

Summary
Small-molecule inhibitors targeting growth factor receptors have failed to show efficacy for brain cancers, potentially due to inability to achieve sufficient drug levels in the CNS. Targeting non-oncogene tumor co-dependencies provides an alternative approach, particularly if drugs with high brain penetration can be identified. Here we demonstrate that the highly lethal brain cancer glioblastoma (GBM) is remarkably dependent on cholesterol for survival, rendering these tumors sensitive to Liver X receptor (LXR) agonist-dependent cell death. We show that LXR-623, a clinically viable, highly brain-penetrant LXRα-partial/LXRβ-full agonist selectively kills GBM cells in an LXRβ- and cholesterol-dependent fashion, causing tumor regression and prolonged survival in mouse models. Thus, a metabolic co-dependency provides a pharmacological means to kill growth factor-activated cancers in the CNS.
Introduction
Malignant brain tumors are among the most devastating types of cancer. Despite improvements in surgical techniques, development of various cytotoxic chemotherapy regimens, and advances in the delivery of focused radiation, the survival of most brain cancer patients, including those with glioblastoma (GBM), has not appreciably improved over the past 50 years.
The blood-brain barrier prevents many circulating molecules from entering into the brain. This includes most cytotoxic chemotherapies and targeted agents, and contributes to treatment failure (Deeken and Löscher, 2007). Thus, despite actionable drug targets such as the epidermal growth factor receptor (EGFR), which is amplified and/or mutated in nearly 60% of cases (Brennan et al., 2013), EGFR tyrosine kinase inhibitors have so far proven ineffective (Cloughesy et al., 2014; Furnari et al., 2015) in part because they do not reach therapeutic intratumoral drug levels in the central nervous system (CNS) (Vivanco et al., 2012). This suboptimal dosing frequently leads to the appearance of multiple resistance mechanisms (Furnari et al., 2015).
Cancer cells, due to alterations in their biochemical and signaling state, may become dependent on specific enzymes or transcription factors that are not themselves oncogenic, opening up treatment possibilities. This process, known as non-oncogene addiction (Galluzzi et al., 2013; Luo et al., 2009; Solimini et al., 2007) or non-oncogene co-dependency (Raj et al., 2011), may extend the pharmacopeia of cancer drugs to include brain-penetrant compounds that act on non-traditional targets. Importantly, co-dependencies can be shaped both by specific oncogenes that reprogram cellular metabolism and signaling and by the local biochemical environment in which tumor cells grow (Galluzzi et al., 2013).
The unique metabolic environment of the brain may generate actionable vulnerabilities. The brain, for instance, is the most cholesterol rich organ of the body, containing approximately 20% of total body cholesterol (Dietschy, 2009). However, the brain cholesterol pool is virtually separate from cholesterol metabolism in the periphery. Because cholesterol cannot be transported across the blood-brain barrier into the CNS, almost all brain cholesterol is synthesized de novo (Björkhem and Meaney, 2004; Dietschy and Turley, 2001). Astrocytes synthesize the majority of brain cholesterol from glucose, glutamine or acetate-derived acetyl-CoA and supply cholesterol to neighboring cells, including neurons, in the form of ApoE-containing HDL-like lipoprotein particles (Hayashi et al., 2004; Karten et al., 2006; Wahrle et al., 2004). Neurons and astrocytes both produce oxysterols as products of cholesterol metabolism, which act as endogenous ligands for the liver X receptors (LXRs) to decrease excess cellular cholesterol levels by promoting efflux through sterol transporters such as ABCA1 and suppressing uptake through IDOL-dependent degradation of LDLR (Chen et al., 2013; Repa et al., 2000; Venkateswaran et al., 2000; Zelcer et al., 2009). This negative feedback system complements suppression of HMG-CoA reductase (HMGCR), the rate-limiting enzyme for sterol synthesis, when cholesterol levels rise (Björkhem, 2006; Brown and Goldstein, 1980) to maintain cholesterol homeostasis within distinct cell types in the brain. Statins, which block HMCGR, and synthetic LXR agonists have been suggested as anti-cancer agents. A recent study reported that LXR agonist suppressed metastasis in the periphery through an APOE-dependent mechanism (Pencheva et al., 2014). In the brain, the efficacy of targeting either cholesterol synthesis or LXR activation may depend both on the specific tumor genotype and on the tissue-specific context required for cancer cell survival.
We reasoned that tumor cells in the brain may behave in a parasitic fashion with regard to cholesterol, obtaining CNS-derived cholesterol while suppressing endogenous LXR-ligand synthesis, enabling GBM cells to access a nearly limitless supply of cholesterol to fuel their growth. If this hypothesis is correct, GBM cells may be exquisitely vulnerable to synthetic brain-penetrant LXR ligands that would otherwise spare normal brain cell constituents, including astrocytes and neurons.
Results
GBM cells display dysregulated cholesterol metabolism
We previously demonstrated that mutant EGFR signaling in GBM cells upregulates LDLR expression (Guo et al., 2011), suggesting that these tumor cells may rely on exogenous cholesterol for survival. Therefore, to determine the relative dependency of GBM cells on endogenous cholesterol synthesis versus uptake, we examined the status of the de novo cholesterol biosynthetic pathway in GBM and normal brain specimens using data from The Cancer Genome Atlas (TCGA) (Brennan et al., 2013; Cancer Genome Atlas Research Network, 2008). We determined the expression of mRNAs encoding three key enzymes: HMGCS1 that converts acetyl-CoA to HMG-CoA; HMGCR, the rate limiting step in the pathway that converts HMG-CoA to mevalonate, and DHCR24 which ultimately converts desmosterol to cholesterol. Expression of these critical enzymes was coordinately suppressed in GBM clinical samples relative to normal brain (p < 0.001 for each) (Figure 1A). We reasoned that if GBM cells suppress de novo cholesterol synthesis, then they should be relatively resistant to statins, which target HMCGR. In contrast, normal human astrocytes (NHAs), which rely primarily on endogenous synthesis, should be highly vulnerable. Consistent with this hypothesis, both lovastatin and atorvastatin caused significant cell death of NHAs while showing limited activity against two independent models of EGFRvIII-expressing GBMs – U87EGFRvIII cells, which are derived by overexpression of EGFRvIII in an established GBM cell line (Wang et al., 2006), and GBM39, which is a patient-derived GBM line that maintains endogenous EGFR amplification and EGFRvIII expression in neurosphere culture (Sarkaria et al., 2007) (Figure 1B and S1A).
Figure 1. Dysregulated cholesterol metabolism renders GBM cells selectively vulnerable to an exogenous LXR agonist.
