

A strategy for bonding water-rich hydrogels to diverse materials for electronic skins, energy storage, and soft optics is reported.
Keywords: hydrogels, Soft Electronics, Soft Robots, stretchable electronics, Soft Batteries, energy harvesting, Electronic Skins, Adaptive Optics
Abstract
Introducing methods for instant tough bonding between hydrogels and antagonistic materials—from soft to hard—allows us to demonstrate elastic yet tough biomimetic devices and machines with a high level of complexity. Tough hydrogels strongly attach, within seconds, to plastics, elastomers, leather, bone, and metals, reaching unprecedented interfacial toughness exceeding 2000 J/m2. Healing of severed ionic hydrogel conductors becomes feasible and restores function instantly. Soft, transparent multilayered hybrids of elastomers and ionic hydrogels endure biaxial strain with more than 2000% increase in area, facilitating soft transducers, generators, and adaptive lenses. We demonstrate soft electronic devices, from stretchable batteries, self-powered compliant circuits, and autonomous electronic skin for triggered drug delivery. Our approach is applicable in rapid prototyping and in delicate environments inaccessible for extended curing and cross-linking.
INTRODUCTION
Hydrogels are versatile building blocks of life—living beings are in essence gel-embodied soft machines. The intricacy and vast diversity found in nature, from entirely soft Cnidaria or mollusks to hybrid vertebrates, rely on insightful merging of a wide variety of biological materials (1) forming gels, tissues, fibers, muscles, tendons, and skeletal structures (2). Although technologically inimitable in full complexity, the unique properties of engineered, tough hydrogels (3, 4) advance soft machines (5) and electronics (6), sparking a new generation of biomimetic systems. A high level of functionality in the artificial counterparts is achievable through hybrid combinations of hydrogels and soft-to-solid materials, from elastomers to polymers, metals, or mineralized tissue. Without addressing issues of adhesion, impressive demonstrations of tough ionic hydrogels and elastomer membranes for transparent loudspeakers (7) and cables (8), touch panels (7, 9, 10), and stretchable electroluminescent displays (11, 12) were realized. Overcoming the low interfacial toughness when bonding diverse substances to water-rich hydrogels is challenging (13, 14). Recent approaches realizing strong bonds between tough gels and nonporous solids (15) involve surface functionalization via self-assembled monolayers; bonding to elastomers was achieved through ultraviolet (UV)–initiated polymerization (16). However, all of these methods require extended curing processes difficult to implement in the vicinity of sensitive target surfaces. They are not generally applicable over a wide range of antagonistic material classes and lack an instant character, essential in fracture healing and other time-critical applications such as rapid prototyping and high-throughput manufacturing (17).
RESULTS AND DISCUSSION
We introduce here a set of concepts, material approaches, and design rules for wide-ranging classes of soft, hydrogel-based electronic, ionic, and photonic devices based on soft and soft-to-hard hybrid architectures. Robust interfaces between the individual layers prevent delamination and are essential for mechanically tough yet soft, highly functional and compliant systems. By mixing synthetic adhesives that rapidly polymerize in the presence of hydroxyl ions with suitable nonsolvents, we achieve easy-to-use dispersions with tailorable properties. The general principle is outlined in Fig. 1A, where our adhesive dispersion is applied to form tough, instant bonds between (i) separate hydrogels, (ii) hydrogels and elastomers, and (iii) hydrogels and solid surfaces (illustrated here with polymer foils bonded to prestretched gels). The use of a bonding agent where the adhesive is dispersed in a nonsolvent, instead of the pure or solvent-diluted adhesive, overcomes several difficulties. Alignment of the surfaces to bond requires sufficiently delaying the water-triggered polymerization reaction, achievable with the volatile nonsolvent acting as temporal protecting shell. Our dispersion does not reduce the bonding strength, an effect we observe when using simple dilutions, and allows for precise control of bonding agent quantities such that no hard and brittle interfaces form in soft hybrids. With cyanoacrylate-based adhesives dispersed in alkanes (see the Supplementary Materials for details), the bonding process, including application of the dispersion, alignment, and curing, is typically completed in less than a minute, easing design iterations and upscaling. We demonstrate the versatility of our approach on a wide class of fully swollen hydrogels, including conventional [polyacrylamide (PAAm), poly(2-hydroxyethyl methacrylate) (PHEMA), and poly(vinyl alcohol) (PVA)] and double-network PAAm/alginate with internal dissipation (18, 19). The bonding agent polymerizes such that instant tough bonding is achieved, for example, between a tough hydrogel containing covalently and dissipatively cross-linked subnetworks and a single-network elastomer, hydrogel, or polymer (Fig. 1B). Glues of the cyanoacrylate family were previously applied to fixate hydrogels (16, 20) but without investigating the mechanical properties and interfacial bonding strength, likely due to the rapid formation of bulky, brittle acrylic resin when the monomers polymerize in direct contact to the water-containing hydrogels. Our dispersions of cyanoacrylate monomers and 2,2,4-trimethylpentane overcome these issues, because they allow diffusion of the adhesive into the hydrogel and into elastomers, leading to tough bonds but without forming rigid resin interlayers. We evidence these effects with spatially resolved Raman spectroscopy (diffusion of adhesive; figs. S1 and S2) and with practical demonstrations of highly stretchable hydrogel-hydrogel and hydrogel-elastomer heterostructures. We suspect that the synergistic effects of physical entanglement due to adhesive interdiffusion and the ready formation of van der Waals and hydrogen bonds of cyanoacrylates (21) with a wide variety of materials result in the high interfacial toughness of our adhesive interfaces. Here, the hydrophobic nature of the alkanes may additionally support diffusion of the adhesive into the hydrogel due to the formation of a protective shell, sufficiently slowing down the water-induced polymerization of cyanoacrylates. Instant healing of fractures in hydrogels, including ionic conductors, is a clear benefit of our approach. We are able to restore the mechanical and electrical properties almost completely within seconds, even for severed structures (Fig. 1C and video S1). The healed conductors endure large stretch ratios λ with electrical resistance almost identical to the pristine one (Fig. 1D). Although requiring the application of cyanoacrylates as polymerization agent, the instant character of our method is of advantage for time-critical applications, whereas autonomously self-healing, highly stretchable ionic conductors (22) typically require longer times for fully restoring their mechanical properties. Instant tough bonding of elastomers and hydrogels without impairing the overall elasticity is challenging, so far only possible after extensive pretreatment of both elastomer and hydrogel surfaces followed by postcuring processes (16). In contrast, our dispersions facilitate rapid adhesion of hydrogels on elastomers following a simple casting or spin-coating process. The formed interface remains highly elastic and enables the sandwich to be stretched reversibly without delamination by more than 2000% in area, whereas physically attached gels slip (Fig. 1, E to G, and figs. S3 and S4). In our approach, stretchability is essentially only limited by the intrinsic rupture strength of the hydrogel. Enhancing functionality and complexity is, for example, feasible though the integration of flexible (23, 24) and imperceptible (25) electronic foils. We work with prestructured, fully polymerized hydrogels that are mechanically deformable before linking, allowing us to exploit wrinkling instabilities when targeting stretchable hydrogel electronics. This approach is illustrated with a structured conductive copper electrode on a thin polymer foil, intimately bonded to a prestretched PVA hydrogel (Fig. 1, H to J).
Fig. 1. Instant tough bonding of hydrogels to a wide range of materials.
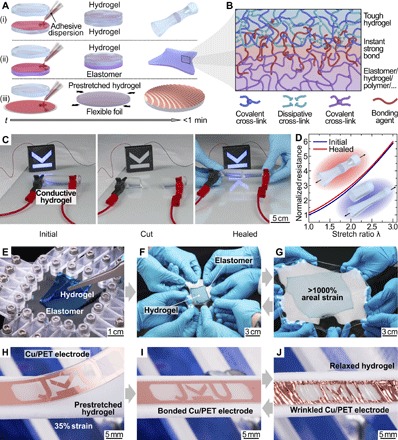
(A) Schematic illustration of the bonding method. An adhesive dispersion is used to instantly tough-bond (i) heterogeneous hydrogels, (ii) hydrogels and elastomers, and (iii) rigid to flexible foils to (prestretched) hydrogels in less than a minute. (B) The bonding agent links tough hydrogels with covalent and dissipative cross-links to elastomers, and the formed interface is instant and tough yet remains stretchable. (C) Instant healing of a conductive hydrogel rod used to light up a light-emitting diode (LED) circuit. Both mechanical and electrical properties are restored after complete incision. (D) Normalized resistance versus uniaxial strain before (blue trace) and after (red trace) healing a hydrogel conductor. (E to G) Hydrogel square (colored blue for visibility) bonded to a transparent elastomer. The soft hybrid is stretched more than 1000% in area without delamination. (H to J) A conducting Cu-coated poly(ethylene terephthalate) (PET) foil is tough-bonded to a prestretched hydrogel. Upon release of the prestrain, out-of-plane wrinkles form in the foil, creating a reversibly stretchable soft-hard hybrid.
