

Abstract
Sodium-activated potassium (KNa) channels contribute to firing frequency adaptation and slow afterhyperpolarization. The KCNT1 gene (also known as SLACK) encodes a KNa subunit that is expressed throughout the central and peripheral nervous systems. Missense mutations of the SLACK C-terminus have been reported in several patients with rare forms of early onset epilepsy and in some cases severely delayed myelination. To date, such mutations identified in patients with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE), epilepsy of infancy with migrating focal seizures (EIMFS) and Otahara syndrome (OS) have been reported to be gain-of-function mutations (Villa & Combi, 2016). An exome sequencing study identified a p.Phe932Ile KCNT1 mutation as the disease-causing change in a child with severe early infantile epileptic encephalopathy and abnormal myelination (Vanderver et al., 2014). We characterized an analogous mutation in the rat Slack channel and unexpectedly found this mutation to produce a loss-of-function phenotype. In an effort to restore current, we tested the known Slack channel opener loxapine. Loxapine exhibited no effect, indicating that this mutation either caused the channel to be insensitive to this established opener or proper translation and trafficking to the membrane was disrupted. Protein analysis confirmed that while total mutant protein did not differ from wild type, membrane expression of the mutant channel was substantially reduced. Although gain-of-function mutations to the Slack channel are linked to epileptic phenotypes, this is the first reported loss-of-function mutation linked to severe epilepsy and delayed myelination.
Keywords: Kcnt1, Slack channel, early infantile epilepsy, leukoencephalopahy, mutation, loss-of-function
Introduction
The SLACK channel (also known as KNa1.1, Slo2.2) is a sodium-gated potassium channel encoded for by the KCNT1 gene. Upon activation by elevated intracellular Na+, high conductance of K+ through Slack channels contributes to firing frequency adaptation and slow afterhyperpolarization (Bhattacharjee & Kaczmarek, 2005; Kaczmarek, 2013). Numerous human genetic studies have identified missense mutations of the SLACK C-terminus and pore region in several patients with rare forms of early onset epilepsy and in some cases severely delayed myelination. To date, sixteen mutations identified in epileptic patients have been characterized as gain-of-function with increased amplitude of macroscopic currents (Villa & Combi, 2016). These mutations were identified in patients with autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE), epilepsy of infancy with migrating focal seizures (EIMFS) and Otahara syndrome (OS), early onset epilepsies that are most often resistant to treatment. Recently, Vanderver et al. (2014) identified a de Novo p.Phe932Ile KCNT1 mutation as the disease-causing change in a child with severe early infantile epileptic encephalopathy and abnormal myelination. Although this C-terminus mutation to Slack is in close proximity to known gain-of-function mutations, the resultant epileptic phenotype does not align with one of the three diagnoses mentioned above. It is, however, described as a case of severe early onset epilepsy and leukoencephalopathy that has resulted in considerable developmental stagnation. Here, we characterize the analogous mutation in the canonical rat Slack-B channel (Brown et al., 2008).