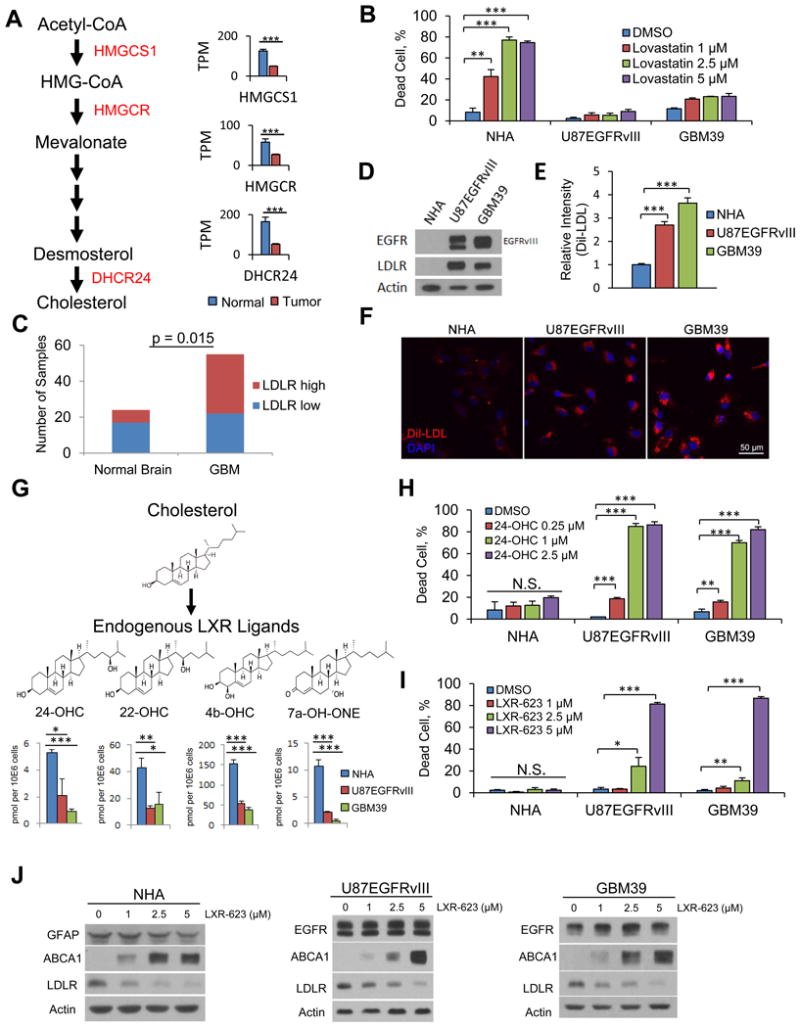
(A) Analysis of cholesterol synthesis genes in GBMs vs. normal brain from TCGA gene expression data. Data are reported as mean ± SEM.
(B) FACS analysis of Annexin V/PI staining comparing NHA to U87EGFRvIII and GBM39 cells after a three-day treatment with the brain penetrant HMGCR inhibitor lovastatin.
(C) Quantification of IHC analysis of tissue microarray samples for LDLR receptor.
(D) Immunoblotting comparing EGFR and LDLR protein levels in NHA, U87EGFRvIII, and GBM39 cells.
(E-F) FACS quantification (E) and representative images (F) of LDL uptake in U87EGFRvIII and GBM39 cells.
(G) Schematic model and molecular structures of cholesterol synthesis into LXR ligands and LC/MS-MS data evaluating the levels of endogenous LXR ligands in GBM cells and astrocytes. Data are reported as mean ± SEM.
(H) Quantification of cell death via FACS analysis of Annexin V/PI staining in response to endogenous LXR agonists at day five of treatment in NHA, U87EGFRvIII, and GBM39 cells.
(I) Quantification of cell death in response to LXR-623 at day five of treatment in NHA, U87EGFRvIII, GBM39 cells.
(J) NHA, U87EGFRvIII, and GBM39 were treated with the indicated concentrations of LXR-623 for 48 hr and immunoblotting was performed.
Unless otherwise stated, data are reported as mean ± SD. *p < 0.05; **p < 0.01; ***p < 0.001; N.S. = not significant. See also Figure S1.
Consistent with the notion that GBM cells obtain cholesterol primarily through uptake, LDLR protein was significantly elevated in GBM clinical samples relative to normal brain in tissue microarray analyses (Figure 1C). LDLR protein was also substantially elevated in GBM cells relative to NHAs in culture (Figure 1D), and GBM cells took up three- to four-fold more LDL than did NHAs (Figure 1E, 1F, S1B). Removal of lipids from the culture media caused extensive GBM cell death while having no effect on the viability of NHAs, (Figure S1C), motivating us to further examine the role of cholesterol uptake in GBM survival.
In non-cancerous cells, excess cholesterol is used to synthesize oxysterols, which act as endogenous LXR ligands to suppress LDL uptake and promote the efflux of cholesterol from the cell (Repa et al., 2000; Venkateswaran et al., 2000). However, the activity of this pathway in GBM cells and in many other cancer types (Lin and Gustafsson, 2015) is not well understood. Therefore, we performed mass spectrometric analysis of GBM cell lines and NHAs to measure endogenous LXR ligand levels and surveyed the mRNA expression of the enzymes that produce these endogenous LXR ligands in these cells, as well as more broadly across GBMS in the TCGA dataset. The levels of 24(S)-hydroxycholesterol (24-OHC), 22(R)-hydroxycholesterol (22-OHC), 4β-hydroxycholesterol (4b-OHC), 27-hydroxycholesterol (27-OHC), and the downstream product of 7α-hydroxycholesterol, 7α-hydroxy-4-cholesten-3-one (7a-OH-ONE), were significantly reduced in GBM cells relative to NHAs (Figure 1G, S1D), as were the mRNA levels of the enzymes that catalyze their production (Figures S1E, S1F). In particular, CYP46A1, the most highly expressed enzyme that catalyzes oxysterol synthesis in the brain, was reduced nearly ten-fold in GBMs in the TCGA dataset (Figure S1F). mRNA expression analysis of the enzymes that catabolize LXR ligands revealed HSD3B7 is expressed at a higher level in GBM cells relative to normal brain in the TCGA data set (Figure S1G), raising the possibility that catabolism of LXR ligands may also play a role in GBM cholesterol homeostasis. Taken together, these data demonstrate that GBM cells have a decreased capacity to produce endogenous LXR ligands.
GBM cells show selective vulnerability to LXR agonists
We hypothesized that the relatively low levels of endogenous LXR ligands in GBM cells would render them selectively sensitive to exogenous LXR ligands. We first tested the effect of exogenous administration of LXR ligands that are normally synthesized by cells in the presence of excess cholesterol. 24-OHC induced a dramatic dose-dependent increase in GBM cell death in vitro (Figure 1H, S1H). Importantly, NHAs, which synthesize 24-OHC (Figure 1G), were insensitive to exogenous 24-OHC administration (Figures 1H, S1H). Therefore, we reasoned that synthetic LXR agonists might also kill GBM cells while sparing NHAs.
We focused our initial investigation on LXR-623, a synthetic LXR agonist originally developed for cardiovascular indications and tested in patients (Hong and Tontonoz, 2014). A phase I clinical trial raised the possibility of brain penetration (Katz et al. J Clin Pharmacol. 2009), motivating our focus on this drug. Consistent with our proposed model, LXR-623 potently killed U87EGFRvIII and GBM39 cells in vitro while completely sparing NHAs (Figure 1I and S1I-L). LXR-623 also increased ABCA1 protein and decreased LDLR protein levels in all three cell lines (Figure 1J). These results prompted a deeper examination of LXR-623 as a potential therapy for GBM.
LXR-623 achieves therapeutic levels in GBM cells in the brain
In a healthy participant trial of LXR-623 (2-(2-chloro-4-fluorobenzyl)-3-(4-fluorophenyl)-7-(trifluoromethyl)-2H-indazole) (Figure S2A), subjects experienced mild to moderate CNS side effects, suggesting that this compound crosses the blood-brain barrier (Katz et al., 2009). Consistent with this premise, LXR-623 levels in the brains of nude mice treated daily by oral gavage were higher than plasma levels at two or eight hours after dosing (Figure 2A, S2B).
Figure 2. LXR-623 crosses the blood brain barrier, induces target gene expression, and achieves therapeutic levels in GBM cells in the brain with minimal activity in the periphery.

