

Abstract
Na+/H+ Exchanger Regulatory Factor-1 (NHERF1) is a scaffolding protein containing two PSD95/discs large/ZO1 (PDZ) domains that modifies signaling, trafficking, and function of the parathyroid hormone receptor (PTHR), a Family B G-protein coupled receptor. PTHR and NHERF1 bind through a PDZ-ligand recognition mechanism. We show that PTH elicits phosphorylation of Thr591 in the canonical-ETVM binding motif of PTHR. Conservative substitution of Thr591 with Cys does not affect PTH(1–34) induced cAMP production or PTHR binding to NHERF1. The findings suggested the presence of additional sites upstream of the PDZ-ligand motif through which the two proteins interact. Structural determinants outside the canonical NHERF1 PDZ-PTHR interface influence binding have not been characterized. We used Molecular Dynamics simulation to predict residues involved in these interactions. Simulation data demonstrate that the negatively charged Glu side chains at positions −3, −5 and −6 upstream of the PDZ-binding motif are involved in PDZ-PTHR recognition. Engineered mutant peptides representing the PTHR C-terminal region were used to measure the binding affinity with NHERF1 PDZ domains. Comparable micromolar affinities for peptides of different length were confirmed by fluorescence polarization, isothermal titration calorimetry, and surface plasmon resonance. Binding affinities measured for Ala variants validate MD simulations. The linear relation between the change in enthalpy and entropy following Ala substitutions at upstream positions −3, −5 and −6 of the PTHR peptide provide a clear example of the thermodynamic compensation rule. Overall, our data highlight sequences in PTHR that contribute to NHERF1 interaction and can be altered to prevent phosphorylation-mediated inhibition.
TOC Image
Na+/H+ Exchanger Regulatory Factor-1 (NHERF1, SLC9A3R1) is a scaffolding protein that coordinates the assembly, signaling and trafficking of transmembrane receptors and ion channels. NHERF1, also known as the 50-KDa ezrin-binding protein EBP50, contains two PDZ (PSD95, discs large protein, ZO1) domains and an ezrin-binding domain (EBD). NHERF1 associates with a variety of signaling receptors through specific PDZ-ligand interactions. The C-termini of potential target partners contain so-called PDZ recognition motifs that bind to the PDZ domains in a pocket, or groove, located between the α2 helix and β2 sheet.
The parathyroid hormone receptor (PTHR) a Family B G-protein coupled receptor (GPCR) that plays a key role in mineral-ion metabolism and bone physiology1,2 binds to the PDZ domains of NHERF1 through its C-terminal PDZ-binding motif (-ETVM).3–5 This sequence corresponds to a Class I PDZ-binding motif with the consensus –[D/E]-[S/T]-X-ϕ, where X is promiscuous and ϕ is a hydrophobic amino acid. By convention, the PDZ-recognition motif is numbered from the carboxy-terminal residue starting as zero and upstream residues designated as −1, −2, −3, −4, etc. Crystallographic or NMR structures for the complex between the PDZ domains of NHERF1 and PTHR have not been solved. Biochemical studies6 and Molecular Dynamics simulations (MD)7 predict that the both PDZ domains engage in canonical PDZ-ligand interactions with the carboxy-terminal Met0 and Thr−2 of PTHR. Although these canonical PDZ1/PDZ2-PTHR interactions are essential components of the binding, previous work indicates that upstream PTHR sequences enhance formation of the binary [NHERF1-PTHR] complex.6 Interactions outside the PDZ-ligand binding site, thus, contribute to NHERF1 PDZ-ligand specificity. These considerations underscore the need to characterize the structural determinants contributing to PDZ domain–protein interactions. To accomplish such an assessment we applied complementary experimental and unbiased computational modeling. MD simulations were used to predict residues involved in dynamic peptide interactions between NHERF1 PDZ domains and the PTHR C-terminus, and these computational predictions were experimentally validated by fluorescence polarization (FP), surface plasmon resonance spectroscopy (SPR), and isothermal titration calorimetry (ITC) measurements. Our data suggest a model wherein the negatively charged residues upstream of the PTHR C-terminus form an expanded electrostatic network with the PDZ domains outside the core-binding pocket. In addition to these key electrostatic interactions, we also examined the role of Thr−2 (Thr591), which we show is phosphorylated upon PTH treatment. Replacement with Cys for strictly conserved Thr−2 (Thr591), a defining feature of Class I PDZ binding motifs, does not interfere with receptor binding or activation. Because Thr591 phosphorylation negatively regulates NHERF1 binding, this mutation provides critical information how PTH-induced phosphorylation modulates PTHR-NHERF1 association.
MATERIALS AND METHODS
Constructs and Protein Purification
The expression plasmids pET16-N1P1 encoding PDZ1 (1–140) and pET16-N1P2 encoding PDZ2 (133–300) of NHERF1 were kindly provided by Dr. Dale F. Mierke (Department of Chemistry, Dartmouth College, Hanover, NH, USA). Plasmid fidelity was confirmed by DNA sequencing (ABI PRISM 377, Applied Biosystems, Foster City, CA) and subsequent sequence alignment (NCBI BLAST) with human NHERF1 (GenBank AF015926) to ensure the accuracy of the constructs. Recombinant proteins were expressed in E. coli BL21 (DE3) cells (Novagen) and purified using Ni-NTA agarose (Qiagen).8 The resulting proteins were divided into aliquots and stored in phosphate buffer (25 mM NaH2PO4, 10 mM NaCl, pH 7.4) at −80°C until used for FP experiments.
Cys-PTHR (-ECVM), where Thr591 is replaced by Cys was generated from wild-type human PTHR (-ETVM)_using the QuikChange (Agilent) site-directed mutagenesis kit following the manufacturer’s instructions. All constructs were confirmed by DNA sequencing.
cAMP
HEK293 cells stably expressing the GloSensor™ cAMP reporter were transfected with wild-type TAP-PTHR or a variant of PTHR, where Thr591 in the C-terminus was mutated to Cys. Cells were transferred to a 96-well plate after 48 h post-transfection and pretreated with 1 mM luciferin in the dark at room temperature for 30 min. Bioluminescence was measured at 2-min intervals for 30 min using a Mithras LB 940 multimode microplate reader (Berthold) in the absence or presence of 100 nM human Nle8,18Tyr34-PTH(1–34). Each PTHR constructs was analyzed in quadruplicate.
