. Finally, the internalized E-cadherin was calculated as percentage of that of initial surface labeling before internalization. The resulting data indicated that E-cadherin internalization was less efficient in cells expressing Dsg3ΔC mutant or with Dsg3 knockdown, compared to the respective control samples (Fig. 6A and C). The E-cadherin recycling was also analyzed and in this case, the internalized antibodies were allowed to recycle back to the surface by incubating cells at 37°C for 30 min followed by another surface antibody quenching.21 Finally, the percentage of E-cadherin recycling was calculated based on the reduction in the amount of internalized proteins relative to that of pro-recycling step. The results of this analysis indicated that the cadherin recycling was also affected in mutant cells and showed a trend of reduction in cells with Dsg3 depletion (Fig. 6B and C).
Figure 6.
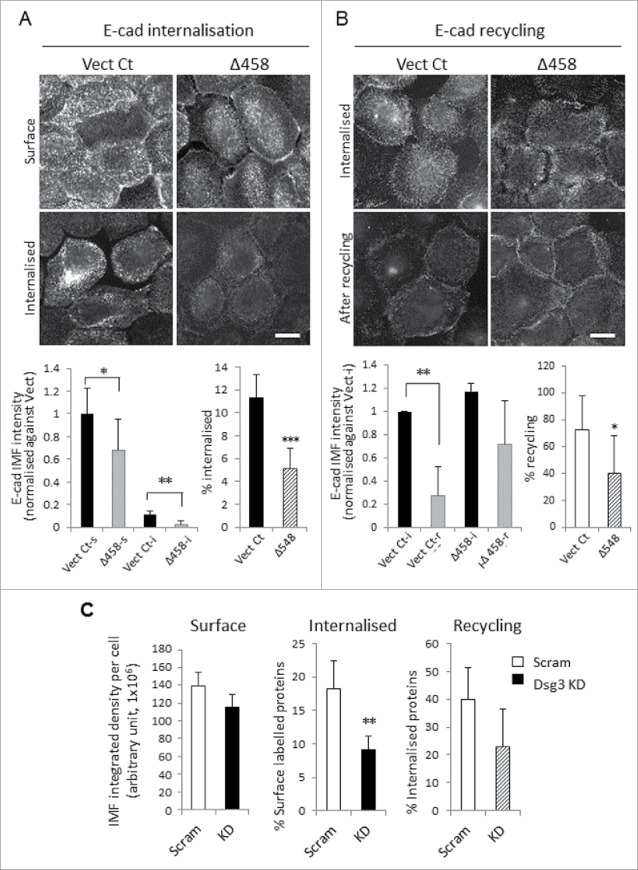
Altered E-cadherin internalization and recycling in cells with either Dsg3ΔC expression or Dsg3 depletion. (A, B) Analysis of E-cadherin internalization (A) and recycling (B) in live SqCC/Y1 Vect Ct and Δ458 lines. Cells seeded on coverslips were subjected to calcium switching (1.8 mM, keratinocyte growth medium) for 2 hrs prior to the internalization and recycling assays (details in Materials and Methods). For internalization, cells were surface labeled for E-cadherin with HECD-1 and then A488 conjugated mouse IgG on ice. A set of samples were fixed for determination of surface labeled proteins (Vect Ct-s and Δ458-s). Another set was incubated at 18°C for 30 min to allow surface labeled proteins to be internalized. Then, the remaining antibodies on the surface were quenched before fixation (Vect Ct-i and Δ458-i) and counterstained with DAPI. For recycling, the surface labeled E-cadherins were allowed to be internalized at 18°C for 30 min prior to surface antibodies quenching. A set of samples was fixed for later analysis of the internalized proteins (Vect Ct-i and Δ458-i) and the other was further incubated at 37°C for 30 min to allow internalized proteins to recycle back to the surface before another quench of the surface antibodies prior to fixation and DAPI staining (Vect Ct-r and Δ458-r). Images were acquired at arbitrary fields (≥ 5 fields/coverslip) and analyzed with ImageJ. Data were a representative of at least 3 independent experiments for both internalization and recycling assays (> 1000 cells per sample were included in the analysis, mean ± SD, *p < 0.05, **p < 0.01). Scale bar, 10 µm. (C) Reduced E-cadherin internalization and recycling were also detectable in SqCC/Y1 parent cells with Dsg3 knockdown (pooled of 2 experiments, >1000 cells per sample were included in the analysis, mean ± s.e.m, **p < 0.01).
In parallel, the membrane trafficking for native Dsg3 was also analyzed in MDCK and SqCC/Y1 stable lines and similar results were obtained showing a reduction of Dsg3 internalization and recycling (Fig. S5). In accord, an enhanced Dsg3 internalization was observed in MDCK FL cells compared to control. Moreover, in an attempt to gain the evidence of Dsg3 and E-cadherin association in the endocytic process we performed the Dsg3 internalization assay in conjunction with direct immunofluorescence for E-cadherin with rabbit E-cadherin-A555 in MDCK cells. In this case, after steps of surface labeling, internalization and surface quenching, the cells containing the internalized Dsg3 were fixed and immune-stained with anti-E-cadherin antibody conjugated with Alexa 555 for 30 min followed by counterstaining with DAPI (Fig. S6). Fluorescent microscopy showed colocalisation of 2 cadherins in area adjacent to the plasma membrane, especially in regions with multi-cellular junctions suggesting a likelihood of their dimerization (heterodimer) in the endocytic process (Fig. S6 arrows).
Rab GTPases serve as master regulators in membrane trafficking along the endocytic pathway that are requisite for correct sorting and targeting of E-cadherin to the basolateral membrane during epithelial polarization and morphogenesis. Since the activation of Rab5 and Rab7 have been implicated in E-cadherin endocytosis and shuttling from endosome to lysosome,12 whereas active Rab11 are required for correct sorting and polarized delivery of E-cadherin to the basolateral cell surface during epithelial morphogenesis 15 we decided to examine the Rab protein expression and to explore whether changes in cadherin trafficking were accompanied with any alteration for Rab5, Rab7 and Rab11 in cells with the loss of Dsg3 function. Western blotting analysis of total cell lysates was performed and showed all 3 Rabs, Rab5, Rab7 and Rab11 were reduced in Dsg3ΔC mutant cells compared to control (Fig. 7A, left panel). Correspondingly, Dsg3 overexpressing cells showed increase in the expression of all 3 Rabs relative to control cells. Similarly, a reduction of Rabs was also detectable in SqCC/Y1 parental cells with Dsg3 knockdown compared to those transfected with scrambled siRNA (Fig. 7A, right panel). In parallel, the Rab activity assay was performed and it was not surprising that the GTP bound form of Rab5 or Rab11 also showed markedly decreased in mutant cells compared to control, especially for GTP-loaded Rab11 that was almost undetectable (Fig. S7). Furthermore, to test whether exogenous expression of Rab11-GFP could rescue the defect in E-cadherin trafficking, we transfected SqCC/Y1 Vect Ct and Dsg3Δ458 cells with Rab11-GFP construct and then performed the E-cadherin recycling assay 2 d later. Cells without the transfection were analyzed alongside as a comparison. After cadherin recycling, all the fixed cells were labeled with anti-GFP and counterstained with DAPI. Compared to Dsg3Δ458 cells without transfection, overexpression of Rab11-GFP in these mutant cells resulted in a significant reduction of E-cadherin with its fluorescence intensity almost comparable to the matched Vect Ct cells with the transfection (Fig. 7B). In summary, this result indicated an enhanced cadherin recycling in Dsg3Δ458 cells after transfection of Rab11-GFP, or in other words, transfection of Rab11-GFP was able to restore the reduced E-cadherin recycling in Dsg3Δ458 mutant, implying that the defect of cadherin trafficking in mutant cells was indeed partly associated with altered expression of Rab11.