The Slack gene produces multiple transcripts differing in their amino terminals; in fact 3 amino terminals for the rodent Slack gene have been identified (Brown et al, 2008). In higher mammals, including all primates, there is a putative 4th transcript lying approximately 10 kB upstream of the human SLACK-A (Brown et al., 2008) promoter. We are raising this point because functional studies and sequence comparisons of the human SLACK channel have been utilizing a clone where there are apparently two amino terminals fused together: the SLACK-A amino terminal plus the novel amino terminal (accession number NM_020822.2). When comparing this human sequence to original cloned Slack channel (Joiner et al., 1998) the phenylalanine at position 932 occurs at position 911 in the rat Slack-B gene. We designated the mutated rat channel as F932(911)I. The functions of the different amino terminals of Slack channels are not completely understood, although may involve heteromultimerization (Chen et al., 2009). It is possible that the amino terminals target channels to different subcellular locale. The portion of channel where gating occurs lays within the transmembrane domains and the expansive C-terminal containing the RCK domains. Sequence comparisons of phenylalanine 932(911) and this region in general between various species indicated that this segment of the channel is highly conserved (Fig. 1). Between human and rat Kcnt1, amino acid sequence similarity is 94%. The human KCNT1 clone commonly used in Xenopus oocyte expression studies encodes a variant with two amino termini spliced together and two potential start methionines. Translation of the resultant cRNA will likely produce two channels differing in N termini. To avoid any differences in expression or trafficking in a mammalian system and because its amino terminal has been shown to be important for trafficking channels to the membrane (Chen et al., 2009), we studied the canonical rKcnt1 channel originally described by Joiner et al. (1998) and used in the original publication that first described epilepsy-linked KCNT1 mutations (Barcia et al., 2012). In addition, the Slack monoclonal antibody used in this study has been validated by multiple labs for biochemical analyses. (Bansal & Fisher, 2016; Gururaj et al., 2016; Lu et al., 2013; Martinez-Espinosa et al., 2015; Rizzi et al., 2016). This antibody only weakly reacts with the human channel due to amino acid differences in the epitope, necessitating the use of the rat variant.
Figure 1. Mutated region of Slack is highly conserved across species.

Shown is the alignment of nucleotide sequences of the Slack gene in human (h), rat (r), chicken (c) and xenopus laevis (x), zebrafish (z), nematode Slo2 and human Slick.
Experimental Procedures
Site-directed mutagenesis
Site-directed mutagenesis of the rat Slack pTracer construct was performed to obtain the analogous mutation. Primers GGTTCATGCAGATCCGGGCCAAGG and CCTTGGCCCGGATCTGCATGAACC were used. Mutation and construct fidelity were confirmed by DNA sequencing. The analogous mutation is designated as F911I with the rat amino acid numbering system.
Transient transfection
CHO cells were grown in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% FBS, 1% hypoxanthine/thymidine (HT) supplement and 1% Penstrep antibiotic. On the day before transient transfection, cells were plated onto 35mm dishes (Corning) at approximately 50% confluency. Cells were transiently transfected with pTracer vector containing either wildtype or F911I mutant rat Slack. Lipofectamine Reagent and 1 μg of Slack pTracer DNA were premixed with Opti-MEM Reduced Serum Medium. If not stated otherwise, all reagents were purchased from Life Technologies. Cells from each experimental group were plated from the same batch of cells and underwent transfection and data collection concurrently.
Whole cell recording
Cells expressing green fluorescent protein (GFP) were recorded 24–48 hours after transfection. Electrodes had a resistance of 5–8 MΩ. The bath (extracellular) solution consisted of 140 mM NaCl, 1.0 mM CaCl2, 3 mM KCl, 29 mM glucose and 25 mM HEPES (pH 7.4) and the pipette (intracellular) solution contained 32.5 mM KCl, 97.5 mM potassium gluconate, 5 mM EGTA and 10 mM HEPES (pH 7.2) (Joiner et al., 1998). Macroscopic currents were recorded at voltages ranging from −120 to 120 mV. CHO cells were clamped at −70mV and then voltage steps of 20mV were applied for a duration of 200 milliseconds. Capacitance measurements were also collected and used to compute current density. Capacitance (pF) did not significantly differ between groups (wildtype 11.73+/−0.837, mutant 12.15+/−1.716). Based on previous studies (Biton et al., 2012, Yang et al., 2006), loxapine, bithionol and niclosamide were dissolved in DMSO and added to the bath solution at a concentration of 10μM. Dilutions were performed to ensure that the percentage of DMSO in the bath solution never exceeded 0.05% and any possible effect of DMSO was controlled for in the bath solution for control recordings. Cells resided in bath solution containing drug for at least 30 minute before recordings were collected.
