

Abstract
Studies of the new tetra-amido macrocyclic ligand (TAML) activator [FeIII{(Me2CNCOCMe2NCO)2CMe2}OH2]− (4) in water in the pH range of 2–13 suggest its pseudo-octahedral geometry with two nonequivalent axial H2O ligands and revealed (i) the anticipated basic drift of the first pKa of water to 11.38 due to four electron-donating methyl groups alongside (ii) its counterintuitive enhanced resistance to acid-induced iron(III) ejection from the macrocycle. The catalytic activity of 4 in the oxidation of Orange II (S) by H2O2 in the pH range of 7–12 is significantly lower than that of previously reported TAML activators, though it follows the common rate law (v/[FeIII] = kIkII[H2O2][S]/(kI[H2O2] + kII[S]) and typical pH profiles for kI and kII. At pH 7 and 25 °C the rate constants kI and kII equal 0.63 ± 0.02 and 1.19 ± 0.03 M−1 s−1, respectively. With these new values for pKa, kI and kII establishing new high and low limits, respectively, the rate constants kI and kII were correlated with pKa values of all TAML activators. The relations log k = log k0 + α × pKa were established with log k0 = 13 ± 2 and 20 ± 4 and α = −1.1 ± 0.2 and −1.8 ± 0.4 for kI and kII, respectively. Thus, the reactivity of TAML activators across four generations of catalysts is predictable through their pKa values.
Graphical abstract
Introduction
Designed over the last three decades,1,2 tetra-amido macrocyclic ligand (TAML) activators 1, 2, and 3 (Chart 1), which are catalysts of the first, fourth, and fifth generations,3 respectively, are particularly effective for the catalyzed degradation of micropollutants by hydrogen peroxide in aqueous media and therefore are extremely promising for the purification of environmental and drinking waters. These catalysts function at the parts per billion (ppb) to parts per trillion (ppt) concentration range and direct the oxidizing power of H2O2 at a wide spectrum of environmental threats,4 among which endocrine disruptors such as steroid hormone derivatives are particularly hazardous.5 Additionally, TAML activators are successfully used in fine organic synthesis.6–9
Chart 1. TAML Activators Applicable to Water Treatment.
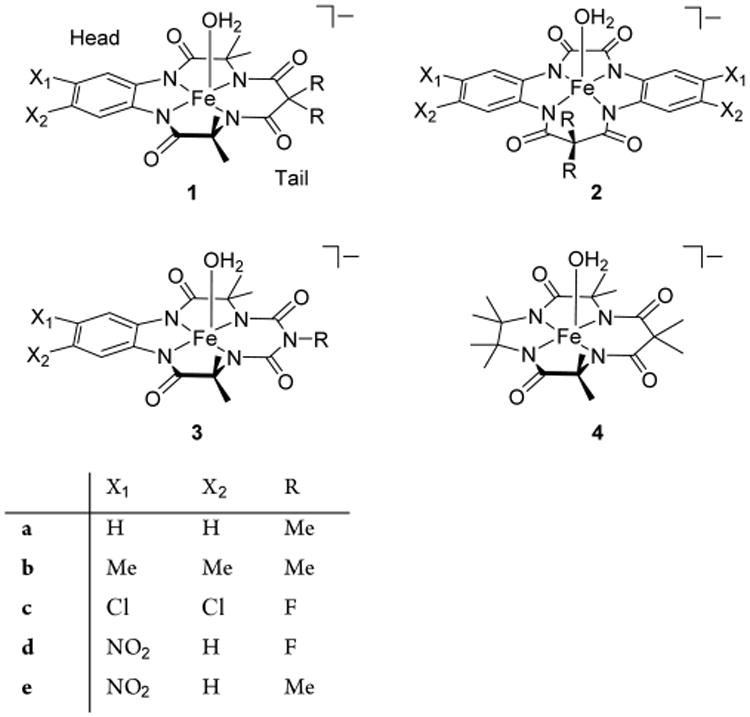
Catalysis by TAML activators is kinetically and mechanistically similar to that by peroxidase enzymes including issues related to operational instability of fragile biomolecules.10–12 The recently emphasized importance of understanding the inactivation of man-made biorelevant catalysts in terms of “causes, avoidance, and cure” has been among our top TAML research priorities.13–15 On the basis of the results of earlier work14 we hypothesized that suicidal degradation of oxidized forms of TAML activators could start with electron transfer from the aromatic ring or “head” of 1, because electron-withdrawing aryl substituents enhanced the operational stability of TAML activators. The mechanistic knowledge acquired14 has been successfully applied for the synthesis of “ideal” TAML activator 1d.16 Even though TAML activators of later generations, specifically, of fourth (2)17,18 and fifth (3),3,19 have been designed, the first generation activator 1d remains one of the most efficient catalysts for environmental5 and other20 applications.
In the pursuit of an exceedingly operationally stable activator, it has been decided to eliminate all aryl rings from the TAML ligand system, that is, to design a “beheaded” catalyst with a nonexisting electron transfer pathway from the aromatic moiety. This is how the idea of making new TAML activator 4 emerged, and here we report on the characterization, properties, and reactivity of 4 in aqueous solution. A correlation was found between the pKa of the axially ligated water molecule of TAML activators across all generations and the rate constants kI and kII. This allows for the effortless and straightforward estimation of catalytic activity of TAML activators for the oxidative degradation of organic substrates at neutral pH. A part of this work has been recently communicated.21
Results and Discussion
Properties of 4 in the Solid State and in Solution
TAML activator 4 with different counter cations (tetramethylammonium and bis(triphenyphosphine)iminium (PNP)) readily forms yellow needlelike crystals on diffusion of diethyl ether into acetonitrile. Unfortunately, these do not diffract X-rays well enough for structural characterization. The absence of a rigid aromatic moiety in the ligand system could increase nonplanarity of the complex with respect to the four deprotonated amide nitrogens. In turn, enhanced nonplanarity results in instability of iron TAML activators,22 which compromises catalysis. Therefore, the structural features of 4 were tested by density functional theory (DFT) calculations, and the results are presented in Figure 1.
Figure 1.