Tough hydrogel bonding characterization
We substantiate our initial findings with systematic 90° peeling tests (Fig. 2A; see details in Materials and Methods) (15, 26) on various hydrogels (PHEMA, PAAm/alginate, and PVA), where tough versions dissipate a substantial amount of mechanical energy through the breakup of noncovalent bonds in their bulk. Following initial elastic deformation of hydrogel and, where applicable, elastomer, a stable plateau region in the displacement versus normalized peeling force (identical to interfacial energy; Fig. 2B) evidences bulk rupture of the hydrogel without bonding failure at the gel/target surface interface (see also video S2). We observe this behavior for all investigated interface classes [polymers, elastomers, leather, bone, and chromium-coated metals and glass (27)] and thus find an average fracture toughness of 736 ± 112 J/m2 for PHEMA, 1427 ± 89 J/m2 for PAAm/alginate, and 2208 ± 186 J/m2 for PVA (Fig. 2C). PAAm hydrogels, although having bonding mechanisms similar to their tough counterparts, typically rupture at around 30 J/m2 because of their much lower bulk toughness (fig. S5A). The observed independence of fracture toughness on substrate material for all hydrogels further corroborates that the interfacial strength achieved with our method exceeds, in any case, the intrinsic fracture toughness of the constituent gels. Consequently, peeling tests on hydrogel-hydrogel (PHEMA-PHEMA and PHEMA-PVA) heterostructures result in bulk rupture of the weaker gel (fig. S5B). These results are comparable with the bulk fracture toughness of 489 ± 47 J/m2 for our PHEMA and 1472 ± 440 J/m2 for our PVA hydrogels (see fig. S6) obtained from pure shear notch tests (3, 28). The deviations of bulk and interfacial toughness stem from differences in sample and test setup geometries. We find that the interfacial bonding strength is over a wide range nearly independent on the mixing ratio of the cyanoacrylate with the nonsolvent (1:1 to 1:15 in volume; fig. S5C). However, the optical and elastic properties of the composite depend on the total amount of applied adhesive per unit area, where stretchable, transparent hydrogel-elastomer heterojunctions are achieved with mixtures of 1:10 to 1:15. In a similar manner, the choice of alkane has little influence on the interfacial toughness (fig. S5D); here, the dynamics of the bonding process is influenced by the volatility of the alkane. High–vapor pressure 2,2,4-trimethylpentane will more rapidly evaporate, and longer-chain 1-octadecene or paraffin oil is nonvolatile and delays (on the order of seconds) the polymerization of cyanoacrylates, providing more time for specimen alignment. Despite its simplicity, our approach leads to bonding strengths surpassing previously reported methods. The limits are not yet reached; designing hydrogels with higher internal fracture toughness is expected to further increase resistance to fracture and tear of these soft systems. We additionally visualize the instant character of our bonding method by assembling a model vertebral column with three-dimensional (3D)–printed vertebrae and hydrogel intervertebral discs (fig. S7 and video S3), and the toughness of the interface by lifting a 1-kg metal weight instantly attached to a tough hydrogel rod (fig. S8 and video S4). Adaptive optics, soft photonics, and camouflaging benefit from near-perfect transparency in the visible range (29), an intriguing feature of some hydrogels and certain elastomers (7). Not impairing this advantage by the bonding method is paramount; we demonstrate this for PAAm/VHB double layers that remain perfectly transparent throughout the visible spectrum from 400 to 800 nm. Near-zero absorption within the materials and approximately 93.5% transmission through the stack (Fig. 2D), where scattering at rough surfaces and reflection due to refractive index mismatch on the air/hydrogel and elastomer/air interfaces cause the main losses (for reflectance measurements, see fig. S9), is maintained even when the soft structures are undergoing areal extension by more than 2000% (Fig. 2E). Here, using our adhesive dispersions is crucial, because pure cyanoacrylates will result in the formation of a diffuse scattering layer at the bonding interface (fig. S10).
Fig. 2. Interfacial toughness and optically transparent, stretchable bonds.
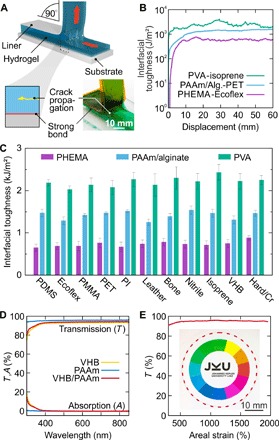
(A) Illustration of the 90° peeling test, with the hydrogel instantly tough-bonded to the bulk substrate. A liner serves as stiff backing. Crack propagation in the hydrogel occurs perpendicular to the peeling direction (detail and photograph with colored hydrogel). (B) Measured interfacial toughness for PVA-isoprene (turquoise trace), PAAm/alginate–PET (light blue trace), and PHEMA-Ecoflex (purple trace) instant tough bonds. (C) Interfacial toughness of PHEMA (purple), PAAm/alginate (light blue), and PVA (turquoise) hydrogels instantly tough-bonded to plastics, elastomers, leather, bone, and chromium-coated metals (aluminum and copper) and glass. Mean values and variance for at least three individual peel tests are shown. PDMS, poly(dimethylsiloxane); PI, polyimide. (D) Transmission and absorption spectra of the acrylic elastomer VHB (yellow trace), PAAm hydrogel (blue trace), and PAAm hydrogel instantly bonded to VHB (red trace). Bonding does not deteriorate the optical properties of the soft hybrid. (E) Nearly strain-independent transmission (red trace) at 600 nm for a biaxially stretched VHB/PAAm stack up to 2000% areal expansion. Inset: Photograph of the sandwich at 1000% strain with a color wheel in the background. The dashed red line outlines the PAAm hydrogel.
