

Abstract
The most common rearrangement in childhood precursor B-cell acute lymphoblastic leukemia is the t(12;21)(p13;q22) translocation resulting in the ETV6-AML1 fusion gene. A frequent concomitant event is the loss of the residual ETV6 allele suggesting a critical role for the ETV6 transcriptional repressor in the etiology of this cancer. However, the precise mechanism through which loss of functional ETV6 contributes to disease pathogenesis is still unclear. To investigate the impact of ETV6 loss on the transcriptional network and to identify new transcriptional targets of ETV6, we used whole transcriptome analysis of both pre-B leukemic cell lines and patients combined with chromatin immunoprecipitation. Using this integrative approach, we identified 4 novel direct ETV6 target genes: CLIC5, BIRC7, ANGPTL2 and WBP1L. To further evaluate the role of chloride intracellular channel protein CLIC5 in leukemogenesis, we generated cell lines overexpressing CLIC5 and demonstrated an increased resistance to hydrogen peroxide-induced apoptosis. We further described the implications of CLIC5’s ion channel activity in lysosomal-mediated cell death, possibly by modulating the function of the transferrin receptor with which it colocalizes intracellularly. For the first time, we showed that loss of ETV6 leads to significant overexpression of CLIC5, which in turn leads to decreased lysosome-mediated apoptosis. Our data suggest that heightened CLIC5 activity could promote a permissive environment for oxidative stress-induced DNA damage accumulation, and thereby contribute to leukemogenesis.
Introduction
ETV6 is a known transcriptional repressor1 involved in hematopoiesis.2 ETV6 rearrangements are frequently observed in multiple hematological diseases, including precursor B-cell acute lymphoblastic leukemia (pre-B ALL), the most common pediatric cancer.3 In fact, the t(12;21)(p13;q22) translocation, which generates an inframe ETV6-AML1 fusion product,4 is the most frequent chromosomal abnormality in childhood pre-B ALL, present in 20% of cases.5 The expression of ETV6-AML1 is systematically observed in t(12;21)-positive pre-B ALL,6,7 indicating a possible function for this chimeric protein in pre-B ALL etiology. However, it was shown that the frequency of the t(12;21) translocation is 100 times greater than that of pre-B ALL,8 suggesting that its presence alone is insufficient to induce leukemia. It has been demonstrated that the second non-rearranged ETV6 allele is frequently deleted or inactivated in t(12;21)-positive pre-B ALL,7,9–11 and recent studies have reported germline ETV6 loss-of-function mutations that were shown to be associated with familial hematological disorders, including ALL.12 Although these data suggest that ETV6 plays a key tumor suppressor role and that its complete inactivation may be required for leukemogenesis,13 little is known about the function of ETV6 in normal hematopoiesis and leukemic transformation.
Given its role in transcriptional repression, we postulated that loss of ETV6 could result in deregulated expression of downstream target genes and perturb key cellular processes and pathways leading to oncogenesis. Only two transcriptional targets of ETV6 have been identified to date: the MMP3 matrix metallopeptidase14 and the anti-apoptotic protein BcL-xL.15 To comprehensively identify novel ETV6 target genes, we combined whole transcriptome analysis and chromatin immunoprecipitation (ChIP) assays. Using an in vitro cell-based system combined with data from childhood pre-B ALL patient tumors, we identified 4 genes (CLIC5, BIRC7, ANGPTL2 and WBP1L) whose expression was directly regulated by ETV6. Functional interrogation of CLIC5 revealed its implication in lysosome-mediated cell death, possibly by regulating iron homeostasis through the transferrin receptor with which it colocalizes intracellularly. In this study, we provide the first evidence of a role for CLIC5-mediated resistance to oxidative stress that may contribute to leukemogenesis.
Methods
Complete methods can be found in the Online Supplementary Methods section.
Expression profiling by RNA-sequencing
Total RNA from two different Reh clones (generated in methylcellulose media) each stably expressing ETV6-His and ETV6ΔETS_NLS-His (and pLENTI control) were processed through the TruSeq Stranded Total RNA protocol and sequenced on the HiSeq 2500 system (Illumina). Reads for each sample were mapped to the hg19 reference genome using STAR with default settings,16 and read counts per genes were determined using HTSeq-count.17 To identify differentially expressed genes (DEGs), we used the R bioconductor package edgeR18 with Benjamini-Hochberg P-value adjustment. The two clones were considered as biological replicates.
The patient cohort used for RNA sequencing was composed of 9 hyperdiploid and 9 t(12;21) patients. Total RNA was extracted from leukemic bone marrow samples of all patients and from control pre-B cells (CD19+CD10+) isolated from healthy cord blood samples. cDNA libraries were prepared using the SOLiD Total RNA-seq kit and sequenced on the SOLiD 4/5500 System (Life Technologies). Reads were aligned to the hg19 reference genome and read counts per gene obtained using LifeScope Genomic Analysis Software with default parameters.
Quantitative real-time PCR
350ng of total RNA were retro-transcribed with M-MLV reverse transcriptase (Life Technologies). cDNA was then subjected to quantitative real-time PCR using the primer sets listed in the Online Supplementary Table S1. Relative expression was determined by the 2-(ΔΔCt) comparative method19 using GAPDH as the reference gene.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed on 10×106 transduced Reh cells cross-linked directly in cell medium for 10min with 1% methanol-free formaldehyde (Polysciences, Inc.). Immunoprecipitation of sheared chromatin was carried out using anti-HA magnetic beads (Thermo Fisher Scientific). Beads were eluted twice with HA peptides (Thermo Fisher Scientific) before reverse cross-linking. DNA was purified twice by standard phenol/chloroform/isoamyl alcohol (Sigma-Aldrich) extraction prior to qRT-PCR analysis (primers are listed in the Online Supplementary Table S2).
Apoptosis assays
Apoptosis was induced by treating cells for 20h with hydrogen peroxide (PRXD; Sigma-Aldrich), camptothecin (CPT; Tocris Bioscience) or doxorubicin (DOXO; Sigma-Aldrich) and assayed by Alexa Fluor 488-coupled Annexin V and propidium iodide (PI) double staining. 1×104 stained cells were analyzed by flow cytometry. Total apoptosis includes Annexin V+/PI − (early apoptotic), Annexin V+/PI + (late apoptotic) and Annexin V−/PI+ (necrotic) cells.