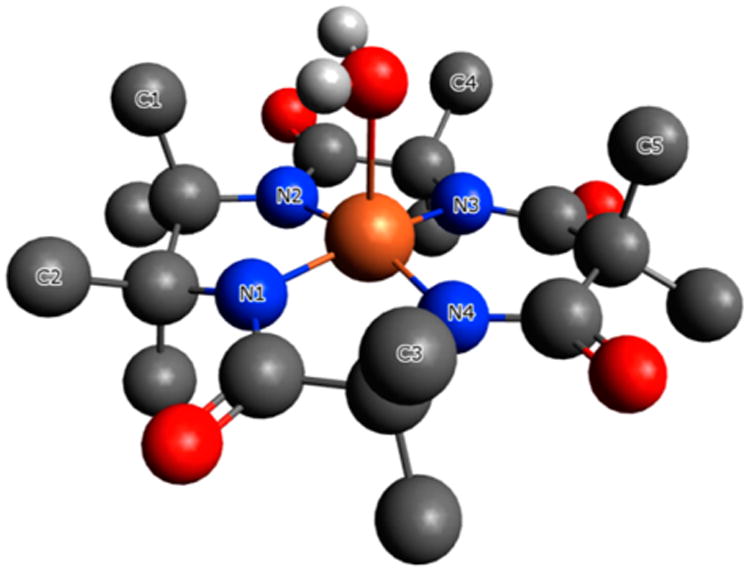
Results of DFT optimization of the structure of 4 in the form [FeL(OH2)]− in vacuum. H atoms other than those of H2O ligand are omitted for clarity. Selected bond distances: Fe–O, 2.285; Fe–N(1–4) 1.878, 1.872, 1.886, and 1.888 Å, respectively.
It was particularly interesting to analyze the effects brought about by the (CMe2)2 unit of 4, which replaced the traditional phenylene ring in TAML activators of previous generations (cf. structures 1–4). The aromatic ring is referred to as the “head” part of TAML activators.22 The calculated geometry of beheaded 4 was compared with the X-ray structural data collected for 1a.22 There is a noticeably elongated Fe–O bond 2.285 Å in 4 versus 2.097 Å in 1a perhaps due to the electron-donating effect of the (CMe2)2 unit. The C(Me2)2 units are rotated around the Me2C–CMe2 bond, the dihedral angle C(1)C2C(2) being 36.9°. Iron lies just 0.20 Å above the average plane of the four amide nitrogens versus 0.36 Å in 1a. In turn, this places the C(1)H3 group very close to the oxygen of the axial water ligand, the C(1)…O separation being 3.415 Å. On the one hand, the latter value matches the sum of the van der Waals radii of CH3 (2.0) and O (1.4 Å). On the other hand, the average Fe–N distance in 4 (1.881) is close to that in 1a (1.885 Å) suggesting that the (CMe2)2 unit translates its influence primarily at the axial ligand. As it will be shown below, the Fe–O bond elongation agrees perfectly with a pronounced increase in the pKa of coordinated water.
It is worth noting that the carbon atom periplanar to C1 sits close to Fe, the separation being 3.415 Å. This suggests a steric repulsion between the C atom and the sixth (aqueous) ligand in water, where previous TAML activators are octahedral with two axial H2O ligands ([FeL(OH2)2]−).22 Optimization of [FeL(OH2)2]− for 4 by DFT in vacuum confirmed this hypothesis. The calculated Fe–O bonds appeared very different, specifically, of 2.273 and 3.418 Å, suggesting that one of the water ligands should be much more weakly bound to iron(III).
Compound 4 is moderately soluble in methanol and water, where its properties were tested in some detail. In methanol, 4 has an absorption maximum at 379 nm (ε = 7.8 × 103 M−1 cm−1), and Beer's law holds in the concentration range of (0.11 – 1.2) × 10−4 M ruling out significant aggregation under these conditions. In pure water the maximum is at 368 nm (ε = 7.2 × 103 M−1 cm−1) in the concentration range of (0.13–5.4) × 10−4 M. As mentioned above, iron TAML activators are octahedral diaqua or aqua/hydroxo species in aqueous solution,22 which was also confirmed for 4 by electron paramagnetic resonance (EPR) measurements (see below). The diaqua species typically dominate under neutral conditions and transform into aqua/hydroxo derivatives under basic conditions. The acid–base equilibria are accompanied by profound spectral changes in the UV–vis region, which for 4 are demonstrated in Figure 2. The spectral changes are fully pH-reversible, occur within two pH units, and there are isosbestic points at 343 and 407 nm. This all is consistent with the single-proton acid–base equilibrium:
Figure 2.
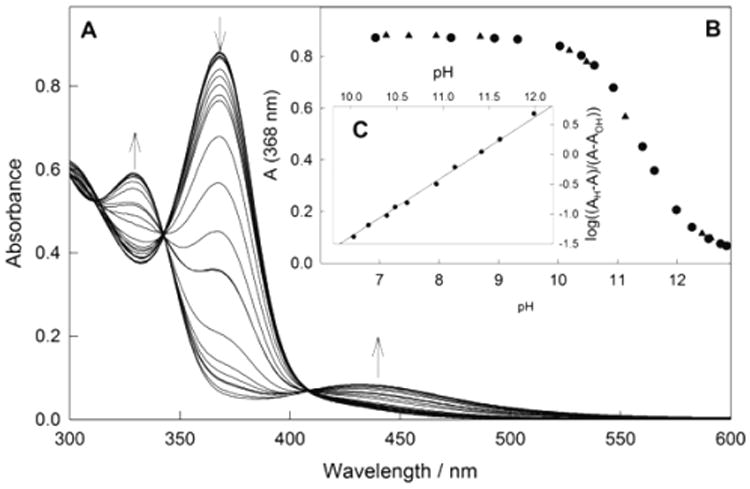
(A) Spectral changes of 4 at pH 7–13. Circles and triangles are the data obtained by changing the pH from 7 to 13 and from 13 to 7, respectively. (B) Absorbance (368 nm) vs pH. (C) Linearization of the data using eq 2. Conditions: 1.2 × 10−4 M 4, 25 °C, Carmody buffer.
| (1) |
The pKa of 4 (11.38 ± 0.01) was calculated using eq 2, where AH, AOH, and A are absorbances at neutral, basic, and intermediate pH, respectively, (Figure 2C).
| (2) |
The pKa value of 11.38 for 4 is the highest ever reported for iron TAML activators. The low acidity of the coordinated water reflects a profound decrease of the Lewis acidity of the central metal due to the combined donor effect of four methyl groups in the beheaded part of 4. It agrees also with predictions by DFT. The pKa value closest to 11.38, of 10.32, has been recently measured for N-tailed “biuret” TAML activator 3a.3 Such distinct variance prompted us to measure pKa for 1b. A similar approach as described above for 4 (see Figure S1) resulted in the value of 10.35 ± 0.02, which is not significantly higher than that for 3a and 1a (pKa values of both equal 10.3). The latter value indicates that the phenylene ring is a decent “buffer” that neutralizes the electron-donating properties of the methyl groups.