Western blot protein analysis
Approximately 48 hours after transient transfection, cells were lysed, proteins denatured, then separated on a 4% – 15% Mini-PROTEAN TGX Precast Gel (Bio-Rad) and transferred to a 0.45μm nitrocellulose membrane (BioRad). Membranes were probed overnight at 4°C with Mouse-Anti Slack (1:2000; Antibodies Inc.) in 5% Milk prepared in 1X tris-buffered saline tween (TBST). On the following day, the membrane was washed three times for five minutes in 1XTBST before being incubated for 1 hour at room temperate in 1:5000 Anti-Mouse IgG horseradish peroxidase conjugate (Promega) and 0.1% bovine serum albumin (BSA) prepared in 1X phosphate buffered saline (PBS). The membrane was again washed 3 times for 5 minutes before being developed and imaged. Blots were quantified using ImageJ (NIH) and plotted as average Slack expression relative to housekeeping genes actin or βtubulin.
Biotinylation assay
Approximately 48 hours after transient transfection, biotinylation assays were performed using the Pierce Cell Surface Protein Expression Kit (Thermo Scientific). Cells were washed twice with ice cold PBS and then incubated in EZ-Link Sulfo-NHS-SS-Biotin for 30 min at 4°C. Cells were harvested, washed in TBS and lysed on ice for 30 minutes in a lysis buffer/protease inhibitor cocktail. Lysates were collected and incubated with 500 μL of NeutrAvidin Agarose for 60 minutes at room temperature with rotation. Following incubation, the column underwent centrifugation to collect the unbiotinylated protein. To elute biotinylated proteins, sodium dodecyl sulfate (SDS) and dithiothreitol (DDT) were added to column membrane and incubated with rotation for 1 hour at room temperature. Biotinylated and unbiotinylated samples were probed for Slack and Actin protein by western blot, as described above.
Immunocytochemistry
CHO cells were plated onto glass coverslips and transiently transfected, as described above. Approximately 36 hours after transfection cells were fixed for 10 minutes in 4% paraformaldehyde. Membranes were then permeablized for 5 minutes in 1X PBS containing 0.2% Triton X-100. Following permeablization and a wash in 1XPBS, cells were incubated for 1 hour in 3% BSA and Rabbit Anti-Slack (1:500). Cells were again washed in 1XPBS before a 20 minute incubation with secondary antibody Goat Anti-Rabbit AlexaFluor 546 (1:1000, Molecular Probes). After a last washing step in 1X PBS, coverslips were mounted onto glass microscope slides with dapi containing VectaShield mounting medium (Vector Laboratories).
Statistical Analysis
Statistics were computed using GraphPad Prism software. Comparisons of current density at −120mV to 120mV between groups were made using a Two-way ANOVA. At voltages greater than 0mV comparisons between wildtype and mutant groups (with or without) drug yielded p values <0.01. Capacitance measurements and western blot quantifications were compared using a T-test and were both found to be non-significant.
Results
F932(911)I Mutation of Slack Produces a Loss-of-function Channel Phenotype
Macroscopic currents of CHO cells expressing wildtype and mutant rat Slack channels were recorded using whole-cell voltage-clamp. Cells expressing GFP, a marker indicative of successful transient transfection, were selected for recording. Average current density (pA/p F) was measured at various voltages and compared, revealing significant difference between wildtype and mutant Slack at all positive voltage steps (Fig. 2A). Representative current traces illustrate that F932(911)I in Slack results in a complete loss-of-function channel phenotype (Fig. 2B). This loss of current was observed in every F932(911)I Slack expressing CHO cell recorded throughout the study (mutant (circles) n=18, wildtype (squares) n=13; p<0.001 at 20–120mV). In an effort to model the heterozygous genotype of the patient described by Vanderver et al. (2014), wildtype and F932(911)I Slack were co-expressed. Co-expression resulted in more than 50% reduction of current density differing significantly from both WT and F932(911)I alone (Fig. 2A, n=12, p<0.01 at 40–120V).This finding is consistent with the notion that the mutant allele codes for a protein that confers a loss-of-function channel phenotype.
