

Abstract
Iron overload due to hemochromatosis or chronic blood transfusions has been associated with the development of osteoporosis. However, the impact of changes in iron homeostasis on osteoblast functions and the underlying mechanisms are poorly defined. Since Wnt signaling is a critical regulator of bone remodeling, we aimed to analyze the effects of iron overload and iron deficiency on osteoblast function, and further define the role of Wnt signaling in these processes. Therefore, bone marrow stromal cells were isolated from wild-type mice and differentiated towards osteoblasts. Exposure of the cells to iron dose-dependently attenuated osteoblast differentiation in terms of mineralization and osteogenic gene expression, whereas iron chelation with deferoxamine promoted osteogenic differentiation in a time- and dose-dependent manner up to 3-fold. Similar results were obtained for human bone marrow stromal cells. To elucidate whether the pro-osteogenic effect of deferoxamine is mediated via Wnt signaling, we performed a Wnt profiler array of deferoxamine-treated osteoblasts. Wnt5a was amongst the most highly induced genes. Further analysis revealed a time- and dose-dependent induction of Wnt5a being up-regulated 2-fold after 48 h at 50 μM deferoxamine. Pathway analysis using specific inhibitors revealed that deferoxamine utilized the phosphatidylinositol-3-kinase and nuclear factor of activated T cell pathways to induce Wnt5a expression. Finally, we confirmed the requirement of Wnt5a in the deferoxamine-mediated osteoblast-promoting effects by analyzing the matrix mineralization of Wnt5a-deficient cells. The promoting effect of deferoxamine on matrix mineralization in wild-type cells was completely abolished in Wnt5a−/− cells. Thus, these data demonstrate that Wnt5a is critical for the pro-osteogenic effects of iron chelation using deferoxamine.
Introduction
Iron is an essential nutrient for life as it plays a key role in several physiological processes, including the transport of oxygen in erythrocytes, generating ATP as a source of energy, and controlling innate immune responses to combat bacteria.1,2 However, the otherwise desirable redox potential of iron can also generate cellular toxicity in conditions of iron overload, as it produces reactive oxygen species intermediates that damage lipids, DNA, and proteins.3 Therefore, iron concentrations within the body are tightly controlled to maintain an optimal range.
Bone remodeling is susceptible to changes in iron homeostasis, and both iron deficiency and iron excess affect bone health in humans and rodents. Dietary iron is positively associated with bone mineral density in healthy postmenopausal women.4,5 Accordingly, rats rendered iron deficient display low bone mass due to a reduced bone turnover.6,7 On the other hand, elderly women with low bone mass and ovariectomized rats have increased iron levels in the bone, and by chelating iron in ovariectomized rats, bone loss is partially prevented.8,9 Consistent with this, patients with iron overload due to hemochromatosis, β-thalassemia and sickle-cell anemia have been shown to have a higher incidence of osteoporosis.10–13 Importantly, bone mineral density was shown to increase again in patients with β-thalassemia after iron chelation with deferasirox.14 Similar to humans, rats fed with a high iron diet or injected with colloidal iron also experience bone loss, mainly due to an increased osteoclast activation and bone resorption.15–17 Collectively, these data show that certain iron levels are required to maintain bone homeostasis.
Despite the clear evidence that bone homeostasis is tightly regulated by iron, the underlying cellular and molecular mechanisms are not fully understood. In osteoclasts, iron excess stimulates osteoclastogenesis by supporting mitochondrial respiration,18–20 while sequestering iron from osteoclasts inhibits their maturation and function.21–22 In contrast, osteoblast function is suppressed by iron excess in osteoblast cell lines,23–25 while iron chelation using deferoxamine (DFO) seems to exert positive effects on osteoblast maturation.26–28 In addition, bone regeneration during distraction osteogenesis and after radiation therapy has been shown to be promoted in the presence of DFO.29–30 However, some studies have reported an inhibitory effect of DFO on osteoblasts and a suppression of alkaline phosphatase (ALP).31 Thus, further investigations are required to conclusively address the effects of iron excess and chelation on osteoblast function and to assess the underlying mechanisms.
One of the most important pathways for maintaining bone homeostasis is the Wnt signaling pathway.32 Both canonical (β-catenin-dependent) and non-canonical (β-catenin-independent) pathways regulate bone turnover, mainly by promoting osteoblast differentiation and indirectly controlling osteoclastogenesis.33–34 So far, the modulation of Wnt signaling by iron has not been investigated in vivo or in vitro.
Herein, we set out to examine the effects of iron excess and iron chelation via DFO on osteoblast differentiation and function using human and murine primary cells. Moreover, we investigated the iron-dependent regulation of Wnt signaling to assess whether Wnt signaling plays a role in mediating the effects of iron and/or DFO on osteoblasts.
Methods
Mice
Primary murine bone marrow stromal cells (BMSCs) were collected from 10–12 week old C57BL/6J mice of both genders. Also, Wnt5afl/fl mice that were crossed with ROSA26-Cre-ERT2 mice (B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J, Jax Mice, Stock number: 8463) to produce mice in which Wnt5a can be globally deleted upon the administration of tamoxifen.35 Wnt5afl/fl mice without the cre transgene were used as littermate controls. Transferrin receptor 2 knockout mice on a 129XI/SvJ background (male, 10–12 weeks of age) were used as a model of iron overload.36 Wild-type 129XI/SvJ mice served as controls. The sacrifice of the animals for the collection of organs was approved by the institutional animal care committee and the Landesdirektion Sachsen. All mice were fed a standard diet with water ad libitum and were kept in groups of 5 animals per cage. Mice were exposed to a 12 h light/dark cycle and an air-conditioned room at 23 °C.
DFO treatment in mice and μCT analysis
Twelve week old male 129/Sv wild-type mice were treated for three weeks with 250 mg/kg body weight deferoxamine (daily i.p. injections). Micro-CT of the vertebrae was performed ex vivo using the vivaCT 40 (Scanco Medical AG, Brüttisellen, Switzerland) with an X-ray energy of 70 kVp, 114 mA, 200 msec integration time, and an isotropic voxel size of 20 μm. Pre-defined scripts from Scanco were used for the reconstruction and evaluation of the trabecular bone.