Hydrogels for soft actuators, generators, and power sources
Encouraged by these results, we use our methods in demonstrating tough, stretchable adaptive optics, soft machines, and generators. Soft lenses with electrically tunable focal length (29, 30) may have applications ranging from consumer electronics to biomimetic robots (31–33). Our approach (Fig. 3A) uses transparent PAAm hydrogel sheets as ionic conductors, bonded to a convex lens formed from acrylic elastomer filled with NaCl solution. Applying a dc voltage causes the whole lens to thin, thereby changing the focal length by up to 110% at a voltage of 6.5 kV (Fig. 3, B and C). Here, the voltage drop across the hydrogel-dielectric-water capacitors is large, preventing electrochemical side reactions at the high-capacitance interface between hydrogel and metal wiring to the power supply (7). The response time of this centimeter-scale soft, tunable lens is in the 300-ms range, demonstrated by diverting a laser beam guided through a dye-filled water basin (Fig. 3, D and E, and video S5). Here, the mass of the lens dominates the time constant and is readily reducible by downscaling the dimensions to form microlenses. Crucial not only for powering soft electronics and machines, energy generation from mechanical sources on small (that is, walking) and large (that is, ocean waves) scales is currently pursued with soft, dielectric elastomer generators (34) due to their promise of high specific energy of conversion (35). New concepts for operating such devices in contact with seawater are required (29), rendering ionic hydrogels a promising electrode material. Dielectric elastomer actuators and generators are highly deformable capacitors (Fig. 3F), allowing the conversion of electrical to mechanical energy and vice versa (36). We have developed test setups and methodologies for assessing key material parameters (37). Electromechanical coupling is most efficient if the soft capacitor is deformed equibiaxially, that is, if the capacitance C scales with the fourth power of stretch ratio λ: C ∝ λ4. The capacitance of our strongly bonded hydrogel-VHB-hydrogel membranes scales accordingly (Fig. 3H). A simple method of achieving equibiaxial stretch, at least in good approximation at the apex, is realized by inflating a balloon. We exploit this for our soft hydrogel generator (Fig. 3G) and transform approximately 500 mJ of mechanical into 54 mJ electrically usable energy per cycle (Fig. 3, I and J), yielding an overall conversion efficiency of 11%. Improvements are expected with low-viscosity elastomers and the use of stacked multilayer capacitors with shared hydrogel electrodes. Energy storage is an equally central issue in the quest for fully autonomous soft machines to date, which is, for example, beautifully addressed through the catalytic decomposition of onboard fuel (5). Powering embedded electronics including logic and sensors will, however, require current sources. Soft batteries are a natural choice; we have developed the first concepts for such intrinsically stretchable electrochemical cells (38). Briefly, realizing stretchable batteries requires all electrochemical components (anode and cathode materials, electrolyte, and current collectors) in the form of pastes or gels embedded in an elastomeric casing. These early concepts required lateral separation of anode and cathode chambers to enable mechanical compliance, in turn reducing power density and short-circuit currents. Now state-of-the-art (39–42), here we radically redesign this approach using tough, electrolyte-containing hydrogels as stretchable separator (Fig. 4A). Tough bonding to the elastomer matrix enables a “top configuration” of anode, separator, and cathode, greatly reducing internal resistance (Fig. 4B and fig. S11) and increasing specific capacity to 1.6 mA·hour/cm2 (fig. S12) when discharging to 0.9 V for a zinc-manganese dioxide primary battery. In conjunction with imperceptible electrodes as current collectors, not only are our tough batteries highly stretchable, but also their performance improves when stretched as the electrolyte resistance decreases with thinning of the separator gel (Fig. 4C). We use our soft batteries for self-powered stretchable circuits directly integrated atop the compliant power source (Fig. 4D). Imperceptible circuit boards (43) equipped with off-the-shelf surface mounted device (SMD) electronics (fig. S11) including dc-dc step-up converters and LEDs are laminated to the prestretched battery, forming an out-of-plane wrinkle structure when the strain is released (Fig. 4E). These devices are tough soft-hard hybrids that endure repeated twisting and stretching without impairing performance (Fig. 4F and fig. S11). Future work should focus on tough, secondary batteries, a fully biocompatible chemistry (44, 45), and integrated wireless or solar charging (41).