EPR spectra of the TAML species in solutions with pH above and below the pKa reveal the difference in speciation between the protonated and deprotonated forms of 4. Figure 3 shows the EPR spectra of samples prepared in pH 2, 10.5, and 13 buffer solution with 10% glycerol. All samples revealed intermediate spin, S = 3/2, iron(III) complexes. The spectra recorded for the samples at pH below the pKa of 4 are nearly identical. The simulation overlaid for the pH 10.5 spectrum is calculated for S = 3/2, D = 1.2 cm−1, and E/D = 0.22. The g-values for the ±1/2 doublet and the gz-value of the ±3/2 doublet are indicated in Figure 3. The spectrum of the pH 13 sample showed a significant change in symmetry to a more axial species. The spectrum at pH 13 showed a 50% conversion to the higher pH species. The difference spectrum of pH 13 minus pH 10.5 is also shown in Figure 3. The simulation overlaid on the difference spectrum is calculated for S = 3/2, D = 1.3 cm−1, and E/D = 0.11. The g-values for the ±1/2 doublet and the gz-value of the ±3/2 doublet are indicated on Figure 3. For a pKa value of 11.4, 95% of the complex should be the deprotonated species at pH 13. The different ratio of the deprotonated species is attributed to a change in pKa and/or pH of aqueous solutions upon freezing of the EPR sample.23,24
Figure 3.
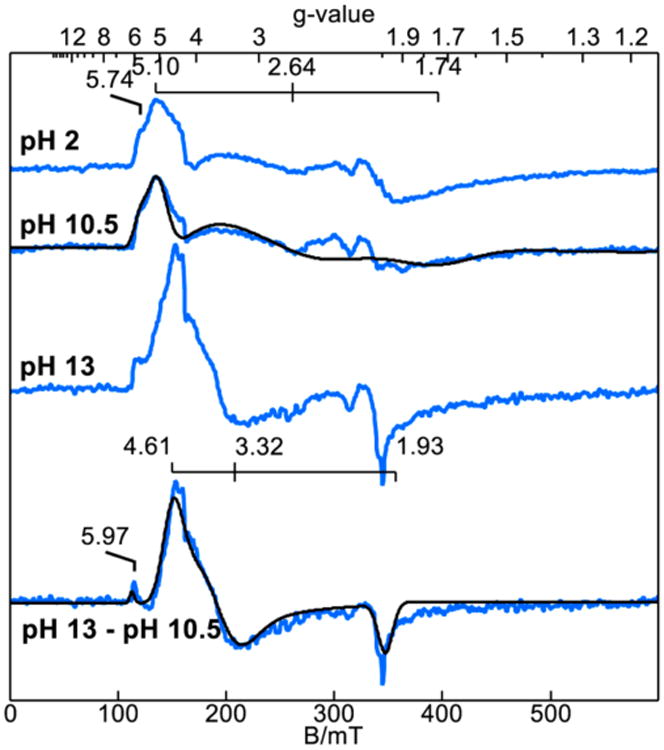
EPR spectra of iron(III) complex 4 (5 × 10−4 M) at pH 2, 10.5, and 13. Samples were prepared in Carmody buffer with 10% glycerol.
Mössbauer spectra of the pH 2 and 13 samples were recorded at 100 K. For this temperature the magnetic spectra essentially collapsed to doublets. At pH 2, the parameters of the doublet were the isomer shift (δ) 0.14 mm s−1 and the quadrupole splitting (ΔEQ) 3.59 mm s−1. At pH 13, the doublets are broad due to residue magnetic effects, but a doublet was observed from the deprotonated species with δ = 0.11 mm s−1 and ΔEQ = 2.94 mm s−1.
Kinetics of Acid-Induced Iron Ejection from 4
At pH 4 and below TAML acctivator 1a undergoes proton-induced demetalation, which occurs according to eq 3.22 The process is kinetically interesting due to its rate law, specifically, kobs = k1[H+] + k3[H+]3.22 The third-order term in H+ was rationalized by the peripheral protonation of the tail amide oxygens of 1a, because its 1c analogue with the fluorine tail appeared to be 9 orders of magnitude more resistant to demetalation.22
| (3) |
As far as 4 is concerned, the increased electron-donating capacity of the four methyl units should greatly increase the rate of demetalation. Therefore, it has been thrilling to probe this concept through the kinetic investigation of reaction 3 with complex 4, as we have found just the opposite effect. It should be emphasized first that 4 is markedly more resistant to H+ than 1a, since we were able to manipulate 4 at pH 2 for a span of 8 h without any evidence for its collapse. Correspondingly, the acid-induced demetalation of 4 was studied in the range of [H+] 0.003–0.250 M by following the exponential decrease in absorbance at 368 nm at 25 °C. Pseudo-first-order rate constants kobs depend strictly linearly on [H+] with the corresponding second-order rate constant k1 of (3.41 ± 0.05) × 10−4 M−1 s−1 (Figure 4). There is no evidence for higher-order terms in the acid concentration suggesting that peripheral protonation does not assist the iron ejection from 4 and the rate-limiting step involves a proton attack at one the Fe–N bonds as previously proposed.22 Since the beheaded (tetramethylated) part of 4 could be more susceptible to the electrophilic attack compared to its “tail” part, it is likely that proton attacks Fe–N(1) or Fe–N(2) bond (Figure 1). Peripheral protonation of 1 was considered to involve the tail part of TAML activators.22 If similar tail protonation occurs in the case of 4, the actual reactive Fe–N(1) or Fe–N(2) sites are too far away from the protonation site, and therefore the peripheral phenomena do not affect the speed of reaction 3 in the case of 4.
Figure 4.