For mRNA expression analysis, the mice were sacrificed and primary murine bone BMSCs were yielded by flushing the femora and tibiae with PBS. After centrifugation at 300 × g for 5 min, 1 ml TriFast (Peqlab, Erlangen, Germany) was added to the cell pellet. For the isolation of RNA from the cortical bone tissue of iron-overloaded mice (transferrin receptor 2-deficient mice and wild-type controls), bone marrow was flushed and the cortical bone was crushed in liquid nitrogen. The bone powder was recovered in TriFast. Total RNA from the cells was isolated according to the manufacturer’s protocol. Five-hundred ng RNA were reverse transcribed using Superscript II (Invitrogen, Darmstadt, Germany), and subsequently used for SYBR green-based real-time PCRs using the standard protocol (Applied Biosystems Inc., Carlsbad, CA, USA).
Culture of murine bone marrow stromal cells
Femurs and tibias were dissected from the mice and cells were obtained by flushing the bone marrow with DMEM (supplemented with 10% FCS and 1% penicillin/streptomycin, all from Invitrogen).37,38 Cells were maintained in growth medium (DMEM with 10% FCS and 1% penicillin/streptomycin) until 70% confluence before they were switched to differentiation media (growth medium supplemented with 100 μM ascorbate phosphate and 5 mM β-glycerol phosphate, both from Sigma-Aldrich) for up to 21 days. To delete Wnt5a expression in vitro, cells were treated with 1 μM tamoxifen (Sigma-Aldrich) for 3 days prior to starting the differentiation process.
For some experiments, iron (III) chloride (FeCl3) or deferoxamine (DFO, both from Sigma-Aldrich) was added at different concentrations either for the entire differentiation period or for 48 h. Where indicated, ferric ammonium citrate or holo-transferrin were used as alternative iron sources (both from Sigma-Aldrich). During the 21 day differentiation period, the medium as well as the supplemented FeCl3 and DFO were replaced three times per week. For the signaling studies, cells were pre-treated for 1 h with the specific pathway inhibitors BAY-11-7082 (I-κB phosphorylation inhibitor, 1 μM, Calbiochem), UO126 (MEK1 inhibitor, 10 μM, Calbiochem), LY-294002 (PI3K inhibitor, 10 μM, Cell Signaling), and VIVIT (NFAT inhibitor, 1 μM, Calbiochem). Gene and protein expression of Wnt5a were analyzed after another 48 h of treatment with DFO and the inhibitors.
Culture of human BMSCs
Primary human BMSCs were collected from healthy donors (aged 22–49 years, mixed sex); following Institutional Review Board approval and having obtaining written informed consent; and cultured according to previously reported methods.39 Briefly, cells at passages 2–4 were used. Cells were grown in DMEM with 10% FCS and 1% penicillin/streptomycin. Osteogenic differentiation was induced using growth medium supplemented with 100 μM ascorbate phosphate, 5 mM β-glycerol phosphate, and 10 nM dexamethasone (all from Sigma-Aldrich) for 21 days. Cells were treated with 25 μM FeCl3 or 25 μM DFO for 21 days or for 48 h.
Cell viability assay
Osteoblast cultures were differentiated for 7 days before starting the 48 h treatment with FeCl3 and DFO in a medium with a reduced FCS content (1%) at varying concentrations. Cell viability was measured using the CellTiter-Blue® assay according to the manufacturer’s protocol (Promega, Mannheim, Germany). The fluorescent signal was measured using the FLUOstar Omega (BMG labtech, Jena, Germany).
Alizarin red staining
Osteoblast cultures were fixed in 70% ethanol for 30 min and stained with 1% alizarin red S (pH 5.5, Sigma-Aldrich) for 10 min at room temperature. Excess dye was removed by repeatedly washing the plates with distilled water. The amount of incorporated calcium was eluted with 100 mM cetylpyridinium chloride (Sigma-Aldrich) for 10 min at room temperature. Aliquots were taken and measured photometrically at 540 nm in duplicates.
Alkaline phosphatase activity assay
Cells were lysed in 100 μl lysis buffer (1.5 mM Tris-HCl pH 7, 1 mM ZnCl2, 1 mM MgCl2, 1% triton X-100) and centrifuged for 20 min at 6,000 rpm at 4 °C. Aliquots of each sample were incubated with 100 μl alkaline phosphatase (ALP) substrate buffer (100 mM diethanolamine, 0.1% triton X-100 supplemented with 1:10 37 mM p-nitrophenyl phosphate) for 30 min at 37 °C. The enzymatic reaction was stopped with 40 mM NaOH, measured at a wavelength of 405 nm, and normalized to the total protein content determined by the BCA method from the same protein extracts.
RNA isolation, RT, and real-time RT-PCR
Total RNA from cell culture was isolated using the High Pure RNA Isolation kit (Roche, Mannheim, Germany) according to the manufacturer’s protocol. Five-hundred ng RNA were reverse transcribed using Superscript II (Invitrogen, Darmstadt, Germany) and subsequently used for SYBR green-based real-time PCRs using standard protocol (Applied Biosystems Inc., Carlsbad, CA, USA). The primer sequences for mice were: β-actin: ATCTGGCACCACACCTTCT, β-actin as: GGGGTGTTGAAGGTCTCAAA; Runx2: CCCAGCCACCTTTACCTACA, Runx2 as: TATGGAGTGCTGCTGCTGGTCTG; Alp as: CTACTTGTGTGGCGTGAAGG, Alp: CTGGTGGCATCTCGTTATCC; Ocn: GCGCTCTGTCTCTCTGACCT, Ocn as: ACCTTATTGCCCTCCTGCTT; Wnt5a as: CCAACTGGCAGGACTTTCTC, Wnt5a: GCATTCCTTGATGCCTGTCT. The primer sequences for the human genes were: β-ACTIN: CCAACCGCGAGAAGATGA, β-ACTIN as: CCAGAGGCGTACAGGGATAG; ALP: CAACCCTGGGGAGGAGAC, ALP as: GCATTGGTGTTGTACGTCTTG; RUNX2: CACCATGTCAGCAAAACTTCTT, RUNX2 as: TCACGTCGCTCATTTTGC, WNT11: CAAGTTTTCCGATGCTCCTATGAA, WNT11 as: TTGTGTAGACGCATCAGTTTATTGG; WNT5a: CCTGCCAAAAACAGAGGTGT. WNT5a as: CCTGCCAAAAACAGAGGTGT. The results were calculated using the ΔΔCT method and are presented in x-fold increase relative to β-actin mRNA levels.