Figure 7.
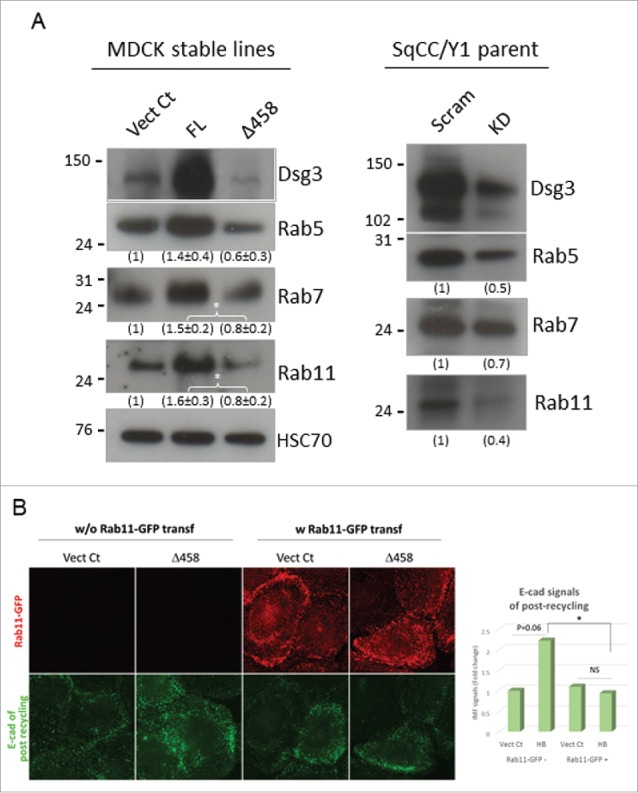
Altered expression of Rab GTPases was shown in cells with either Dsg3ΔC expression or Dsg3 depletion. (A) Western blotting analysis of Rab5, Rab7 and Rab11 in total lysates of MDCK-Vect Ct, FL and Δ458 lines and also SqCC/Y1 parental cells treated with scrambled or Dsg3 siRNA. A trend of reduction of 3 Rabs was shown in Dsg3ΔC cells or cells with Dsg3 knockdown compared to the respective control cells. By contrast, an increase of all 3 Rabs was detected in MDCK FL cells with overexpression of Dsg3, compared to control. The densitometry data were shown underneath each blot (left: n = 3, *p < 0.05; right: n = 2). (B) E-cadherin recycling in SqCC/Y1-Vect Ct and Δ458 cells without or with transfection of Rab11-GFP. The result was a representative of 2 independent attempts (*p < 0.05) and showed a reduction of E-cadherin signals in Δ458 cells with transfection of Rab11-GFP compared to that without Rab11-GFP transfection.
Altogether, these findings support our hypothesis that Dsg3 plays a role in regulating E-cadherin trafficking, and disruption of native Dsg3 would cause retardation of this process leading to a defect in E-cadherin and desmosomal cadherin junction assembly and stabilization and ultimately a failure in cell-cell adhesion. Our results also suggest that the observed retardation in cadherin trafficking could be, at least in part, due to the altered expression and activity of Rab5, Rab7 and Rab11, the key regulators for cadherin membrane trafficking.12,15
Both Dsg3ΔC mutant expression and Dsg3 knockdown caused enhanced cell spreading due to defect of the cortical actin formation
E-cadherin plays an essential role in intercellular junction formation, cell polarization and epithelial morphogenesis, via a link to the cortical actin cytoskeleton. If E-cadherin adhesion is affected the other intercellular junctions, such as desmosomes and tight junctions, will not form properly and thus epithelial cell polarization and morphogenesis cannot be achieved, resulting in cell flattening and abnormal spreading. To confirm this is the case in the Dsg3ΔC mutant, we analyzed cell size (area) in various epithelial lines, including SqCC/Y1, MDCK and A431 stable lines and also in cells with Dsg3 knockdown. Cells were seeded on coverslips and labeled for F-actin with A488 conjugated phalloidin. Figure 8A showed the results of SqCC/Y1 and MDCK (data for A431 not shown) and consistently, both Dsg3 knockdown and the Dsg3ΔC expression caused a significant increase of cell size (≥ 3-fold), relative to the respective control cells. Furthermore, to verify whether this effect was cell density dependent, MDCK-Vect Ct and Δ458 cells were seeded at a series of densities with the highest approaching a confluent monolayer. Cells were fixed and stained with A488-phalloidin before analysis of cell size. Result indicated that except for the most sparse cultures where there was little cell-cell contact, cells in mutant populations exhibited increased cell size at higher densities compared to counterparts of control cells (Fig. 8A), suggesting that the retardation in E-cadherin junction formation in Dsg3ΔC cells had a negative impact on cortical actin organization, epithelial polarization and morphogenesis. Indeed, quantitation analysis of peripheral fluorescent intensities of F-actin staining (Fig. 8B) demonstrated a decrease of the cortical actin formation in mutant cells in both sub-confluent and confluent cultures (data not shown), similar to that shown in Figure S2B as well as the cells with Dsg3 depletion.10
Figure 8.

Dsg3 knockdown and Dsg3ΔC expression caused an increase of cell size and abnormal spreading. (A) Quantitation of cell size (area) in SqCC/Y1 and MDCK cells with Dsg3 knockdown (KD) in parental cells or with stable expression of Dsg3Δ458 protein. A significant increase of cell size (≥ 3-fold) was consistently observed in both KD and Dsg3ΔC cells compared to the respective controls. Right graph shows result of MDCK Vect Ct and Δ458 cells that were seeded at a series of densities with the highest reaching confluent monolayer. All cells were subject to calcium switch for 3 hrs before F-actin staining with A488-phalloidin. Significantly increased cell size was readily detectable in Δ458 at all seeding densities except for the sparsest cultures, compared to controls. (n > 100 cells/sample, mean ± SD, *p < 0.05, **p < 0.01, ***p < 0.001) (B) Fluorescent images of F-actin staining in sub-confluence (Sub-conf) and confluence (Conf) MDCK cells subjected to calcium switch for 3 hrs. Augmented cell size coupled with reduced F-actin periphery staining (quantitation data not shown) was shown in Δ458 cells compared to control in both cell densities. Overall, an enhanced cortical F-actin formation was seen in confluent cultures relative to sub-confluence in both conditions. Scale bar, 10 µm.
There are 2 distinct populations of actin (junctional actin and peripheral thin bundles) that are detectable at the early phase of junction formation.22 To examine whether the Dsg3ΔC expression causes any defect in these 2 populations of actin, we analyzed actin assembly in various SqCC/Y1 lines that were subject to a calcium switch for 1 hr. Cells were double labeled for F-actin and Dsg3 and the fluorescence intensities of both protein staining at the interface of cell-cell contacts were quantified in ImageJ using a line scan across the junctions that extended into the peripheral actin bundles. The profiles of 12 line scans for both protein staining in each sample were measured and the result showed that a pool of Dsg3 (extra-desmosome) colocalized with the junctional actin in control cells and this appeared to be significantly increased in FL cells (Fig. 9A). In remarkable contrast, such a colocalization was vastly attenuated in Dsg3Δ mutants compared to control cells. To further verify that the actin assembly was indeed affected by Dsg3Δ mutant expression, we performed the G-actin incorporation assay in live cells. In this case, cells were saponin-permeabilized in buffer containing 0.45 µM Alexa 488-conjugated G-actin in live and this procedure favors the barbed end incorporation.23 Finally, cells were fixed and stained for F-actin with Rhodamine-phalloidin. Analysis of both newly formed G-actin and steady state of cortical F-actin at cell periphery showed a marked decrease in G-actin incorporation at the barbed end and a reduction of cortical F-actin in Dsg3ΔC mutant compared to control cells (Fig. 9B). Collectively, the findings in this current study and also from our previous publications 8-10 support the notion that a pool of Dsg3 (extra-desmosome) cross talks with E-cadherin and regulates E-cadherin and its associated cortical actin cytoskeleton. Loss of Dsg3 not only affects E-cadherin junction but also it's associated cortical actin filaments that in turn has a negative impact on other junction formation, such as desmosomes, and thus ultimately causing a defect in epithelial cell polarity.