(A) Mice were treated with a single dose of LXR-623 by oral gavage (PO) 400 mg/kg (mpk). Plasma and brain were extracted from mice at 2 or 8 hr after gavage. LXR-623 plasma concentrations are reported (n = 5 for each time point).
(B-D) Mice were treated with LXR-623 or GW3965 (40 mg/kg) by oral gavage daily for three days. RNA was extracted from cerebral cortex (B), liver (C), and epididymal white adipose tissue, eWAT (D), and qPCR was performed for the indicated genes. n = 4 for each treatment condition.
(E) U87EGFRvIII turbo FP 635 orthotopic mouse xenograft. Mice were treated with vehicle or LXR-623 400 mpk PO daily. Tumor size was assessed via fluorescence molecular tomography (FMT) on day five of treatment. Tumors were excised from mice and the intratumor concentration of LXR-623 was assessed via liquid chromatography-tandem mass spectrometry (LC-MS/MS). Intratumoral LXR-623 concentrations are indicated within parentheses. n = 3 for vehicle and n = 4 for LXR-623 treated mice.
(F) Representative FMT images of mice from (E). Scale bar, 3.6 mm.
Data are reported as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001; N.S. = not significant. See also Figure S2.
Many of the undesirable hyperlipidemic effects of synthetic LXR agonists are mediated through LXRα in the liver and adipose tissue (Bradley et al., 2007; Joseph et al., 2002a, 2002b; Schultz et al., 2000). Previous studies had indicated that LXR-623, unlike structurally distinct LXR agonists such as GW3965, does not activate hepatic lipogenesis (Quinet et al., 2009) because it is an LXRα-partial/LXRβ-full agonist with a unique transcriptional cofactor recruitment profile (Quinet et al., 2009; Wrobel et al., 2008). Therefore, we compared the abilities of LXR-623 and GW3965 to induce target gene expression in the brain and peripheral tissues. LXR-623 significantly induced the expression of LXR target genes Abcg1 and Idol in cerebral cortex tissue in a similar fashion to GW3965, and also induced Abca1, although not as effectively as GW3965. LXR-623 did not induce Srebp1c in cerebral cortex (Figure 2B). LXR-623 also did not induce LXR target gene expression in the liver and epididymal white adipose tissue (eWAT) (Figure 2C-D). These data suggested that at least two special properties of LXR-623 – high CNS penetrance and reduced activity on LXRα – may provide a therapeutic window for treating GBM, and possibly other brain cancers.
We next performed a pilot study of LXR-623 treatment in an in vivo intracranial GBM xenograft model. We examined the potential relationship between intracranial GBM growth, as monitored by non-invasive fluorescence molecular tomography (FMT) imaging, and intratumoral LXR-623 levels. As shown in Figure 2E, LXR-623 reached low-μM concentrations within intracranial GBMs, meeting or exceeding the concentrations required to kill GBM cells in culture (Figure 1I), and significantly reduced GBM tumor growth in vivo (Figure 2F). These data demonstrate favorable brain penetration and potent anti-GBM activity for LXR-623 in vivo.
LXR-623 induces cell death in established and patient-derived GBMs
We extended our analysis of LXR-623 effects to a panel of established GBM cell lines (Figures 3A-B, S3A-B) and patient-derived GBM neurosphere cultures (Figure 3C-D) and found that LXR-623 suppressed LDLR expression, increased expression of the ABCA1 efflux transporter, and induced substantial cell death in all of the GBM samples tested. The brain metastatic breast cancer cell line MDA-MB-361, which harbors ERBB2 amplification, was also highly sensitive to LXR-623-dependent cell death in a concentration-dependent manner, raising the possibility that the drug could also have activity against systemic cancers that metastasize to the brain (Figure S3C-D).
Figure 3. LXR-623 induces cell death in established and patient-derived GBM cells.
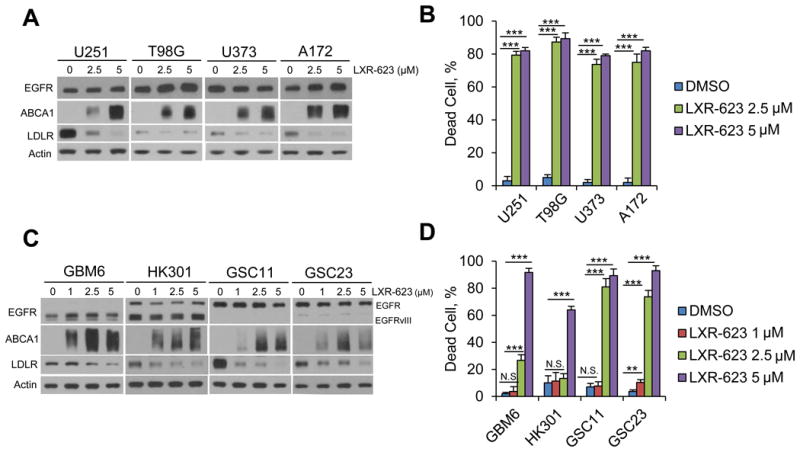
(A) Established GBM cell lines U251, T98G, U373 and A172 were treated with the indicated concentration of LXR-623 for 48 hr and immunoblotting was performed with the indicated antibodies.
(B) Trypan blue exclusion assay was carried out in parallel to (A) after three days of LXR-623 treatment.
(C) Patient derived GBM neurosphere lines GBM6, HK301, GSC11 and GSC23 were treated with the indicated concentration of LXR-623 for 48 hr and immunoblotting was performed as in (A).
(D) Trypan blue exclusion assay of cells from (C) after five days of treatment.
Data are reported as mean ± SD. **p < 0.01; ***p < 0.001; N.S. = not significant. See also Figure S3.
LXR-623 Induces GBM cell death through activation of LXRβ
To determine whether LXR-623 promotes its effects on GBM cells through activation of LXR, we first examined the effect of LXR-623 on gene expression. Gene microarray analysis demonstrated upregulation of mRNAs encoding the ABCA1 cholesterol efflux transporter and the E3 ligase IDOL, which targets the LDLR for degradation (Figure 4A), and gene set enrichment analysis (GSEA) revealed the anticipated transcriptional effects on lipid metabolism that are characteristic of LXR activation (Figure 4A). In contrast to GW3965, which induced ApoE-mediated suppression of metastasis in melanoma (Pencheva et al., 2014), we did not observe induction of APOE expression in GBM cells or NHA treated with LXR-623 (Figure 4A), indicating an ApoE-independent mechanism. Validation of our microarray results via qPCR and immunoblotting revealed concentration-dependent increases in ABCA1 and IDOL expression, and enhanced LDLR degradation (Figures 4B, 1J). Although LXR forms a heterodimer with retinoid X receptors (RXRs), which are expressed in patient GBM samples (Figure S4A), treatment of U87EGFRvIII cells with the RXR agonist bexarotene did not induce cell death or LXR target gene expression, despite inducing RXR target gene expression (Figures S4B-S4D). This suggests a specific role for LXR activation in LXR-623 induced cell death.
Figure 4. LXR-623 kills GBM cells through activation of LXRβ, the dominant subtype in brain tumors.

(A) Microarray analysis of LXR target genes in U87EGFRvIII and NHA cells treated with LXR-623 5 μM for 24 hr. Triplicates are shown. Gene Set Enrichment Analysis (GSEA) for Gene Ontology (GO) pathways for microarray data in (A) shown in the table below.
(B) U87EGFRvIII cells were treated with LXR-623 for 48 hr and qPCR was performed for LXR target genes.
(C) RNA was extracted from U87EGFRvIII, GBM39, and qPCR was performed for LXRα and LXRβ.