Wnt profiler PCR array
Cells were differentiated with osteogenic medium for 7 days and treated with 25 μM DFO for 48 h. Afterwards, RNA was isolated as described above, reverse transcribed using the RT2 First Strand Kit (SABiosciences) and 500 ng cDNA, and were subjected to the Wnt profiler PCR array, containing 84 Wnt-related genes, according to the manufacturer’s protocol (SABiosciences). Genes were normalized to the mean of five housekeeping genes (β-actin, Gapdh, Hsp90ab1, Gusb, b2m).
Western blot analysis
For the Western Blot analysis, cells were either stimulated with 25 μM DFO for 30 min (signaling studies) or 48 h (Wnt5a expression). Cells were lysed in lysis buffer containing 20 mM Tris/HCl pH 7.4, 1% SDS and a protease inhibitor (complete mini, Roche). Lysates were processed through a 24-gauge needle and centrifuged at 20,000 × g for 20 min. The protein content was measured using the BCA method. For electrophoresis, 20 μg protein was loaded on a 10% SDS-PAGE and transferred onto a 0.2 μm nitrocellulose membrane (Whatman). After blocking for 1 h with 5% nonfat dry milk (Wnt5a) or 1% BSA (signaling antibodies) in Tris-buffered saline with 1% Tween-20 (TBS-T), membranes were incubated with an anti-WNT5a antibody (Santa Cruz Biotech, 1:1,000) or the respective signaling antibodies (AKT, pAKT, ERK, pERK, p38, pp38, NF-κB, pNF-κB, all from Cell Signaling, 1:1,000; NFATc1: Thermo Fischer Scientific, 1:500) overnight. GAPDH was used as a loading control (1:2,000, Santa Cruz Biotech). Membranes were washed and incubated with the appropriate HRP conjugated secondary antibodies for 1 h at RT. After washing again, membranes were incubated with an ECL substrate (Pierce, Thermo Fisher Scientific) to visualize the proteins using the MF-ChemiBIS 3.2 bioimaging system (Biostep, Jahnsdorf, Germany).
Statistical analysis
Data are presented as mean ± standard deviation (SD). Statistical evaluations of two group comparisons were performed using a two-sided Student’s t-test. One-way analysis of variance (ANOVA) was used for experiments with more than two groups or time/dose experiments.
Results
Excess iron and iron chelation exhibit opposing effects on osteogenic differentiation
To determine the effects of iron excess and iron deficiency on the osteogenic differentiation, we treated mBMSC with increasing concentrations of iron (III) chloride or DFO for the entire differentiation period. Iron excess led to a marked decrease in osteoblast differentiation as measured by the amount of mineralized matrix reaching a maximum suppression of 67% at a concentration of 50 μM (Figure 1A). Similar results were also seen when ferric ammonium citrate or holo-transferrin were used as different iron sources (Figure 1B). In contrast, DFO stimulated osteogenic differentiation showing the most potent effect at a concentration of 25 μM (3.7-fold) (Figure 1C). To address whether the osteoblast-promoting effect of DFO is specifically due to chelating iron, DFO-treated cells were incubated with increasing concentrations of iron chloride. Indeed, higher doses of iron dose-dependently suppressed the DFO-induced mineralization (Figure 1D). Cell viability was not altered by iron concentrations up to 50 μM (50606 ± 11205 vs. 48175 ± 9875 fluorescence units, not significant), while DFO started to inhibit osteoblast viability at concentrations of 50 μM (50606 ± 11205 vs. 42671 ± 14690 fluorescence units, P<0.05). The inhibitory effects of iron excess and stimulatory effects of iron chelation were further demonstrated by analyzing the gene expression levels of the osteoblast markers Runx2, Alp and osteocalcin (Bglap) as well as by measuring the enzyme activity of ALP (Figures 1E–H). In all cases, iron excess suppressed osteoblast markers, while DFO increased their expression. The DFO-mediated induction of the osteoblastic genes was reversed when cells were concomitantly exposed to iron (Figures 1E–G).
Figure 1.
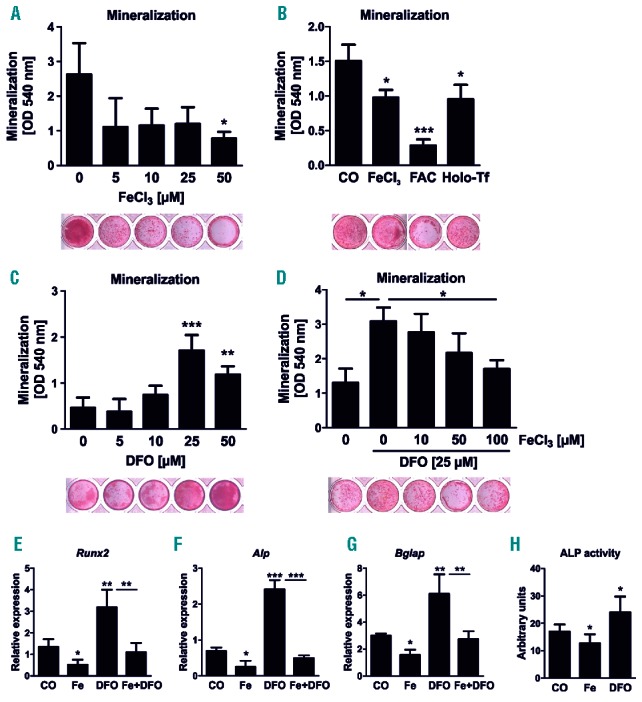
Iron excess inhibits and iron chelation supports osteogenic differentiation. (A–D) Bone marrow stromal cells were differentiated towards osteoblasts in the presence of (A) various concentrations of iron chloride (FeCl3), (B) 25 μM FeCl3, 1 μM ferric ammonium citrate (FAC), and 250 μM holo-transferrin (Holo-Tf), (C) the iron-chelator deferoxamine (DFO) as well as (D) combinations of FeCl3 and DFO for 21 days. The mineralization was visualized with alizarin red S staining and quantified after elution with cetylpyridinium chloride. (E–G) Gene expression analysis of Runx2, alkaline phosphatase (Alp) and osteocalcin (Bglap) using real-time polymerase chain reaction (PCR) after treating day 10 differentiated cells with 5 μM FeCl3 (Fe), 25 μM DFO or Fe+DFO for an additional 48 hours. (H) Enzyme activity of ALP in day 10 osteoblasts after 48 hours of Fe (5 μM) and DFO (25 μM) treatment. N=4–6. *P<0.05, **P<0.01, ***P<0.001 vs. control (CO).