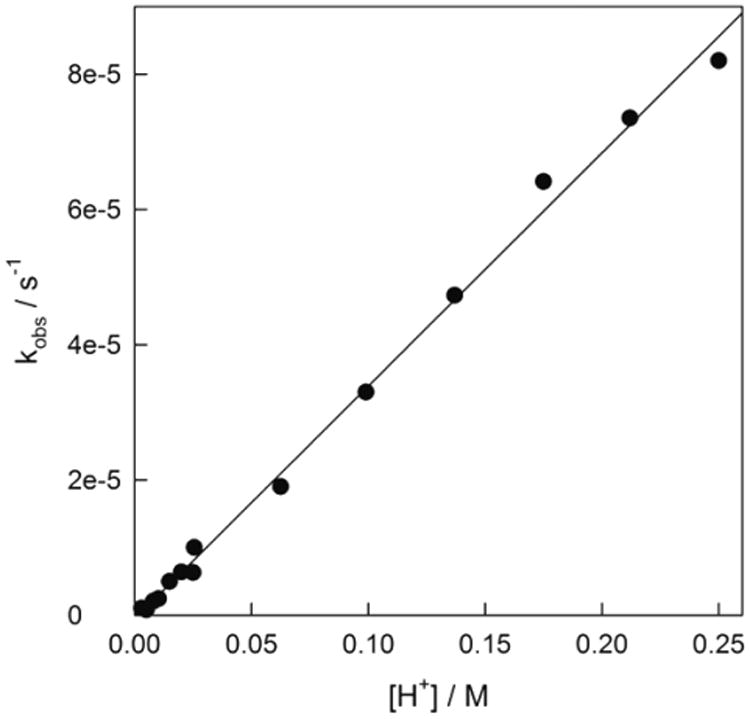
Pseudo-first-order rate constants kobs vs [H+] for the demetalation of 4 at 25 °C.
The value of k1 displays 10 000 and 10-fold resistance of 4 to acid compared to TAML activators 1a and 3a, with k1 values of 2.2 ± 0.7 and (3.7 ± 0.5) × 10−3 respectively.19,22 This increased resistance to acid-induced demetalation is counter-intuitive, because 4 is more electron-rich than 1a or 3a and therefore should be more prone to electrophilic demetalation. Therefore, the retardation might have steric origin. The methyl groups of the (CMe2)2 unit may function as a fence that prevents the proton attack at Fe–N(1) or Fe–N(2) sites revealing a rare case of steric retardation of the reaction involving specific acid catalysis.25 This may also explain why there is no indication of third-order dependence on [H+] in this concentration range.
Catalytic Activity of 4 versus Lewis Acidity
The catalytic intrigue was how the high value of pKa for 4 would affect its activity in catalyzed oxidations by hydrogen peroxide. We3,17,18,26 and others27–29 employ Orange II dye for catalyzed oxidations by H2O2 and other primary oxidants. Measurements were initiated at pH 7 because of our long-term goal of using TAML activators for purification of water in the environment. The steady-state rate of fading of the Orange II band at 484 nm is a linear function of [4] in a range from 1 × 10−5 to 1 × 10−4 M at [H2O2] = 5 × 10−3 M and [Orange II] = 4 × 10−5 M. The rate law is typical of catalysis by TAML activators. There is a hyperbolic dependence on concentration of H2O2 (Figure 5A), which agrees with common rate law 4 (with negligible kI) and is consistent with the general two-step mechanism of catalysis shown in Scheme 1.4,12
Figure 5.
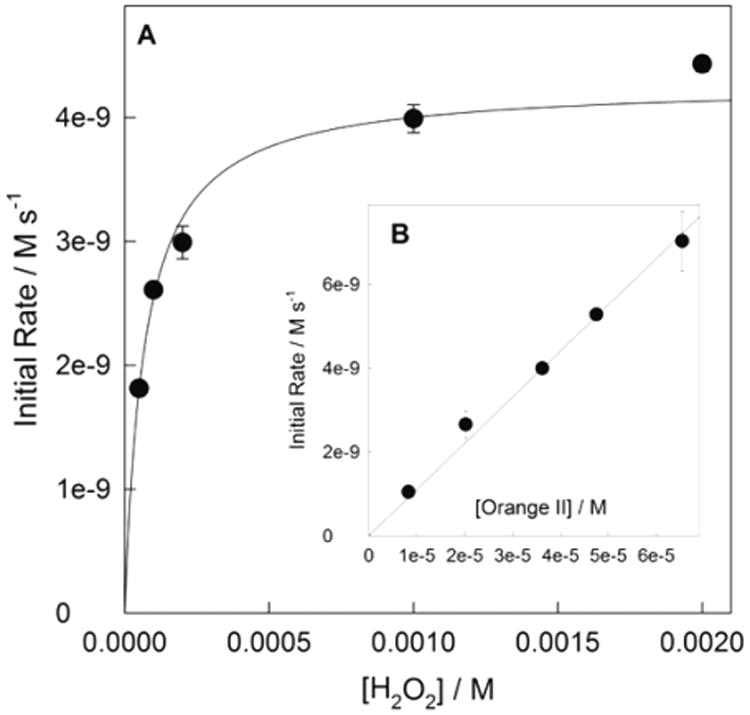
(A) Initial rate of 4-catalyzed Orange II degradation by H2O2 as a function of H2O2 concentration. Conditions: pH 7 (0.01 M phosphate buffer), 1.0 × 10−4 M 4, 3.6 × 10−5 M Orange II, 25 °C. (B) Linear dependence of initial rate on [Orange II] at 1 × 10−3 M H2O2 and 1 × 10−4 M 4 (see text for details).
Scheme 1. General Stoichiometric Mechanism of Catalysis by TAML Activators4,12.

| (4) |
Equation 4 implies that when the reaction rate levels off at high H2O2 concentrations, it should be a linear function of [Orange II], which is confirmed in Figure 5B. Though the kinetic data in Figure 5 are normal for TAML activators, the absolute concentrations of 4 used to generate the data are by ca. 3–4 orders magnitude higher than usually employed implying that the rate constants kI and kII should be markedly lower. In fact, the values kI and kII obtained by fitting the data in Figure 5A to eq 4 equal 0.63 ± 0.02 and 1.19 ± 0.03 M−1 s−1, respectively, and kII calculated from the slope of the line in Figure 5B equals 1.10 ± 0.02 M−1 s−1. These values are significantly lower than those for the prototype TAML activator 1a (31.4 and 4.95 × 103 M−1 s−1, respectively).19,22 An additional unusual feature of 4 is that kI and kII are very close to each other, whereas the relation kI ≪ kII usually holds for TAML activators and other man-made iron-containing peroxidase mimetics, though the opposite relation is typical of peroxidase enzymes.4,12
To check whether or not the relation kI ≈ kII is valid at pH other than 7, the corresponding rate constants were obtained at pH from 7 to 12 (Figure 6). The normal trend, that is, kI ≪ kII, reappears at pH above 9, though the gap between kI and kII is not as large as for 1–3 TAML activators. If compared with the data for 1–3, the bell-shaped pH profile for kI is moved to the alkaline region with the maximum around 11.75, which reflects the highest pKa value of 4. Its quantification is based on the mechanistic model in Scheme 2, which leads to the pH dependence of kI (eq 5).