Mass Spectrometry
Cell lysates were prepared and analyzed as described9 using a Thermo Scientific LTQ Orbitrap XL mass spectrometer coupled with a Thermo Finnigan Nanospray II electrospray ionization source. Each sample was loaded onto a 75 μm × 150-mm BEH C18 column (particle size 1.7 μm; Waters) and separated using a Waters nano-ACQUITY Ultra Performance LC™ (UPLCTM) system (Waters, Milford, MA). The measurements and further analyses were performed as detailed previously.10
Peptide Synthesis
The carboxy-terminal PTHR (PTHRct) 9-residue human peptide (-585LQEEWETVM593) (hereafter PTHRct-9) was synthesized by the University of Pittsburgh Peptide Core Facility and labeled at the N-terminus with fluorescein isothiocyanate of (FITC). The final product was characterized by electron spray mass spectrometry.
Biotinylated carboxy-terminal 22-amino acid PTHR peptide (biotin-572DEEASGPERPPAL-LQEEWETVM593) (hereafter b-PTHRct-22), the Thr−2/Cys−2 mutant of PTHRct-9 (hereafter Cys-PTHRct 9), as well as phosphorylated Thr−2-PTHRct-9 (hereafter PTHR-pThrct-9) were prepared by solid phase synthesis using standard Fmoc (N-(9 fluorenyl)methoxycarbonyl) chemistry (0.1 mmol scale) on an Applied Biosystems AB433 peptide synthesizer. For PTHR-pThrct-9, N-α-Fmoc-O-benzyl-L-phosphothreonine (Fmoc–Thr(PO(OBzl)OH)-OH) was used. For the biotinylated PTHR peptide, the peptidyl resin was treated overnight with 10 eq of biotin in the presence of HBTU/HOBt/DIEA in N-methylpyrrolidone. Following standard trifluoroacetic acid cleavage, the peptides were purified by HPLC on a Vydac C-18 reverse phase column and lyophilized. The final products were characterized by electron spray mass spectrometry. FITC-labeled b-PTHRct-22 was prepared and characterized as above. All peptides were dissolved and serially diluted in storage buffer (25 mM NaH2PO4, 10 mM NaCl, pH 7.4).
Fluorescence Polarization (FP) Assays
Two types of fluorescence polarization measurements were used in this study. Solution phase, direct binding assays (henceforth FP saturation binding assays) were performed to measure affinity (KD) between the PDZ domains of NHERF1 and FITC-labeled PTHR peptides following the protocol described by Madden and co-workers.11 Assays were performed in FP buffer (storage buffer, supplemented to a final concentration of 0.1 mg/ml with bovine IgG (Sigma) and 1 mM DTT) at pH 7.4, 7.0, or 6.4 by applying increasing amounts of the recombinant proteins to a fixed concentration of the FITC-labeled peptide (0.5 μM). Indicated FP experiments were performed in FP buffer supplemented with 300 mM NaCl.
In FP competition assays the binding affinity (Ki) of unlabeled peptide (competitor) corresponding to the concentration that inhibits 50% binding of the FITC-labeled peptide was measured. Competition assays were performed in FP buffer (DTT was avoided for Cys-PTHRct-9) containing fixed concentrations of both fluorescent labeled peptide and protein following the protocol described by Madden and co-workers11. This mixture was equilibrated for 20 min in the dark at room temperature. Unlabeled competitor peptide was dissolved and serially diluted in storage buffer supplemented with 5% DMSO (Sigma). Each serial dilution was aliquoted at 1/10 final volume, to which was added 9/10 volume of the protein:peptide mixture. All FP assays were performed in a 96-well format. Polarized fluorescence intensities were measured at 23 °C with a Perkin Elmer Wallac Victor3 multilabel counter using excitation and emission wavelengths of 485 nm and 535 nm, respectively, for the FITC-labeled peptide. Experimental data were analyzed using Prism (GraphPad). All measurements are reported as fluorescent anisotropy (FA) rather than polarization. Anisotropy was computed using equation 1 from the measured fluorescence emission intensities that are polarized parallel (I║) and perpendicular (I┴) to the plane of the incident light12:
| (1) |
The equilibrium dissociation constant (KD) for interaction between the indicated PDZ domain and labeled peptide was determined by fitting the FA data to a quadratic equation12 as we described previously.13 The KD obtained from direct binding experiments was used to calculate the dissociation constant of the interaction between unlabeled peptide (competitor) and PDZ domain (Ki) using equation 17 described previously.12 FP saturation and competition binding assays were performed in triplicate and repeated independently at least three times unless otherwise noted. The mean values were plotted. Error bars represent the S.E.M.
Surface Plasmon Resonance (SPR)
SPR measurements were performed with a Biacore X100 (GE Healthcare Life Sciences, NJ) at 15.0 °C. Biotinylated PTHRct-22 ligand was immobilized on an SA streptavidin sensor chip (GE Healthcare Life Sciences) using the manufacturer’s protocol until the desired response target was reached.
PDZ1 or PDZ2 analyte, dissolved in 10 mM HEPES buffer, pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.005% surfactant polysorbate 20, was injected on the PTHRct-coated sensor surface at increasing concentrations from 10 nM to 100 μM. At the end of each ligand injection-dissociation cycle, the sensor chip was regenerated with 4.0 M MgCl2, 50 mM triethylamine (pH 9.15), and HBS-EP buffer. The response curves were obtained by subtracting the background signal, generated from a control cell injected with the same analyte but without ligand coating of the hydrogel matrix to remove the effects of bulk refractive index and non-specific binding. The data were further corrected by subtracting the signal generated from buffer alone. Background corrected response curves were fit to the non-linear equation: Req=C Rmax/(C+ KD), where Rmax is the maximum binding response, and KD is the dissociation constant.
Isothermal Titration Calorimetry (ITC)
ITC measurements were conducted on a Microcal iTC200 (Malvern Instruments) in buffer consisting of 25 mM Tris (pH 8.0) and 100 mM NaCl. Proteins were exchanged into ITC buffer by gel filtration using a Superdex200 column. Wild-type modified synthetic peptides were purchased from Genscript (USA) and described in Results. 1 mM PTHR peptides (wild-type and mutant variants) were titrated into 100 μM of individual proteins, PDZ1 or PDZ2, at 25 °C. Data were processed using ORIGIN (OriginLab, Northampton, MA) to extract the thermodynamic parameters ΔH, KD (1/Ka), and the stoichiometry, n. ΔG and ΔS were derived from the relationship: ΔG = −RTlnKa and ΔG = ΔH−TΔS.