Fig. 3. Soft adaptive lens and energy harvester with instantly tough-bonded hydrogel electrodes.
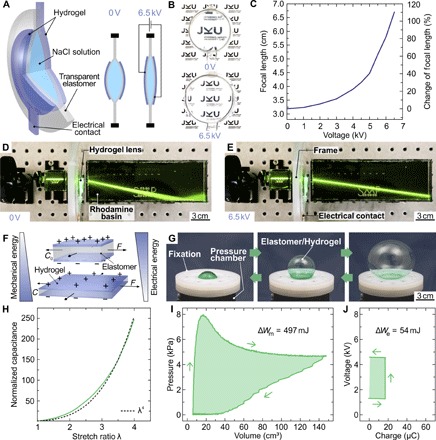
(A) Working principle of the tunable lens. Transparent hydrogel electrodes were used to electrically deform the lens, defined by a liquid reservoir embedded within a VHB elastomer. (B) Photograph of the soft lens without and with applied voltage, demonstrating electrical tuning of focal length. (C) Focal length and change of focal length versus voltage, illustrating a large voltage-induced focal length change of 110% at 6.5 kV. (D and E) Visualization of the voltage-controlled focal length change by showing laser light traces in a basin filled with a diluted rhodamine/water solution. (F) Illustration of mechanical into electrical energy conversion with a deformable elastomer/hydrogel capacitor. (G) Photographs of the deformable balloon-shaped elastomer/hydrogel capacitor. (H) Normalized capacitance of the deformable capacitor versus stretch ratio λ, demonstrating a λ4 behavior. (I and J) Exemplary energy harvesting cycle in pressure-volume and voltage-charge work conjugate plots. The enclosed areas in the two diagrams illustrate the mechanical energy input (497 mJ) and electrical energy output (54 mJ) of the cycle, resulting in a conversion efficiency of more than 10%.
Fig. 4. Tough, stretchable battery and self-powered stretchable circuit.
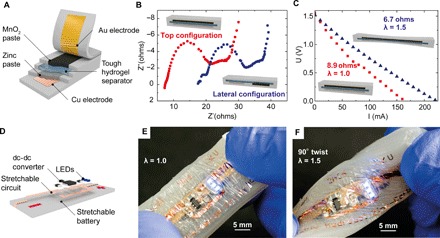
(A) Scheme of a stretchable battery in top configuration, enabled by the tough hydrogel separator instantly tough-bonded to the elastomer matrix. Imperceptible 1.4-μm-thick electrodes serve as current collector. Cu-coated PET foil with Zn paste is the anode, and MnO2 paste contacted with Au-coated PET is the cathode. (B) Nyquist plot for top (red dots) and lateral (blue dots) configuration of a stretchable battery, showing reduced internal resistance due to shorter ionic paths through the separator in top configuration. (C) Voltage versus discharge current at 0% (red squares) and 50% (blue triangles) strain. Internal resistance decreases from 8.9 to 6.7 ohms with stretching due to thinning of the hydrogel separator. (D) Illustration of the self-powered stretchable LED circuit with a 6-μm PET circuit board and SMD elements. A dc-dc converter boosts the voltage of the battery to power three LEDs. (E and F) Stretchable circuit atop a hydrogel-based battery in relaxed (E) and strained and twisted state (F), without impairing function.