Figure 2. F932(911)I Mutation of Slack results in a loss-of-function channel phenotype.
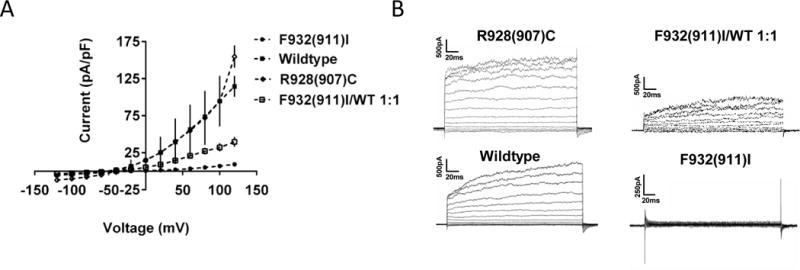
(A) Mean current voltage relationship with SEM recorded from CHO cells transfected with F932(911)I, wildtype, R928(907)C and 1:1 F932(911)I/wildtype. (B) Representative current traces for each group in A.
This loss-of-function finding is in contrast with every other epilepsy-associated KCNT1 mutation characterized, all of which are gain-of-function. Indeed, the R928(907)C KCNT1 mutation associated with ADNFLE epilepsy lying in very close proximity to F932 produced a gain of function phenotype (Milligan et al., 2014) and in our hands engineering the same mutation in the rat Slack channel produced a similar gain of function phenotype when expressed in CHO cells. A kinetic change resulting in rapid channel activation, previously shown by Milligan et al. (2014) was also observed.
F932(911)I of Slack Renders Channel Insensitive to Slack Channel Openers
In an effort to restore channel function, we tested Loxapine, a known channel opener and clinically approved antipsychotic agent. In vitro studies confirm channel activation by loxapine (Biton et al., 2012), and a recent behavioral study shows that it acts in a Slack-dependent manner to reduce pain (Lu et al, 2015). Loxapine significantly increased the current density of wildtype channels but was unable to restore current from mutant channels (Fig. 3A, B & C). In addition, we tested bithionol and niclosamide, anthelmintic drugs known to open Slack channels (Yang et al., 2006; Biton et al., 2012). Like loxapine, mutant currents did not significantly differ in the presence or absence of bithionol (n=6, p=0.653) or niclosamide (n=10, p=0.154) (Data not shown). Based on this finding, it may be expected that mutations in this region are likely to have effects on gating. Nonetheless, we wanted to further test if insufficient protein translation, or changes in trafficking could underlie the complete loss of Slack current.
Figure 3. F932(911)I mutant channel is resistant to established slack channel openers.
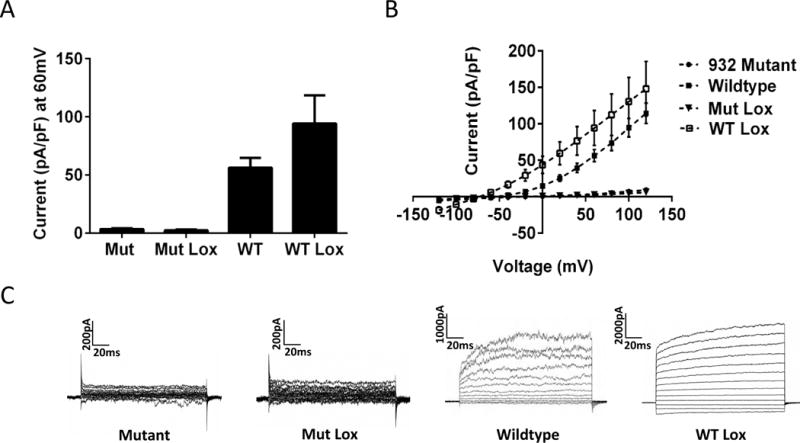
(A) Mean current density at 60mV with SEM in wildtype (n=13) and mutant (n=18) slack. Loxapine applied to the mutant channel at a concentration of 10 μM increases current in the wildtype (n=3) but not mutant (n=10) channel. (B) Mean current voltage relationship and SEM for the groups describes in A. (C) Representative traces for each group in A & B
F932(911)I of Slack Reduces Membrane Expression
To determine whether the loss-of-function channel phenotype results from disrupted Slack protein translation, we compared protein expression level and distribution following transient transfection by immunocytochemistry and western blot analysis. Immunocytochemistry experiments revealed similar levels of expression (Figure 4A), and quantitative western blot analysis confirmed that levels of wildtype and mutant protein did not significantly differ (Figure 4B & C, n=7, p=0.21). We also performed biotinylation assays to examine