Figure 9.
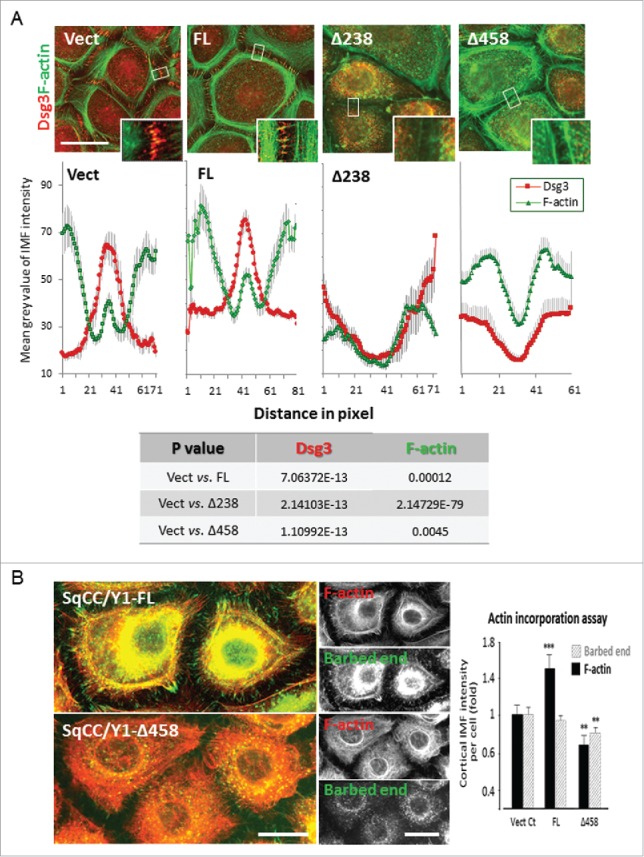
Dsg3 is required for cortical F-actin assembly. (A) Quantitation of Dsg3 and F-actin staining at the junctions in cells with 1 hr calcium switch with ImageJ by a line scan tool across the junctions (the line width set at 50 pixels and the line length at 60 pixels as indicated by small white boxes that cover the junctional and part of the peripheral bundles of actin). Dsg3 was found to be relocated at the interface between cell-cell contacts (large peak) along with the junctional actin (small peak) in Vect control cells and these were significantly enhanced in FL cells. In striking contrast, a significant reduction of both Dsg3 and actin was detected in mutants. Data were mean±s .e.m and the results of Student t-test is shown in the table below. Scale bar, 20 µm. (B) Actin incorporation assay indicated a significant decrease of newly formed G-actin near the junctions in mutant compared to control and FL cells (**p < 0.01). Live cells subjected to 1 hr calcium switch were saponin-permeabilized followed by incorporation of Alexa 488-G-actin as described in Materials and Methods. Total F-actin was labeled with Rhodamine-phalloidin after fixation. Again, FL cells showed an enhanced F-actin staining near the cell periphery (***p < 0.001) and in contrast, this was significantly reduced in mutant cells (**p < 0.01).
Discussion
This study adopted an alternative approach of expressing dominant negative mutants in addition to RNAi mediated knockdown of Dsg3 and demonstrates that loss of Dsg3 function in epithelial cells causes defects in E-cadherin trafficking and cell-cell adhesions that ultimately result in a failure of cell polarity (Fig. 10). We showed that changes in several aspects of cellular processes may contribute to these defects: 1) alteration in the E-cadherin and Dsg3 junctional complex formation, 2) aberrant expression of Rab proteins and retardation of E-cadherin and Dsg3 membrane trafficking (internalization and recycling), and 3) disruption of cortical F-actin formation and organization. Thus, this report provides several lines of evidence that the modulation of Dsg3 can have detrimental effect in E-cadherin adhesion and stabilization, a prerequisite for desmosome formation that confers strong cell-cell adhesion. As a consequence, the intercellular junctions cannot form properly and the cells failed to polarize with a result of abnormal flattening. Together, the results from this study are consistent with the notion that Dsg3 cross talks with E-cadherin and regulates its adhesive function.
Figure 10.

A model that summarizes the major findings of the study. E-cadherin trafficking is required for normal cell-cell adhesions and junction stability, and this process seemed to be Dsg3-dependent. In cells with loss of Dsg3 function, either by RNAi mediated knockdown or by the expression of Dsg3ΔC dominant negative mutant, cell-cell adhesion is disrupted due to a lack of surface junction protein assembly, impaired cadherin complex formation and their association with the actin cytoskeleton, and a retardation of cadherin trafficking that is caused, at least in part, by the altered expression of Rab proteins. As a consequence, cell-cell junction cannot form properly resulting in augmented cell flattening and a failure in cell polarization.
The important role of Dsg3 in cell-cell adhesion has been underscored in the study of pemphigus, an autoimmune blistering disease in which circulating autoantibodies in patients bind to Dsg3 on keratinocytes in oral mucosa and skin, and cause disruption of cell-cell adhesion leading to severe mucosal erosion and epidermal blistering.20,24 It has been demonstrated that autoantibodies targeting Dsg3 induce rapid internalization of such an immune complex resulting in depletion of Dsg3 from the desmosomes as well as the surface of keratinocytes, indicating a coordinated response of Dsg3 endocytosis and disruption of cell-cell adhesions.25,26 In line with this, we have shown that RNAi mediated Dsg3 depletion in epithelial cells, including HaCaT and MDCK, induces disruption of cell cohesion accompanied with a reduction of cell height.10,19 However, the underlying mechanism remains not fully elucidated. To further verify the role of Dsg3 in cell-cell adhesion, as a supplementary approach in addition to RNAi mediated Dsg3 depletion, in the present study we stably expressed the C-terminally truncated mutants of Dsg3 with dominant negative action in various epithelial cell systems and analyzed the cell-cell adhesion, junctional protein expression and dynamic process, such as cadherin membrane trafficking, using a combination of immunofluorescence and biochemical methods. A similar approach with the truncated form of classical cadherins has been successfully demonstrated before in an attempt to study their function in Xenopus development, osteoblast differentiation, junction formation of epithelial cells and cardiomyocytes.27-30
We showed that the Dsg3ΔC mutant expression in epithelial cells also caused weakening in cell-cell adhesion strength (Fig. 1), analogous to that of Dsg3 depletion.19 Although the actual mechanisms of junction disruption in cells with Dsg3 knockdown and mutant expression may differ, both situations elicit loss and disruption of native Dsg3, leading to similar consequences. Apparently, RNAi mediated Dsg3 depletion causes defect in protein complex formation and junction assembly,7,9,10,19 the expression of Dsg3 mutant might be more detrimental due to the disruption not only inside the cells but also on the surface through binding of Dsg3 by secreted mutant as demonstrated in Figure S1C. It is speculated that the dominant negative function of Dsg3ΔC is probably achieved by competing for dimerization or clustering with native Dsg3 after synthesis in the cytoplasm therefore causing impairment of the junctional complex formation. As Dsg3ΔC lacks the transmembrane domain it is plausible that the dimerization or clustering with native Dsg3 was likely via a type of oligomer formation with native Dsg3 through the extracellular domain. To elucidate the underlying mechanism, we analyzed Dsg3ΔC mutant expression in conjunction with native Dsg3 by immunofluorescence and observed aberrant Dsg3 distribution at the junctions (Fig. 2A). Importantly, the colocalization of Dsg3ΔC mutant and endogenous Dsg3 was indeed detectable within the broad zone of punctate staining, consistent with the notion of dimerization between Dsg3ΔC mutant and native Dsg3 (homodimer formation via extracellular domain). If such an oligomer formation exists in cells, one would expect that Dsg3ΔC mutants could compete with native Dsg3 in the process of junction assembly and thus have a negative impact on Dsg3 associated junctional complex formation that involves both Pg and p120. Indeed, we have provided several lines of evidence in this study that support this notion. First, we showed a decrease in cytoskeleton association of Pg and p120, in addition to endogenous