MD Simulation
The PTHRct-9 peptide (-585LQEEWETVM593) was computationally generated using the Leap program (AMBER 9)14 and fitted to the binding pocket of PDZ1 or PDZ2 using PDZ1/PDZ2-PTHR (-589WETVM593) as a template.7 The PTHRct-9 peptide was superposed over the template PDZ1/PDZ2-WETVM structure using backbone atoms. The template-WETVM fragment was then removed from the system. The final complex includes PDZ1/PDZ2 and PTHRct-9 with the sequences corresponding to residues 0 to −8 according to the PDZ-ligand nomenclature (hereafter [PDZ1-PTHR] or [PDZ2-PTHR] complex). The complexes were solvated with TIP3P water molecules in a periodically replicated box, neutralized with four Na+ ions, and energy minimized over 500 steps including 100 steps of steepest descent minimization using the AMBER 9 sander module.
The simulations were performed using the AMBER 1115 software package with AMBER ff99SB force field. Equilibration and production simulations were performed as detailed previously.7 Briefly, equilibration runs of 20 ns were carried out in the NVT ensemble with weak harmonic restrains (ks = 0.1 kcal/mol/Å2) applied to the N-terminal backbone atoms of the ligand and the PDZ1 domain to prevent diffusion. The root-mean-square deviations (RMSDs) calculated for the Cα atoms of PDZ1/PDZ2 and peptide ligand relative to their position in the initial structure were used to assess system equilibration (Supplementary text). Production simulations of up to 80 ns were conducted for each system at 300 K in the canonical (constant NVT) ensemble, with configurations saved every 2 fs for analysis. Analysis of hydrogen bonds, salt bridges and hydrophobic interactions was performed using HARLEM (HAmiltonian for Response properties of LargE Molecules)16 and Python scripts on 5-ns trajectories selected from the last 20 ns of MD simulation for each system. The geometric criteria were applied to identify hydrogen bonds, salt bridges, and hydrophobic contacts.17,18 A similar protocol of equilibration and production simulation was repeated for the [PDZ2-PTHR] complex.
RESULTS
Phosphorylation within Canonical PTHR PDZ-recognition Motif Impairs Binding
The C-terminal PTHR harbors Thr591(Thr−2) as part of a canonical -WETVM motif that forms a hydrogen bond with conserved His72 (PDZ1) or His212 (PDZ2) from the α2 helix7 (Figure 1). This bond is required for PDZ-ligand recognition.19 Engineered phospho-modification of Ser/Thr at the −2 residue of class I PDZ-recognition motifs impairs binding.20 We applied mass spectrometry to identify naturally occurring sites of PTHR phosphorylation. The results demonstrate that PTH(1–34) treatment promoted phosphorylation of Thr591 in the activated PTHR tail (Figure 2A). To explore the functional consequence of Thr591 phosphorylation, we synthesized a PTHRct-9 peptide incorporating phosphothreonine at position-2. Binding was probed by FP competition assay. Weak binding with a Ki of 121 μM was observed (Figure 2B; Table 1). Notably, although introduction of a phosphate group nearly abolished the interaction, compared to wild-type PTHRct-9 (Ki of 1.4 ± 0.9 μM) conservative substitution of Thr−2 with Cys only modestly reduced binding affinity (Ki of 11.0 μM) (Figure 2B; Table 1). These findings suggest that Thr-to-Cys modification should not affect PTHR activity, and that sites upstream of the canonical PTHR PDZ ligand and outside the NHERF1 PDZ-binding pocket contribute to or stabilize PTHR interactions with NHERF1. To test these hypotheses, we first generated a full-length PTHR construct with a Thr591Cys substitution and compared its signaling fidelity to wild-type PTHR. As shown in Figure 2C, PTHR-Cys591 exhibited similar cAMP formation in response to challenge with PTH(1–34) as did wild-type PTHR. Thus, wild-type PTHR and PTHR-Cys591 display comparable signaling. These observations are consistent with the similar binding of Cys-PTHRct-9 to PDZ1/PDZ2 with that of wild-type PTHR (Figure 2B; Table 1). Taken together, the results are compatible with the view that additional, noncanonical interactions stabilize PTHR binding to NHERF1.
Figure 1.
The structure of PDZ1 and PDZ2 in complex with PTHRct-9. The PDZ1 and PDZ2 domains are highlighted in wheat cartoon, whereas the peptide is represented in green sticks. The key residues stabilizing the complex are highlighted and labeled on the structure. Plausible electrostatic interactions involved Glu−3, Glu−5 and Glu−6 are discussed in the text. The dotted line represents a hydrogen bond or salt bridge between a hydrogen atom and acceptor with a distance labeled in Å. Hydrogen atoms are white, oxygens are red, and nitrogens are blue. N-terminal Leu and Gln of PTHRct-9 are not shown for simplicity.
Figure 2.
(A) MS-MS spectrum of the C-terminus of PTHR. The peak heights show the relative abundances of the corresponding fragmentation ions, with the annotation of the identified matched amino terminus-containing b ions in blue and the C-terminus-containing y ions in red. (B) In FP competition assays a mixture of FITC-PTHRct-9 (0.5 μM) and PDZ1 (7 μM) was incubated with increasing concentrations of the unlabeled PTHRct-9, Cys-PTHRct-9 or PTHR-pThrct-9 (competitors). (C) PTHR-Cys591 exhibited similar cAMP formation in response to challenge with PTH(1–34) as wild-type PTHR.
Table 1.
Ki values for Wild-type and Modified PTHR peptidesa
| PDZ | Peptide | FP
|
|
|---|---|---|---|
| Ki μM | ΔG° kcal/mol | ||
| PDZ1 | PTHRct-9 | 1.4 ± 0.9 | −8.0 ± 0.5 |
| PDZ1 | PTHR-pThrct-9 | 121b | −5.3 |
| PDZ1 | Cys-PTHRct-9 | 11.0b | −6.7 |
determined by FP competition assay as detailed in Materials and Methods
single determination in triplicates
Computational Prediction of PTHR Binding to NHERF1
A detailed description of canonical PDZ-PTHR interactions was published previously.7 Briefly, canonical PDZ-PTHR interactions were observed with apparent hydrogen bonding between backbone amides of GYGF loop of PDZ1 (Gly23-Tyr24-Gly25-Phe26) and PDZ2 (Gly163-Tyr164-GLy165-Phe166) and Met0 of PTHR, as well as between the side chain of His72 in PDZ1 and the analogous His212 in PDZ2 and the PTHR peptide OH group of Thr−2. We extended this analysis by performing comprehensive Molecular Dynamics characterizations of the PDZ-PTHRct-9 assembly. The details of the calculation of hydrogen bonds and salt bridges are included in Materials and Methods. We constructed a network of potential residues involved in donor-acceptor interactions along the simulation time. Representative structures of the [PDZ1-PTHRct-9] and [PDZ2-PTHRct-9] complexes at the end of 100-ns MD simulations are shown in Figure 1. The general structural stability for all systems was assessed by the root-mean-square deviation (RMSD) over the Cα atoms of the PDZ domains, which provides a measurement of the deviation of the atoms from their initial positions over the simulation time. The Cα atom RMSD values stabilized after approximately 20 ns (results not shown), with the average RMSD values of 1.3–1.6 ± 0.1 Å and 0.5–0.9 ± 0.2 Å for the PDZ domains and PTHRct-9, respectively, over the last 20 ns of the simulation (text in supporting information; Table S1). The absence of the backbone conformational changes for the core of the PDZ domains, as well as for the bound peptide during the equilibration and production simulation, is evident from the RMSD values (Table S1), indicating that the resulting complexes are stable and closely approximate the initial structure. Analysis of the structures evaluated from MD simulations confirmed the docking position of the C-terminal portion of PTHRct-9 is similar to other PDZ-ligand systems.7,13,19
The present MD simulations strongly support the hypothesis that the carboxylate group of Glu−3 forms a salt bridge with the positively charged guanidine group of Arg40 in PDZ1 and the analogous Arg180 in PDZ2. The distances (2.5 Å or less) (Figure 1) between donor-hydrogen-acceptor pairs indicate that these interactions could be strong and play a significant role in the PDZ1/PDZ2-PTHR association.