Hydrogel electronics
Tough hydrogels currently find applications in soft transducers, exploiting their good ionic conductivity and high stretchability (7–12). However, advanced soft systems, from prosthetic skins (23) to smart medical implants (46–49), benefit from the pluripotency of electronic sensors, actuators, and logic circuits. Pairing such systems with hydrogels is in an early state, with initial attempts toward basic circuit elements including stretchable serpentine-based conductors and SMD LEDs, as well as designs for microparticles and microfluidic channels allowing drug delivery (6, 16, 50). Relying on our instant tough bonding approach, we demonstrate here design strategies and methods for untethered hydrogel-based electronic patches including power supply, control/readout electronics, wireless communication, and stretchable actuators and sensors (Fig. 5A). In a modular architecture, we assemble a reusable power, logic, and radio-containing unit on a flexible circuit board that connects to the stretchable, imperceptible transducer array consisting of four temperature sensors and heater elements. The electronic unit is then bonded to a tough, biocompatible PVA hydrogel matrix, serving as interface to biological tissue (Fig. 5B and fig. S13, with details on the circuit layout). In contrast to thin polymer foils (24, 25) or elastomers (51) commonly used for electronic skins (23, 52, 53), hydrogels more closely mimic mammalian skin, with the possibility of providing nourishment, waste removal, or drug delivery through convection and diffusion throughout the gel matrix or embedded diffusive microchannels (6, 16). In particular, thermally triggered release of drugs stored in lipid-coated reservoirs within the hydrogel body may turn out advantageous for chronic wound treatment, easily controllable via smartphone apps or computers. We therefore initially design an array of four heaters and resistive-type temperature sensors on 1.4-μm PET foil, strongly bonded to a sheet of a 2-mm-thick PVA hydrogel. Finite element simulations of the heat distribution throughout such a patch (Fig. 5C) agree well with IR measurements (Fig. 5D). We then integrate the stretchable transducer array with the flexible power, communication, and control unit. This wireless hydrogel electronic skin is stretchable to about 20% (fig. S14) and intimately conforms to human skin, with the biocompatible PVA gel at the biotic/abiotic interface. We demonstrate untethered, fully autonomous operation including data logging with a smartphone app (Fig. 5E and fig. S15) while, in addition, monitoring the heater function with an IR camera (Fig. 5F and video S6). The temperature response of the four heater elements is precisely monitored by the integrated temperature sensors and agrees excellently with IR camera control measurements (Fig. 5, G and H). Physical attachment is sufficient for wearing our devices cutaneously. However, it is possible to directly bond hydrogel electronics to mammalian tissue with U.S. Food and Drug Administration–approved (54) octyl cyanoacrylate (fig. S16A); we initially assess the biocompatibility of this approach with cell viability studies (fig. S16B). Thermally triggered release of drugs becomes possible through lipid-coated reservoirs atop a heater element (Fig. 5I). Once breaching of the lipid shell is induced by a short temperature increase, the water-soluble drug is enabled to diffuse throughout the hydrogel (Fig. 5J) with an initial diffusion constant of 2.1 × 10−9 m2/s (see fig. S17), visualized here with a green food colorant. Water loss in hydrogel electronics may be an issue for prolonged operation. Several options exist to address this, from adding hygroscopic salts to designing (stretchable) encapsulations (16, 55). The latter is challenging; however, our hydrogel electronic skins are in direct contact with skin on one side and have a PET encapsulation bearing the sensor electronics on the other side. In addition, a modular approach that allows replacement of the hydrogel part after use may alleviate these issues and additionally addresses concerns of hygiene.
Fig. 5. Hydrogel electronic skin.
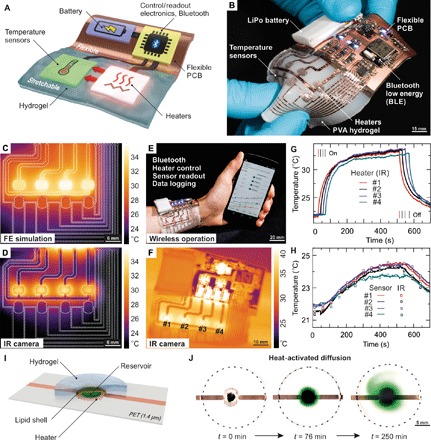
(A) Concept of a hydrogel smart skin, with a flexible unit bearing power supply, control, readout and communication units, and a stretchable transducer batch. PCB, printed circuit board. (B) Photograph of an untethered electronic hydrogel with four stretchable heating elements and adjoined temperature sensors strongly bonded to a PVA hydrogel. Battery, control, readout, and Bluetooth low energy communication electronics are hosted on a flexible circuit board. (C) Finite element (FE) simulation of the transducer batch with four heating elements enabled. (D) Corresponding infrared (IR) thermography image of the freestanding device. Dashed lines in (C) and (D) outline the heater and sensor metal traces. (E) Autonomous hydrogel electronic skin controlled and read out continuously via mobile phone, worn on a human wrist with all heaters activated. (F) Corresponding IR image after ~450 s with all heater elements activated. (G) Measured temperature evolution of the heater elements measured via IR thermography and (H) the sensor elements. Switching of the heating elements is indicated by vertical lines. Recorded temperature traces of the sensor elements (solid lines) are in excellent agreement with data taken from IR images (open squares). (I) Concept of thermally triggered drug delivery where the substance is enclosed in a lipid shell. Temperature increase melts the shell, releasing the drug into the hydrogel. (J) Thermally triggered diffusion of a green food colorant throughout the hydrogel matrix.
CONCLUSION
In summary, we introduce here a facile, universally applicable method for instant tough bonding of hydrogels to a wide variety of materials—from soft to hard—with unprecedented interfacial toughness exceeding the intrinsic fracture strength of the gels. We apply our approach to create a new set of soft machines and electronics and to demonstrate instant healing, adaptive optics, soft actuators and generators, tough batteries, and hydrogel electronic skins. Applications range from robotics, energy harvesting from renewable sources, consumer electronics, and wearables to a new class of medical tools and health monitors.