(D) Boxplot of The Cancer Genome Atlas (TCGA) RNASeq data from patients with GBM showing LXRβ is the primary LXR subtype expressed in GBM. The box extends from the 25th to 75th percentiles and the middle line inside indicates median. Whiskers are drawn down to the 10th percentile and up to the 90th.
(E) U87EGFRvIII cells were transfected with siRNA pools targeting LXRα or LXRβ. After 48 hr RNA was extracted and qPCR was performed. *Gene expression was normalized for scramble control.
(F) Cells were transfected with siRNA as in (E) and treated with LXR-623 for 24 hr. Immunoblotting was performed with the indicated antibodies.
(G) U87EGFRvIII cells were transfected with siRNA as in (E) and treated with LXR-623. Trypan blue exclusion assay was performed after three days of treatment.
Data are reported as mean ± SD. **p < 0.01; ***p < 0.001; N.S. = not significant. See also Figure S4.
The two LXR isotypes – LXRα and LXRβ – display different tissue distributions and have been shown to vary among some tumor types, but their relative expression in many cancers, including glioblastoma, remains, to date, unknown (Lin and Gustafsson, 2015). We found that LXRβ expression was nearly eightfold greater than LXRα in U87EGFRvIII cells and nearly fourfold greater in the patient-derived GBM39 neurosphere culture (Figure 4C). Analysis of the TCGA dataset similarly showed that LXRβ is the predominant LXR isotype in GBM (Figure 4D) and that its expression is unaffected by EGFR mutational status (Figure S4E).
To determine whether LXR-623 mediates its effects in GBM through LXRβ, we knocked down both LXRs by RNA-interference and assessed the impact on LXR-623 biochemical and functional responses. siRNA-mediated knockdown of LXRβ, but not LXRα, in GBM cells inhibited the upregulation of ABCA1 and IDOL expression, limited degradation of LDLR, and prevented cell death in response to LXR-623 (Figures 4E-4G, S4F-J). Further, the LXR inverse agonist SR9243, which has been shown to promote tumor cell death in prostate, colon, and lung cancer models (Flaveny et al., 2015), failed to cause GBM cell death despite robust inhibition of LXR target genes (Figure S4K-L). These results show that LXR-623 promotes its anti-tumor activity in GBM specifically by activating LXRβ.
Depletion of cholesterol drives LXR-623-induced killing of GBM cells
Our finding that LXRβ activation suppressed LDLR and induced expression of the ABCA1 efflux transporter and that this was concomitant with promotion of tumor cell death, raised the possibility that LXR-623 kills GBM cells by reducing cellular cholesterol. To test this idea, we measured the effect of LXR-623 on LDL uptake and cholesterol efflux. LXR-623 inhibited LDL uptake and induced cholesterol efflux in GBM cells (p < 0.001 for each) (Figures 5A, 5B, S5A-C), resulting in a significant reduction in cellular cholesterol content (Figure 5C, S5D).
Figure 5. LXR-623 Depletes GBM Cells of cholesterol.
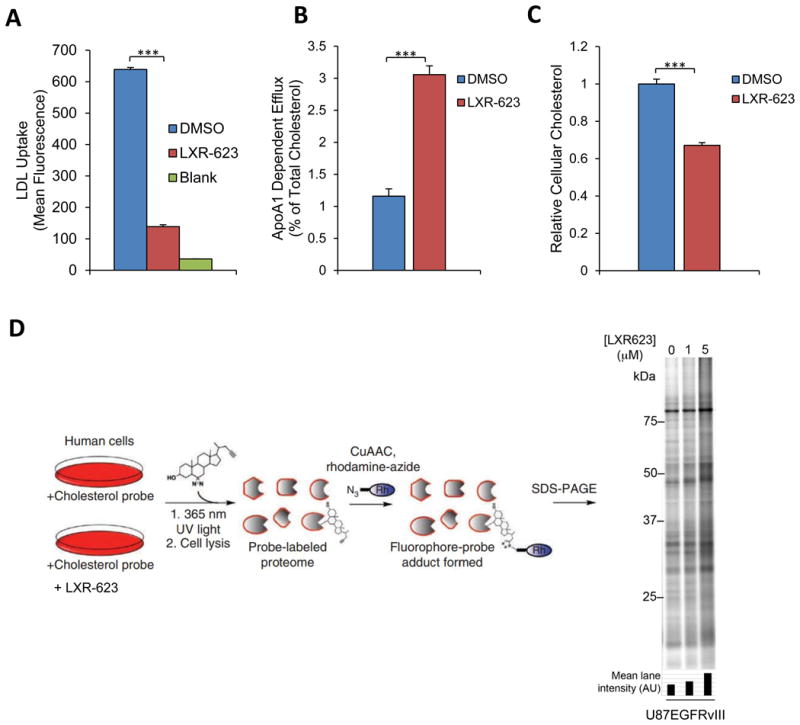
(A) U87EGFRvIII cells were treated with LXR-623 for a total of 48 hr. Cells were incubated with fluorescently labeled LDL for four hr and LDL uptake was determined via flow cytometry.
(B) U87EGFRvIII cells were loaded with 3H-cholesterol and treated with LXR-623 5 μM. Cholesterol efflux was determined by scintillation counting.
(C) U87EGFRvIII cells were treated as in (A) and total cholesterol levels were assessed by LC/MS.
(D) Sterol probe labeling profile (10 μM probe, 30 min) of U87EGFRvIII cells pre-treated with DMSO or the indicated concentrations of LXR-623 for 48 hr.
Data are reported as mean ± SD. ***p < 0.001. See also Figure S5.
We have described a chemical proteomic assay that uses a photoreactive, clickable, sterol-based probe to survey cholesterol-binding proteins in human cells (Hulce et al., 2013). Using this methodology, we found that LXR-623 induced a concentration-dependent increase in sterol probe binding to proteins in U87EGFRvIII cells (Figures 5D). Because the sterol probe competes with endogenous cholesterol for interaction with cholesterol-binding proteins (Hulce et al., 2013), these results provide further evidence that LXR-623 depletes intracellular cholesterol and, by doing so, enhances sterol probe-protein interactions in GBM cells.
We reasoned that if LXR-623 kills GBM by depleting cholesterol, the addition of exogenous cholesterol should prevent LXR-623-induced cell death. Consistent with this hypothesis, adding cholesterol that was complexed to methyl-β-cyclodextrin, which enables cholesterol to permeate into cells, to the cultures fully rescued LXR-623-induced cell death in both established and patient-derived GBM cell lines (Figure 6A). Cholesterol repletion did not affect LDLR or ABCA1 levels (Figure 6B), indicating that the rescue effect of cholesterol was not mediated by altering LXR-623 activity. Further, exposure to methyl-β-cyclodextrin (MβCD), a chemical means of decreasing cellular cholesterol levels (Caliceti et al., 2012; Christian et al., 1997), also reduced cholesterol levels (Figure S6A) and caused GBM cell death, an effect that was considerably greater in GBM cells than NHAs (Figures 6C, S6B). Taken together, these results demonstrate that GBM cells have an enhanced reliance on cholesterol for survival and that LXR-623 induces GBM cell death by depleting cellular cholesterol.
Figure 6. LXR-623 kills GBM Cells by Depleting Cholesterol.

(A) U87EGFRvIII and GBM39 cells were treated with LXR-623 in the presence or absence of methyl-β-cyclodextrin complexed to cholesterol at the indicated concentrations (final concentration of cholesterol is shown). Cell death was assessed via Annexin/PI staining on day three for U87EGFRvIII and via trypan blue staining on day five for GBM39.