whether mutant Slack channels were being successfully trafficked and inserted into the cell membrane. Quantification of membrane Slack revealed significantly decreased levels of mutant channels (Figure 3D, n=4, p<0.05). Levels of actin were consistent between groups in unbiotinylated samples and as expected, were absent in biotinylated samples. Taken together, this data indicated that when compared to wildtype, similar levels of mutant Slack channels were translated, but substantially less of these mutant channels were successfully inserted into the membrane, accounting for the reduction in observed current and lack of loxapine sensitivity.
Figure 4. F932(911)I Mutation of Slack results in reduced membrane expression.
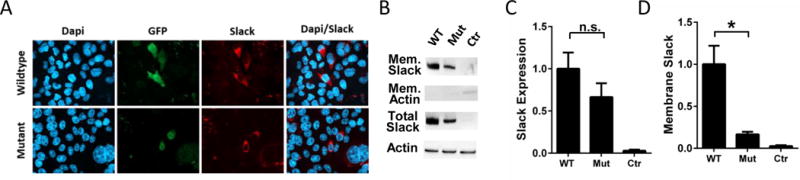
(A) Immunocytochemical analysis of wildtype (top panels) and mutant (bottom panels) slack distribution in CHO cells. (B) Representative western blot analysis verifying biotinylated membrane and total Slack (140 kDa) protein expression from wildtype and mutant slack in CHO cells. Quantification of total (C, n=7, p=0.21) and biotinylated membrane (D, n=4, p<0.05) Slack expression.
Discussion
Together, the results presented here establish the channel phenotype for the analogous mutation to the Slack channel identified by Vanderver et al. (2014). During mammalian heterologous expression, F932(911)I disrupts trafficking and/or insertion into the membrane. Importantly, the co-expression of mutant and wildtype alleles results in close to a 70% reduction in current density at 80mV (Fig. 2A,B). Such a finding further strengthens the argument that the mutant allele confers a loss-of-function channel phenotype and that co-expression of mutant and wildtype subunits produces a dominant negative phenotype. The Slack channel opener loxapine potentiated wildtype currents but did not restore function of mutant channels. It should be noted that potentiation of rat wildtype currents by loxapine was modest compared to the 10-fold increase of human currents shown previously by Biton et al. (2010). Identical recording conditions are needed to clearly discern any potential differences in sensitivity to loxapine between rat and human Slack channels, currents recorded by Biton et al. (2012) included 10 mM sodium in the pipette solution, while we used a sodium-free intracellular solution described by Joiner et al. (1998).
The approximately 75% reduction in membrane expression of the mutant channel strongly suggests that F932(911)I interferes with trafficking and/or insertion to the membrane. It is possible that protein folding is altered in such a way that prevents membrane insertion. Such a circumstance may explain the consistently observed decrease in total membrane expression of the mutant channel. The F932(911)I mutation resides in the C-terminal, a region containing various regulatory sites. In a recent study, Gururaj et al. (2016) illustrate the importance of the C-terminal in membrane trafficking by showing that p38 phosphorylation is required for Slack channel membrane expression.