Dsg3 (Triton insoluble fraction) in mutant cells, compared to control, during the process of calcium induced junction formation (Fig. 4A). Secondly, we demonstrated a reduction of surface expression of Pg and p120, along with several cadherins (Dsg2/3 and E-cadherin) analyzed, in mutant cells (Fig. 4B), indicating a defect of surface junction protein assembly. Thirdly, an attenuated expression of Pg and/or p120 was shown in the immune complex of mutant cells purified with anti-Dsg3 and anti-E-cadherin antibodies, respectively (Fig. 5). The reduced expression of Pg and p120 at the cell borders with concomitant cytoplasmic accumulation was also readily detectable in mutant cells in both SqCC/Y1 and MDCK by immunofluorescence. The reduced expression of Pg, but not β-catenin, in the E-cadherin complex of mutant cells suggests that Pg, but not β-catenin that associates with E-cadherin, is affected by the Dsg3 mutant expression and its competition with native Dsg3 for dimerization with E-cadherin. Given that Pg is preferentially binding to Dsg3 with high affinity,31 the Dsg3Δ458 mutant expressions in cells can compete with native Dsg3 for binding to E-cadherin and thus causes a reduction of the association of Pg with E-cadherin. Although we have previously reported that Dsg3 forms a complex with E-cadherin and in addition, we showed that both Pg and p120 were required for this complex formation,8,9 the exact nature of their interaction remains not fully understood. In the present study, we provide evidence suggesting a possibility that the interaction between these 2 cadherins could well be via their extracellular domains, which warrants further investigation. Evidence of inter-cadherin interaction between E-cadherin and Dsg, via EC3 and EC4, has been demonstrated before in A431 cells but it was shown to be induced by EGTA.32 In addition, our study does not rule out a possibility of Dsg3ΔC mutant secretion into the culture media that potentially bind to cadherins on the surface leading to the disruption of cell-cell adhesion by steric hindrance as we observed here (Fig. S1). Although the definite proof of mutant secretion awaits further study, the observed disruption in Dsg3 and E-cadherin adhesions in HaCaTs treated with the conditioned media of Dsg3Δ458 implies that this mechanism might exist. The detected strong band of ∼95 kDa in conditioned media from Dsg3Δ458 culture by co-IP analysis is larger than expected (∼67 kDa) and this could well be due to the protein glycosylation of the extracellular domain (Fig. S1B).
The effect of Dsg3ΔC expression on junction assembly for E-cadherin and native Dsg3 was analyzed by immunofluorescence in cells subjected to a time course of calcium switching. In control cells, we observed that E-cadherin was assembled at the junctions within 1 hr of calcium switch and enhanced over time for up to 6 hours (Fig. 3). In contrast, this process was modulated in mutant cells, especially at early time points of 1 hr of calcium addition. Similar findings were also seen for native Dsg3 in mutant cells (Fig. S2), suggesting their coordination in junction assembly. The complex formation of Dsg3 and E-cadherin along with Src has been shown previously from our studies8,9 and it is worth noting that recently, another independent study has also demonstrated the existence of such a complex in HaCaT cells and its requirement for desmosome assembly and adhesion.7 Therefore, the cooperation of these 2 cadherins seems to be a key in epithelial junction formation. During the process of junction assembly and cell polarization newly synthesized cadherins are transported to cell surface while coupled to its associated catenins in the Triton insoluble, cytoskeleton associated pool in cells. To further verify the defective junction formation in mutant cells, we performed Western blotting analysis in Triton soluble and insoluble fractions (cytoskeleton associated pool), respectively, and showed a reduction of junctional proteins including Dsg3, Pg and p120 in Triton insoluble fraction of mutant cells, compared to control (Fig. 4A and B), indicating a perturbation of their junction assembly as well as the association with cytoskeleton. Furthermore, the analysis of surface biotinylated proteins revealed a significant reduction of surface cadherins, including Dsg3 and E-cadherin, as well as Pg and p120-catenins in mutant cells compared to control, and such a reduction was accompanied with increased levels of internal unbound proteins, indicating accumulation and redundancy of these proteins in the cytoplasm (Fig. 4C). Finally, our co-IP analysis of E-cadherin and Dsg3 complexes demonstrated deficiency of Pg/p120-catenins associated with cadherins in mutant cells (Fig. 5). Altogether, these results suggest that the Dsg3ΔC mutant expression has a negative impact on the function of native Dsg3 and E-cadherin, the evidence that supports the notion that Dsg3 regulates E-cadherin adhesion. The cross talk between Dsg3 and E-cadherin is still an issue under debate, however, our data8,9 as well as that of another independent study7 supports the notion that there is a pool of Dsg3 involved in the association with E-cadherin that is crucial for junction formation and stabilization.
The adherens junction formation is a prerequisite for other junction assembly, including desmosomes,7 and has been demonstrated to be the key in establishing cell polarity and inhibition of cell spreading such as that in endothelial cells.33 Here we analyzed cell size in various epithelial cell lines, SqCC/Y1, MDCK and A431, with Dsg3 depletion or the Dsg3ΔC mutant expression. Consistently, we observed marked increase of cell size in all epithelial lines with the loss of Dsg3 function as opposed to the respective control cells (Fig. 8A). It is worth noting that this augmented cell size is cell density-dependent with a significant increase in populations with relatively high densities where cell-cell contacts occur with high frequencies, suggesting that Dsg3 is required for proper function of E-cadherin and its mediated other junction formation. This finding was in line with defect in cortical actin formation associated with the loss of Dsg3 (Fig. 9A),10 and this was further supported by the actin incorporation assay that demonstrated a reduced G-actin incorporation at the barbed ends during actin polymerization (Fig. 9B). Correspondingly, enhanced G-actin incorporation at the barbed ends was detected in Dsg3 overexpressing cells. We have reported that non-junctional Dsg3 regulates Src and Rac1/Cdc42 that control the F-actin organization and dynamics and thus it is likely that the activities of these signaling pathways were affected in Dsg3ΔC mutant cells.8-10 Collectively, these observations suggest strongly that loss of Dsg3 has a significant impact on contact inhibition of cell spreading and induction of cell polarity, the processes that require coordination of adherens junctions and desmosomes as well as proper organization of cortical actin cytoskeleton.