The Oε1/Oε2 atom of Glu−3, a hydrogen bond acceptor, additionally forms a bifurcated hydrogen bond with the NHδ1 atom of the imidazole ring hydrogen bond donors His27 and His29. In contrast to PDZ1, PDZ2 domain has Asn167 and His169 at the analogous positions. As shown by the MD simulations, the carboxylate group of Glu−3 fits between the side chains of Asn167 and Arg180 (Figure 1). The Oε1/Oε2 atom of Glu−3 makes a hydrogen bond with the amide group (NHδ21) of Asn167. Also, Oε1/Oε2 could accept the hydrogen atom from the positively charged guanidine group of Arg180.
Apart from the interactions described above, Glu−5 and Glu−6 showed a strong tendency to be involved into the extensive network of electrostatic interactions with Lys32 and Lys172 of the β2-β3 loop of PDZ1 and PDZ2, respectively (Figure 1). The proximity of negatively charged carboxylate moieties of Glu−5 and Glu−6 along the MD simulations is enhanced by the positively charged β2-β3 loop of PDZ1/PDZ2. The combined positive potential at both Lys32 and Lys34 (Figure S1) augment the affinity of PDZ1/PDZ2 for PTHRct-9. Calculation of the donor-acceptor pairs confirmed that Oδ1 and Oδ2 atoms of Glu−5 and Glu−6 are involved in salt bridge interactions with the amide hydrogens (NHζ1, NHζ2, and NHζ3) of Lys32/Lys172 of PDZ1/PDZ2. However, the stability of these salt bridges appeared to be influenced by the highly flexible side chain of Lys32/Lys172 at the top of the β2-β3 loop (Figure 1). As shown in Figure S2, the atomic distances fluctuate between 2 and 10 Å, indicating that salt bridge formation is dynamic and dispersive along the time scale of the MD simulations (100-ns).
Experimental Assessment of PTHR Binding Affinity to PDZ1/PDZ2
We applied independent biochemical approaches to characterize PTHR binding to NHERF1 PDZ domains. Fluorescence polarization (FP) is a convenient means to measure equilibrium binding affinities between relatively short peptides and PDZ domains.11,13 We also employed label-free procedures using isothermal calorimetry (ITC) and surface plasmon resonance (SPR). PTHRct-9 was synthesized, labeled with FITC, and probed with the individual PDZ domains. Increasing concentrations of the PDZ domains were used at a fixed concentration (0.5 μM) of PTHRct-9. KD values calculated from the binding experiments are presented in Table 2 and Table S2. The two PDZ domains demonstrated comparable binding affinities for PTHRct-9 with observed KD values of 1.0 ± 0.2 μM and 0.6 ± 0.2 μM for PDZ1 and PDZ2, respectively (Table S2; Figure S3A). Binding was also measured by ITC and SPR (Table 2). Again, PDZ1 and PDZ2 displayed similar affinities to one another but marginally weaker (approximately 1.5 kcal/mol) than those determined by direct saturation binding of FITC-labeled PTHRct-9 with PDZ1/PDZ2 (Table 2; Table S2). However, when reciprocal competition assays were performed using FITC-labeled PTHRct-9 and unlabeled b-PTHRct-22 (competitor), the Ki of 7.0 ± 0.5 dM (−7.0 ± 0.1 kcal/mol) for unlabeled b-PTHRct-22 and PDZ2 from FP measurements (Table 2) favorably agrees with the KD of 9.7 ± 1.1 dM (6.9 ± 0.4 kcal/mol) and the KD of 11.5 ± 0.4 dM (−6.5 ± 0.0 kcal/mol) determined by ITC and SPR, respectively (Table 2).
Table 2.
Binding Affinity of PDZ domains for C-terminal PTHR peptides determined by FP, ITC, and SPR
| PDZ | FPa | ITCb | SPRa | |||
|---|---|---|---|---|---|---|
|
| ||||||
| Ki μM | ΔG° kcal/mol | KDμM | ΔG° kcal/mol | KDμM | ΔG° kcal/mol | |
| PDZ1 | nm | nm | 11.7 ± 0.8 | −6.7 ± 0.2 | 16.0 ± 0.5 | −6.3 t 0.0 |
| PDZ2 | 7.0 ± 0.5 | −7.0 ± 0.1 | 9.7 ± 1.1 | −6.9 ± 0.4 | 11.5 t 0.4 | −6.5± 0.0 |
b-PTHRct-22;
-PTHRct-8; nm – not measured. The KD and Ki represent the mean ± S.E.M. (n = 3)
pH and Ionic Strength Modulate PDZ Binding Affinity
The effects of pH and ionic strength were assessed by FP. Experiments performed at pH 7.0 and 7.4 showed that both PDZ domains responded similarly. However, at pH 6.4 binding affinity increased (lower KD) (Table 3; Figure S3B). Binding also displayed the expected strong salt dependency with an attendant reduction of KD when Na+ was increased from 10 mM to 300 mM (Table 3; Figure S3B) suggesting that long-range electrostatic forces steer the binding reaction.