Figure 6.
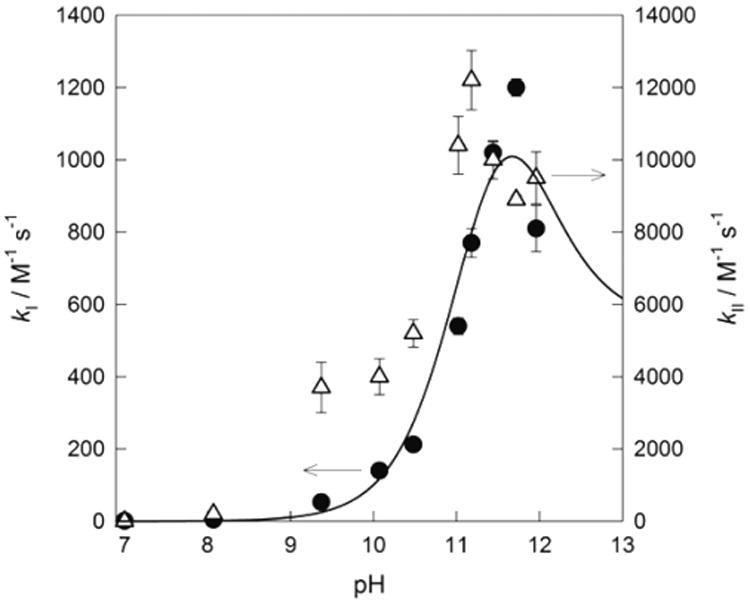
Values of kI (●, kI axis) and kII (Δ, kII axis) as a function of pH. The solid line was calculated for kI (see text for details).
Scheme 2. Typical TAML Activator Mechanism for kI Step that Accounts for the pH Profile as in Figure 6a.
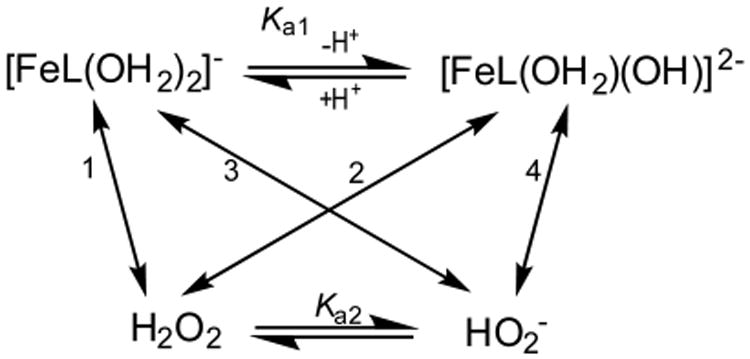
aNumerals at the two-sided arrows 1–4 correspond to the rate constants k1–k4, respectively (eq 5).
| (5) |
It is important to note that in the case of 4 eq 5 reduces to eq 6, because (i) pKa of 4 (pKa1) is essentially equal to pKa of H2O2 (pKa2) and (ii) one of the kinetically indistinguishable rate constants (k2 or k3) is considerably larger than the other.10
| (6) |
Here Ka = Ka1 = Ka2 and k2/3 = k2 or k3. The data in Figure 6 were fitted to eq 6 assuming Ka = 1 × 10−11.5 M. The fitting procedure was similar to such used by us previously.3,10 The value of 0.63 ± 0.03 M−1 s−1, which was measured at pH 7, can be assumed to be the upper limit for the intrinsic rate constant k1, as fitting the data gives greater than 100% error for this parameter. The low value of this intrinsic rate constant means that the reaction between [FeL(OH2)2]− and HOOH is by far the slowest of the four possible reaction pathways. The value of k4, for the reaction between [FeL(OH2)(OH)]2− and HOO−, was determined to be 550 ± 70 M−1 s−1, which is 3 orders of magnitude higher than k1, due to the higher electron density of iron species. The value for k2/3, 1600 ± 200 M−1 s−1 is, in turn, threefold higher than k4 indicating that perhaps the electrostatic repulsion of the two deprotonated species at higher pH mitigates the higher reactivity due to increased electron density. The dominant reaction pathway described by k2/3, either [FeL(OH2)2]− + HOO− or [FeL(OH2) (OH)]2− + HOOH, is kinetically indistinguishable; however, the increased electron density on the metal center of the [FeL(OH2) (OH)]2− complex would probably favor the k2 pathway. The comparison with the values of 4.0 × 102, 1.8 × 104, and 1.5 × 103 M−1 s−1 for k1, k2, and k4 respectively, previously reported for 1c,10 suggests that even though the reactivity of 4 in terms of kI does increase under alkaline conditions, it is nevertheless much lower than that of TAML activators of earlier generations. For the reasons outlined previously, no attempt was made to quantify the pH profile for kII in Figure 6. Semiquantitatively, kII for 4 is ca. 10 times lower than that for 3a.3
The results of our recent work30 on characterization of iron(V)oxo species derived from 4 in pure water allow to hypothesize that iron(V)oxo is most likely the reactive intermediate in catalysis by 4. It has been demonstrated that iron(IV)oxo species is stable at pH ≈ 13. If the solution pH is decreased to 10.6 and below, iron(IV)oxo disproportionates rapidly to form iron(III) and iron(V)oxo derivatives of 4.30 Therefore, iron(V)oxo is the only plausible oxidized species in the pH range shown in Figure 6. This feature is unique for TAML activators, because just iron(IV) state could be generated in pure water for all previously known activators (generations 1–5).