Our observation of reduced surface protein expression in mutant cells suggests that the protein kinetic might be altered. Having established Dsg3 as a prerequisite in junction formation and cell polarization, we investigated cadherin trafficking and to determine whether E-cadherin trafficking was indeed modulated in cells with the loss of Dsg3. E-cadherin trafficking has been emerging as one of the key regulators in junction formation and homeostasis and is characterized to be coordinated with many physiological processes, such as cell polarization and morphogenesis.15,18 Alteration of E-cadherin trafficking has been independently implicated in various diseases including cancers and EMT.12,18,34 Here, we analyzed cadherin trafficking in both SqCC/Y1 and MDCK cells with respect to Dsg3 expression and showed that both E-cadherin internalization and recycling were affected with retardation in cells that had disrupted Dsg3 function (Fig. 6). Similar finding was also observed for native Dsg3 in Dsg3ΔC cells (Fig. S5), suggesting a possibility of the coordinated process for both cadherins. In line with this, we observed their colocalisation in the endocytic pathway in control cells (Fig. S6). Importantly, we found that these alterations are likely caused, at least in part, by reduced expression and activity of Rab5, Rab7 and Rab11 (Figs. 7A and S7), the master regulators of protein membrane trafficking,14 and the transfection of Rab11-GFP construct in mutant cells appeared to rescue the defect of E-cadherin trafficking (Fig. 8B). Accordingly, we showed an increased expression of Rab proteins in FL cells compared to control cells (Fig. 9), and this could well be due to the enhanced Src signaling induced by overexpression of Dsg3, as reported previously.8,9,12 Alternatively, this finding may also suggest that Dsg3 somehow is involved in the regulation of Rab expression and thus could have a more global effect on protein trafficking. Although this requires further investigation, our findings provide the first evidence that Dsg3 may play a role in regulating the Rab protein expression. We have reported recently that Dsg3 is capable of regulating the transcription factor c-Jun/AP-1 11 and the evidence for JNK/AP-1 in regulating the Rab gene expression has been documented in Theileria-transformed B cells.35 Thus, it is possible that our observed change in the Rab protein expression was caused by JNK/AP-1 activation induced by Dsg3 and loss of Dsg3 function could consequently cause a reduction in the Rab expression and therefore the defects in cadherin trafficking, as we showed here in cells with Dsg3 knockdown or with its mutant expression. Rab proteins are small GTPases that cycle between GTP-and GDP bound status and serve as the scaffolds to integrate both the membrane trafficking and intracellular signaling in a temporally and spatially sensitive manner.36 The 4 major membrane trafficking steps, such as vesicle budding, cytoskeletal transport, vesicle tethering and fusion, all are involved in GTP-bound activated Rab proteins.37 Additionally, abnormality in the actin formation and organization can also contribute to the defect of cadherin trafficking in Dsg3ΔC mutant or cells with Dsg3 depletion as the actin cytoskeleton is involved in the process of membrane protein endocytosis and transport, including disassembly of desmosomes.38
Conclusions
Using a combination approach of RNAi mediated knockdown and dominant negative action of Dsg3ΔC mutant, this study demonstrates that Dsg3 acts as an important regulator in cell-cell adhesion and cell polarization, a prerequisite for epithelial differentiation and morphogenesis. This regulation involves E-cadherin membrane trafficking, junction complex formation and stability as well as the organization of actin cytoskeleton. Disruption of the Dsg3 function could result in detrimental effect in epithelial cells, as highlighted in pemphigus disease. Finally, our data also suggest that Dsg3ΔC mutants may serve as an alternative research tool in furthering our understanding of the role of Dsg3 in epithelial biology, including cell adhesion, differentiation, morphogenesis as well as migration.
Materials and methods
Antibodies and reagents
The following mouse monoclonal and rabbit polyclonal antibodies (Abs) were used: 5H10, mouse Ab against the N-terminus of Dsg3 (Santa Cruz); H145, rabbit anti-C-terminus of Dsg3 (Santa Cruz); 33–3D, mouse IgM against Dsg2 (gift from Professor Garrod); HECD-1, mouse anti-N-terminus of E-cadherin (Abcam); monoclonal Ab to mouse E-cadherin (Takara); A555 conjugated E-cadherin (24E10) rabbit mAb (Cell Signaling); rabbit anti-Myc tag (Cell signaling); PG 5.1, mAb to Plakoglobin (Progen); 6F9, monoclonal anti-β-Catenin mouse ascites fluid (Sigma); p120 (H-90), a rabbit polyclonal Ab (Santa Cruz Biotechnology, Inc.); Rabbit polyclonal to β-Actin - Loading Control (Abcam); GAPDH (14C10) rabbit mAb (Cell Signaling); rabbit anti-A488 Ab (Sigma); sampler kit of Rab Abs containing C8B1, rabbit mAb against Rab5; D95F2, rabbit mAb against Rab7; D4F5, rabbit mAb against Rab11 (Cell Signaling); Rab5 and Rab11 activity assay kit (Abcam).
Generation of retroviral constructs of Dsg3 C-terminally truncated mutants
The retroviral construct of full-length human Dsg3 cDNA tagged with a Myc epitope at C-terminus (pBABE-hDsg3.myc), as shown in Figure 1A, was generated before.8 Three C-terminally truncated mutants of Dsg3 (Dsg3Δ238, Dsg3Δ458 and Dsg3Δ560) (Fig. 1A) were generated by cutting out different length sequences in pBABE-hDsg3.myc construct at restriction sites between Bam HI and BstZ17I (Dsg3Δ238), Bam HI and HpaI (Dsg3Δ458), or Bam HI and BsgI (Dsg3Δ560), respectively. The cutting ends were either blunted with Klenow fragment and low concentration of dNTP (Dsg3Δ238) or with Klenow fragment (Dsg3Δ458 and Dsg3Δ560) and ligated with T4 DNA ligase. All three constructs were verified by sequencing.
Cell culture, calcium switch and siRNA transfection
Three epithelial cell types, SqCC/Y1 human oral buccal SCC line, A431 vulva SCC line and MDCK (Madin Darby canine kidney) simple epithelial cells, were included in the study and cell lines with stable transduction of full length (hDsg3.myc: FL) and truncated mutants (Dsg3Δ238, Dsg3Δ458 and Dsg3Δ560) of Dsg3 were generated in the laboratory. The matched control was the cells transduced with empty vector (pBABE-puro). All cell lines were generated following procedures as described previously.8 The SqCC/Y1 lines were maintained in EpiLife medium (with 60 μM calcium concentration) supplemented with a growth supplement EDGS (Epilife Defined Growth Supplement) and MDCK lines were routinely cultured in DMEM containing 10% FCS. The exogenous protein expression was determined by both mAb 5H10 directed to N-terminus of Dsg3 and the rabbit Myc tag Ab against to the Myc tag epitope located at C-terminus (Fig. 1A). Calcium induction of the junction formation was used routinely in almost all experiments to synchronize junction assembly. As for MDCK cells, when the cells reached to the desired confluence in growth medium (DMEM containing 10% FCS), they were treated with calcium-free medium (Ca2+ and Mg2+ free DMEM plus 5% decalcified FCS) for 30–60 minutes until they rounded up before replacement with growth medium for 2–4 hrs. For SqCC/Y1 cells, EpiLife was replaced with modified keratinocyte growth medium (DMEM:Ham's F12 3:1 supplemented by insulin (5 μg/ml) and hydrocortisone (1.4 μM) plus 10% FCS) for 2–4 hrs. Transient siRNA transfection was conducted by following the protocol described previously.9,10
Dispase dissociation assay
The cell-cell adhesive strength was analyzed by the dispase dissociation assay as reported previously.19 Briefly, cells were seeded at equal confluent density in 6-well plates. Once they reached confluent monolayers wells were washed with PBS twice and then incubated in 2 ml PBS containing dispase II (2.4 U/ml, Invitrogen) at 37°C for 20–30 minutes till the epithelial sheets began detaching. They were then subjected to mechanical stress by pipetting 5 times with a 1 ml pipette to break the epithelial sheets. Images were taken and the number of fragments for each sample was determined with ImageJ software. Five independent experiments were performed and pooled data was used for statistical analysis. P values less than 0.05 was reported as statistically significant.
ELISA assay
Ninety six-well plate was coated with anti-Dsg3 mAb 5H10 (1:1000 dilution) for overnight prior to blocking with 1% BSA. Then, 100 µl of conditioned media, harvested from 24 hr cultures of Vect Ct and mutant cells, in a series of dilutions were added into the wells alongside a negative (medium alone) and a positive control (mutant cell lysate), and incubated for 2 hrs at room temperature. After 2 washes with PBS, wells were incubated with rabbit anti-Myc Ab conjugated with Alkaline Phosphatase (Sigma, 1:1000) for 2 hrs before additional 2 washes. Finally, wells were incubated with 100 ul per well of the substrate p-Nitrophenyl phosphate (Sigma) for 20 min in the dark before the reaction was stopped by adding 50 ul of 3 M NaOH to each well. The optical density was measured by a plate reader (Fluostar Optima/BMG LABTECH) at 405 nm wavelength.