Table 3.
pH and Salt Dependence of Binding Affinity for the [PDZ-FITC-PTHRct-9] complexa
| PDZ domain |
KD, μM
|
|||
|---|---|---|---|---|
| pH 6.4 | pH 7.0 | pH 7.4 | 300 mM NaCl | |
| PDZ1 | 0.4 ± 0.1 | 1.0 ± 0.1a | 1.0 ± 0.2 | 4.7 ± 0.4 |
| PDZ2 | 0.4 ± 0.1 | 0.6b | 0.6 ± 0.2 | 2.4 ± 0.4 |
determined by FP saturation binding assay as described in Materials and Methods. The KD represents the mean ± S.E.M. (n = 3)
single determination
Steady-state FP equilibrium binding determinations were extended by applying SPR solid-state analysis with immobilized PTHRct to allow measurement of both association and dissociation rates. For SPR we tagged the N-terminus of the 22-residue PTHR peptide with biotin (b-PTHRct-22). We validated the use of b-PTHRct-22 by analyzing its interaction with NHERF1 PDZ2 domain by competition assay. The results (Table 2) confirm that biotinylation did not change the binding properties of the peptide. b –PTHRct-22 was immobilized on carboxymethylated dextran biosensors coated with streptavidin. Increasing concentrations of the PDZ domains were injected. Figure 3 illustrates a typical binding curve and sensorgram for the interaction between b-PTHRct-22 and the PDZ2. The binding isotherms yield KD values of 16.0 ± 0.5 μM and 11.5 ± 0.4 μM for PDZ1 and PDZ2, respectively (Table 2) and are generally consistent with the KD from ITC measurements of PTHRct-8 binding to PDZ1 and PDZ2 (Table 2). The SPR data were fit to a range of the analyte concentrations (100 to 500 nM) using a monovalent binding algorithm. The results yielded association rate constants for PDZ1 of ka = 1.47×104 M−1s−1 and 5.7 ×104 M−1s−1 and dissociation rate constant kd = 0.606 s−1 and 0.393 s−1 for b-PTHRct-22 bound to PDZ1 and PDZ2, respectively. These data are summarized in Table S3. These findings are in good agreement with previous values published for NHERF1 PDZ1 interacting with the peptide contained the C-terminal 9 residues of CFTR.11
Figure 3.
Binding curve (left) and sensorgrams (right) from SPR experiments for b-PTHRct-22 bound PDZ1 and PDZ2 with the affinity reported in Table 2. SPR experiments were carried out three times; standard errors are presented in Table 2.
Contribution of Charged Residues Upstream of PTHR-Recognition Motif on PDZ Domain Interactions
The PTHRct harbors Glu residues at positions −3, −5, and −6. The contribution of Glu−3, Glu−5, and Glu−6 on PDZ1/PDZ2 binding was measured by ITC using PTHRct with single or double Ala substitutions in the PTHRct-8 peptide. Thermograms and binding isotherms for wild-type PTHRct-8 and Ala-modified peptides are displayed in Figure 4 and Figure S4, respectively. The thermodynamic binding parameters are shown in Table 4. As expected, all Ala substitutions in the PTHR peptide reduce the binding affinity (higher KD) for both PDZ1 and PDZ2 domains. Mutation of Glu−5 alone or in combination with Glu−6 reduced association, whereas double mutation at positions −3 and −5 virtually abrogated the interaction with the PDZ domains. Experimentally determined ΔH° values indicate that Ala mutations make the interaction less favorable. A positive change in entropy (TΔS°) for the various Ala substitutions (Table 4) could be related to solvent effects. Overall, the binding energy (ΔG°) changed modestly for the Ala variants, whereas the most dramatic effect on binding was observed for the double Ala substitutions at position −3 and −5 (Figure S4-3; Table 4).
Figure 4.
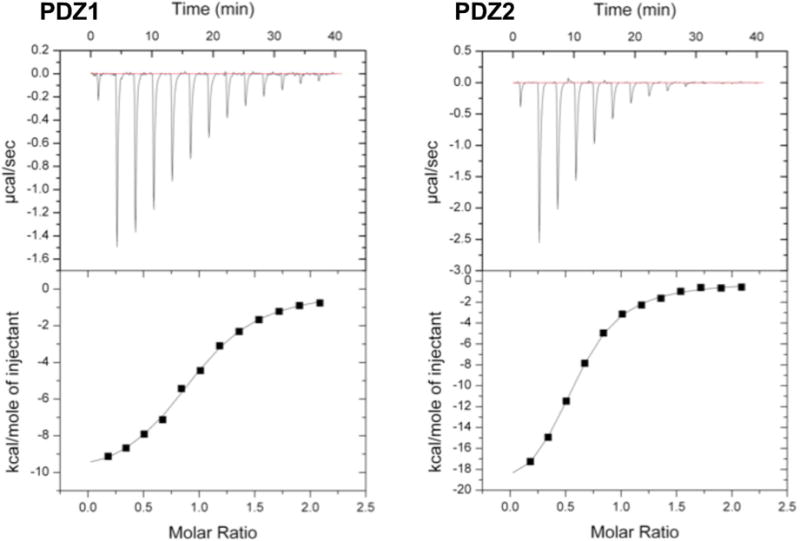
Thermograms (top) and binding isotherms (bottom) from the ITC experiments measuring the binding of the PTHRct-8 peptide with PDZ1 and PDZ2 with the affinity reported in Table 2.
Table 4.