Immunofluorescence microscopy
Reh cells were seeded at 2×106 cells/mL in a 96 well glass plate (Whatman) and fixed in a 3.7% formaldehyde solution. Immunostaining was performed overnight with CLIC5A antibody (ab191102 dil. 1:1000; Abcam) and transferrin receptor antibody (ab84036 dil. 1:200; Abcam). Goat anti-Mouse Alexa Fluor 488 (dil. 1:500; Thermo Fisher Scientific) was used to detect anti-CLIC5A, and Goat anti-Rabbit Alexa Fluor 546 (dil. 1:500; Thermo Fisher Scientific) was used to detect anti-transferrin receptor. Hoechst 33258 DNA stain (dil. 1:500; Thermo Fisher Scientific) was included to stain nuclei.
Statistical tests
The significance of observations was assessed using one or two-tailed Fisher’s exact test or Mann-Whitney U test when appropriate.
Ethics statement
The CHU Sainte-Justine Research Ethics Board approved the protocol. Informed consent was obtained from the parents of the patients to participate in this study and for publication of this report and any accompanying images.
Results
ETV6 represses the expression of 6 genes in pre-B ALL cell lines and patient samples
To identify novel direct ETV6-regulated genes, we carried out a transcriptome analysis in both cell lines and patient tumor samples (Figure 1). Based on the expression profiles of transduced t(12;21)-positive pre-B ALL Reh clones (Online Supplementary Figure S1), we found 331 genes repressed in His-tagged ETV6 (ETV6-His) cells compared to control cells (pLENTI empty vector; P-value≤0.05), of which 88 remained significant after multiple testing corrections (FDR≤0.1; Online Supplementary Table S3). 18 genes were significantly repressed by ETV6-His compared to its DNA-binding deficient mutant ETV6ΔETS_NLS-His (FDR≤0.1; Online Supplementary Table S4), of which 11 were both confidently expressed (logCPM≥1) and also present in the above-mentioned list of 88 genes. These genes are thus more likely to be direct targets of ETV6 since their repression depends on ETV6’s DNA-binding domain. However, only 7 of these genes absolutely required the DNA-binding domain for repression (ETV6ΔETS_NLS-His vs. pLENTI; P-value≥0.05 or logFC≥−0.5), further confirming their direct regulation by ETV6: CLIC5, BIRC7, DDIT4L, ANGPTL2, WBP1L, LRRC4, and SLC51A. We then assessed whether the ETV6-dependent transcriptional repression observed in vitro translated to childhood pre-B ALL patient tumor samples. Expression of the 331 genes repressed by ETV6 in vitro was evaluated in transcriptome data from 9 t(12;21)-positive samples (ETV6 negative) and compared to 9 hyperdiploid cases as well as to 3 normal pre-B cell (CD19+/CD10+) samples (ETV6 positive).
Figure 1.
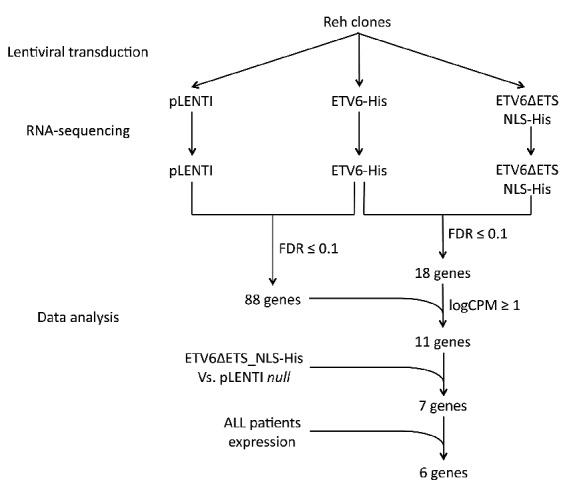
Schematic representation of the transcriptome-based design to detect putative direct ETV6 target genes. To identify direct targets of ETV6, we first designed an in vitro RNA-seq experiment using ETV6−/− Reh-derived clones. Cells were transduced with lentiviral constructs to express ETV6-His and ETV6ΔETS_NLS-His. Total RNA was extracted from stable cell populations and RNA-seq libraries were sequenced. Expression profiles were analyzed using EdgeR. Gene expression profiles in ETV6-His cells were first compared with ETV6ΔETS_NLS-His and pLENTI cells to identify repressed genes (FDR ≤ 0.1). We then included data from the ETV6ΔETS_NLS-His vs. pLENTI comparison and further considered genes whose expression remains constant (P-value≥0.05 or logFC≥−0.5) which are more likely to be direct ETV6 targets. Finally, only genes that showed a specific overexpression in t(12;21)-positive childhood pre-B ALL (pre-B acute lymphoblastic leukemia) patients were considered. ALL: acute lymphoblastic leukemia; FDR; false discovery rate; logCPM: log counts per million reads.
We identified 45 genes that were downregulated in vitro (13.6%) and that were also specifically overexpressed in t(12;21)-positive patients (Figure 2). Interestingly, these include 6 of the 7 genes identified as putative direct ETV6 targets in vitro (CLIC5, BIRC7, ANGPTL2, WBP1L, LRRC4 and SLC51A, but not DDIT4L), further supporting a role for ETV6 in their regulation.
Figure 2.
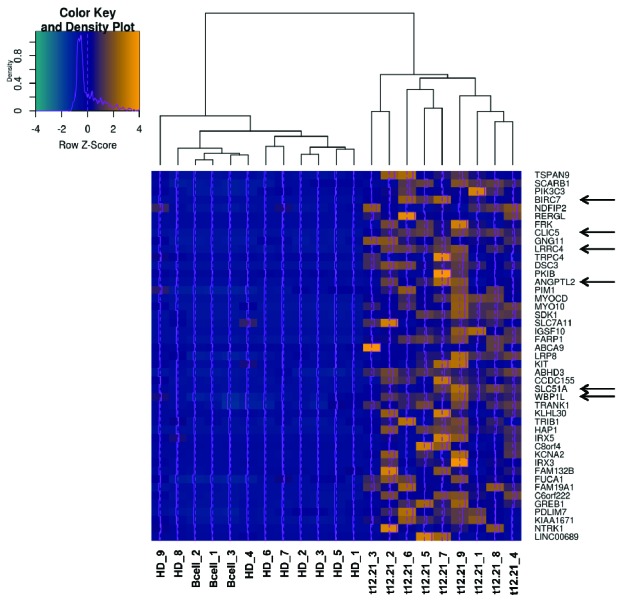
Expression of putative ETV6 targets in pediatric pre-B ALL (pre-B acute lymphoblastic leukemia) patients. An unsupervised clustering heatmap was generated with the 45 genes specifically overexpressed (yellow) in t(12;21)-positive ALL (acute lymhoblastic leukemia) patients (t12.21, n=9) compared to hyperdiploid ALL patients (HD, n=9) and normal B-cells controls (B-cell, n=3). Among the 7 genes identified as putative direct ETV6 targets in our cell line model, CLIC5, BIRC7, ANGPTL2, WBP1L, LRRC4 and SLC51A were specifically overexpressed in t(12;21)-positive pediatric pre-B ALL patient tumor samples (Arrows).