(B) U87EGFRvIII (top panel) or GBM39 (bottom panel) were treated with LXR-623 for 48 hr in the presence of absence of MβCD-Cholesterol. Immunoblotting was performed with the indicated antibodies.
(C) NHA and U87EGFRvIII were treated with the indicated concentrations of methyl-β-cyclodextrin (MβCD) for 1 hr and cell death was assessed by trypan blue exclusion assay.
Data are reported as mean ± SD. ***p < 0.001. See also Figure S6.
LXR-623 efficacy in intracranial patient-derived GBM xenograft models
The relative efficacy (Figure 3) and specificity (Figure 1I, 2B-D) of LXR-623 against GBM cells, as well as the high brain penetrance of the drug (Figure 2A), inspired us to assess the potential efficacy and toxicity of this drug in a relevant pre-clinical animal model of GBM. Therefore, we investigated the therapeutic impact of LXR-623 in mice bearing patient-derived GBM orthotopic xenografts. Oral administration of LXR-623 (400 mg/kg) to mice bearing GBM39 cells engineered to express the infrared fluorescent protein 720 (IRFP720) caused a striking inhibition of tumor growth (Figure 7A-B) and highly significant prolongation of survival (Figure 7C).
Figure 7. LXR-623 inhibits tumor growth, promotes tumor cell death and prolongs the survival of mice bearing intracranial patient-derived GBMs.

(A) GBM39 patient derived neurosphere cells, engineered to stably express the infrared protein 720 (IRFP 720), were orthotopically injected into five-week old nu/nu mice. Mice were treated with vehicle or LXR-623 400 mpk PO daily (n = 8 for each group). Representative FMT images of mice at week five of treatment. Scale bar, 3.8 mm.
(B) Tumor size was assessed via fluorescence molecular tomography (FMT) weekly.
(C) Kaplan-Meier curves assessing overall survival of mice from (A). Log-rank (Mantel-Cox) test: p = 0.0001, Gehan-Breslow-Wilcoxon test: p = 0.0002.
(D) Tumors were excised and immunohistochemistry analysis was performed with the indicated antibodies. Scale bar, 50 μm.
(E) Quantification of the immunohistochemistry performed in (D).
Data are reported as mean ± SEM. **p < 0.01; ***p < 0.001. See also Figure S7.
Immunohistochemical analysis revealed a significant increase in ABCA1 protein expression (p < 0.01) and a significant decrease in LDLR protein expression, (p < 0.001) in LXR-623-treated GBM tumors (Figure 7D-E). LXR-623 induced a greater than ten-fold increase in TUNEL staining, (p < 0.001) (Figure 7D-E), demonstrating that it induces substantial apoptosis in GBM tumors. Importantly, no change in the number of neurons in the brain was seen response to LXR-623 and no drug-induced cell death was detected in normal brain (Figures S7A-B). Further, mice bearing GBMs maintained their body weights throughout LXR-623 treatment (Figure S7C), and showed no evidence of fatty liver (Figure S7D), a known consequence of LXR-target gene activation in the periphery (Chisholm et al., 2003). These findings are consistent with the high penetration of the LXR-623 into the brain relative to the periphery (Figures 2A-B) and the relative lack of LXR-target gene induction in peripheral tissues (Figures 2C-D). Taken together, these results demonstrate that LXR-623 selectively kills GBM cells with excellent efficacy in vivo and prolongs survival in a clinically-relevant, patient-derived GBM model.
Discussion
The brain is biochemically separated from the rest of the body by the blood-brain barrier, which prevents many compounds and molecules from entering the CNS. The blood-brain barrier presents challenges to achieving therapeutic concentrations of drugs in the brain without causing serious dose-limiting toxicities in peripheral organs, which likely contributes to the lack of clinical activity for most brain cancer treatments tested to date, including those that act on compelling drug targets. GBM, for instance, is one of the most deeply sequenced human cancers (Brennan et al., 2013; Cancer Genome Atlas Research Network, 2008; Lawrence et al., 2014; Parsons et al., 2008). However, this information had yet to improve the outcome for GBM patients.
Motivated by the persistent clinical failure and the lack of therapeutic options for brain cancers, we adopted an alternative strategy designed to take advantage of a targetable metabolic co-dependency using a highly brain penetrant drug. We focused on cholesterol homeostasis because: 1) GBM cells are highly dependent on cholesterol for survival (Bovenga et al., 2015); 2) EGFR mutations can regulate cholesterol homeostasis in GBM and possibly in other types of cancers (Gabitova et al., 2015; Guo et al., 2011); 3) the brain has a unique mechanism of cholesterol regulation that may expose a targetable vulnerability specific to tumor cells in this organ (Dietschy and Turley, 2001); and 4) anecdotal evidence from a Phase 1 clinical trial raised the possibility that a highly brain penetrant compound, LXR-623, could be used to exploit this vulnerability (Katz et al., 2009).
Most of the cells in the brain are dependent on astrocyte-synthesized cholesterol for survival (Nieweg et al., 2009). Here we show that GBM cells exploit this tissue-specific physiology to fuel their growth. By upregulating LDLR to enhance cholesterol uptake while concurrently suppressing de novo synthesis of cholesterol and oxysterols, GBMs evade two key negative feedback mechanisms of cholesterol homeostasis: 1) feedback inhibition of HMGCR, the rate limiting enzyme in cholesterol synthesis, and 2) activation of LXRs by oxysterols to inhibit LDL uptake and induce cholesterol efflux. It is remarkable that CYP46A1, the main oxysterol-synthesizing enzyme in the brain, was suppressed ten-fold in GBM tissue relative to normal brain. These coordinated metabolic adaptations give tumor cells access to an abundant and uninterrupted source of cholesterol. De novo cholesterol synthesis consumes 26 reducing equivalents of NADPH (Lunt and Vander Heiden, 2011), and it is tempting to speculate that the reliance of GBM cells on CNS-derived cholesterol enables them to direct their cellular NADPH, a key reducing agent in relatively short supply (Pavlova and Thompson, 2016), towards buffering ROS and synthesizing other macromolecules (Vander Heiden et al., 2009). This coordinated set of metabolic adaptations likely enhances the growth of GBMs in the brain, but also makes them exquisitely and selectively vulnerable to exogenous LXR agonists.
A strong case has been made for nuclear hormone receptor modulators as potential cancer drugs (Gronemeyer et al., 2004), including LXR modulators that show activity against melanoma, intestinal cancers, squamous cell cancers, prostate cancers cells and breast cancer cell lines (Flaveny et al., 2015; Gabitova et al., 2015; Lin and Gustafsson, 2015; Lo Sasso et al., 2013; Nguyen-Vu et al., 2013; Pencheva et al., 2014; Pommier et al., 2013). However, what is unanticipated and important from our studies is the critical role that tissue type and organ context seems to play in determining the activity of a specific LXR modulator. In peripheral melanomas, activation of LXRβ by GW3965 potently suppresses metastasis by transcriptional induction of tumoral and stromal ApoE (Pencheva et al., 2014). In the same study, however, GW3965 did not kill melanoma cells. In contrast, we show that LXR-623 kills GBM cells specifically by depleting them of cholesterol, independent of any effect on ApoE. We also found that the LXR inverse agonist SR9243, which was shown to have activity against a variety of cancer cell lines through its ability to inhibit the Warburg effect (Flaveny et al., 2015), has no anti-tumor effect on GBM cells, despite robust LXR target gene inhibition. Of course, differences in the transcriptional regulation/cofactor recruitment profiles of specific LXR modulators and the expression of LXR isoforms may contribute to the gamut of responses to LXR agonists and inverse agonists observed thus far in cancer models. It is also alternatively plausible that individual cancers respond differently to the same drugs as a consequence of the interplay between their specific genotype, metabolic state, and the tissue context to which they are exposed. Importantly, actionable metabolic co-dependencies are likely to be shaped both by specific tumor cell oncogenes that reprogram cellular metabolism and by the local biochemical environment to which the tumor cells must adapt their metabolic circuitry for growth and survival (Galluzzi et al., 2013).