To assess whether altering the iron balance also affects osteogenic differentiation in human cells, BMSCs isolated from human donors were treated with iron (III) chloride and DFO. Similar to murine cells, iron excess inhibited osteoblast function by 42%, while DFO stimulated matrix mineralization by 2.6-fold (Figure 2A). Also, corresponding effects were seen on the gene expression of RUNX2 and ALP (Figure 2B,C). Thus, iron excess suppresses osteogenic differentiation while iron chelation has pro-osteogenic effects in human and murine BMSC.
Figure 2.

Osteogenic differentiation of human bone marrow stromal cells is impaired by iron excess and increased by iron chelation. (A) Bone marrow stromal cells were obtained from healthy donors (hBMSCs) and differentiated towards osteoblasts in the presence of 5 μM iron chloride (Fe) and 25 μM deferoxamine (DFO). Mineralization was determined using alizarin red S staining and subsequent elution and quantification. (B–C) Quantitative gene expression analysis of Runx2 and alkaline phosphatase (ALP) after treating day 10 differentiated cells with Fe (5 μM) or DFO (25 μM) for an additional 48 hours. N=4. *P<0.05 vs. control (CO).
Wnt5a expression is induced by deferoxamine
As DFO exerted potent pro-osteogenic effects and Wnt signaling is required for osteoblast differentiation, we analyzed the Wnt profile of osteoblasts after treatment with 25 μM DFO for 48 h. As indicated in Figure 3A, three distinct Wnt inhibitors were significantly down-regulated after DFO treatment (Sfrp1, Sfrp2 and Sfpr4) along with the canonical Wnt ligands Wnt2b, Wnt4, Wnt7b, Wnt8 and Wnt16 and the transcription factor Tcf7 and Lef1 (Figure 3A). In addition, the canonical Wnt target genes axin 2, cyclin D1 (Ccnd1) and cyclin D2 (Ccnd2) were significantly down-regulated by DFO suggesting an overall down-regulation of canonical Wnt signaling (Figure 3A). With regard to non-canonical Wnt signaling, Wnt5a and Wnt11 as well as the downstream mediators Jun and Fosl1 were up-regulated 2.5-6-fold (Figure 3A).
Figure 3.

Deferoxamine (DFO) modulates the expression of components of the Wnt pathway. (A) Bone marrow stromal cells were differentiated towards osteoblasts for 10 days and treated with 25 μM DFO for 48 hours. A Wnt profiler polymerase chain reaction (PCR) array was performed using real-time PCR. Genes are separated into three categories: Wnt ligands/receptors, Wnt target genes and Wnt inhibitors. Genes in green indicate down-regulated genes (lighter green means more intensively down-regulated); red ones are up-regulated. All genes presented in the bar graph were significantly regulated (P<0.05). N=3. (B–C) Day 10 differentiated cells were stimulated with various concentrations and durations of DFO. Afterwards, Wnt5a expression was determined using real-time PCR. N=4. (D–E) Western blot analysis of WNT5a in osteoblasts after DFO treatment (25 μM) for 48 hours. Three independent experiments are shown. Graph indicates quantification experiments using ImageJ. (F) Human bone marrow stromal cells (hBMSCs) were obtained from healthy donors and differentiated towards osteoblasts for 10 days. Afterwards, cells were treated with Fe (iron chloride) (5 μM) or DFO (25 μM) for an additional 48 hours to assess WNT5a expression using real-time PCR. N=4. (G) Murine bone marrow stromal cells were differentiated towards osteoblasts for 10 days and treated with 5–50 μM FeCl3 for 48 hours. Afterwards, Wnt5a expression was determined using real-time PCR. N=3. *P<0.05, **P<0.01 vs. control (CO).
The induction of Wnt5a was further validated using time-and dose-response curves. Wnt5a expression was dose-dependently increased after DFO treatment reaching a maximum at 100 μM (2.3-fold) after 48 h (Figure 3B,C). The induction of Wnt5a expression by DFO was finally also confirmed by Western blot analysis showing a 3.7-fold induction (Figure 3D,E). In addition, WNT5a expression was also increased after 48 h of DFO treatment in human BMSC (Figure 3F). Iron stimulation did not alter Wnt5a expression (Figure 3G). Hence, DFO targets several components of the Wnt signaling pathway, with the robust induction of Wnt5a representing a pro-osteogenic Wnt ligand that may contribute to the osteoblast-supporting effects of DFO.
The PI3K and NFAT pathways mediate the induction of WNT5a expression by DFO
To examine the pathways involved in the induction of Wnt5a levels after DFO treatment, we analyzed the activation of different pathways that have previously been reported to be activated by DFO.40–43 After 30 min stimulation, DFO (50 μM) activated several pathways, most notably NFATc1, AKT, and NF-κB. The MAPK downstream pathways ERK and p38 were not activated by DFO (Figure 4A). To verify the necessity of the NFATc1, AKT, and NF-κB pathways, we used specific pathway inhibitors prior to stimulation with DFO. Only the PI3K/AKT inhibitor (LY-294002) and the NFAT inhibitor (VIVIT) were able to block the DFO-induced Wnt5a expression, as shown in Figure 4B,C. Inhibition of the NF-κB (BAY-11-7082) and ERK pathways (UO126) did not interfere with the induction of Wnt5a expression after 48 h of DFO treatment (Figure 4B,C). Thus, DFO signals through the PI3K/AKT as well as the NFATc1 pathways to up-regulate Wnt5a expression in osteoblasts.
Figure 4.