CLIC5, BIRC7, ANGPTL2 and WBP1L are direct targets of ETV6
To validate ETV6-dependent expression of these 6 genes (Table 1), we used quantitative real-time PCR (qRT-PCR) in both Reh clones and the original Reh population overexpressing ETV6 WT, ETV6-HA or GFP as a control (Online Supplementary Figure S2). Both ETV6 WT and ETV6-HA efficiently repressed expression of these genes except for LRRC4 (Figure 3A). To assess the physical interaction between ETV6 and the proximal promoters of these 5 putative ETV6 targets we performed ChIP experiments in Reh cells overexpressing ETV6-HA or ETV6 WT as a negative control. Importantly, ETV6-HA behaves similarly to ETV6 WT in our qRT-PCR experiments (Figure 3A), indicating that the epitope tag does not negatively interfere with normal ETV6 repressor function. As shown in Figure 3B, we successfully enriched the proximal promoters of CLIC5, BIRC7, ANGPTL2 and WBP1L, but not SLC51A, further confirming that these 4 genes are indeed direct targets of ETV6.
Table 1.
Expression status of the 6 putative direct ETV6 target genes.
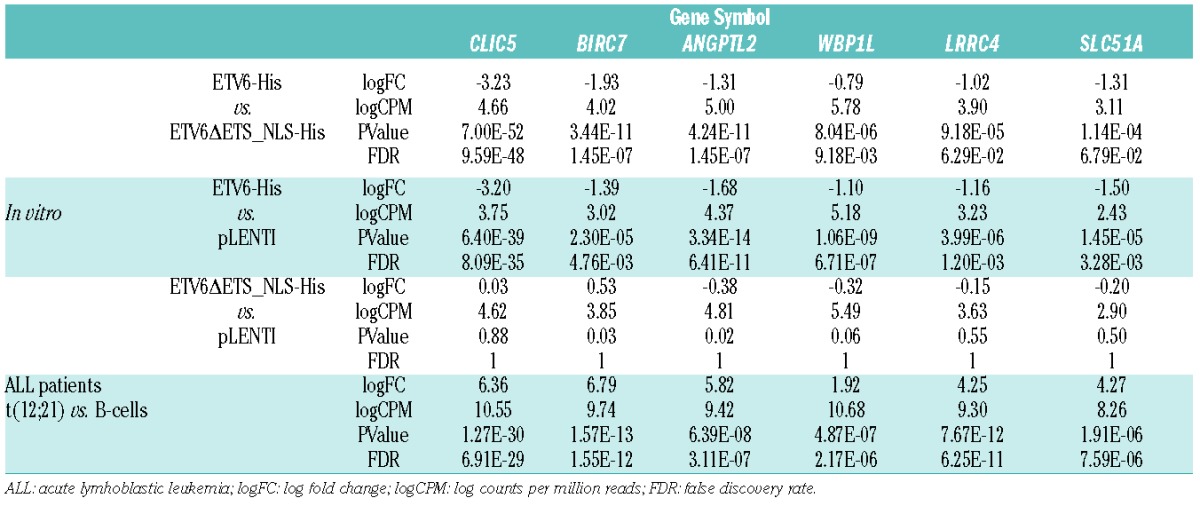
Figure 3.
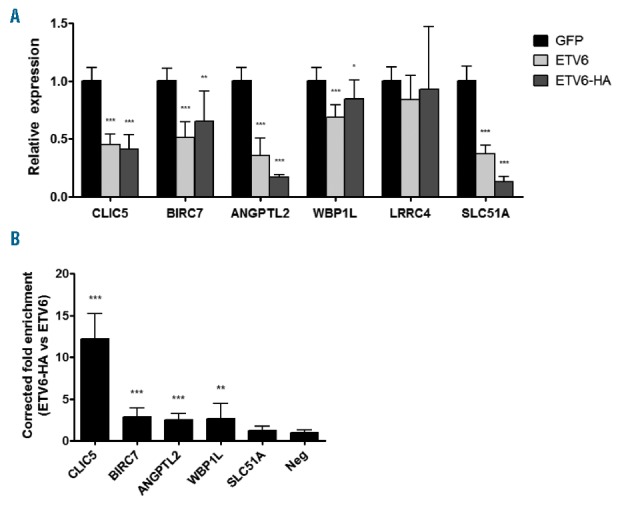
Quantitative real-time PCR and chromatin immunoprecipitation validation of putative ETV6 target genes. Reh cells together with the two Reh derivated clones were infected with pCCL GFP, ETV6 WT or ETV6-HA. (A) Total RNA was extracted from these cells and complementary DNA was generated. This cDNA was submitted to qRT-PCR (quantitative real time PCR) analysis to quantify relative expression of putative ETV6 targets. Expression of all genes but LRRC4 is repressed by ETV6 WT (wild-type) and ETV6-HA. (B) ChIP (chromatin immunoprecipitation) experiments were performed in ETV6-WT and ETV6-HA cells. Putative ETV6 target gene proximal promoter enrichment was determined by qRT-PCR using promoter-specific primers. Results are presented as the ratio of the input percentage obtained in ETV6-HA cells compared to ETV6 WT cells, corrected by the background enrichment obtained with an unbound region (Neg). CLIC5, BIRC7, ANGPTL2 and WBP1L promoters are enriched. For both qRT-PCR and ChIP experiments, 4 values were calculated for each of the 3 cell lines and were merged for a total of 12 values (n=12). Error bars represent the standard deviation. Statistical significance is calculated by two-tailed and one-tailed Mann-Whitney U test for qRT-PCR and ChIP analysis, respectively.