LXR-623 also showed a remarkably specific activity against GBMs in vivo, sparing normal cells, including neurons, in the brain and failing to elicit obvious toxicity in peripheral tissues. Three features may contribute to the specificity we found in our in vitro and in vivo models. First, the intact cellular cholesterol homeostasis machinery that is present in neurons and glia, but absent in GBM cells, may render normal brain cells less sensitive to LXR-623. Astrocytes rely on de novo cholesterol synthesis for survival and they are not dependent on cholesterol uptake, rendering them vulnerable to statins, but not to LXR-623. Similarly, no decrease in neuron number was seen and no neuronal cell death was detected in LXR-623-treated mice in vivo, indicating that neurons were less sensitive to the LXR-623 than were GBM cells. Thus, normal brain cell insensitivity to LXR-623 may be due to reliance on endogenous synthesis of cholesterol and intact negative feedback through synthesis of endogenous oxysterols. This is in contrast to GBMs, which rely primarily on uptake of cholesterol, rather than de novo production, and do not synthesize endogenous oxysterols. Second, the relatively high LXR-623 brain/plasma ratio limits the dose needed to reach therapeutic levels in tumor cells in the brain, thus limiting peripheral drug exposure. Third, LXR-623 is an LXRα-partial/LXRβ-full agonist (Wrobel et al., 2008) and many of the undesirable effects of synthetic LXR agonists are mediated through LXRα, including hepatic lipogenesis (Bradley et al., 2007; Joseph et al., 2002a, 2002b; Schultz et al., 2000). On this note, we did not detect fatty livers in tumor-bearing mice treated with LXR-623 at a dose that dramatically shrunk GBMs in the brain.
Taken together, our data suggest that LXR-623 could offer a viable pharmacological therapy for GBM patients who currently have no effective treatment options. Similar to GBMs, tumors that metastasize to the brain also commonly contain amplification and gain-of-function mutations of EGFR and erbB family members, including HER2 (encoded by ERBB2) (Brastianos et al., 2015). LXR-623 potently killed ERBB2-amplified MDA-MB-231 breast cancer cells that were derived from a brain metastasis, raising the possibility that LXR-623 might further be used to treat systemic tumors that migrate to the brain. Future studies are warranted to determine the potential efficacy of this drug for the treatment of an array of brain cancers.
Experimental Procedures
Additional details are available in Supplemental Experimental Procedures.
Cell Culture
The human glioma cell line U87 (or U-87MG) was purchased from ATCC and the U87EGFRvIII isogenic GBM cell line was obtained as described previously (Wang et al., 2006). U87EGFRvIII, U251, T98, U373 and A172 GBM cell lines were cultured in Dulbecco's Modified Eagle Media (DMEM, Cellgro), and PC9 cells were cultured in RPMI1640 (Gibco), supplemented with 10% fetal bovine serum (FBS, Hyclone) and 1% penicillin/streptomycin/glutamine (Invitrogen) in a humidified 5% CO2 incubator at 37°C. MDA-MB-361 cells were cultured in Leibovitz's L-15 media (Cellgro) supplemented with 20% FBS in a humidified 0% CO2 incubator at 37°C. Normal human astrocytes were purchased from Lonza and cultured per manufacturer's guidelines. GBM39, GBM6, HK301, GSC11, and GSC23 were cultured in Neurocult media (Stemcell Technologies) supplemented with EGF (Sigma), FGF (Sigma) and heparin (Sigma) in a humidified 5% CO2 incubator at 37°C.
Tissue Microarray
Tissue microarrays (TMA) were constructed as reported previously (Guo et al., 2009), and immunohistochemical staining was performed, as described under immunohistochemistry methods, to analyze the expression of LDLR in 55 GBM samples and 24 normal brain samples. All of the tumor samples included in the GBM tissue microarray were obtained and approved under the UCLA IRB# 10-000655 (formerly known as UCLA IRB# 99-07-061). Patients who were alive at the time that the TMA was constructed, provided written consent. Patients who had expired by that time, were exempt from this requirement, in accordance with the UCLA IRB policies. Images of LDLR-stained tissue sections were reviewed in a blinded fashion and dichotomized as either undetectable or barely detectable – “low”, or as readily detectable or abundant – “high”.
TCGA Data Analysis
Processed TCGA data were downloaded through the TCGA data portal. The EGFR status of TCGA GBM samples are designated as euploid, regionally amplified, focally amplified, or EGFRvIII based on annotations previously published (Brennan et al., 2013). Specifically, EGFRvIII samples are samples with non-zero Δ2–7 values, and euploid / regionally amplified / focally amplified samples are non-EGFRvIII samples labeled as “Euploid” / “Regional gain” / “Focal Amplification.” The EGFR status of TCGA LUAD samples are designated as “EGFR Euploid” or “EGFR Amplification” based on annotations previously published (Lawrence et al., 2014).
Pharmacokinetic Analysis
Plasma and brain concentrations of LXR-623 (0.5% methylcellulose, 2% Tween-80 in water) in female nu/nu mice after oral dosing (day 1 and 7) were determined by standard LC-MS/MS-based methods by WuXi AppTec (Shanghai, China).
Intracranial Xenograft
Five week old female athymic nu/nu mice were purchased from Harlan Sprague Dawley Inc. 1 × 105 U87EGFRvIII iRFP720 or GBM39 IRFP720 cells in 5 μl of phosphate-buffered saline (PBS) were intracranially injected into the mouse brain as described previously (Ozawa and James, 2010). Tumors were allowed to establish over the course of 7-10 days and engraftment of tumors was quantitatively confirmed via fluorescence molecular tomography (FMT) signal intensity. Tumor growth was monitored using an FMT 2500 Fluorescence Tomography System (PerkinElmer). For drug treatment studies, vehicle (0.5% methylcellulose, 2% Tween-80 in water) or LXR-623 (400 mg/kg) resuspended in vehicle were administered to mice via oral gavage daily starting at day seven post-injection. All procedures were reviewed and approved by the Institutional Animal Use and Care Committee at University of California, San Diego.
Data Analysis
The Fischer's exact test was used for statistical analysis of tissue microarray staining. The Mantel-Cox log-rank and Gehan-Breslow-Wilcoxon tests were used for statistical comparisons in survival analyses. All other statistical comparisons were carried out using Student's t tests. Throughout all figures, *p < 0.05, **p < 0.01, and ***p < 0.001, N.S. = not significant. Significance was concluded at p < 0.05.
Supplementary Material
Significance.
Adult brain cancers are almost universally fatal in part because of the physicochemical segregation and biochemical composition of the central nervous system (CNS). We exploit these unique features by showing glioblastoma (GBM) cells suppress cholesterol and liver X receptor (LXR) ligand synthesis, making them dependent on CNS-derived cholesterol for survival. This pathophysiological property renders GBMs highly vulnerable to a brain penetrant LXR agonist, thus illuminating a pharmacologically viable means to kill GBMs, and possibly brain metastases.