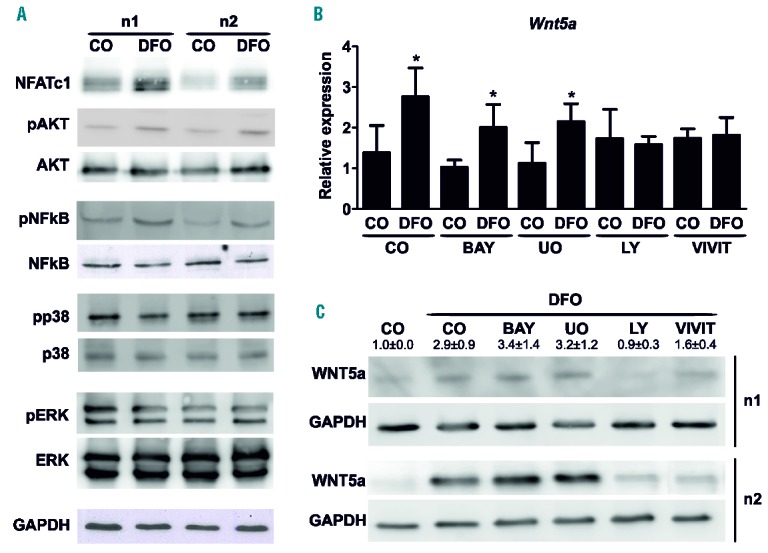
Deferoxamine (DFO) modulates the expression of Wnt5a via the PI3K and NFAT pathways. (A) Day 10 differentiated bone marrow stromal cells (BMSCs) were treated with 50 μM DFO for 30 min. Western blot analysis was performed for various signaling pathways. Protein amounts were normalized to GAPDH. Three independent experiments were performed. Two are shown. (B–C) BMSCs were treated with the following inhibitors prior to the addition of 50 μM DFO for 48 hours: BAY-11-7082 (I-κB phosphorylation inhibitor, 1 μM), UO126 (MEK1 inhibitor, 10 μM), LY-294002 (PI3K inhibitor, 10 μM), and VIVIT (NFAT inhibitor, 1 μM). Afterwards, Wnt5a expression was determined using either real-time polymerase chain reaction (PCR) (B) or Western Blot analysis (C). All three Western blots were quantified using ImageJ. N=3–4. *P<0.05 vs. control (CO).
Wnt5a mediates the pro-osteogenic effect of DFO
As Wnt5a is a known pro-osteogenic Wnt ligand and its expression is highly induced by DFO, we next assessed whether the pro-osteogenic effect of DFO is mediated via Wnt5a. Therefore, we isolated BMSC from Wnt5afl/fl:ROSA26-Cre-ERT2 or their littermate controls (Wnt5afl/fl), and deleted Wnt5a expression in osteogenic cells in vitro using tamoxifen for 3 consecutive days. After 7 days, the recombination efficiency was analyzed using real-time PCR analysis and revealed a 75% deletion of Wnt5a expression levels (Wnt5afl/fl: 1.17 ± 0.22; Wnt5afl/fl:ROSA26-Cre-ERT2: 0.28 ± 0.08 relative expression, P<0.001). Next, we treated Wnt5a proficient and Wnt5a deficient cells with increasing concentrations of DFO throughout the entire differentiation period of 21 days. Alizarin red staining showed that the pro-osteogenic effect of DFO at the concentrations of 25 and 50 μM were completely abrogated in Wnt5a deficient cells (Figure 5A,B). Therefore, WNT5a mediates the osteoinductive effect of DFO.
Figure 5.

Wnt5a mediates the pro-osteogenic effect of deferoxamine (DFO). (A–B) Bone marrow stromal cells were isolated from wild-type (WT) or Wnt5a:ROSA26-ERT2Cre mice. Deletion of Wnt5a was induced by treating the cells with 1 μM tamoxifen for 3 consecutive days. After deletion, cells were differentiated for 21 days with or without the addition of increasing concentrations of DFO. (A) Mineralization was assessed using alizarin red S. (B) Quantification of alizarin red S after elution with cetylpyridinium chloride. N=4. (C–G) Twelve-week-old male 129Sv mice were treated with 250 mg/kg DFO or phosphate buffered saline (PBS) daily for three weeks. Thereafter, mice were sacrificed to assess the expression of Wnt5a in the bone marrow using real-time polymerase chain reaction (PCR). (C) (D) Bone volume/total volume (BV/TV), (E) trabecular thickness (Tb.Th), (F) trabecular number (Tb.N), and (G) trabecular separation (Tb.Sp) at the fourth lumbar vertebrae were determined using μCT. N=5. *P<0.05 vs. control (CO).
Finally, we treated healthy 12-week-old male mice with DFO for three weeks to assess whether Wnt5a is also regulated in vivo and determine whether DFO alters bone mass. As shown in Figure 5C, Wnt5a expression was induced 2-fold in bone marrow cells isolated from DFO-treated animals. Moreover, DFO tended to increase the bone volume fraction in the fourth lumbar vertebrae (Figure 5D). In addition, the trabecular thickness and the trabecular number also tended to be increased, whereas trabecular separation was decreased, suggesting bone-promoting effects (Figure 5E–G). Similar to the in vitro experiments, Wnt5a expression was not altered in the bones derived from iron-overloaded mice (wild-type: 37.3 ± 10.0 transferrin receptor 2 knockout: 27.3 ± 11.9, relative expression, P=0.14). Thus, DFO also modulates Wnt5a expression in a complex in vivo system.
Discussion
Herein we show that altering the exogenous iron concentration in osteoblast cultures critically influences their differentiation capacity, with iron excess inhibiting and iron chelation promoting osteoblast differentiation. Moreover, we have identified Wnt5a as a target gene of DFO that contributes to its pro-osteogenic effects.
Even though there is strong evidence that bone remodeling is affected by iron concentrations which are too high or too low,6–7,10–13 the cellular effects, in particular on osteoblast biology, are not fully understood. The results of our study confirm the inhibitory effect of iron excess on osteoblast function as indicated by a decreased matrix production and a reduced expression of osteoblast markers.23–25,43 Wnt5a expression was not reduced by the iron treatment, suggesting that the suppression of Wnt5a expression may not be an underlying mechanism. Our study is in line with a number of other studies that have shown an osteoblast-supporting effect of DFO.26–28,44,45 Interestingly, osteoblasts treated with 50 μM DFO showed a somewhat decreased mineralization potential as compared to 25 μM DFO, indicating that this concentration might already exert toxic effects. In fact, we found a reduced cell viability in murine cells treated with 50 μM DFO, while cell vitality was not affected at 25 μM. Depending on the cell type, other studies have also shown toxic effects of DFO in concentrations above 30 μM.27,28 In contrast, human periodontal ligament cells do not seem to be particularly sensitive to DFO, as even concentrations of up to 200 μM still exerted pro-osteogenic effects.26 In our hands, DFO reduced human BMSC viability at similar concentrations as murine BMSC (i.e., 50 μM, data not shown). However, it should be noted that the impact on cell viability did not seem to be of biological significance, as osteoblasts treated with 50 μM still differentiate better than untreated cells. Therefore, at non-toxic concentrations, the iron-chelating effect of DFO seems to promote osteoblast differentiation.