CLIC5A reduces hydrogen peroxide-induced apoptosis
We pursued functional interrogation of our strongest candidate, the chloride intracellular channel CLIC5, to investigate its cellular function and potential contribution to childhood pre-B ALL. The CLIC5 locus encodes two major isoforms, CLIC5A and CLIC5B20 (Online Supplementary Figure S3A), transcribed by two alternative promoters and differing only from their first exon. Using ChIP experiments (as above), we showed that the CLIC5A promoter was specifically enriched, whereas the CLIC5B promoter showed no significant enrichment compared to the negative control region (Online Supplementary Figure S3B). ETV6 overexpression was also shown to lead to a marked decrease of the CLIC5A protein, whereas CLIC5B levels remained constant (Online Supplementary Figure S3C). Together, these results confirm specific ETV6-mediated repression of CLIC5A.
In light of these results, we overexpressed the CLIC5A isoform in Reh cells (Figure 4A) in order to investigate its role on B-lymphoblast function. Importantly, the overexpression of CLIC5A in our Reh cells is highly similar to that observed in a validation cohort of t(12;21) ALL patients (Online Supplementary Figure S4). Given that changes in migration were observed upon silencing of CLIC5A,21 we first evaluated this phenotype. We found no particular difference in migration toward stromal cell-derived factor 1 (SDF-1, also known as CXCL12) in a classic transwell experiment between control and CLIC5A overexpressing cells (Online Supplementary Figure S5). Although CLIC5A had never been shown to be associated with apoptosis, suppression of its closely related family member CLIC4 had previously been shown to enhance hydrogen peroxide-induced apoptosis.22 To test this hypothesis in our cell model, we treated CLIC5A overexpressing cells with hydrogen peroxide, camptothecin, or doxorubicin, and evaluated apoptosis. Of note, peroxide induces an apoptotic cell death in these conditions (Figure 4B), rather than necrosis. We observed a modest yet consistent reduction in hydrogen peroxide-induced apoptosis compared to control cells (Figure 4C), suggesting a potential role in the intracellular response to free radicals. A similar reduction in apoptosis following peroxide treatment was observed with CLIC5A overexpression in the IM9 B-lymphoblastoid cell line (Figure 4D–F) endogenously expressing wild-type ETV6, further confirming that CLIC5A overexpression specifically reduces hydrogen peroxide-induced apoptosis across cellular backgrounds.
Figure 4.

CLIC5A protects cells from hydrogen peroxide-induced apoptosis. (A) pLENTI control and CLIC5A stably infected Reh cells were challenged for 20h with 35μM hydrogen peroxide (PRXD), 250nM camptothecin (CPT), or 150nM doxorubicin (DOXO) and analyzed by flow cytometry with Alexa Fluor 488-coupled Annexin V and propidium iodide (PI) staining. (B) A representative example of staining following PRXD treatment is presented with PI on the x axis and Alexa Fluor 488 on the y axis. (C) Total apoptosis is calculated for each sample. CLIC5A overexpressing cells displayed reduced apoptosis compared to control only when treated with PRXD. Each experiment was performed 3 times in triplicates (n=9). (D) IM9 cells transduced with CLIC5A or pLENTI empty vector control were challenged with 50μM PRXD and processed similarly to assess apoptosis. (E) A representative example of staining and (F) total apoptosis is shown for the IM9 cell line. Again, CLIC5A overexpression leads to reduced apoptosis. Two independent experiments carried out in triplicate and an additional single test were performed (n=7). Error bars in (C) and (F) represent the standard deviation. Statistical significance is calculated by two-tailed Mann-Whitney U test. For (A) and (D) adjustments of brightness and contrast were applied to the whole image. DMSO: dimethyl sulfoxide.
CLIC5A is an endosomal ionic channel involved in lysosome-mediated apoptosis
Given that hydrogen peroxide is known to trigger lysosomal membrane permeabilization (LMP) and initiate the lysosomal-mediated apoptosis pathway (Figure 5A),23,24 we hypothesized that CLIC5A’s role in protecting cells against apoptosis may function through the modulation of LMP. Since LMP has a direct impact on mitochondrial outer membrane permeabilization (MOMP) that can be assessed through mitochondrial membrane potential (MMP), we evaluated MMP in our Reh cellular model treated with hydrogen peroxide. We observed a significant reduction of MMP loss correlated with CLIC5A overexpression compared to control, indicating that substantially more mitochondria remained intact following peroxide exposure when CLIC5A was overexpressed (Online Supplementary Figure S6). This result supports a role for CLIC5A in protecting cells against peroxide-induced apoptosis and suggests that it functions upstream of MOMP, which indeed corroborates a possible implication of CLIC5A in LMP regulation.
Figure 5.
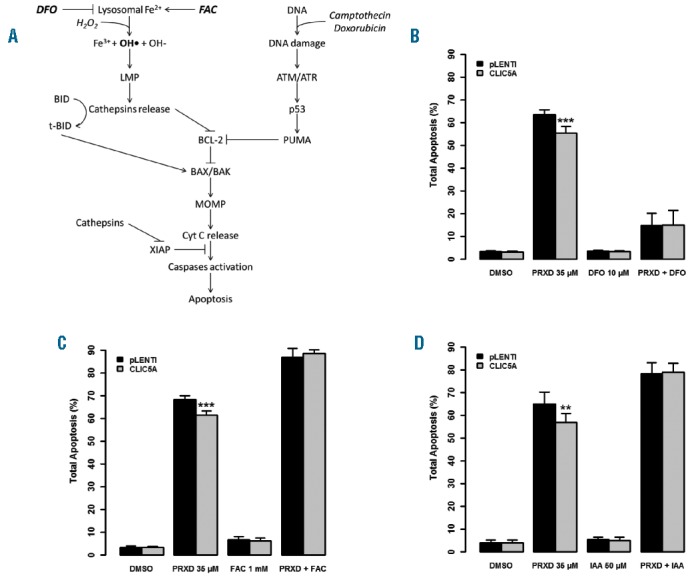
CLIC5A is implicated in lysosome-mediated cell death. (A) Schematic illustration of the lysosomal apoptosis pathway and DNA damage pathway. DFO: deferoxamine mesylate salt; FAC: ferric ammonium citrate; LMP: Lysosomal membrane permeabilization; MOMP: Mitochondrial outer membrane permeabilization. (B) pLENTI control and CLIC5A overexpressing Reh cells were pre-treated with 10μM DFO to chelate lysosomal ferrous iron and apoptosis was induced using 35μM PRXD for 20h followed by flow cytometry quantification. PRXD-induced apoptosis was greatly reduced with DFO treatment. (C) Similarly, 1mM FAC was used to increase ferrous iron concentration which led to higher PRXD-induced apoptosis with no protective effect of CLIC5A overexpression. (D) Cells were pre-treated with 50μM IAA-94 CLIC-specific ion channel inhibitor and apoptosis was assayed after a subsequent PRXD treatment. In these conditions, CLIC5A overexpression did not reduce apoptosis. Each experiment was carried out 3 times in triplicate (n=9). Error bars represent the standard deviation. Statistical significance is calculated by two-tailed Mann-Whitney U test.