Contribution of PTHR Glu−3, Glu−5 and Glu−6 to the Affinity for PDZ1 or PDZ2a
| PDZ | Sequence | n |
KD (μM) |
ΔH° (kcal/mol) |
TΔS° (kcal/mol) |
ΔG° (kcal/mol) |
|---|---|---|---|---|---|---|
| PDZ1 | QEEWETVM | 1.11 ± 0.01 | 11.7 ± 0.8 | −10.6 ± 0.2 | −3.9 ± 0.1 | −6.7 ± 0.2 |
| QAEWETVM | 1.10 ± 0.04 | 13.1 ± 1.7 | −4.3 ± 0.3 | 2.4 ± 0.2 | −6.7 ± 0.2 | |
| QEAWETVM | 1.21 ± 0.01 | 25.5 ± 1.1 | −7.6 ± 0.1 | −1.3 ± 0.2 | −6.3 ± 0.3 | |
| QAAWETVM | 1.26 ± 0.05 | 24.4 ± 0.6 | −3.80 ± 0.2 | 2.5 ± 0.2 | −6.3 ± 0.3 | |
| QEEWATVM | 1.15 ± 0.02 | 34.2 ± 0.5 | −8.5 ± 0.2 | −2.5 ± 0.3 | −6.1 ± 0.4 | |
| QEAWATVM | 1.12 ± 0.03 | ns | ns | ns | ns | |
| PDZ2 | QEEWETVM | 1.13 ± 0.01 | 9.7 ± 1.1 | −21.6 ± 0.3 | −14.7 ± 0.5 | −6.9 ± 0.4 |
| QAEWETVM | 1.12 ± 0.03 | 18.1 ± 2.4 | −8.1 ± 0.3 | −1.6 ± 0.1 | −6.5 ± 0.3 | |
| QEAWETVM | 0.98 ± 0.02 | 26.8 ± 2.1 | −10.3 ± 0.2 | −4.1 ± 0.3 | −6.3 ± 0.4 | |
| QAAWETVM | 1.26 ± 0.04 | 57.8 ± 2. | −7.2 ± 0.4 | −1.4 ± 0.3 | −5.8 ± 0.5 | |
| QEEWATVM | 0.88 ± 0.02 | 35.5 ± 2.0 | −8.6 ± 0.3 | −2.5 ± 0.1 | −6.1 ± 0.2 | |
| QEAWATVM | 1.04 ± 0.02 | ns | ns | ns | ns |
affinity of [PTHRct-8-PDZ] complex determined by ITC. The n, KD, ΔH°, TΔS°, and ΔG° values represent the mean ± S.E.M. (n = 3)
ns – no significant binding
DISCUSSION
To investigate the structural determinants outside the canonical PDZ-PTHR interaction, we applied complementary approaches including MD simulations to predict residues involved in the interaction with PDZ1/PDZ2 from the upstream sequence of the C-terminal peptide of PTHR and biochemical authentication by FP, ITC and SPR. Peptide-sequence specificity was analyzed by characterizing the ability of the C-terminal 22-residue peptide of PTHR to interact with the PDZ domains. The FITC-labeled peptide incubated with PDZ1/PDZ2 failed to show a significant shift in fluorescence polarization. We also experienced additional technical challenges to measure the affinity of the [PTHRct-22-PDZ1/PDZ2] complex using SPR. These obstacles likely stem from the high degree of flexibility of the PTHRct-22 peptide observed along the MD simulations (data not shown), together with the large negative charge of the peptide (isoelectric point 3.6) that may affect the FP and SPR measurements. In contrast, the covalently tagged b-PTHRct-22 peptide behaved predictably upon SPR analysis (Figure 3 and Table 2) and, moreover, yielded similar binding affinities in competition experiments (Table 2). Because the Ki and KD determined for b-PTHRct-22 by FP competition assay and SPR, respectively, are comparable to KD determined for unlabeled PTHRct-8 by ITC (Table 2), the influence of biotin or residues upstream of −8 position do not appear to have a significant effect on the measured binding affinity. The precise values of KD showed some variation between the different analytical methods. Variations in KD/Ki can arise from multiple sources, including protein preparation, different assay conditions (buffer, temperature, pH, presence of detergent). However, the changes in ΔG between FP competition assay, ITC and SPR provide are negligible and provide reasonable and consistent estimates (Table 2). A larger discrepancy was observed between the KD and Ki determined by FP saturation and competition binding assays, respectively. We attribute this difference to nonspecific binding in the presence of the FITC moiety that increased fluorescence intensity, thereby leading to overestimation of the KD’s.21
The present results disclosed that the binding of the C-terminal motif of the PTHR to NHERF1 involves regions outside the canonical core-binding PDZ boundaries. The model we advance suggests that Arg40/Arg180 from the β3 strand, as well as Lys32/Lys172 of the β2-β3 loop of the PDZ1/PDZ2 domain, form an electrostatic network with Glu−3, Glu−5, and Glu−6 of the upstream C-terminal motif of PTHR. Thermodynamic data show that the contribution of ΔH° and TΔS° to the interaction of PDZ1/PDZ2 to PTHRct-8 and to the Ala variants differ. Binding of PDZ1/PDZ2 to the wild-type peptide depends on a large, favorable ΔH°, offset by the unfavorable TΔS°, whereas binding to the Ala variants displays a relatively less favorable (positive) change in ∆H° and a less unfavorable (positive) change in T∆S° (Table 4). These data fully corroborate the MD simulations. Given an average salt bridge energy of −4 to −5 kcal mol−1,22 the enthalpy changes (∆H°) indicate that roughly one or two salt bridges are disrupted when Ala replaces Glu. However, the observed favorable change in entropy (T∆S°) compensates for the decreased enthalpy (∆H°) as seen in the linear relationship of ∆H° versus ∆S° (Figure 5). The plot was constructed using the data for the wild type and the ensemble of Ala variants of PTHRct-8 bound to PDZ1 and PDZ2 domains as listed in Table 4. The slope of the plot represents the temperature at which Ala substitutions have a negligible effect on the binding. Overall, enthalpy entropy compensation results in a modest lowering of the free energy of the binding (∆G°) for the individual Ala replacements at position −3, −5, or −6. However, combined Ala substitutions at positions −3 and −5 result in a nearly complete loss of binding. Combined with our modeling studies, we propose that electrostatic interactions involving Glu−3 and Glu−5 contribute both to the binding strength and specificity in a cooperative manner. The described electrostatic forces are likely to be central to the molecular recognition mechanism and consequently play a notable role in defining PDZ-PTHR specificity. Previous work highlights the importance of negatively charged residues of upstream sequences promoting the coordinated binding of peptides to the extended binding groove.20 The involvement of His27 and His29/His169 of PDZ1/PDZ2 in the hydrogen bond network may impart an additional impact on the binding under acidic conditions, where the protonated Nε2H or Nδ1H group can act as a strong donor. As suggested by several studies, the pH may vary locally between 6.0 and 7.4 among cellular organelles.23 These observations, supported by our FP results, lead us to propose that local changes in pH may potentially control the association between PDZ domains and PTHR. We note that the pH-dependent profile for the [PDZ-PTHR] complex is unique. For instance, the opposite pH dependence was observed for the association between NHERF1 PDZ1 and the type II sodium-dependent phosphate co-transporter (NPT2A).13 These data are consistent with earlier findings that the interaction between NHERF1 and NPT2A occurs only at the apical membrane of polarized epithelial cells,24–26 where the pH is close to 7.4. The impact of pH sensitive His residues in the recognition and regulation of PDZ-PTHR association will require further analysis.
Figure 5.
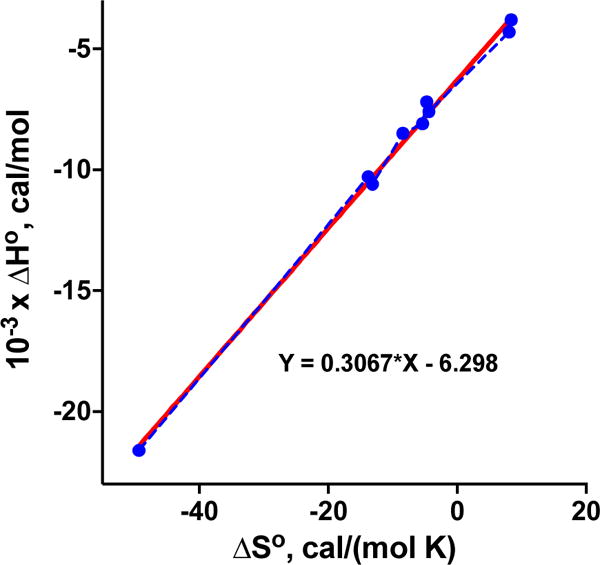
Linear plot of ΔH° vs. ΔS° for the binding of the wild-type and the ensemble of Ala variants of PTHRct-8 to the PDZ1 and PDZ2 domains (using the data from Table 4). Experimental data and linear regression line are blue and red, respectively.