pKa of TAML Activators Is What Controls Reactivity
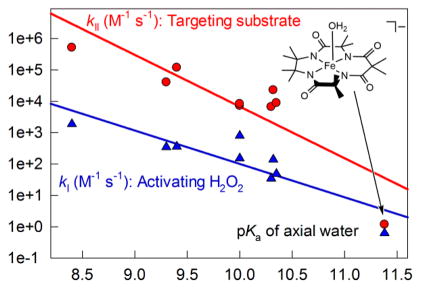
Regardless of their structural variations, TAML activators of generation 1 to 53 all include iron(III) coordinated by four deprotonated amide nitrogens, which form a pseudo plane. The same is true for a new activator 4, the properties of which in aqueous media are described in this work. It is always a challenge to find a parameter (property) of a catalyst that assists in predicting its catalytic activity. The highest value of pKa of the axial water ligand in the family of all TAML activators reported here (11.35) and the lowest of 8.4 for 2e reported previously18 extend the available gap in pKa to three units, which is trustworthy for linear free energy correlations. Both rate constants kI and kII (eq 4) are essential for the oxidative catalysis by TAML activators, and therefore the dependencies of log kI and log kII versus pKa are demonstrated in Figure 7. Here the rate constants kI and kII at pH 7 and 25 °C correspond to reactions with H2O2 and Orange II, respectively. As seen, both kI and kII increase with a decrease in pKa. The rate constants kII are more sensitive than kI to variations in pKa. This agrees with earlier results that revealed poor sensitivity of the kI values to electronic effects.4,12 Dependencies of both rate constants on pKa could be satisfactorily linearized as log k = log k0 + α × pKa with log k0 = 13 ± 2 and 20 ± 4 and α = −1.1 ± 0.2 and −1.8 ± 0.4 for kI and kII, respectively. There is a single data point that deviates more noticeably from the straight line, and this is kII for 4. This we attribute to the steric effect imposed by the tetramethylated part of 4. The steric retardation for kI is obviously less evident than for kII, because the H2O2 molecule is smaller than that of Orange II.
Figure 7.
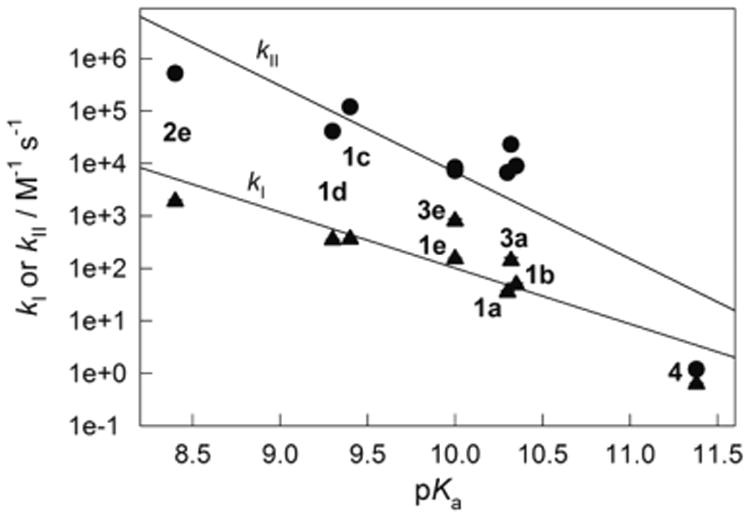
Linear free energy relationships for kI and kII (measured at pH 7 and 25 °C) for selected TAML activators 1–4 as a function of their pKa (see text for details). The data for activators other than 4 and pKa of 1b are reported elsewhere: 2e;18 3a and 3e;3 1a and 1e,3 1d,16 1f;22 other values of kII are from ref 21.
A decrease in pKa parallels the increasing Lewis acidity of the iron(III) center of TAML activators. Correspondingly, both kI and kII increase with increasing Lewis acidity. Because of this it is possible to predict the rate constants kI and kII for TAML activators using the equations log k = log k0 + α × pKa provided the pKa is known. And since the pKa values could be effortlessly measured using the UV–vis spectroscopy as it is shown in Figure 2, estimating the catalytically relevant rate constants kI and kII becomes a painless routine.
Operational Stability of 4
Oxidized forms of TAML activators (active catalyst in Scheme 1) undergo suicidal inactivation, which is quantified by first-order rate constant ki.14 This catalyst inactivation is an attribute of all oxidative catalysts including enzymes.4,13 Values of ki are available through the universal eq 7 provided an oxidant is used in excess with respect to a substrate, and the substrate conversion should be incomplete, which is easily achieved by using low catalyst concentrations.15
| (7) |
Here S0 and S∞ are substrate concentrations at time t = 0 and ∞, that is, degradation of S is incomplete. The value of ki was obtained using kII = 1.19 M−1 s−1 at pH 7 and 25 °C of (4.1 ± 0.1) × 10−7 s−1. It is the lowest measured value to date. However, the ratio kII/ki is comparable to the other TAML activators.14 This implies that the “beheading” of TAML activators to produce 4 does not bring about additional operational stability. This argues against the main degradation pathway by aromatic oxidation, making more probable the degradation via tail group units, as it was postulated earlier.31
Conclusion: Structural and Reactivity Consequences
The novel beheaded TAML activator 4 added several new features to the already rich chemistry of this family of catalysts.1,2,4,11,12 These new features, the most prominent being the lack of phenylene unit in the ligand architecture, the highest pKa of coordinated water, enhanced resistance to the acid-induced iron ejection plus unprecedented aqueous iron-(V)oxo chemistry, which is currently under investigation, allow us to classify 4 as a Generation 6 TAML activator.
Experimental Section
Materials
Synthesis of 4 has recently been communicated.21 Orange II was obtained from Sigma-Aldrich and purified by recrystallization. Hydrogen peroxide was obtained from Fisher Scientific and standardized using the absorbance at 230 nm.32 All other reagents used in this study of at least ACS reagent grade were used as received.
Spectrophotometric Titration
Carmody buffer, consisting of 0.1 M boric acid, 0.025 M citric acid, and 0.05 M K3PO4, was used to prepare a 0.12 mM solution of 4 at pH 7.33 Concentrated KOH and H3PO4 were used for further pH adjustments. The spectral data were collected using a double-beam Shimadzu UV-1800 instrument.