Acknowledgments
G.R.V. would like to dedicate this manuscript to the loving memory of his father Genaro M. Villa. We thank Ms. Jennifer Tsoi for technical assistance with TCGA and microarray data. This work was supported by the National Cancer Institute F31CA186668 (G.R.V) and by grants from National Institute for Neurological Diseases and Stroke (NS73831 and NS080939), the Defeat GBM Program of the National Brain Tumor Society, the Ben and Catherine Ivy Foundation, and generous donations from the Ziering Family Foundation in memory of Sigi Ziering (P.S.M).
Footnotes
Accession Numbers: The microarray gene expression data from U87EGFRvIII and NHA treated with LXR-623 are deposited at the Gene Expression Omnibus (GEO) under the accession number data GSE78703.
Supplemental Information: Supplemental information includes Supplemental Experimental Procedures and seven figures.
Author Contributions: G.R.V., B.F.C., and P.S.M. conceived the project and designed the research. G.R.V., J.J.H., C.Z., J.B., S.I., G.L.C., Y.G., K.M.L., K.M., H.Y., X.R., C.H., K.M.T., F.L., G.C.H., D.J., M.M. and A.M.A. performed experiments and analyzed data. O.Q., T.F.C., F.B.F., W.K.C., P.T., T.C.G., A.K.S., and B.F.C., provided new reagents and reagents and analytic tools and provided intellectual contributions to the design of experiments and interpretation of data. G.R.V. and P.S.M. wrote the original manuscript. G.R.V., J.J.H., C.Z., S.I., Y.G., J.B., K.M.T., F.L., G.C.H, O.Q., T.F.C., F.B.F., W.K.C., P.T., T.C.G., A.K.S., B.F.C., and P.S.M. revised and edited the manuscript. All authors discussed the results and commented on the manuscript.
References
- Björkhem I. Crossing the barrier: oxysterols as cholesterol transporters and metabolic modulators in the brain. J Intern Med. 2006;260:493–508. doi: 10.1111/j.1365-2796.2006.01725.x. [DOI] [PubMed] [Google Scholar]
- Björkhem I, Meaney S. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. 2004;24:806–815. doi: 10.1161/01.ATV.0000120374.59826.1b. [DOI] [PubMed] [Google Scholar]
- Bovenga F, Sabbà C, Moschetta A. Uncoupling nuclear receptor LXR and cholesterol metabolism in cancer. Cell Metab. 2015;21:517–526. doi: 10.1016/j.cmet.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Bradley MN, Hong C, Chen M, Joseph SB, Wilpitz DC, Wang X, Lusis AJ, Collins A, Hseuh WA, Collins JL, et al. Ligand activation of LXR beta reverses atherosclerosis and cellular cholesterol overload in mice lacking LXR alpha and apoE. J Clin Invest. 2007;117:2337–2346. doi: 10.1172/JCI31909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brastianos PK, Carter SL, Santagata S, Cahill DP, Taylor-Weiner A, Jones RT, Van Allen EM, Lawrence MS, Horowitz PM, Cibulskis K, et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015;5:1164–1177. doi: 10.1158/2159-8290.CD-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RGW, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. The Somatic Genomic Landscape of Glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980;21:505–517. [PubMed] [Google Scholar]
- Caliceti C, Zambonin L, Prata C, Vieceli Dalla Sega F, Hakim G, Hrelia S, Fiorentini D. Effect of plasma membrane cholesterol depletion on glucose transport regulation in leukemia cells. PloS One. 2012;7:e41246. doi: 10.1371/journal.pone.0041246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang X, Kusumo H, Costa LG, Guizzetti M. Cholesterol efflux is differentially regulated in neurons and astrocytes: implications for brain cholesterol homeostasis. Biochim Biophys Acta. 2013;1831:263–275. doi: 10.1016/j.bbalip.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm JW, Hong J, Mills SA, Lawn RM. The LXR ligand T0901317 induces severe lipogenesis in the db/db diabetic mouse. J Lipid Res. 2003;44:2039–2048. doi: 10.1194/jlr.M300135-JLR200. [DOI] [PubMed] [Google Scholar]
- Christian AE, Haynes MP, Phillips MC, Rothblat GH. Use of cyclodextrins for manipulating cellular cholesterol content. J Lipid Res. 1997;38:2264–2272. [PubMed] [Google Scholar]
- Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: From Molecular Pathology to Targeted Treatment. Annu Rev Pathol Mech Dis. 2014;9:1–25. doi: 10.1146/annurev-pathol-011110-130324. [DOI] [PubMed] [Google Scholar]
- Deeken JF, Löscher W. The Blood-Brain Barrier and Cancer: Transporters, Treatment, and Trojan Horses. Clin Cancer Res. 2007;13:1663–1674. doi: 10.1158/1078-0432.CCR-06-2854. [DOI] [PubMed] [Google Scholar]
- Dietschy JM. Central nervous system: cholesterol turnover, brain development and neurodegeneration. Biol Chem. 2009;390:287–293. doi: 10.1515/BC.2009.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietschy JM, Turley SD. Cholesterol metabolism in the brain. Curr Opin Lipidol. 2001;12:105–112. doi: 10.1097/00041433-200104000-00003. [DOI] [PubMed] [Google Scholar]
- Flaveny CA, Griffett K, El-Gendy BEDM, Kazantzis M, Sengupta M, Amelio AL, Chatterjee A, Walker J, Solt LA, Kamenecka TM, et al. Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell. 2015;28:42–56. doi: 10.1016/j.ccell.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furnari FB, Cloughesy TF, Cavenee WK, Mischel PS. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat Rev Cancer. 2015;15:302–310. doi: 10.1038/nrc3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabitova L, Restifo D, Gorin A, Manocha K, Handorf E, Yang DH, Cai KQ, Klein-Szanto AJ, Cunningham D, Kratz LE, et al. Endogenous Sterol Metabolites Regulate Growth of EGFR/KRAS-Dependent Tumors via LXR. Cell Rep. 2015;12:1927–1938. doi: 10.1016/j.celrep.2015.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Heiden MGV, Kroemer G. Metabolic targets for cancer therapy. Nat Rev Drug Discov. 2013;12:963–963. doi: 10.1038/nrd4145. [DOI] [PubMed] [Google Scholar]
- Gronemeyer H, Gustafsson JA, Laudet V. Principles for modulation of the nuclear receptor superfamily. Nat Rev Drug Discov. 2004;3:950–964. doi: 10.1038/nrd1551. [DOI] [PubMed] [Google Scholar]
- Guo D, Reinitz F, Youssef M, Hong C, Nathanson D, Akhavan D, Kuga D, Amzajerdi AN, Soto H, Zhu S, et al. An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR-dependent pathway. Cancer Discov. 2011;1:442–456. doi: 10.1158/2159-8290.CD-11-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Campenot RB, Vance DE, Vance JE. Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J Biol Chem. 2004;279:14009–14015. doi: 10.1074/jbc.M313828200. [DOI] [PubMed] [Google Scholar]
- Hegi ME, Diserens AC, Bady P, Kamoshima Y, Kouwenhoven MCM, Delorenzi M, Lambiv WL, Hamou MF, Matter MS, Koch A, et al. Pathway analysis of glioblastoma tissue after preoperative treatment with the EGFR tyrosine kinase inhibitor gefitinib--a phase II trial. Mol Cancer Ther. 2011;10:1102–1112. doi: 10.1158/1535-7163.MCT-11-0048. [DOI] [PubMed] [Google Scholar]
- Hong C, Tontonoz P. Liver X receptors in lipid metabolism: opportunities for drug discovery. Nat Rev Drug Discov. 2014;13:433–444. doi: 10.1038/nrd4280. [DOI] [PubMed] [Google Scholar]