Immunofluorescence and microscopy
Cells seeded on coverslips and subjected to calcium switch for different time periods were fixed routinely in 4% formaldehyde for 10 min and then permeabilized with 0.1% Triton X–100 for 1–5 min at room temperature, according to the experiments. After two washes with PBS, the nonspecific binding sites were blocked for 15-30 minutes with 10% goat serum before the primary and then the secondary antibody incubation, each lasted for 1 hr at room temperature. For F-actin staining, Alexa Fluor 488 conjugated phalloidin was incubated in conjunction with the secondary antibodies. All antibodies were diluted in 10% goat serum. Coverslips were washed 3 times with washing buffer (PBS containing 0.2% Tween 20) after each antibody incubation. Finally, coverslips were counterstained with DAPI for 8–10 minutes. After a last wash, coverslips were mounted on slides. Images of fluorescent staining were acquired with a Leica epi-fluorescence microscope or confocal Zeiss710 and analyzed with ImageJ software.
Western blotting and co-immunoprecipitation
Protein extraction, Triton soluble and insoluble fractionation, Western blotting and co-immunoprecipitation were carried out as described previously.8,9
Protein concentration was determined by Bio-Rad DC protein assay as a routine. For Western blotting, 5–10 µg of total cell protein were resolved by SDS–PAGE. For co-immunoprecipitation, 1 mg protein lysate per sample or the 24 hrs cultured condition media, was used to purify protein complex and to incubate with 3–5 µg of primary antibody overnight at 4°C on rotation. Then, ∼1.7 × 105 protein-A conjugated beads (Invitrogen) were added and incubated for 1 hr at 4°C to capture the immune complex. After washing thoroughly (4x in RIPA buffer and 1x in TTBS) the precipitate was re-suspended in 2x Laemmli sample buffer and heated for 3 minutes at 95°C before resolved by SDS-PAGE followed by Western blotting procedures.
Antibody-based internalization and recycling assays
The internalization assay was carried out following the protocol reported by others21 with some modifications. In brief, cells were seeded on coverslips in 24-well plate at sub-confluent densities and after 1 day they were subjected to a calcium switch for 2–4 hours. The plate was removed from incubator, placed on ice and allowed to cool down. After a brief wash the surface E-cadherin or Dsg3 was labeled by incubating coverslips on ice with primary antibody HECD-1 or 5H10 for 30 min followed by anti-mouse A488 IgG for further 30 min. Antibodies were diluted (1:100) in growth medium supplemented with 30 mM HEPES (pH 7.4). Coverslips were washed before fresh growth medium supplemented with 30 mM HEPES (pH 7.4) was added. At this point one set of coverslips was fixed in 4% formaldehyde for 10 min for later analysis of surface antibody labeling. Another set was placed back in water bath at 18°C to allow internalization of E-cadherin or Dsg3 for 30 minutes. It has been described in previous studies that temperature of 18°C at this point enables the accumulation of internalized proteins in early or sorting endosomes by preventing them from progressing further down the endocytic or recycling pathways.17,39 After internalization, plates were lifted and cooled down on ice. The remaining surface antibodies were quenched by incubating coverslips, first in a low pH washing buffer (PBS containing 100 mM glycine, 20 mM magnesium acetate, and 50 mM potassium chloride, 3% bovine serum albumin (BSA), pH 2.2)5 for 30 minutes and then in rabbit anti-Alexa Fluor 488 antibody (1:100 dilution in growth medium) for 30 min.21 We found that this combined quenching procedure offered the best result in cultures we studied. Cells with the endocytosed antibodies were then fixed in 4% formaldehyde for 10 min at room temperature. All coverslips were permeabilized with 0.1% Triton X-100 buffer for 1 min followed by washing with PBS twice and nucleus counter stain with DAPI before mounting on slides. Besides, 2 negative controls were included here: to ensure that the surface quenching procedures efficiently removed all surface bound antibodies, a coverslip subjected to surface labeling followed by quenching straightaway was fixed. Another coverslip was subjected to surface labeling with the secondary antibody only to ensure that antibody staining was specific.
The recycling assay was carried out as described above for cadherin surface labeling, internalization and surface antibody quenching. After that, one set of coverslips was fixed for later quantitation of the internalized proteins. Another set was incubated in a water bath at 37°C for 30 min to allow the accumulated, internalized proteins in early or sorting endosomes to recycle back to surface. Then, the surface signals of recycled proteins were quenched again following the same procedures as described above. Finally, they were fixed and all coverslips were subject to permeabilization, washing and nucleus DAPI staining before mounting.
Images on each coverslip (> 4 images per sample) were acquired in arbitrary fields at the same exposure with a Leica epi-microscope. The fluorescence intensities were measured by ImageJ. The quantitation of internalized and recycled proteins was determined by taking the ratio of internalized fluorescence signals relative to mean surface signals (% internalized = 100*(IMFinternalised/ IMFsurface) or by calculating the reduction of internalized fluorescence signals after recycling and surface quenching procedures (% recycling = 100*(IMFpre-recycling – IMFpost-recycling)/ IMFpre-recycling).
Cell surface protein isolation
The surface protein analysis was carried out by using the Pierce Cell Surface Protein Isolation Kit (Thermo Scientific) and the protocol provided by the manufacturer. Briefly, cells were grown in 4x T75 cm2 tissue culture flasks until ∼90% confluent. Then, they were subjected to a calcium switch for 3 hrs. After washed twice with ice-cold PBS cells were incubated with 10 ml of biotin solution (Sulfo-NHS-SS-Biotin in ice cold PBS) per flask on a rocking platform for 30 min at 4°C. The biotinylation reaction was quenched by adding 500 μl of Quenching Solution. Cells were washed with ice-cold PBS, harvested by gentle scraping and pelleted by centrifugation at 500 × g for 3 min. Cell pellet was lysed on ice for 30 min with 500 µl of lysis buffer containing protease inhibitor cocktail (Sigma) with intermittent vortexing and sonicating for 5×2 sec bursts. The lysate was then centrifuged at 10,000 × g for 2 min at 4°C and the clarified lysate was used for purification of biotinylated proteins on NeutrAvidin Agarose. Before use, 500 μl of NeutrAvidin Agarose slurry was washed 3 times with wash buffer before incubation with lysate for 2 hrs at 4°C to allow the biotinylated proteins to bind to the NeutrAvidin Agarose. The unbound lysate, representing the intracellular proteins, was collected by centrifugation of the column at 1,000 × g for 2 min and stored at −20°C. The captured surface proteins by NeutrAvidin Agarose were washed repetitively to remove unbound proteins and finally were eluted from the Agarose by incubation in SDS-PAGE sample buffer plus 50 mM DTT for 60 min at room temperature on a rocking platform. After a final centrifugation for 2 min at 1000 × g, a trace amount of bromophenol blue was added to elute. Finally, both surface biotinylated and internal unbiotinylated proteins were analyzed side by side by Western blotting.