To gain more insights into the molecular mechanisms that contribute to the osteoblast-promoting effects of DFO, we investigated whether Wnt signaling, a critical pathway for osteoblast differentiation, is involved. Canonical and non-canonical Wnt pathways both positively affect bone mass. While canonical signaling increases osteoblast function and suppresses osteoclast function, non-canonical WNT5a signaling increases both osteoblast and osteoclast function. This mechanism nonetheless also results in an increased bone mass.32 Using a Wnt profiler array we found that several genes involved in canonical Wnt signaling were down-regulated, including Wnt2b, Wnt7b, Tcf7, Fzd5, Fzd6 and axin-2. Previously, Qu et al. demonstrated that DFO promotes the stabilization of β-catenin through phosphorylating glycogen synthase kinase-3β, and that the pro-osteogenic effect of DFO is dependent on β-catenin.27 Other studies, however, report an inhibition of Wnt target genes by DFO in osteoblasts46 and a suppression of canonical Wnt signaling in neuronal cells.47,48 As Wnt signaling is strongly context- and cell type-dependent, it would not be surprising if DFO would exert opposing effects on Wnt signaling in different cell types. Nevertheless, more research is required to address the discrepancies on the regulation of canonical Wnt signaling by DFO.
In contrast to the canonical Wnt genes, the non-canonical ligands Wnt5a and Wnt11 were up-regulated after DFO treatment. In particular Wnt5a was robustly induced by DFO in a time- and dose-dependent manner and required the activation of the PI3K/AKT and NFAT pathways. Both the PI3K/Akt and NFAT pathways have also previously been shown to be activated upon stimulation with DFO in epidermal and pheochromocytoma cells.40,41 Even though the ERK and p38 pathways have also been reported to be activated by DFO, these pathways were not necessary to induce Wnt5a expression in osteoblasts.42,43 Besides directly activating specific signaling pathways, DFO is also known to inhibit prolyl hydroxylases (PHDs) by sequestering the iron that is required for their activity. Inhibition of PHDs leads subsequently to the stabilization of hypoxia-inducible factors (HIFs).49 Although the exact role of HIF proteins on osteoblast differentiation is not fully understood, the majority of studies show that HIF activation reduces osteoblast differentiation and bone mass.49–52 While we cannot exclude that hypoxia-dependent pathways contribute to the effects of DFO on osteoblasts, it seems unlikely to mediate its osteoblast supportive effects.
As Wnt5a promotes osteoblast differentiation,34 we further examined whether the induction of Wnt5a is necessary for the stimulation of osteoblast differentiation by DFO. Even though the ablation of Wnt5a expression in osteoblasts did not affect the differentiation potential in vitro, it completely abolished the pro-osteogenic effect of DFO, indicating that this is a crucial pathway of DFO to stimulate osteoblast differentiation in vitro. Likewise, DFO treatment in mice led to an increased expression of Wnt5a in the bone marrow, underlining its significance in vivo. Finally, bone volume fraction was almost significantly increased, suggesting bone-promoting effects of DFO in vivo.
Our study is potentially limited by the use of only one iron chelator (DFO) and one type of iron source (FeCl3) in most experiments. In other studies, different sources of iron are found, for example ferric ammonium citrate.28 However, all studies have shown suppressive effects of iron on osteoblast differentiation, suggesting that the exact chemical composition may not be of relevance for in vitro studies. This is further supported by our study showing that ferric ammonium citrate and holo-transferrin also suppressed osteoblast differentiation. Furthermore, considering that DFO also binds to aluminum, copper and zinc, although with a lower affinity,53 we could verify that the osteoblast-promoting effects of DFO are specific for iron, as the addition of iron to DFO-treated cultures inhibited the mineralization capacity of the osteoblasts.
In conclusion, our results demonstrate that certain iron concentrations are necessary for proper osteoblast differentiation and that Wnt5a is required for the pro-osteogenic effect of DFO. Thus, in addition to its potent effect in reducing the iron load in patients with hemochromatosis or sickle cell anemia, treatment with DFO and perhaps also novel iron chelating agents may also improve bone health in those patients. However, this assumption needs to be verified in further studies.
Supplementary Material
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/100/12/1499
Funding
This work was supported by Start-up grants of the Medical Faculty of the Technische Universität Dresden (Meddrive) to UB and MR, the German Research Foundation (SFB655/P13) to UP and LCH, the Josè Carreras foundation to MR and UP, and funding from the Excellence Initiative by the German Federal and State Governments” (Institutional Strategy, measure “Support the best”) to MR and LCH.
DFG funding SFB-655, project B13 to UP and LCH; Start-up grants from the Medical Faculty of the TUD to UB and MR; Jose Carreras foundation to MR and UP (DJCLS R 13/15); Excellence Initiative by the German Federal and State Governments” (Institutional Strategy, measure “Support the best”) to MR and LCH.