Notably, His72/His212 side-chains from the top of the α2 helix of PDZ1/PDZ2 are strongly conserved in PDZ structures.19 The imidazole ring of this His residue is exposed to the PDZ binding pocket and involved in the canonical interactions with Thr−2/Ser−2 of Class I PDZ targets7,13 (Figure 1). To avoid an inhibitory effect of phosphorylation on Thr591 (Thr−2), we substituted Cys591 theorizing that the thiol side chain (SH) would be able to maintain the structural basis for Class I specificity, whereas Ala substitution would eliminate binding because it lacks the hydrogen bond to the conserved His72/His212 side chain. Glu replacement would mimic phosphorylation and prevent binding due to steric occlusion. The competition experiments confirmed that Cys modification does not inhibit the interaction between PTHRct-9 and the PDZ domains. An important practical observation is that the Cys-modified receptor is fully biologically active in terms of cAMP production. Thus, Cys alteration permits conservative modification of the phosphorylation site without inhibiting receptor activity.
Collectively, the MD simulations supported by the experimental data provide new insights and understanding of the regulatory mechanism governing engagement of the PDZ domains of NHERF1 with the carboxy-terminus of PTHR. The importance of this interaction is underscored by the mineral-ion wasting and osteopenia that occur in humans harboring NHERF1 mutations.27,28 The molecular determinants beyond the canonical binding site outline a distinct electrostatic network playing a specific role in the recognition of the PTHR carboxy-terminus by the PDZ domains. The results highlight the synergy of applying molecular modeling and experimental approaches to predict the dynamic PDZ-PTHR interactions and define unique molecular determinants as a framework for future work.
Supplementary Material
Acknowledgments
The authors are grateful to Dr. D. F. Mierke for NHERF1 PDZ1 (1–140) and PDZ2 (133–270) plasmids. The MD simulations were performed with resources at the Pittsburgh Supercomputing Center (PSC) through XSEDE Grant MCB110024. We thank Dr. M. Kurnikova for providing resources to analyze MD simulation data. We appreciate Dr. A. Ryabov’s valuable discussion of the thermodynamic data.
T.M., P.A.F. designed the research project. T.M. ran MD simulations, analyzed simulation data, and performed FP measurements. E.S. purified PDZ2 and collected the SPR data. Z.B. designed the SPR experiments and data analyses, Q.Z. generated NHERF1 PDZ constructs. A.B. synthesized Cys-and PTHR-pThrct-9 as well as PTHRct-22 peptides. B.C and M.C. designed/performed the ITC assays and data analyses, T.M., P.A.F., and B.M.C wrote the manuscript and prepared graphs.
Funding
This work was supported by grant DK105811-01A1 from the National Institutes of Health. BMC was supported by a National Health and Medical Research Council (NHMRC) Career Development Fellowship (APP1061574) and Project Grant (APP1099114). The calculations were performed with resources at the Pittsburgh Supercomputing Center (XSEDE) through Grant MCB110024.
ABBREVIATIONS
- NHERF1
Na+/H+ Exchange Regulatory Factor-1
- PTHR
parathyroid hormone receptor
- PDZ
postsynaptic density 95/disc large/zona occludens
- CBL
carboxylate-binding loop
- MD
Molecular Dynamics
- NPT
constant pressure, temperature, and number of particles
- NVT
constant volume, temperature, and number of particles
- RMSD
root mean square deviation
- FP
fluorescence polarization
- ITC
isothermal titration calorimetry
- SPR
surface plasmon resonance
Footnotes
Supporting Information
The supporting information is available free of charge on the ACS Publication website at DOI…. Additional details concerning analysis of MD simulations (RMSD); additional figures and tables as noted in text.
The authors declare that they have no financial conflicts of interest that might be construed to influence the results or interpretation of our manuscript.
References
- 1.Wang B, Bisello A, Yang Y, Romero GG, Friedman PA. NHERF1 regulates parathyroid hormone receptor membrane retention without affecting recycling. J Biol Chem. 2007;282:36214–36222. doi: 10.1074/jbc.M707263200. [DOI] [PubMed] [Google Scholar]
- 2.Ardura JA, Friedman PA. Regulation of G protein-coupled receptor function by Na+/H+ exchange regulatory factors. Pharmacol Rev. 2011;63:882–900. doi: 10.1124/pr.110.004176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mahon MJ, Donowitz M, Yun CC, Segre GV. Na+/H+ exchanger regulatory factor 2 directs parathyroid hormone 1 receptor signalling. Nature. 2002;417:858–861. doi: 10.1038/nature00816. [DOI] [PubMed] [Google Scholar]
- 4.Voltz JW, Weinman EJ, Shenolikar S. Expanding the role of NHERF, a PDZ-domain containing protein adapter, to growth regulation. Oncogene. 2001;20:6309–6314. doi: 10.1038/sj.onc.1204774. [DOI] [PubMed] [Google Scholar]
- 5.Wheeler D, Garrido JL, Bisello A, Kim YK, Friedman PA, Romero G. Regulation of parathyroid hormone type 1 receptor dynamics, traffic, and signaling by the Na+/H+ exchanger regulatory factor-1 in rat osteosarcoma ROS 17/2.8 cells. Mol Endocrinol. 2008;22:1163–1170. doi: 10.1210/me.2007-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahon MJ, Segre GV. Stimulation by parathyroid hormone of a NHERF-1-assembled complex consisting of the parathyroid hormone I receptor, phospholipase C beta, and actin increases intracellular calcium in opossum kidney cells. J Biol Chem. 2004;279:23550–23558. doi: 10.1074/jbc.M313229200. [DOI] [PubMed] [Google Scholar]