Kinetic Studies
Kinetic studies were conducted at 25 °C in 0.01 M phosphate buffer. Stock solutions of 4, Orange II, and H2O2 were prepared in HPLC-grade water. Appropriate volumes of the buffer, compound 4, and Orange II were added to polystyrene cuvettes, and the reactions were initiated by the addition of an aliquot of H2O2. Reaction progress was assayed by measuring a decrease in absorbance at 484 nm (λmax for Orange II, ε = 2.1 × 104 M−1 cm−1 at pH 7–10) using an HP 8453 diode array spectro-photometer with a thermostated eight-cell changer. Above pH 10 the following values of ε were used: 17 180, 14 850, 12 750, and 11 230 M−1 cm−1 at pH 10.5, 11.0, 11.4, and 11.7, respectively. The initial rates were calculated from linear plots of Orange II concentration versus time when the conversion of the dye did not exceed 10%. Each data point reported is a mean value of at least three measurements.
Electron Paramagnetic Resonance and Mössbauer Measurements
EPR X-band EPR spectra were recorded on a Bruker spectrometer equipped with an Oxford ESR-910 liquid helium cryostat. The signal was quantified relative to a CuEDTA spin standard. For both instruments, the microwave frequency was calibrated with a frequency counter, and the magnetic field was calibrated with an NMR gaussmeter. A modulation frequency of 100 kHz was used for the EPR spectra. The EPR simulation software (Spin Count) was written by one of the authors.34 The software diagonalizes the spin Hamiltonian. The quantitative simulations were least-squares fits of the experimental spectra generated with consideration of the intensity factor, which allows the computation of simulated spectra for a specified sample concentration. Mössbauer spectra were recorded with a spectrometer using a Janis Research dewar. The isomer shifts were reported relative to Fe metal. Mössbauer spectra were obtained as described previously.35
Density Functional Theory Calculations
Density functional theory calculations were performed using Becke's three parameter hybrid functional (B3LYP) and basis set 6-311G provided by the Gaussian 09 software package.36 The geometry optimization for complex 4 was terminated upon reaching the default convergence criteria. The optimizations did not impose any symmetry. Geometry optimizations for each species were performed in the absence of a solvent (in vacuum).
Supplementary Material
Acknowledgments
T.J.C. thanks the Heinz Endowments for support. M.R.M thanks the R. K. Mellon Foundation for support through a Presidential Fellowship.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorg-chem.6b01988.
Spectrophotometric pH titration of 1b (PDF)
Notes: The authors declare no competing financial interest.
References
- 1.Collins TJ. Designing ligands for oxidizing complexes. Acc Chem Res. 1994;27:279–285. [Google Scholar]
- 2.Collins TJ. TAML Oxidant activators: a new approach to the activation of hydrogen peroxide for environmentally significant problems. Acc Chem Res. 2002;35:782–790. doi: 10.1021/ar010079s. [DOI] [PubMed] [Google Scholar]
- 3.Warner G, Mills MR, Enslin C, Pattanayak S, Panda C, Sen Gupta S, Ryabov AD, Collins TJ. Reactivity of N-tailed (‘biuret’) TAMLs in water: Kinetics of the catalyzed oxidation of Orange II by H2O2. Synthesis and X-ray characterization of an N-phenyl biuret TAML. Chem Eur J. 2015;21:6226–6233. doi: 10.1002/chem.201406061. [DOI] [PubMed] [Google Scholar]
- 4.Ryabov AD. Green challenges of catalysis via iron(IV)oxo and iron(V)oxo species. Adv Inorg Chem. 2013;65:118–163. [Google Scholar]
- 5.Mills MR, Arias-Salazar K, Baynes A, Shen LQ, Churchley J, Beresford N, Gayathri C, Gil RR, Kanda R, Jobling S, Collins TJ. Removal of ecotoxicity of 17-ethinylestradiol using TAML/peroxide water treatment. Sci Rep. 2015;5:10511. doi: 10.1038/srep10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Napoly F, Jean-Gérard L, Goux-Henry C, Draye M, Andrioletti B. Fe(TAML)Li/(diacetoxyiodo)benzene-mediated oxidation of alcohols: evidence for mild and selective C–O and C–C oxidative cleavage in lignin model transformations. Eur J Org Chem. 2014;2014:781–787. [Google Scholar]
- 7.Napoly F, Kieffer R, Jean-Gerard L, Goux-Henry C, Draye M, Andrioletti B. Fe(TAML)Li/tert-butyl hydroperoxide as a new combination for benzylic C-H oxidation. Tetrahedron Lett. 2015;56:2517–2520. [Google Scholar]
- 8.Do Pham DD, Kelso GF, Yang Y, Hearn MTW. Studies on the oxidative N-demethylation of atropine, thebaine and oxycodone using a FeIII-TAML catalyst. Green Chem. 2014;16:1399–1409. [Google Scholar]
- 9.Do Pham DD, Kelso GF, Yang Y, Hearn MTW. One-pot oxidative N-demethylation of tropane alkaloids with hydrogen peroxide and a FeIII-TAML catalyst. Green Chem. 2012;14:1189–1195. [Google Scholar]
- 10.Ghosh A, Mitchell DA, Chanda A, Ryabov AD, Popescu DL, Upham E, Collins GJ, Collins TJ. Catalase-peroxidase activity of iron(III)-TAML activators of hydrogen peroxide. J Am Chem Soc. 2008;130:15116–15126. doi: 10.1021/ja8043689. [DOI] [PubMed] [Google Scholar]
- 11.Collins TJ, Khetan SK, Ryabov AD. Chemistry and applications of iron-TAML catalysts in green oxidation processes based on hydrogen peroxide. In: Anastas PT, Crabtree RH, editors. Handbook of Green Chemistry. Wiley-VCH Verlag GmbH & KgaA; Weinheim, Germany: 2009. pp. 39–77. [Google Scholar]
- 12.Ryabov AD, Collins TJ. Mechanistic considerations on the reactivity of green FeIII-TAML activators of peroxides. Adv Inorg Chem. 2009;61:471–521. [Google Scholar]