References
- 1.Cassat JE, Skaar EP. Iron in infection and immunity. Cell Host Microbe. 2013;13(5):509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117(3):285–297. [DOI] [PubMed] [Google Scholar]
- 3.Koskenkorva-Frank TS, Weiss G, Koppenol WH, Burckhardt S. The complex interplay of iron metabolism, reactive oxygen species, and reactive nitrogen species: insights into the potential of various iron therapies to induce oxidative and nitrosative stress. Free Radic Biol Med. 2013;65:1174–1194. [DOI] [PubMed] [Google Scholar]
- 4.Abraham R, Walton J, Russell L, et al. Dietary determinants of post-menopausal bone loss at the lumbar spine: a possible beneficial effect of iron. Osteoporos Int. 2006;17(8):1165–1173. [DOI] [PubMed] [Google Scholar]
- 5.Harris MM, Houtkooper LB, Stanford VA, et al. Dietary iron is associated with bone mineral density in healthy postmenopausal women. J Nutr. 2003;133(11):3598–3602. [DOI] [PubMed] [Google Scholar]
- 6.Katsumata S, Katsumata-Tsuboi R, Uehara M, Suzuki K. Severe iron deficiency decreases both bone formation and bone resorption in rats. J Nutr. 2009;139(2):238–243. [DOI] [PubMed] [Google Scholar]
- 7.Medeiros DM, Plattner A, Jennings D, Stoecker B. Bone morphology, strength and density are compromised in iron-deficient rats and exacerbated by calcium restriction. J Nutr. 2002;132(10):3135–3141. [DOI] [PubMed] [Google Scholar]
- 8.Kenner GH, Brik AB, Liu G, et al. Variation of long-lived free radicals responsible for the EPR native signal in bone of aged or diseased human females and ovariectomized adult rats. Radiat Meas. 2005; 39(3):255–262. [DOI] [PubMed] [Google Scholar]
- 9.Liu G, Men P, Kenner GH, Miller SC. Age-associated iron accumulation in bone: implications for postmenopausal osteoporosis and a new target for prevention and treatment by chelation. Biometals. 2006;19(3):245–251. [DOI] [PubMed] [Google Scholar]
- 10.Guggenbuhl P, Deugnier Y, Boisdet JF, et al. Bone mineral density in men with genetic hemochromatosis and HFE gene mutation. Osteoporos Int. 2005;16(12):1809–1814. [DOI] [PubMed] [Google Scholar]
- 11.Skordis N, Toumba M. Bone disease in thalassaemia major: recent advances in pathogenesis and clinical aspects. Pediatr Endocrinol Rev. 2011;Suppl2: 300–306. [PubMed] [Google Scholar]
- 12.Sarrai M, Duroseau H, D’Augustine J, Moktan S, Bellevue R. Bone mass density in adults with sickle cell disease. Br J Haematol. 2007;136(4):666–672. [DOI] [PubMed] [Google Scholar]
- 13.Valenti L, Varenna M, Fracanzani AL, et al. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporos Int. 2009; 20(4):549–555. [DOI] [PubMed] [Google Scholar]
- 14.Casale M, Citarella S, Filosa A, et al. Endocrine function and bone disease during long-term chelation therapy with deferasirox in patients with -thalassemia major. Am J Hematol. 2014;89(12):1102–1106. [DOI] [PubMed] [Google Scholar]
- 15.Isomura H, Fujie K, Shibata K, et al. Bone metabolism and oxidative stress in postmenopausal rats with iron overload. Toxicology. 2004;15:197(2):93–100. [DOI] [PubMed] [Google Scholar]
- 16.Kudo H, Suzuki S, Watanabe A, et al. Effects of colloidal iron overload on renal and hepatic siderosis and the femur in male rats. Toxicology. 2008;246(2–3):143–147. [DOI] [PubMed] [Google Scholar]
- 17.Tsay J, Yang Z, Ross FP, et al. Bone loss caused by iron overload in a murine model: importance of oxidative stress. Blood. 2010;116(14):2582–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishii KA, Fumoto T, Iwai K, et al. Coordination of PGC-1beta and iron uptake in mitochondrial biogenesis and osteoclast activation. Nat Med. 2009; 15(3):259–266. [DOI] [PubMed] [Google Scholar]
- 19.Jia P, Xu YJ, Zhang ZL, et al. Ferric ion could facilitate osteoclast differentiation and bone resorption through the production of reactive oxygen species. J Orthop Res. 2012;30(11):1843–1852. [DOI] [PubMed] [Google Scholar]
- 20.Xiao W, Beibei F, Guangsi S, et al. Iron overload increases osteoclastogenesis and aggravates the effects of ovariectomy on bone mass. J Endocrinol. 2015;226(3):121–134. [DOI] [PubMed] [Google Scholar]
- 21.Xie W, Lorenz S, Dolder S, Hofstetter W. Extracellular iron is a modulator of the differentiation of osteoclast lineage cells. Calcif Tissue Int. 2016;98(3):275–283. [DOI] [PubMed] [Google Scholar]
- 22.Guo JP, Pan JX, Xiong L, et al. Iron Chelation Inhibits Osteoclastic Differentiation In Vitro and in Tg2576 Mouse Model of Alzheimer’s Disease. PLoS One. 2015;10(11):e0139395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doyard M, Fatih N, Monnier A, et al. Iron excess limits HHIPL-2 gene expression and decreases osteoblastic activity in human MG-63 cells. Osteoporos Int. 2012;23(10):2435–2445. [DOI] [PubMed] [Google Scholar]
- 24.Yamasaki K, Hagiwara H. Excess iron inhibits osteoblast metabolism. Toxicol Lett. 2009;191(2–3):211–215. [DOI] [PubMed] [Google Scholar]
- 25.Messer JG, Kilbarger AK, Erikson KM, Kipp DE. Iron overload alters iron-regulatory genes and proteins, down-regulates osteoblastic phenotype, and is associated with apoptosis in fetal rat calvaria cultures. Bone. 2009;45(5):972–979. [DOI] [PubMed] [Google Scholar]
- 26.Chung JH, Kim YS, Noh K, et al. Deferoxamine promotes osteoblastic differentiation in human periodontal ligament cells via the nuclear factor erythroid 2-related factor-mediated antioxidant signaling pathway. J Periodontal Res. 2014;49(5):563–573. [DOI] [PubMed] [Google Scholar]
- 27.Qu ZH, Zhang XL, Tang TT, Dai KR. Promotion of osteogenesis through beta-catenin signaling by desferrioxamine. Biochem Biophys Res Commun. 2008;370(2):332–337. [DOI] [PubMed] [Google Scholar]
- 28.Zhao GY, Zhao LP, He YF, et al. A comparison of the biological activities of human osteoblast hFOB1.19 between iron excess and iron deficiency. Biol Trace Elem Res. 2012;150(1–3):487–495. [DOI] [PubMed] [Google Scholar]