- 7.Mamonova T, Kurnikova M, Friedman PA. Structural basis for NHERF1 PDZ domain binding. Biochemistry. 2012;51:3110–3120. doi: 10.1021/bi201213w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang B, Yang YM, Friedman PA. Na/H exchange regulatory factor 1, a novel AKT-associating protein, regulates extracellular signal-regulated kinase signaling through a B-Raf-mediated pathway. Mol Biol Cell. 2008;19:1637–1645. doi: 10.1091/mbc.E07-11-1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jean-Alphonse FG, Wehbi VL, Chen J, Noda M, Taboas JM, Xiao K, Vilardaga JP. β2-adrenergic receptor control of endosomal PTH receptor signaling via Gβγ. Nat Chem Biol. 2016 doi: 10.1038/nchembio.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGarvey JC, Xiao K, Bowman SL, Mamonova T, Zhang Q, Bisello A, Sneddon WB, Ardura JA, Jean-Alphonse F, Vilardaga JP, Puthenveedu MA, Friedman PA. Actin-sorting nexin 27 (SNX27)-retromer complex mediates rapid parathyroid hormone receptor recycling. J Biol Chem. 2016;291:10986–11002. doi: 10.1074/jbc.M115.697045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cushing PR, Fellows A, Villone D, Boisguerin P, Madden DR. The relative binding affinities of PDZ partners for CFTR: a biochemical basis for efficient endocytic recycling. Biochemistry. 2008;47:10084–10098. doi: 10.1021/bi8003928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roehrl MHA, Wang JY, Wagner G. A general framework for development and data analysis of competitive high-throughput screens for small-molecule inhibitors of protein-Protein interactions by fluorescence polarization. Biochemistry. 2004;43:16056–16066. doi: 10.1021/bi048233g. [DOI] [PubMed] [Google Scholar]
- 13.Mamonova T, Zhang Q, Khajeh JA, Bu Z, Bisello A, Friedman PA. Canonical and Noncanonical Sites Determine NPT2A Binding Selectivity to NHERF1 PDZ1. PLoS One. 2015;10:e0129554. doi: 10.1371/journal.pone.0129554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Case DA, D T, Cheatham IIITE, Simmerling CL, Wang J, Duke RE, Luo R, Merz KM, Pearlman DA, Crowley M, Walker RC, Zhang W, Wang B, Hayik S, Roitberg A, Seabra G, Wong KF, Paesani F, Wu X, Brozell S, Tsui V, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Beroza P, Mathews DH, Schafmeister C, Ross WS, Kollman PA. AMBER. 2006:9. [Google Scholar]
- 15.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, Debolt S, Ferguson D, Seibel G, Kollman P. Amber, a Package of Computer-Programs for Applying Molecular Mechanics, Normal-Mode Analysis, Molecular-Dynamics and Free-Energy Calculations to Simulate the Structural and Energetic Properties of Molecules. Comp Phys Comm. 1995;91:1–41. [Google Scholar]
- 16.Kurnikov IV. HAmiltonian to Research LargE Molecules (HARLEM) http://harlem.chem.cmu.edu/
- 17.Mamonova T, Yonkunas MJ, Kurnikova MG. Energetics of the cleft closing transition and the role of electrostatic interactions in conformational rearrangements of the glutamate receptor ligand binding domain. Biochemistry. 2008;47:11077–11085. doi: 10.1021/bi801367d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mamonova T, Hespenheide B, Straub R, Thorpe MF, Kurnikova M. Protein flexibility using constraints from molecular dynamics simulations. Phys Biol. 2005;2:S137–S147. doi: 10.1088/1478-3975/2/4/S08. [DOI] [PubMed] [Google Scholar]
- 19.Karthikeyan S, Leung T, Ladias JA. Structural determinants of the Na+/H+ exchanger regulatory factor interaction with the ß2 adrenergic and platelet-derived growth factor receptors. J Biol Chem. 2002;277:18973–18978. doi: 10.1074/jbc.M201507200. [DOI] [PubMed] [Google Scholar]
- 20.Clairfeuille T, Mas C, Chan AS, Yang Z, Tello-Lafoz M, Chandra M, Widagdo J, Kerr MC, Paul B, Merida I, Teasdale RD, Pavlos NJ, Anggono V, Collins BM. A molecular code for endosomal recycling of phosphorylated cargos by the SNX27-retromer complex. Nat Struct Mol Biol. 2016;23:921–932. doi: 10.1038/nsmb.3290. [DOI] [PubMed] [Google Scholar]
- 21.Lea WA, Simeonov A. Fluorescence polarization assays in small molecule screening. Expert Opin Drug Discov. 2011;6:17–32. doi: 10.1517/17460441.2011.537322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar S, Nussinov R. Relationship between ion pair geometries and electrostatic strengths in proteins. Biophys J. 2002;83:1595–1612. doi: 10.1016/S0006-3495(02)73929-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mitsui K, Koshimura Y, Yoshikawa Y, Matsushita M, Kanazawa H. The endosomal Na+/H+ exchanger contributes to multivesicular body formation by regulating the recruitment of ESCRT-0 Vps27p to the endosomal membrane. J Biol Chem. 2011;286:37625–37638. doi: 10.1074/jbc.M111.260612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shenolikar S, Voltz JW, Minkoff CM, Wade JB, Weinman EJ. Targeted disruption of the mouse NHERF-1 gene promotes internalization of proximal tubule sodium-phosphate cotransporter type IIa and renal phosphate wasting. Proc Natl Acad Sci U S A. 2002;99:11470–11475. doi: 10.1073/pnas.162232699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wade JB, Liu J, Coleman RA, Cunningham R, Steplock DA, Lee-Kwon W, Pallone TL, Shenolikar S, Weinman EJ. Localization and interaction of NHERF isoforms in the renal proximal tubule of the mouse. Am J Physiol Cell Physiol. 2003;285:C1494–1503. doi: 10.1152/ajpcell.00092.2003. [DOI] [PubMed] [Google Scholar]
- 26.Wang B, Means CK, Yang YM, Mamonova T, Bisello A, Altschuler DL, Scott JD, Friedman PA. Ezrin-anchored protein kinase A coordinates phosphorylation-dependent disassembly of a NHERF1 ternary complex to regulate hormone-sensitive phosphate transport. J Biol Chem. 2012;287:24148–24163. doi: 10.1074/jbc.M112.369405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Karim Z, Gerard B, Bakouh N, Alili R, Leroy C, Beck L, Silve C, Planelles G, Urena-Torres P, Grandchamp B, Friedlander G, Prie D. NHERF1 mutations and responsiveness of renal parathyroid hormone. N Engl J Med. 2008;359:1128–1135. doi: 10.1056/NEJMoa0802836. [DOI] [PubMed] [Google Scholar]
- 28.Courbebaisse M, Leroy C, Bakouh N, Salaun C, Beck L, Grandchamp B, Planelles G, Hall RA, Friedlander G, Prie D. A new human NHERF1 mutation decreases renal phosphate transporter NPT2a expression by a PTH-independent mechanism. PLoS One. 2012;7:e34764. doi: 10.1371/journal.pone.0034764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.