- 13.Crabtree RH. Deactivation in homogeneous transition metal catalysis: causes, avoidance, and cure. Chem Rev. 2015;115:127–150. doi: 10.1021/cr5004375. [DOI] [PubMed] [Google Scholar]
- 14.Chanda A, Ryabov AD, Mondal S, Alexandrova L, Ghosh A, Hangun-Balkir Y, Horwitz CP, Collins TJ. The activity-stability parameterization of homogeneous green oxidation catalysts. Chem Eur J. 2006;12:9336–9345. doi: 10.1002/chem.200600630. [DOI] [PubMed] [Google Scholar]
- 15.Emelianenko M, Torrejon D, Denardo MA, Ryabov AD, Collins TJ, et al. Estimation of rate constants in nonlinear reactions involving chemical inactivation of oxidation catalysts. J Math Chem. 2014;52:1460–1476. [Google Scholar]
- 16.Popescu DL, Chanda A, Stadler MJ, Mondal S, Tehranchi J, Ryabov AD, Collins TJ. Mechanistically inspired design of FeIII-TAML peroxide-activating catalysts. J Am Chem Soc. 2008;130:12260–12261. doi: 10.1021/ja805099e. [DOI] [PubMed] [Google Scholar]
- 17.Ellis WC, Tran CT, Denardo MA, Fischer A, Ryabov AD, Collins TJ. Design of more powerful iron-TAML peroxidase enzyme mimics. J Am Chem Soc. 2009;131:18052–18053. doi: 10.1021/ja9086837. [DOI] [PubMed] [Google Scholar]
- 18.Ellis WC, Tran CT, Roy R, Rusten M, Fischer A, Ryabov AD, Blumberg B, Collins TJ. Designing green oxidation catalysts for purifying environmental waters. J Am Chem Soc. 2010;132:9774–9781. doi: 10.1021/ja102524v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Panda C, Ghosh M, Panda T, Banerjee R, Sen Gupta S. Fe(III) complex of biuret-amide based macrocyclic ligand as peroxidase enzyme mimic. Chem Commun. 2011;47:8016–8018. doi: 10.1039/c1cc12686e. [DOI] [PubMed] [Google Scholar]
- 20.Ren Q, Guo Y, Mills MR, Ryabov AD, Collins TJ. On the iron(V) reactivity of an aggressive tail-fluorinated tetraamido macrocyclic ligand (TAML) activator. Eur J Inorg Chem. 2015;2015:1445–1452. [Google Scholar]
- 21.DeNardo MA, Mills MR, Ryabov AD, Collins TJ. Unifying evaluation of the technical performances of iron-tetra-amido macrocyclic ligand oxidation catalysts. J Am Chem Soc. 2016;138:2933–2936. doi: 10.1021/jacs.5b13087. [DOI] [PubMed] [Google Scholar]
- 22.Ghosh A, Ryabov AD, Mayer SM, Horner DC, Prasuhn DE, Jr, Sen Gupta S, Vuocolo L, Culver C, Hendrich MP, Rickard CEF, Norman RE, Horwitz CP, Collins TJ. Understanding the mechanism of H+-induced demetalation as a design strategy for robust iron(III) peroxide-activating catalysts. J Am Chem Soc. 2003;125:12378–12378. doi: 10.1021/ja0367344. [DOI] [PubMed] [Google Scholar]
- 23.Williams-Smith DL, Bray RC, Barber MJ, Tsopanakis AD, Vincent SP. Changes in apparent pH on freezing aqueous buffer solutions and their relevance to biochemical electron-paramagnetic-resonance spectroscopy. Biochem J. 1977;167:593–600. doi: 10.1042/bj1670593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson EC, Niem T, Doolphin D. Electron transport via metalloporphyrins. Can J Chem. 1978;56:1381–1388. [Google Scholar]
- 25.Bender ML, Bergeron RJ, Komiyama M. The Bioorganic Chemistry of Enzymatic Catalysis. John Wiley & Sons; New York: 1984. [Google Scholar]
- 26.Chahbane N, Popescu DL, Mitchell DA, Chanda A, Lenoir D, Ryabov AD, Schramm KW, Collins TJ. FeIII-TAML-catalyzed green oxidative degradation of the azo dye Orange II by H2O2 and organic peroxides: products, toxicity, kinetics, and mechanisms. Green Chem. 2007;9:49–57. [Google Scholar]
- 27.Theodoridis A, Maigut J, Puchta R, Kudrik EV, van Eldik R. Novel Iron(III) porphyrazine complex. Complex speciation and reactions with NO and H2O2. Inorg Chem. 2008;47:2994–3013. doi: 10.1021/ic702041g. [DOI] [PubMed] [Google Scholar]
- 28.Ember E, Rothbart S, Puchta R, van Eldik R. Metal ion-catalyzed oxidative degradation of Orange II by H2O2. High catalytic activity of simple manganese salts. New J Chem. 2009;33:34–49. [Google Scholar]
- 29.Rothbart S, Ember EE, van Eldik R. Mechanistic studies on the oxidative degradation of Orange II by peracetic acid catalyzed by simple manganese(II) salts. Tuning the lifetime of the catalyst. New J Chem. 2012;36:732–748. [Google Scholar]
- 30.Mills MR, Weitz AC, Hendrich MP, Ryabov AD, Collins TJ. NaClO-generated iron(IV)oxo and iron(V)oxo TAMLs in pure water. J Am Chem Soc. 2016;138:13866–13869. doi: 10.1021/jacs.6b09572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bartos MJ, Gordon-Wylie SW, Fox BG, Wright LJ, Weintraub ST, Kauffmann KE, Munck E, Kostka KL, Uffelman ES, Rickard CEF, Noon KR, Collins TJ. Designing ligands to achieve robust oxidation catalysts. Iron based systems. Coord Chem Rev. 1998;174:361–390. [Google Scholar]
- 32.George P. The chemical nature of the secon hydrogen peroxide compound fromed by cytochrome c peroxidase and horseradish peroxidase. Biochem J. 1953;54:267–276. doi: 10.1042/bj0540267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carmody WR. An easily prepared wide-range buffer series. J Chem Educ. 1961;38:559–560. [Google Scholar]
- 34.Petasis DT, Hendrich MP. Quantitative interpretation of multifrequency multimode EPR spectra of metal containing proteins, enzymes, and biomimetic complexes. Methods Enzymol. 2015;563:171–208. doi: 10.1016/bs.mie.2015.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang LL, Gunderson WA, Weitz AC, Hendrich MP, Ryabov AD, Collins TJ. Activation of dioxygen by a TAML activator in reverse micelles: Characterization of an FeIIIFeIV dimer and associated catalytic chemistry. J Am Chem Soc. 2015;137:9704–9715. doi: 10.1021/jacs.5b05229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd J, Brothers EN, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian. 09. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.