Deleterious effects of hydrogen peroxide on lysosome membranes are strongly dependent on lysosomal Fe2+ availability since it dictates its conversion to the highly reactive hydroxyl radicals.25 We thus investigated CLIC5A’s impact on lysosome-mediated cell death by modulating lysosomal Fe2+ availability prior to hydrogen peroxide exposure by pre-treating cells with the iron chelator deferoxamine mesylate salt (DFO).26,27 As shown in Figure 5B, cells treated with DFO prior to peroxide showed a drastic reduction in apoptosis. The residual apoptotic activity observed in DFO-treated cells could be driven by DNA damage,28 which appears to be CLIC5A-independent given the results obtained with the two DNA damaging agents camptothecin and doxorubicin (Figure 4C).
Inversely, we used ferric ammonium citrate (FAC) to positively modulate Fe2+ concentration in lysosomes.29 Increased lysosomal Fe2+ concentration favors hydroxyl radical production from hydrogen peroxide and hence is expected to increase LMP and apoptosis. Accordingly, cells pre-treated with FAC showed increased peroxide-induced apoptosis and the protective effect of CLIC5A overexpression was completely lost (Figure 5C), indicating that CLIC5A plays a role upstream of LMP. This specific role further corroborates CLIC5A’s inability to protect cells against DNA damage.
To evaluate CLIC5A’s ion channel activity30 in this context we pre-treated CLIC5A overexpressing Reh cells with indanyloxyacetic acid 94 (IAA-94), a known CLIC-specific ion channel inhibitor.30,31 IAA-94 treatment increased peroxide-induced apoptosis and completely prevented CLIC5A-mediated protection (Figure 5D), similar to that which was observed in the presence of increased Fe2+ availability following FAC treatment (Figure 5C). Endogenous CLIC5A inhibition contributes to this phenotype and explains the increased apoptosis level of control cells. Together, these data confirm that CLIC5A’s ion channel activity is required to protect cells against peroxide-induced apoptosis, perhaps by limiting Fe2+ availability in the lysosomal pathway.
To further investigate the functional implications of CLIC5A in lysosomal apoptosis, we examined the intracellular localization of CLIC5A using immunofluorescence. Although we did not observe colocalization of CLIC5A with lysosomes (Online Supplementary Figure S7), we did show positive colocalization with transferrin receptor (Figure 6), which is in line with CLIC5A’s postulated role in modulating lysosomal Fe2+ availability. Transferrin receptors (TFRC gene) are responsible for cellular iron intake through the binding and internalization of its iron-bound ligand transferrin (TF gene).32,33 CLIC5A could perturb this process and thereby impact cellular iron concentrations and modulate lysosome sensitivity to hydrogen peroxide. Interestingly, double positive staining appears to be particularly present in distinct vesicle-like structures that are likely transferrin receptor-containing recycling endosomes, further suggesting that CLIC5A may interfere with normal iron homeostasis, thus reducing lysosome sensitivity to oxidative stress.
Figure 6.
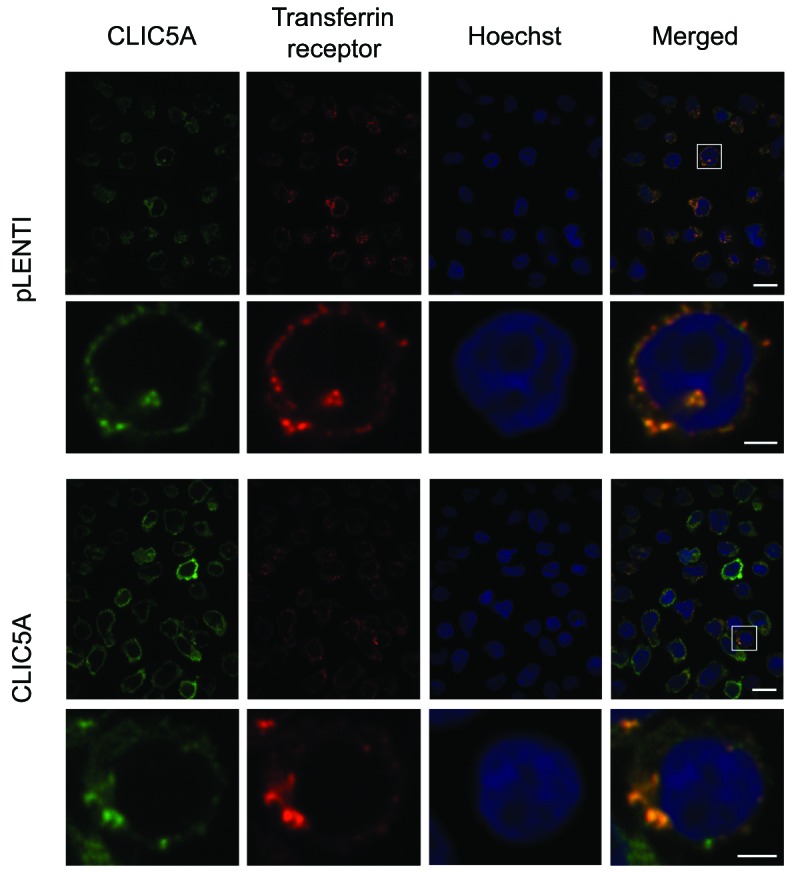
Colocalization of CLIC5A and transferrin receptors. pLENTI control and CLIC5A overexpressing Reh cells were used for colocalization studies. Immunostaining of both CLIC5A and transferrin receptor were performed in the same cells simultaneously with Hoechst DNA staining. Results were obtained at 100× magnification (upper panels; scale bar = 10μm). A strong colocalization was observed between CLIC5A and transferrin receptors. Additional enlargement for the marked region of the initial image is presented in lower panels (scale bar = 2μm). A merged image was generated (right panels).