Our findings are in contrast to all other epilepsy-linked mutations to the Slack channel, which have been characterized as gain-of-function. Studies characterizing other epilepsy-linked Slack mutations have commonly used Xenopus oocytes. A recent report by Tang et al. (2016) studied F932(911)I expression in Xenopus oocytes and report that the mutation slightly increases sodium sensitivity. It is possible that the different cellular environments provided by the oocyte and CHO expression systems are responsible for this inconsistency. Here, we report that when the F932(911)I mutant channels are expressed in a mammalian expression system, we see a substantial decrease in membrane targeting. Kim et al. (2014) found that the increased open probability of KCNT1 mutants was the result of increased cooperative interactions or clustering of multiple channels at the membrane. Our finding of reduced membrane expression does not align with the mechanism proposed by Kim et al. (2014) as it would limit opportunity for cooperative-enhancement. This inconsistency may be attributed to a difference in expression systems, Xenopus oocytes allow for a much greater level of protein expression.
We used CHO cells because they are a mammalian heterologous expression system that can support the conductance of both wildtype and mutant gain-of-function channels (Fig. 2B). In addition, we determine the mutations effects on protein translation and trafficking for the first time. Vanderver et al. (2014) conclude that this mutation is disease causing based on their genome sequencing results and its location immediately adjacent to two gain-of-function KCNT1 mutations thought to cause epileptogenesis (Barcia et al., 2012; Heron et al., 2012). The patient in this case does not fit into any of the three diagnoses associated with Slack channel gain-of-function and in addition to epilepsy is reported to have encephalopathy and delayed myelination. Interestingly, KCNT1 mutations are associated with focal epilepsies (Møller et al., 2015) and the case study by Vanderver et al. (2014) identifies the F911I mutation in a child suffering from generalized seizures. Mutations to various types of ion channels are associated with early infantile epileptic encephalopathies (Gardner, 2006), and it is possible that alteration of another gene is contributing to the phenotype seen here. As stated by Møller and colleagues, the genotype-phenotype relationship of KCNT1 mutations is not straightforward (Møller et al., 2015). Perhaps a loss-of-function channel phenotype results in a more generalized epilepsy and disrupts the proper development of myelin. Bhattacharjee et al. (2002) showed that Slack channels are localized to axons where they could influence proper myelin development.
Very recent studies have demonstrated that in a Slack null mouse line, locomotor activity and spatial learning memory are disturbed (Baush et al., 2015). It does not appear that mice produce spontaneous seizure activity, although how mice respond to induction of seizure (e.g. by kainic acid) was not tested. Still the phenotype of these mice does not compare to that of the patient described by Vanderver. It could be that in generating these Slack null mice, as with many transgenic animal models, functional compensation occurred to mask or distort the phenotype (Picciotto & Wickman, 1998). Based on the severity of the patient’s condition described by Vanderver, KCNT1, homozygous loss-of-function mutations in humans are likely to be lethal. It remains to be seen if any other patients exhibit KCNT1 loss of function mutations.
Conclusion
The present study characterizes the analogous mutation to that identified by Vanderver et al. (2014) as the disease causing mutation is a child with severe epilepsy, leukoencephalopathy and delayed myelination. In a mammalian expression system, this mutated Slack channel produces a loss-of-function channel phenotype and protein analysis reveals that this complete loss of current is likely the result of disrupted membrane expression.
Engineering an F932I mutation in KCNT1(Slack) channels results in a loss of current when expressed in mammalian cells
F932I mutated channels failed to respond to known Slack channel openers.
F932I mutated channels have substantially reduced membrane surface expression compared to wildtype channels
Acknowledgments
Funding
The research was supported by the National Institute of Health, Grant NS078184 (to A.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
None of the authors have any conflicts of interest to disclose. We confirm that we have read the Journal’s position on ethical publication and affirm that this communication is consistent with those guidelines.