MATERIALS AND METHODS
Synthesis of hydrogels
All chemicals were used as received without further purification. Where needed, LiCl (VWR Chemicals, 25012) was introduced as highly hydratable salt to enhance conductivity and reduce dehydration. For PAAm, acrylamide (0.1564 g/ml) (AAm; Sigma-Aldrich, A8887) monomer was dissolved in N2-degassed distilled water (2 hours). N,N′-methylenebisacrylamide (AppliChem, A1096) was dissolved in aqueous solution (0.00154 g/ml) and added to the AAm precursor. N,N,N′,N′-tetramethylethylenediamine (Alfa Aesar, A12536) was added to the monomer solution (0.8 μl/ml) as accelerator with subsequent N2 degassing (1 hour). Ammonium persulfate (Sigma-Aldrich, 215589) [0.18 weight % (wt %) of AAm] was dissolved in an aqueous solution (0.0457 g/ml) and added to trigger the thermal polymerization process. Following quick stirring, the mixture was poured in a glass mold and cured for 24 hours, and the ready-made hydrogel sheets were immersed in distilled water for 3 days to remove residual monomers. For PAAm/alginate, sodium alginate (0.01955 g/ml) (Sigma-Aldrich, A2033) was dissolved in PAAm precursor solution (prepared as described) by stirring for 3 hours at room temperature. 2,2-Dimethoxy-2-phenylacetophenone (Sigma-Aldrich, 196118) (0.5 wt % of monomer) was added as a photoinitiator. Calcium sulfate dihydrate (CaSO4·2H2O; Sigma-Aldrich, C3771) as cross-linker for the alginate (6.65 wt % of alginate) was added, and the solution was poured in a quartz glass mold and illuminated with UV-A and UV-C lamps (20 min) in a UV chamber. Ready-made gel sheets were immersed in 0.5 M calcium chloride (CaCl2; Sigma-Aldrich, C8106) solution to enhance the cross-linking of the alginate network and to remove the residual monomers. For PVA, 15 wt % of PVA (Sigma-Aldrich, 563900) aqueous solution was obtained by dissolving PVA in distilled water at 85°C while sonicating for 10 hours. The solution was poured in a glass mold and physically cross-linked by five subsequent freezing-thawing cycles between −12° and 23°C. For PHEMA, 6 μl of ethylene glycol dimethacrylate (Acros Organics, 409922500) as cross-linker and 13 mg of diphenyl(2,4,6-trimethylbenzoyl)phosphine oxide (Sigma-Aldrich, 415952) as photoinitiator were dissolved in 6 ml of 2-hydroxyethyl methacrylate (Sigma-Aldrich, 128635), yielding 10 ml of the precursor solution. Deionized water (4 ml) was added and degassed with N2 for 1 hour. The final solution was injected in a quartz glass mold, UV-photopolymerized for 10 min, and immersed in distilled water for 24 hours.
Bonding to diverse materials
All substrate materials (except hydrogels) were rinsed with isopropanol to remove impurities. Either ethyl cyanoacrylate (Loctite 406, Henkel) or octyl cyanoacrylate (Dermabond, Ethicon) dispersed in 2,2,4-trimethylpentane (Sigma-Aldrich, 360066), 1-octadecene (Sigma-Aldrich, O806), or paraffin oil (Sigma-Aldrich 18512) was used as bonding agents and applied to the substrates via spin-coating at 3000 rpm for 3 s unless otherwise noted. Immediately after, the hydrogel was pressed onto the substrate. The bonding occurs within seconds as the water (hydroxide ions) in the hydrogel initiates and accelerates the polymerization process of the cyanoacrylate adhesives. Mechanical loading of the samples can follow instantly. We observed that dissolving cyanoacrylate monomers in many common solvents (acetone, γ-butyrolactone, acetic acid, chloroform, and others) markedly reduces or fully diminishes the bonding strength of the adhesive.
We carried out the 90° peeling tests of hydrogels bonded to poly(methyl methacrylate) (PMMA; 3 mm, Evonik Industries), PET (1 mm, Evonik Industries), PI (125 μm, Kapton DuPont), nitrile rubber (VWR nitrile examination gloves), polyisoprene rubber (460 μm, Oppo Band 8016), VHB4905 (500 μm, 3M), PDMS (Dow Corning Sylgard 184), Ecoflex (Ecoflex 00-30, Smooth-On), leather and bone (pork shoulder blade), chromium-coated metals (aluminum and copper), and chromium-coated glass and to hydrogels. We note that silicone elastomers (PDMS and Ecoflex) require surface pretreatment with a commercially available primer (Loctite SF770, Henkel) to promote wetting of the adhesive dispersion; all other materials were used without pretreatment. For the 90° peeling tests, a 75-μm-thick PI foil (Kapton DuPont) bonded to the hydrogel served as stiff backing to prevent excessive tensile stretching.