Taken together, our data strongly support a role for the newly identified ETV6 transcriptional target CLIC5A in modulating lysosome-mediated apoptosis. We propose that CLIC5A overexpression in t(12;21)-positive, ETV6 depleted pre-B cells could contribute to increased resistance to oxidative stress and therefore promote cell survival.
Discussion
Approximately 20% of childhood pre-B ALL patients harbor the t(12;21) translocation, yielding an ETV6-AML1 fusion protein that is, however, insufficient to initiate leukemia.8,34,35 This process often requires further loss of the remaining wild-type ETV6 allele,7,9–11,13 suggesting that deregulation of the ETV6 transcriptional machinery could play an important role in leukemogenesis. Unfortunately, very few ETV6-regulated transcriptional targets are known and the mechanisms through which they are involved in leukemogenesis remain elusive. Herein, we combined both in vitro (cell lines) and ex vivo (pre-B ALL patients) transcriptome data to identify candidate ETV6 target genes, and through additional cellular assays identified 4 novel direct ETV6 target genes: CLIC5, BIRC7, ANGPTL2 and WBP1L.
The CLIC5 gene was previously associated with the t(12;21)-positive ALL molecular signature,36 and our pediatric pre-B ALL expression data corroborated this result with strong CLIC5 overexpression shown to be specific to the t(12;21)-positive subgroup within our cohort. Thus CLIC5 overexpression in these patients is likely due to ETV6 loss. Unfortunately, very little is known about CLIC5’s potential contribution to leukemogenesis. Using engineered cell lines, we demonstrated an increased resistance to hydrogen peroxide-induced apoptosis following overexpression of CLIC5A. Notably, Reh cells already express the endogenous CLIC5A isoform, which could explain the modest effect of the overexpression of CLIC5A (mean=7.46%, P-value=8.32×10−11, merged n=36). Given that normal B-cells show no expression of CLIC5 (mean FPKM<0.1 over our 3 normal CD19+/CD10+ pre-B cell samples isolated from human cord blood), an eventual stronger impact of CLIC5A re-expression in a pre-B cell upon ETV6 depletion can be expected.
We further linked this phenotype to CLIC5A’s ion channel activity in lysosomal-mediated cell death. The presence of CLIC5A on transferrin receptor-containing endosomes potentially negatively impacts lysosomal iron availability, thus reducing lysosome sensitivity to oxidative stress. However, the exact mechanism by which CLIC5A modulates lysosomal iron to prevent peroxide-induced apoptosis remains unclear. It has been demonstrated that changes in the concentration of several ions can modulate endosomal pH through ion dependent H+ pumps.37 With CLIC5A being a poorly selective ion channel,38 we can hypothesize a somewhat similar function: slight differences in endosome acidification could modulate transferrin iron release or trafficking and therefore impact lysosomal iron concentration leading to differential peroxide sensitivity.
Additional experiments should be performed to further dissect CLIC5A’s role in lysosome-mediated apoptosis.
Nonetheless, the observed effects of CLIC5A in peroxide resistance could play a key role in ALL initiation. A recent study highlighted the high oxidative stress levels of leukemic blasts in the bone marrow niche induced by bone marrow stromal cell signaling.39 Furthermore, it has been shown that increased levels of ROS in t(12;21)-positive cells was associated with DNA damage accumulation.40 High oxidative stress levels are thought to trigger apoptotic cell death through the lysosomal pathway, thus preventing DNA damage accumulation. However, in t(12;21)-positive ALL, we have shown that loss of ETV6 expression leads to significant overexpression of CLIC5A. This overexpression leads to decreased lysosome-media-ted apoptosis, which in turn can promote a more permissive environment to heighten ROS levels (Figure 7). Cells evading apoptosis can thus accumulate ROS-induced mutations at a greater rate. Moreover, this mutational process of t(12;21)-positive pre-B cells could be facilitated not only by CLIC5A overexpression, but together with several ETV6 deregulated targets such as the inhibitor of apoptosis BIRC7. Although the overall mutational burden of ALL is low compared to other cancers,41 the difference in ROS-induced DNA damage accumulation can contribute, over time, to promote leukemic transformation when impacting key cancer genes.
Figure 7.

Proposed mechanism of CLIC5A involvement in ETV6-associated childhood pre-B ALL. The t(12;21) translocation occurs early and contributes to an increased level of reactive oxygen species (ROS). Lysosome-mediated apoptosis is sensitive to this excess of ROS and therefore prevents the accumulation of ROS-mediated DNA damage. With the subsequent deletion of the residual ETV6 allele (LOH), CLIC5A expression is drastically upregulated. This overexpression of CLIC5A has a negative impact on the lysosomal apoptosis pathway and thus creates a permissive environment for the accumulation of mutations driven by high oxidative stress. Over time, some of these mutations may impact key cellular biological processes and pathways, which will ultimately lead to full leukemic transformation and development of childhood pre-B acute lymphoblastic leukemia (pre-B ALL).
It remains challenging, however, to evaluate the contribution of this pathway to the total amount of DNA damage found in pre-B ALL patients. Although ROS-mediated alterations of nucleotides are well characterized, their signature in sequencing data remains unclear.42,43 Interrogation of our patient sequencing data did not reveal any particular mutational profile that could undoubtedly be attributed to ROS. However, increased ROS-mediated DNA double strand breaks have been observed in t(12;21)-positive B-cells when assessed by comet assays.40 These alterations do often lead to deletions or rearrangements due to aberrant repair, that are frequent events in leukemia, thus supporting a role for ROS-induced DNA damage in ALL.