Rab GTPase activity assay
The activity of Rab5 and Rab11 were analyzed by using the Rab activity assay kit (Abcam). Cells grown to approximately 80–90% confluence in T75 flasks were washed twice with ice-cold PBS before lysed in 1X lysis buffer on ice. After 10–20 min cells were detached from the flasks by scraping with cell scrapers. Lysates were transferred to a micro-centrifuge tube and centrifuged for 10 min at 12,000 × g at 4°C. Then, supernatant was collected and protein concentration for each sample was determined. One mg protein lysate in 500 µl for each sample was used. For negative and positive controls, 20 µL of 0.5 M EDTA was added to both samples, followed by addition of 5 µl of 100X GDP for negative control and 5 µl of 100X GTP gamma S for positive control. The tubes were incubated for 30 min at 30°C with agitation. Then, loading was stopped by adding 32.5 µl of 1 M MgCl2 to both control samples. For the tested samples, lysates were added with 1 µl of anti-active Rab5 or Rab11 monoclonal antibody and 20 µl of bead slurry (protein A/G agarose beads). All samples were incubated at 4°C for 1 hr with gentle agitation. Beads were pelleted by centrifugation for 1 min at 5,000 × g and washed 3 times. Finally, beads were re-suspended in 20 µl of 2X SDS-PAGE sample buffer, boiled for 5 min and centrifuged briefly at 5,000 × g before Western blotting analysis.
Cell spreading assay
For this assay, the cell plating, fluorescence staining and microscopy were performed as described earlier. For the cell density experiment with MDCK, cells were seeded on coverslips at various densities at a series of dilutions of 1 in 2, with the highest reaching 100% confluence for overnight. They were then subject to calcium depletion and repletion for 3 hours before fixation and F-actin staining with A488-phalloidin and DAPI. F-actin staining was used to measure the cell spreading (area) with ImageJ by setting up a threshold that include all area covered by cells in each image, whereas DAPI channel was used for quantitation of cell number. The average cell area in each image was calculated in an Excel spreadsheet by dividing the total area with cell number for each image. Overall, more than 6 images were acquired in each sample in all experiments, and the resulting data were presented as mean ± SD in the figures.
Statistical analysis
Two tailed Student's t-Test was routinely used for all comparison analyses between different groups, and p < 0.05 was considered to be statistically significant.
Supplementary Material
Abbreviations
- Abs
antibodies
- AP
alkaline phosphatase
- BSA
bovine serum albumin
- co-IP
co-immunoprecipitation
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco's Modified Eagle's medium
- Dsc
desmocollin
- Dsg
desmoglein
- DTT
Dithiothreitol
- ELISA
the enzyme-linked immunosorbent assay
- EMT
epithelial to mesenchymal transition
- FCS
fetal calf serum
- FL
full length
- GDP
Guanosine 5′-diphosphate
- GFP
green fluorescent protein
- GTP
Guanosine-5′-triphosphate
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IMF
immunofluorescence
- mAb
monoclonal antibody
- MDCK
Madin Darby canine kidney
- PBS
phosphate buffer saline
- RNAi
RNA interference
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- TTBS
Tween 20 Tris buffered saline
- Vect Ct
empty vector control
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Angray Kang for providing antibody reagent, Louise Brown and Teck Teh for technique help and Siu Man Tsang for assistance with generation of stable lines. HM was supported by the Libya embassy. All work was conducted in The Institute of Dentistry, Barts and The London School of Medicine and Dentistry, Queen Mary University of London.
Author contributions
Conceived and designed the experiments: HW HM KD LL SK LB. Performed the experiments: HM KD JU HW. Analyzed the data: HW HM KD EHA JU. Contributed reagents/materials/analysis tools: LL SK LB, Wrote the paper: HW HM.
References
- [1].Hennings H, Holbrook KA. Calcium regulation of cell-cell contact and differentiation of epidermal cells in culture. An ultrastructural study. Exp. Cell Res 1983; 143 (1):127-42; PMID:6186504; https://doi.org/ 10.1016/0014-4827(83)90115-5 [DOI] [PubMed] [Google Scholar]
- [2].Watt FM, Mattey DL, Garrod DR. Calcium-induced reorganization of desmosomal components in cultured human keratinocytes. J Cell Biol 1984; 99 (6):2211-5; PMID:6209289; https://doi.org/ 10.1083/jcb.99.6.2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Garrod D, Chidgey M. Desmosome structure, composition and function. Biochim Biophys Acta 2008; 1778 (3):572-87; PMID:17854763; https://doi.org/ 10.1016/j.bbamem.2007.07.014 [DOI] [PubMed] [Google Scholar]
- [4].Kanno M, Isa Y, Aoyama Y, Yamamoto Y, Nagai M, Ozawa M, Kitajima Y. P120-catenin is a novel desmoglein 3 interacting partner: identification of the p120-catenin association site of desmoglein 3. Exp Cell Res 2008; 314(8):1683-92; PMID:18343367; https://doi.org/ 10.1016/j.yexcr.2008.01.031 [DOI] [PubMed] [Google Scholar]
- [5].Nanes BA, Chiasson-MacKenzie C, Lowery AM, Ishiyama N, Faundez V, Ikura M, Vincent PA, Kowalczyk AP. p120-catenin binding masks an endocytic signal conserved in classical cadherins. J Cell Biol 2012; 199 (2):365-80; PMID:23071156; https://doi.org/ 10.1083/jcb.201205029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Perez-Moreno M, Fuchs E. Guilt by association: what p120-catenin has to hide. J Cell Biol 2012; 199(2):211-4; PMID:23071151; https://doi.org/ 10.1083/jcb.201209014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Rotzer V, Hartlieb E, Vielmuth F, Gliem M, Spindler V, Waschke J. E-cadherin and Src associate with extradesmosomal Dsg3 and modulate desmosome assembly and adhesion. Cell Mol Life Sci 2015; 72:4885-97; PMID:26115704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Tsang SM, Liu L, Teh MT, Wheeler A, Grose R, Hart IR, Garrod DR, Fortune F, Wan H. Desmoglein 3, via an interaction with E-cadherin, is associated with activation of Src. PLoS One 2010; 5(12):e14211; PMID:21151980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Tsang SM, Brown L, Lin K, Liu L, Piper K, O'Toole EA, Grose R, Hart IR, Garrod DR, Fortune F, Wan H. Non-junctional human desmoglein 3 acts as an upstream regulator of Src in E-cadherin adhesion, a pathway possibly involved in the pathogenesis of pemphigus vulgaris. J Pathol 2012; 227(1):81-93; PMID:22294297; https://doi.org/ 10.1002/path.3982 [DOI] [PubMed] [Google Scholar]
- [10].Tsang SM, Brown L, Gadmor H, Gammon L, Fortune F, Wheeler A, Wan H. Desmoglein 3 acting as an upstream regulator of Rho GTPases, Rac-1/Cdc42 in the regulation of actin organisation and dynamics. Exp Cell Res 2012; 318 (18):2269-83; PMID:22796473; https://doi.org/ 10.1016/j.yexcr.2012.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Brown L, Waseem A, Cruz IN, Szary J, Gunic E, Mannan T, Unadkat M, Yang M, Valderrama F, O'Toole EA, Wan H. Desmoglein 3 promotes cancer cell migration and invasion by regulating activator protein 1 and protein kinase C-dependent-Ezrin activation. Oncogene 2014; 33 (18):2363-2374; PMID:23752190; https://doi.org/ 10.1038/onc.2013.186 [DOI] [PubMed] [Google Scholar]