References
- Bansal V, Fisher TE. Na(+) -Activated K(+) Channels in Rat Supraoptic Neurones. J Endocrinol. 2016;28 doi: 10.1111/jne.12394. [DOI] [PubMed] [Google Scholar]
- Barcia G, Fleming MR, Deligniere A, Gazula VR, Brown MR, Langouet M, Chen H, Kronengold J, Abhyankar A, Cilio R, Nitschke P, Kaminska A, Boddaert N, Casanova JL, Desguerre I, Munnich A, Dulac O, Kaczmarek LK, Colleaux L, Nabbout R. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat Genet. 2012;44:1255–9. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bausch AE, Dieter R, Nann Y, Hausmann M, Meyerdierks N, Kaczmarek LK, Ruth P, Lukowski R. The sodium-activated potassium channel Slack is required for optimal cognitive flexibility in mice. Learn Mem. 2015;22:323–35. doi: 10.1101/lm.037820.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee A, Gan L, Kaczmarek LK. Localization of the Slack potassium channel in the rat central nervous system. J Comp Neurol. 2002;454:241–54. doi: 10.1002/cne.10439. [DOI] [PubMed] [Google Scholar]
- Bhattacharjee A, Kaczmarek LK. For K+ channels, Na+ is the new Ca2+ Trends Neurosci. 2005;28:422–8. doi: 10.1016/j.tins.2005.06.003. 2005. [DOI] [PubMed] [Google Scholar]
- Biton B, Sethuramanujam S, Picchione KE, Bhattacharjee A, Khessibi N, Chesney F, Lanneau C, Curet O, Avenet P. The antipsychotic drug loxapine is an opener of the sodium-activated potassium channel slack (Slo2.2) J Pharmacol Exp Ther. 2012;340:706–15. doi: 10.1124/jpet.111.184622. [DOI] [PubMed] [Google Scholar]
- Brown MR, Kronengold J, Gazula VR, Spilianakis CG, Flavell RA, von Hehn CA, Bhattacharjee A, Kaczmarek LK. Amino-termini isoforms of the Slack K+ channel, regulated by alternative promoters, differentially modulate rhythmic firing and adaptation. J Physiol. 2008;586:5161–79. doi: 10.1113/jphysiol.2008.160861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Kronengold J, Yan Y, Gazula VR, Brown MR, Ma L, Ferreira G, Yang Y, Bhattacharjee A, Sigworth FJ, Salkoff L, Kaczmarek LK. The N-terminal domain of Slack determines the formation and trafficking of Slick/Slack heteromeric sodium-activated potassium channels. J Neurosci. 2009;29:5654–65. doi: 10.1523/JNEUROSCI.5978-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner M. Molecular genetics of infantile nervous system channelopathies. Early Hum Dev. 2006;82:775–9. doi: 10.1016/j.earlhumdev.2006.09.013. [DOI] [PubMed] [Google Scholar]
- Gururaj S, Fleites J, Bhattacharjee A. Slack sodium-activated potassium channel membrane expression requires p38 mitogen-activated protein kinase phosphorylation. Neuropharmacology. 2016;103:279–289. doi: 10.1016/j.neuropharm.2015.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heron SE1, Smith KR, Bahlo M, Nobili L, Kahana E, Licchetta L, Oliver KL, Mazarib A, Afawi Z, Korczyn A, Plazzi G, Petrou S, Berkovic SF, Scheffer IE, Dibbens LM. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat Genet. 2012;44:1188–90. doi: 10.1038/ng.2440. [DOI] [PubMed] [Google Scholar]
- Joiner WJ, Tang MD, Wang LY, Dworetzky SI, Boissard CG, Gan L, Gribkoff VK, Kaczmarek LK. Formation of intermediate-conductance calcium-activated potassium channels by interaction of Slack and Slo subunits. Nat Neurosci. 1998;1:462–9. doi: 10.1038/2176. [DOI] [PubMed] [Google Scholar]