With a similar CLIC5A-mediated protective effect on peroxide-induced apoptosis obtained in both Reh and IM9 cell lines, we demonstrated a phenotype that is independent from a particular genetic background. Interestingly, CLIC5 expression has been associated with poor prognosis in breast cancer,44 and expression arrays of a broad range of normal and tumoral tissues obtained through the GENT database45 showed an overexpression of CLIC5 in ovarian cancers (Online Supplementary Figure S8). Based on our results, and given the importance of oxidative stress resistance in these solid tumors that require angiogenesis to promote growth, it suggests that CLIC5A protection against oxidative stress may not be limited to pre-B cell leukemia. The unfavorable prognosis associated with CLIC5 deregulation may be due to increased oxidative stress resistance, thus reducing the necessity of angiogenesis and fostering an environment prone to ROS-induced DNA damage. Although ETV6 alterations have not been reported in these solid tumors, other mechanisms could drive CLIC5 overexpression. Interestingly, ETS factor binding sites (EBS) were found to be highly enriched for small non-coding mutations across a wide variety of cancers.46 These mutations disrupt the consensus binding site of ETS factors, such as ETV6, and thus may prevent it’s binding and the repression of its targets. Despite the presence of wild-type ETV6 in these cases, some of its key targets (CLIC5, BIRC7 or ANGPTL2) may be overexpressed, thus leading to a selective advantage.
While we focused on CLIC5, the other identified targets were also of interest. Notably, the caspases inhibitor BIRC7 has previously been shown to be part of a t(12;21)-positive pre-B ALL molecular signature.36 Overexpression of BIRC7 was also observed in a variety of tumors, and contributes to oncogenesis through the inhibition of apoptosis,47 suggesting a similar involvement for BIRC7 in t(12;21)-positive childhood pre-B ALL. The ANGPTL2 gene encodes a secreted protein with, among others, pro-angiogenic and anti-apoptotic properties. Although its expression has been linked to a broad range of diseases, including cancers, it has been shown to increase survival and expansion of hematopoietic stem cells.48 ANGPTL2’s contribution to leukemogenesis remains unknown. Lastly, WBP1L, also known as OPAL1 (Outcome Predictor in Acute Leukemia 1), is associated with the t(12;21) favorable outcome in ALL.49 It is still unclear whether WBP1L plays a role in leukemia, since its molecular function is yet to be characterized.
In conclusion, for the first time, we describe a role for CLIC5-mediated resistance to oxidative stress that could promote cell survival and contribute to leukemogenesis. We propose a mechanism in which complete loss of wild-type ETV6 expression in a pre-leukemic blast leads to CLIC5A overexpression, thus creating a permissive environment for the accumulation of mutations driven by high oxidative stress, eventually giving rise to full leukemic transformation.
Supplementary Material
Acknowledgments
The authors would like to thank the patients and their parents for participating in this study. We are grateful to Dr. Christian Beauséjour who kindly provided lentiviral-related material.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/12/1534
Funding
This study was supported by research funds provided by the Terry Fox Research Institute and the Canadian Institutes of Health Research. BN is the recipient of a Cole Foundation scholarship. JFS is the recipient of a Réseau de médecine génétique appliquée (RMGA) scholarship. DS holds the François-Karl Viau Research Chair in Pediatric Oncogenomics. Whole transcriptome sequencing was performed at the Integrated Clinical Genomic Center in Pediatrics at the CHU Sainte-Justine Research Center. Computations were performed on the Briarée supercomputer at the Université de Montréal, managed by Calcul Québec and Compute Canada. The operation of this supercomputer is funded by the Canada Foundation for Innovation (CFI), NanoQuébec, RMGA and the Fonds de recherche du Québec - Nature et technologies (FRQNT). GH thanks the CFI, the Cole Foundation and the Fonds de recherche du Québec-Santé for infrastructure, transition award and salary support, respectively.
References
- 1.Lopez RG, Carron C, Oury C, Gardellin P, Bernard O, Ghysdael J. TEL is a sequence-specific transcriptional repressor. J Biol Chem. 1999;274(42):30132–30138. [DOI] [PubMed] [Google Scholar]
- 2.Wang LC, Swat W, Fujiwara Y, et al. The TEL/ETV6 gene is required specifically for hematopoiesis in the bone marrow. Genes Dev. 1998;12(15):2392–2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bohlander SK. ETV6: a versatile player in leukemogenesis. Semin Cancer Biol. 2005;15(3):162–174. [DOI] [PubMed] [Google Scholar]
- 4.Golub TR, Barker GF, Bohlander SK, et al. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci USA. 1995; 92(11):4917–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tasian SK, Loh ML, Hunger SP. Childhood acute lymphoblastic leukemia: Integrating genomics into therapy. Cancer. 2015; 121(20):3577–3590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agape P, Gerard B, Cave H, et al. Analysis of ETV6 and ETV6-AML1 proteins in acute lymphoblastic leukaemia. Br J Haematol. 1997;98(1):234–239. [DOI] [PubMed] [Google Scholar]
- 7.Poirel H, Lacronique V, Mauchauffe M, et al. Analysis of TEL proteins in human leukemias. Oncogene. 1998;16(22):2895–2903. [DOI] [PubMed] [Google Scholar]
- 8.Mori H, Colman SM, Xiao Z, et al. Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci USA. 2002;99(12):8242–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel N, Goff LK, Clark T, et al. Expression profile of wild-type ETV6 in childhood acute leukaemia. Br J Haematol. 2003; 122(1):94–98. [DOI] [PubMed] [Google Scholar]
- 10.Lilljebjorn H, Soneson C, Andersson A, et al. The correlation pattern of acquired copy number changes in 164 ETV6/RUNX1-positive childhood acute lymphoblastic leukemias. Hum Mol Genet. 2010; 19(16):3150–3158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Montpetit A, Larose J, Boily G, Langlois S, Trudel N, Sinnett D. Mutational and expression analysis of the chromosome 12p candidate tumor suppressor genes in pre-B acute lymphoblastic leukemia. Leukemia. 2004;18(9):1499–1504. [DOI] [PubMed] [Google Scholar]