- [12].Palacios F, Tushir JS, Fujita Y, Souza-Schorey C. Lysosomal targeting of E-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions. Mol Cell Biol 2005; 25 (1):389-402; PMID:15601859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Funakoshi S, Kong J, Crissey MA, Dang L, Dang D, Lynch JP. Intestine-specific transcription factor Cdx2 induces E-cadherin function by enhancing the trafficking of E-cadherin to the cell membrane. Am J Physiol Gastrointest Liver Physiol 2010; 299(5):G1054-67; PMID:20671195; https://doi.org/ 10.1152/ajpgi.00297.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schwartz SL, Cao C, Pylypenko O, Rak A, Wandinger-Ness A. Rab GTPases at a glance. J Cell Sci 2007; 120 (Pt 22):3905-10; PMID:17989088; https://doi.org/ 10.1242/jcs.015909 [DOI] [PubMed] [Google Scholar]
- [15].Desclozeaux M, Venturato J, Wylie FG, Kay JG, Joseph SR, Le HT, Stow JL. Active Rab11 and functional recycling endosome are required for E-cadherin trafficking and lumen formation during epithelial morphogenesis. Am J Physiol Cell Physiol 2008; 295 (2):C545-56; PMID:18579802; https://doi.org/ 10.1152/ajpcell.00097.2008 [DOI] [PubMed] [Google Scholar]
- [16].Lock JG, Stow JL. Rab11 in recycling endosomes regulates the sorting and basolateral transport of E-cadherin. Mol Biol Cell 2005; 16(4):1744-55; PMID:15689490; https://doi.org/ 10.1091/mbc.E04-10-0867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Le TL, Yap AS, Stow JL. Recycling of E-cadherin: a potential mechanism for regulating cadherin dynamics. J Cell Biol 1999; 146(1):219-32; PMID:10402472; https://doi.org/ 10.1083/jcb.146.1.219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Delva E, Kowalczyk AP. Regulation of cadherin trafficking. Traffic 2009; 10 (3):259-67; PMID:19055694; https://doi.org/ 10.1111/j.1600-0854.2008.00862.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mannan T, Jing S, Foroushania SH, Fortune F, Wan H. RNAi-mediated inhibition of the desmosomal cadherin (desmoglein 3) impairs epithelial cell proliferation. Cell Prolif 2011; 44(4):301-10; PMID:21702856; https://doi.org/ 10.1111/j.1365-2184.2011.00765.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell 1991; 67 (5):869-77; PMID:1720352; https://doi.org/ 10.1016/0092-8674(91)90360-B [DOI] [PubMed] [Google Scholar]
- [21].Arjonen A, Alanko J, Veltel S, Ivaska J. Distinct recycling of active and inactive beta1 integrins. Traffic 2012; 13 (4):610-25; PMID:22222055; https://doi.org/ 10.1111/j.1600-0854.2012.01327.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang J, Betson M, Erasmus J, Zeikos K, Bailly M, Cramer LP, Braga VM. Actin at cell-cell junctions is composed of two dynamic and functional populations. J Cell Sci 2005; 118(Pt 23):5549-62; PMID:16291727; https://doi.org/ 10.1242/jcs.02639 [DOI] [PubMed] [Google Scholar]
- [23].Kovacs EM, Verma S, Ali RG, Ratheesh A, Hamilton NA, Akhmanova A, Yap AS. N-WASP regulates the epithelial junctional actin cytoskeleton through a non-canonical post-nucleation pathway. Nat Cell Biol 2011; 13 (8):934-43; PMID:21785420 [DOI] [PubMed] [Google Scholar]
- [24].Karpati S, Amagai M, Prussick R, Stanley JR. Pemphigus vulgaris antigen is a desmosomal desmoglein. Dermatology 1994; 189(Suppl 1):24-26; PMID:8049558; https://doi.org/ 10.1159/000246923 [DOI] [PubMed] [Google Scholar]
- [25].Calkins CC, Setzer SV, Jennings JM, Summers S, Tsunoda K, Amagai M, Kowalczyk AP. Desmoglein endocytosis and desmosome disassembly are coordinated responses to pemphigus autoantibodies. J Biol Chem 2006; 281(11):7623-34; PMID:16377623; https://doi.org/ 10.1074/jbc.M512447200 [DOI] [PubMed] [Google Scholar]
- [26].Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Invest Dermatol 1999; 112 (1):67-71; PMID:9886266; https://doi.org/ 10.1046/j.1523-1747.1999.00463.x [DOI] [PubMed] [Google Scholar]
- [27].Hertig CM, Eppenberger-Eberhardt M, Koch S, Eppenberger HM. N-cadherin in adult rat cardiomyocytes in culture. I. Functional role of N-cadherin and impairment of cell-cell contact by a truncated N-cadherin mutant. J Cell Sci 1996; 109(Pt 1):1-10; PMID:8834785 [DOI] [PubMed] [Google Scholar]
- [28].Levine E, Lee CH, Kintner C, Gumbiner BM. Selective disruption of E-cadherin function in early Xenopus embryos by a dominant negative mutant. Development 1994; 120 (4):901-9; PMID:7600966 [DOI] [PubMed] [Google Scholar]
- [29].Fujimori T, Takeichi M. Disruption of epithelial cell-cell adhesion by exogenous expression of a mutated nonfunctional N-cadherin. Mol Biol Cell 1993; 4 (1):37-47; PMID:8443408; https://doi.org/ 10.1091/mbc.4.1.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Cheng SL, Shin CS, Towler DA, Civitelli R. A dominant negative cadherin inhibits osteoblast differentiation. J Bone Miner Res 2000; 15 (12):2362-70; PMID:11127201 [DOI] [PubMed] [Google Scholar]
- [31].Andl CD, Stanley JR. Central role of the plakoglobin-binding domain for desmoglein 3 incorporation into desmosomes. J Invest Dermatol 2001; 117(5):1068-74; PMID:11710914; https://doi.org/ 10.1046/j.0022-202x.2001.01528.x [DOI] [PubMed] [Google Scholar]
- [32].Troyanovsky RB, Klingelhofer J, Troyanovsky S. Removal of calcium ions triggers a novel type of intercadherin interaction. J Cell Sci 1999; 112 (Pt 23):4379-87; PMID:10564655 [DOI] [PubMed] [Google Scholar]
- [33].Nelson CM, Pirone DM, Tan JL, Chen CS. Vascular endothelial-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol Biol Cell 2004; 15 (6):2943-53; PMID:15075376; https://doi.org/ 10.1091/mbc.E03-10-0745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ye Y, Tellez JD, Durazo M, Belcher M, Yearsley K, Barsky SH. E-cadherin accumulation within the lymphovascular embolus of inflammatory breast cancer is due to altered trafficking. Anticancer Res 2010; 30(10):3903-10; PMID:21036701 [PubMed] [Google Scholar]
- [35].Lizundia R, Chaussepied M, Naissant B, Masse GX, Quevillon E, Michel F, Monier S, Weitzman JB, Langsley G. The JNK/AP-1 pathway upregulates expression of the recycling endosome rab11a gene in B cells transformed by Theileria. Cell Microbiol 2007; 9 (8):1936-45; PMID:17388783; https://doi.org/ 10.1111/j.1462-5822.2007.00925.x [DOI] [PubMed] [Google Scholar]
- [36].Cavalli V, Corti M, Gruenberg J. Endocytosis and signaling cascades: a close encounter. FEBS Lett 2001; 498(2–3):190-6; PMID:11412855; https://doi.org/ 10.1016/S0014-5793(01)02484-X [DOI] [PubMed] [Google Scholar]
- [37].Stein MP, Dong J, Wandinger-Ness A. Rab proteins and endocytic trafficking: potential targets for therapeutic intervention. Adv Drug Deliv Rev 2003; 55 (11):1421-37; PMID:14597139; https://doi.org/ 10.1016/j.addr.2003.07.009 [DOI] [PubMed] [Google Scholar]
- [38].McHarg S, Hopkins G, Lim L, Garrod D. Down-regulation of desmosomes in cultured cells: the roles of PKC, microtubules and lysosomal/proteasomal degradation. PLoS One 2014; 9 (10):e108570; PMID:25291180; https://doi.org/ 10.1371/journal.pone.0108570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Czekay RP, Orlando RA, Woodward L, Lundstrom M, Farquhar MG. Endocytic trafficking of megalin/RAP complexes: dissociation of the complexes in late endosomes. Mol Biol Cell 1997; 8 (3):517-32; PMID:9188102; https://doi.org/ 10.1091/mbc.8.3.517 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.