- Hulce JJ, Cognetta AB, Niphakis MJ, Tully SE, Cravatt BF. Proteome-wide mapping of cholesterol-interacting proteins in mammalian cells. Nat Methods. 2013;10:259–264. doi: 10.1038/nmeth.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SB, Laffitte BA, Patel PH, Watson MA, Matsukuma KE, Walczak R, Collins JL, Osborne TF, Tontonoz P. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver X receptors. J Biol Chem. 2002a;277:11019–11025. doi: 10.1074/jbc.M111041200. [DOI] [PubMed] [Google Scholar]
- Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, Laffitte BA, Chen M, Noh G, Goodman J, Hagger GN, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci U S A. 2002b;99:7604–7609. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karten B, Campenot RB, Vance DE, Vance JE. Expression of ABCG1, but not ABCA1, correlates with cholesterol release by cerebellar astroglia. J Biol Chem. 2006;281:4049–4057. doi: 10.1074/jbc.M508915200. [DOI] [PubMed] [Google Scholar]
- Katz A, Udata C, Ott E, Hickey L, Burczynski ME, Burghart P, Vesterqvist O, Meng X. Safety, pharmacokinetics, and pharmacodynamics of single doses of LXR-623, a novel liver X-receptor agonist, in healthy participants. J Clin Pharmacol. 2009;49:643–649. doi: 10.1177/0091270009335768. [DOI] [PubMed] [Google Scholar]
- Lassman AB, Rossi MR, Raizer JJ, Razier JR, Abrey LE, Lieberman FS, Grefe CN, Lamborn K, Pao W, Shih AH, et al. Molecular study of malignant gliomas treated with epidermal growth factor receptor inhibitors: tissue analysis from North American Brain Tumor Consortium Trials 01-03 and 00-01. Clin Cancer Res Off J Am Assoc Cancer Res. 2005;11:7841–7850. doi: 10.1158/1078-0432.CCR-05-0421. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, Getz G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature. 2014;505:495–501. doi: 10.1038/nature12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Gustafsson JÅ. Targeting liver X receptors in cancer therapeutics. Nat Rev Cancer. 2015;15:216–224. doi: 10.1038/nrc3912. [DOI] [PubMed] [Google Scholar]
- Lo Sasso G, Bovenga F, Murzilli S, Salvatore L, Di Tullio G, Martelli N, D'Orazio A, Rainaldi S, Vacca M, Mangia A, et al. Liver X receptors inhibit proliferation of human colorectal cancer cells and growth of intestinal tumors in mice. Gastroenterology. 2013;144:1497–1507. 1507.e1–e13. doi: 10.1053/j.gastro.2013.02.005. [DOI] [PubMed] [Google Scholar]
- Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Vu T, Vedin LL, Liu K, Jonsson P, Lin JZ, Candelaria NR, Candelaria LP, Addanki S, Williams C, Gustafsson JÅ, et al. Liver × receptor ligands disrupt breast cancer cell proliferation through an E2F-mediated mechanism. Breast Cancer Res BCR. 2013;15:R51. doi: 10.1186/bcr3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieweg K, Schaller H, Pfrieger FW. Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J Neurochem. 2009;109:125–134. doi: 10.1111/j.1471-4159.2009.05917.x. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JCH, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016;23:27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pencheva N, Buss CG, Posada J, Merghoub T, Tavazoie SF. Broad-spectrum therapeutic suppression of metastatic melanoma through nuclear hormone receptor activation. Cell. 2014;156:986–1001. doi: 10.1016/j.cell.2014.01.038. [DOI] [PubMed] [Google Scholar]
- Pommier AJC, Dufour J, Alves G, Viennois E, De Boussac H, Trousson A, Volle DH, Caira F, Val P, Arnaud P, et al. Liver x receptors protect from development of prostatic intra-epithelial neoplasia in mice. PLoS Genet. 2013;9:e1003483. doi: 10.1371/journal.pgen.1003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet EM, Basso MD, Halpern AR, Yates DW, Steffan RJ, Clerin V, Resmini C, Keith JC, Berrodin TJ, Feingold I, et al. LXR ligand lowers LDL cholesterol in primates, is lipid neutral in hamster, and reduces atherosclerosis in mouse. J Lipid Res. 2009;50:2358–2370. doi: 10.1194/jlr.M900037-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj L, Ide T, Gurkar AU, Foley M, Schenone M, Li X, Tolliday NJ, Golub TR, Carr SA, Shamji AF, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Repa JJ, Turley SD, Lobaccaro JA, Medina J, Li L, Lustig K, Shan B, Heyman RA, Dietschy JM, Mangelsdorf DJ. Regulation of absorption and ABC1-mediated efflux of cholesterol by RXR heterodimers. Science. 2000;289:1524–1529. doi: 10.1126/science.289.5484.1524. [DOI] [PubMed] [Google Scholar]
- Sarkaria JN, Yang L, Grogan PT, Kitange GJ, Carlson BL, Schroeder MA, Galanis E, Giannini C, Wu W, Dinca EB, et al. Identification of molecular characteristics correlated with glioblastoma sensitivity to EGFR kinase inhibition through use of an intracranial xenograft test panel. Mol Cancer Ther. 2007;6:1167–1174. doi: 10.1158/1535-7163.MCT-06-0691. [DOI] [PubMed] [Google Scholar]
- Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solimini NL, Luo J, Elledge SJ. Non-Oncogene Addiction and the Stress Phenotype of Cancer Cells. Cell. 2007;130:986–988. doi: 10.1016/j.cell.2007.09.007. [DOI] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkateswaran A, Laffitte BA, Joseph SB, Mak PA, Wilpitz DC, Edwards PA, Tontonoz P. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc Natl Acad Sci U S A. 2000;97:12097–12102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, Kubek S, Oldrini B, Chheda MG, Yannuzzi N, et al. Differential Sensitivity of Glioma- versus Lung Cancer–Specific EGFR Mutations to EGFR Kinase Inhibitors. Cancer Discov. 2012;2:458–471. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahrle SE, Jiang H, Parsadanian M, Legleiter J, Han X, Fryer JD, Kowalewski T, Holtzman DM. ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J Biol Chem. 2004;279:40987–40993. doi: 10.1074/jbc.M407963200. [DOI] [PubMed] [Google Scholar]
- Wang MY, Lu KV, Zhu S, Dia EQ, Vivanco I, Shackleford GM, Cavenee WK, Mellinghoff IK, Cloughesy TF, Sawyers CL, et al. Mammalian target of rapamycin inhibition promotes response to epidermal growth factor receptor kinase inhibitors in PTEN-deficient and PTEN-intact glioblastoma cells. Cancer Res. 2006;66:7864–7869. doi: 10.1158/0008-5472.CAN-04-4392. [DOI] [PubMed] [Google Scholar]
- Wrobel J, Steffan R, Bowen SM, Magolda R, Matelan E, Unwalla R, Basso M, Clerin V, Gardell SJ, Nambi P, et al. Indazole-based liver X receptor (LXR) modulators with maintained atherosclerotic lesion reduction activity but diminished stimulation of hepatic triglyceride synthesis. J Med Chem. 2008;51:7161–7168. doi: 10.1021/jm800799q. [DOI] [PubMed] [Google Scholar]
- Zelcer N, Hong C, Boyadjian R, Tontonoz P. LXR regulates cholesterol uptake through Idol-dependent ubiquitination of the LDL receptor. Science. 2009;325:100–104. doi: 10.1126/science.1168974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.