- 29.Farberg AS, Jing XL, Monson LA, et al. Deferoxamine reverses radiation induced hypovascularity during bone regeneration and repair in the murine mandible. Bone. 2012;50(5):1184–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wan C, Gilbert SR, Wang Y, et al. Role of hypoxia inducible factor-1 alpha pathway in bone regeneration. J Musculoskelet Neuronal Interact. 2008;8(4):323–324. [PubMed] [Google Scholar]
- 31.Messer JG, Cooney PT, Kipp DE. Iron chelator deferoxamine alters iron-regulatory genes and proteins and suppresses osteoblast phenotype in fetal rat calvaria cells. Bone. 2010;46(5):1408–1415. [DOI] [PubMed] [Google Scholar]
- 32.Baron R, Kneissel M. WNT signaling in bone homeostasis and disease: from human mutations to treatments. Nat Med. 2013;19(2):179–192. [DOI] [PubMed] [Google Scholar]
- 33.Gong Y, Slee RB, Fukai N, et al. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001; 107(4):513–523. [DOI] [PubMed] [Google Scholar]
- 34.Maeda K, Kobayashi Y, Udagawa N, et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. 2012;18(3):405–412. [DOI] [PubMed] [Google Scholar]
- 35.Miyoshi H, Ajima R, Luo CT, Yamaguchi TP, Stappenbeck TS. Wnt5a potentiates TGF-beta signaling to promote colonic crypt regeneration after tissue injury. Science. 2012;338(6103):108–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roetto A, Di Cunto F, Pellegrino RM, et al. Comparison of 3 Tfr2-deficient murine models suggests distinct functions for Tfr2-alpha and Tfr2-beta isoforms in different tissues. Blood. 2010;115(16):3382–3389. [DOI] [PubMed] [Google Scholar]
- 37.Rauner M, Föger-Samwald U, Kurz MF, et al. Cathepsin S controls adipocytic and osteoblastic differentiation, bone turnover, and bone microarchitecture. Bone. 2014;64:281–287. [DOI] [PubMed] [Google Scholar]
- 38.Sinningen K, Albus E, Thiele S, et al. Loss of milk fat globule-epidermal growth factor 8 (MFG-E8) in mice leads to low bone mass and accelerates ovariectomy-associated bone loss by increasing osteoclastogenesis. Bone. 2015;76(25):107–114. [DOI] [PubMed] [Google Scholar]
- 39.Rauner M, Stein N, Winzer M, et al. WNT5A is induced by inflammatory mediators in bone marrow stromal cells and regulates cytokine and chemokine production. J Bone Miner Res. 2012;27(3):575–585. [DOI] [PubMed] [Google Scholar]
- 40.Alvarez-Tejado M, Naranjo-Suarez S, Jiménez C, et al. Hypoxia induces the activation of the phosphatidylinositol 3-kinase/Akt cell survival pathway in PC12 cells: protective role in apoptosis. J Biol Chem. 2001;276(25):22368–22374. [DOI] [PubMed] [Google Scholar]
- 41.Huang C, Li J, Zhang Q, Huang X. Role of bioavailable iron in coal dust-induced acti vation of activator protein-1 and nuclear factor of activated T cells: difference between Pennsylvania and Utah coal dusts. Am J Respir Cell Mol Biol. 2002;27(5):568–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klettner A, Koinzer S, Waetzig V, Herdegen T, Roider J. Deferoxamine mesylate is toxic for retinal pigment epithelium cells in vitro, and its toxicity is mediated by p38. Cutan Ocul Toxicol 2010;29(2):122–129. [DOI] [PubMed] [Google Scholar]
- 43.Markel TA, Crisostomo PR, Wang M, et al. Iron chelation acutely stimulates fetal human intestinal cell production of IL-6 and VEGF while decreasing HGF: the roles of p38, ERK, and JNK MAPK signaling. Am J Physiol Gastrointest Liver Physiol. 2007; 292(4):958–963. [DOI] [PubMed] [Google Scholar]
- 44.Chen B, Yan YL, Liu C, et al. Therapeutic effect of deferoxamine on iron overload-induced inhibition of osteogenesis in a zebrafish model. Calcif Tissue Int. 2014; 94(3):353–360. [DOI] [PubMed] [Google Scholar]
- 45.Naves Díaz ML, Elorriaga R, Canteros A, Cannata Andía JB. Effect of desferrioxamine and deferiprone (L1) on the proliferation of MG-63 bone cells and on phosphatase alkaline activity. Nephrol Dial Transplant. 1998;Suppl3:23–28. [DOI] [PubMed] [Google Scholar]
- 46.Chen D, Li Y, Zhou Z, et al. Synergistic inhibition of Wnt pathway by HIF-1 and osteoblast-specific transcription factor osterix (Osx) in osteoblasts. PLoS One. 2012;7(12):e52948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meng H, Li F, Hu R, et al. Deferoxamine alleviates chronic hydrocephalus after intraventricular hemorrhage through iron chelation and Wnt1/Wnt3a inhibition. Brain Res. 2015;1602:44–52. [DOI] [PubMed] [Google Scholar]
- 48.Ziaei A, Ardakani MR, Hashemi MS, et al. Acute course of deferoxamine promoted neuronal differentiation of neural progenitor cells through suppression of Wnt/-catenin pathway: a novel efficient protocol for neuronal differentiation. Neurosci Lett. 2015;590:138–144. [DOI] [PubMed] [Google Scholar]
- 49.Rauner M, Franke K, Murray M, et al. Increased EPO Levels Are Associated with Bone Loss in Mice Lacking PHD2 in EPO-Producing Cells. J Bone Miner Res. 2016. April 15 (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 50.Hsu SH, Chen CT, Wei YH. Inhibitory effects of hypoxia on metabolic switch and osteogenic differentiation of human mesenchymal stem cells. Stem Cells. 2013;31(12):2779–2788. [DOI] [PubMed] [Google Scholar]
- 51.Liu T, Zou W, Shi G, Xu J, Zhang F, Xiao J, Wang Y. Hypoxia-induced MTA1 promotes MC3T3 osteoblast growth but suppresses MC3T3 osteoblast differentiation. Eur J Med Res. 2015;20:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xu Y, Wang S, Tang C, Chen W. Upregulation of long non-coding RNA HIF 1-anti-sense 1 induced by transforming growth factor–mediated targeting of sirtu-in 1 promotes osteoblastic differentiation of human bone marrow stromal cells. Mol Med Rep. 2015;12(5):7233–7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cuajungco MP, Faget KY, Huang X, Tanzi RE, Bush AI. Metal chelation as a potential therapy for Alzheimer’s disease. Ann N Y Acad Sci. 2000;920:292–304. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.