- Kaczmarek LK. Slack, Slick and Sodium-Activated Potassium Channels. ISRN Neurosci. 2013;pii:354262. doi: 10.1155/2013/354262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GE, Kronengold J, Barcia G, Quraishi IH, Martin HC, Blair E, Taylor JC, Dulac O, Colleaux L, Nabbout R, Kaczmarek LK. Human slack potassium channel mutations increase positive cooperativity between individual channels. Cell Rep. 2015;9:1661–72. doi: 10.1016/j.celrep.2014.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Bausch AE, Kallenborn-Gerhardt W, Stoetzer C, Debruin N, Ruth P, Geisslinger G, Leffler A, Lukowski R, Schmidtko A. Slack channels expressed in sensory neurons control neuropathic pain in mice. J Neurosci. 2015;35:1125–35. doi: 10.1523/JNEUROSCI.2423-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Espinosa PL, Wu J, Yang C, Gonzalez-Perez V, Zhou H, Liang H, Xia XM, Lingle CJ. Knockout of Slo2.2 enhances itch, abolishes KNa current, and increases action potential firing frequency in DRG neurons. Elife. 2015;4(pii):e10013. doi: 10.7554/eLife.10013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan CJ, Li M, Gazina EV, Heron SE, Nair U, Trager C, Reid CA, Venkat A, Younkin DP, Dlugos DJ, Petrovski S, Goldstein DB, Dibbens LM, Scheffer IE, Berkovic SF, Petrou S. KCNT1 gain of function in 2 epilepsy phenotypes is reversed by quinidine. Ann Neurol. 2014;5:581–90. doi: 10.1002/ana.24128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Møller RS, Heron SE, Larsen LH, Lim CX, Ricos MG, Bayly MA, van Kempen MJ, Klinkenberg S, Andrews I, Kelley K, Ronen GM, Callen D, McMahon JM, Yendle SC, Carvill GL, Mefford HC, Nabbout R, Poduri A, Striano P, Baglietto MG, Zara F, Smith NJ, Pridmore C, Gardella E, Nikanorova M, Dahl HA, Gellert P, Scheffer IE, Gunning B, Kragh-Olsen B, Dibbens LM. Mutations in KCNT1 cause a spectrum of focal epilepsies. Epilepsia. 2015;56:e114–2. doi: 10.1111/epi.13071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Wickman K. Using knockout and transgenic mice to study neurophysiology and behavior. Physiol Rev. 1998;78:1131–63. doi: 10.1152/physrev.1998.78.4.1131. [DOI] [PubMed] [Google Scholar]
- Rizzi S, Knaus HG, Schwarzer C. Differential distribution of the sodium-activated potassium channels slick and slack in mouse brain. J Comp Neurol. 2016;524:2093–116. doi: 10.1002/cne.23934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang QY, Zhang FF, Xu J, Wang R, Chen J, Logothetis DE, Zhang Z. Epilepsy-Related Slack Channel Mutants Lead to Channel Over-Activity by Two Different Mechanisms. Cell Rep. 2016;14:129–39. doi: 10.1016/j.celrep.2015.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderver A, Simons C, Schmidt JL, Pearl PL, Bloom M, Lavenstein B, Miller D, Grimmond SM, Taft RJ. Identification of a novel de novo p.Phe932Ile KCNT1 mutation in a patient with leukoencephalopathy and severe epilepsy. Pediatr Neurol. 2014;50:112–4. doi: 10.1016/j.pediatrneurol.2013.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villa C, Combi R. Potassium channels and human epileptic phenotypes: An updated overview. Front Cell Neurosci. 2016;10:81. doi: 10.3389/fncel.2016.00081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Gribkoff VK, Pan J, Damagnez V, Dworetzky SI, Boissard CG, Bhattacharjee A, Yan Y, Sigworth FJ, Kaczmarek LK. Pharmacological activation and inhibition of Slack (Slo2.2) channels. Neuropharmacology. 2006;51(4):896–906. doi: 10.1016/j.neuropharm.2006.06.003. [DOI] [PubMed] [Google Scholar]