- 12.Moriyama T, Metzger ML, Wu G, et al. Germline genetic variation in ETV6 and risk of childhood acute lymphoblastic leukaemia: a systematic genetic study. Lancet Oncol. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderson K, Lutz C, van Delft FW, et al. Genetic variegation of clonal architecture and propagating cells in leukaemia. Nature. 2011;469(7330):356–361. [DOI] [PubMed] [Google Scholar]
- 14.Fenrick R, Wang L, Nip J, et al. TEL, a putative tumor suppressor, modulates cell growth and cell morphology of ras-transformed cells while repressing the transcription of stromelysin-1. Mol Cell Biol. 2000;20(16):5828–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irvin BJ, Wood LD, Wang L, et al. TEL, a putative tumor suppressor, induces apoptosis and represses transcription of Bcl-XL. J Biol Chem. 2003;278(47):46378–46386. [DOI] [PubMed] [Google Scholar]
- 16.Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- 20.Shanks RA, Larocca MC, Berryman M, et al. AKAP350 at the Golgi apparatus. II. Association of AKAP350 with a novel chloride intracellular channel (CLIC) family member. J Biol Chem. 2002;277(43):40973–40980. [DOI] [PubMed] [Google Scholar]
- 21.Flores-Tellez TN, Lopez TV, Vasquez Garzon VR, Villa-Trevino S. Co-Expression of Ezrin-CLIC5-Podocalyxin Is Associated with Migration and Invasiveness in Hepatocellular Carcinoma. PLoS One. 2015;10(7):e0131605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu Y, Kang J, Yuan Z, et al. Suppression of CLIC4/mtCLIC enhances hydrogen peroxide-induced apoptosis in C6 glioma cells. Oncol Rep. 2013;29(4):1483–1491. [DOI] [PubMed] [Google Scholar]
- 23.Turk B, Turk V. Lysosomes as “suicide bags” in cell death: myth or reality? J Biol Chem. 2009;284(33):21783–21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aits S, Jaattela M. Lysosomal cell death at a glance. J Cell Sci. 2013;126(Pt 9):1905–1912. [DOI] [PubMed] [Google Scholar]
- 25.Kruszewski M. Labile iron pool: the main determinant of cellular response to oxidative stress. Mutat Res. 2003;531(1–2):81–92. [DOI] [PubMed] [Google Scholar]
- 26.Kurz T, Leake A, Von Zglinicki T, Brunk UT. Relocalized redox-active lysosomal iron is an important mediator of oxidative-stress-induced DNA damage. Biochem J. 2004;378(Pt 3):1039–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27(50):6434–6451. [DOI] [PubMed] [Google Scholar]
- 28.Oller AR, Thilly WG. Mutational spectra in human B-cells. Spontaneous, oxygen and hydrogen peroxide-induced mutations at the hprt gene. J Mol Biol. 1992;228(3):813–826. [DOI] [PubMed] [Google Scholar]
- 29.Repnik U, Cesen MH, Turk B. The endolysosomal system in cell death and survival. Cold Spring Harb Perspect Biol. 2013;5(1):a008755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berryman M, Bruno J, Price J, Edwards JC. CLIC-5A functions as a chloride channel in vitro and associates with the cortical actin cytoskeleton in vitro and in vivo. J Biol Chem. 2004;279(33):34794–34801. [DOI] [PubMed] [Google Scholar]
- 31.Landry DW, Akabas MH, Redhead C, Edelman A, Cragoe EJ, Jr., Al-Awqati Q. Purification and reconstitution of chloride channels from kidney and trachea. Science. 1989;244(4911):1469–1472. [DOI] [PubMed] [Google Scholar]
- 32.Bresgen N, Eckl PM. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules. 2015;5(2):808–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5(2):121–132. [DOI] [PubMed] [Google Scholar]
- 34.Andreasson P, Schwaller J, Anastasiadou E, Aster J, Gilliland DG. The expression of ETV6/CBFA2 (TEL/AML1) is not sufficient for the transformation of hematopoietic cell lines in vitro or the induction of hematologic disease in vivo. Cancer Genet Cytogenet. 2001;130(2):93–104. [DOI] [PubMed] [Google Scholar]
- 35.van der Weyden L, Giotopoulos G, Rust AG, et al. Modeling the evolution of ETV6-RUNX1-induced B-cell precursor acute lymphoblastic leukemia in mice. Blood. 2011;118(4):1041–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross ME, Zhou X, Song G, et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood. 2003;102(8):2951–2959. [DOI] [PubMed] [Google Scholar]
- 37.Scott CC, Gruenberg J. Ion flux and the function of endosomes and lysosomes: pH is just the start: the flux of ions across endosomal membranes influences endosome function not only through regulation of the luminal pH. Bioessays. 2011;33(2): 103–110. [DOI] [PubMed] [Google Scholar]
- 38.Singh H, Cousin MA, Ashley RH. Functional reconstitution of mammalian ‘chloride intracellular channels’ CLIC1, CLIC4 and CLIC5 reveals differential regulation by cytoskeletal actin. FEBS J. 2007;274(24):6306–6316. [DOI] [PubMed] [Google Scholar]
- 39.Liu J, Masurekar A, Johnson S, et al. Stromal cell-mediated mitochondrial redox adaptation regulates drug resistance in childhood acute lymphoblastic leukemia. Oncotarget. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kantner HP, Warsch W, Delogu A, et al. ETV6/RUNX1 induces reactive oxygen species and drives the accumulation of DNA damage in B cells. Neoplasia. 2013;15(11):1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cooke MS, Evans MD, Dizdaroglu M, Lunec J. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 2003;17(10):1195–1214. [DOI] [PubMed] [Google Scholar]
- 43.Helleday T, Eshtad S, Nik-Zainal S. Mechanisms underlying mutational signatures in human cancers. Nat Rev Genet. 2014;15(9):585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yau C, Sninsky J, Kwok S, et al. An optimized five-gene multi-platform predictor of hormone receptor negative and triple negative breast cancer metastatic risk. Breast Cancer Res. 2013;15(5):R103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shin G, Kang TW, Yang S, Baek SJ, Jeong YS, Kim SY. GENT: gene expression database of normal and tumor tissues. Cancer Inform. 2011;10:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Araya CL, Cenik C, Reuter JA, et al. Identification of significantly mutated regions across cancer types highlights a rich landscape of functional molecular alterations. Nat Genet. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang L, Zhang Q, Liu B, Han M, Shan B. Challenge and promise: roles for Livin in progression and therapy of cancer. Mol Cancer Ther. 2008;7(12):3661–3669. [DOI] [PubMed] [Google Scholar]
- 48.Thorin-Trescases N, Thorin E. Angiopoietin-like-2: a multifaceted protein with physiological and pathophysiological properties. Expert Rev Mol Med. 2014; 16:e17. [DOI] [PubMed] [Google Scholar]
- 49.Holleman A, den Boer ML, Cheok MH, et al. Expression of the outcome predictor in acute leukemia 1 (OPAL1) gene is not an independent prognostic factor in patients treated according to COALL or St Jude protocols. Blood. 2006;108(6):1984–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.