Experiments
Detailed descriptions of all measurements and experiments are included in the Supplementary Materials.
Supplementary Material
Acknowledgments
Funding: This work was partially supported by the European Research Council Advanced Investigators Grant “SoftMap” and the European Union’s Horizon 2020 WETFEET project (grant agreement no. 641334). M.K. acknowledges funding through the Linz Institute of Technology startup grant LIT013144001SEL. Author contributions: M.K. conceived and supervised the research. D.W., R.P., R.G., and F.H. synthesized the hydrogels with contributions from S.H. D.W., R.P., and R.G. characterized the hydrogel bonding. D.W., R.P., M.D., G.K., R.M., E.B., R.K., and S.E.S. designed/performed the experiments and demos. R.G. and S.H. performed and analyzed the Raman measurements. C.M.S. modeled the diffusion process. E.B. carried out cell viability tests. D.W., R.P., M.D., G.K., and R.M. analyzed the data and designed the figures. M.K. and S.B. wrote the manuscript with contributions from all the other authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700053/DC1
Supplementary Materials
Supplementary Methods
fig. S1. Raman spectroscopy of an instantly tough-bonded compound of a 6-μm-thick PET and PHEMA hydrogel using a 1:4 ethyl cyanoacrylate/2,2,4-trimethylpentane dispersion.
fig. S2. Diffusion of bonding agent into PDMS and VHB.
fig. S3. Biaxial stretching of instantly tough-bonded hydrogel.
fig. S4. Uniaxial stretching of physically attached hydrogel versus instantly tough-bonded hydrogels.
fig. S5. Interfacial toughness measurement of hydrogels bonded to various substrates and varying dispersion compositions.
fig. S6. Single-edge notch test of PHEMA and PVA hydrogels.
fig. S7. Model of a human lumbar vertebrae.
fig. S8. Instant tough bonding of PAAm/alginate hydrogel to PMMA and aluminum and instant loading of the bond with more than 1-kg weight.
fig. S9. Reflectance R of PAAm, VHB, and toughly bonded PAAm-VHB sandwich sheets.
fig. S10. Transmission and absorption of PHEMA-PET heterostructures.
fig. S11. Stretchable battery and self-powered stretchable circuit.
fig. S12. Schematic top view and cross section of the stretchable battery in top configuration.
fig. S13. Details for the electronic circuit board of the hydrogel electronic unit.
fig. S14. Illustrating the stretchability of the complex electronic system.
fig. S15. Illustrating the smartphone control of our hydrogel electronic patch.
fig. S16. Interfacial toughness and cell viability studies.
fig. S17. Heat-activated diffusion of a model drug.
video S1. Instant healing of conductive hydrogels.
video S2. Bulk rupture of a tough PAAm/alginate hydrogel undergoing 90° peeling.
video S3. Assembly of a 3D-printed human lumbar vertebrae model.
video S4. Instant tough bonding and instant loading of tough PAAm/alginate hydrogel.
video S5. Visualization of the voltage-controlled focal length change in a soft hydrogel lens.
video S6. Hydrogel electronic skin.
REFERENCES AND NOTES
- 1.Dunlop J. W. C., Weinkamer R., Fratzl P., Artful interfaces within biological materials. Mater. Today 14, 70–78 (2011). [Google Scholar]
- 2.M. A. Meyers, P.-Y. Chen, Biological Materials Science, (Cambridge Univ. Press, 2014). [Google Scholar]
- 3.Sun J.-Y., Zhao X., Illeperuma W. R. K., Chaudhuri O., Oh K. H., Mooney D. J., Vlassak J. J., Suo Z., Highly stretchable and tough hydrogels. Nature 489, 133–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peak C. W., Wilker J. J., Schmidt G., A review on tough and sticky hydrogels. Colloid Polym. Sci. 291, 2031–2047 (2013). [Google Scholar]
- 5.Wehner M., Truby R. L., Fitzgerald D. J., Mosadegh B., Whitesides G. M., Lewis J. A., Wood R. J., An integrated design and fabrication strategy for entirely soft, autonomous robots. Nature 536, 451–455 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Lin S., Yuk H., Zhang T., Parada G. A., Koo H., Yu C., Zhao X., Stretchable hydrogel electronics and devices. Adv. Mater. 28, 4497–4505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keplinger C., Sun J.-Y., Foo C. C., Rothemund P., Whitesides G. M., Suo Z., Stretchable, transparent, ionic conductors. Science 341, 984–987 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Yang C. H., Chen B., Lu J. J., Yang J. H., Zhou J., Chen Y. M., Suo Z., Ionic cable. Extreme Mech. Lett. 3, 59–65 (2015). [Google Scholar]
- 9.Sun J.-Y., Keplinger C., Whitesides G. M., Suo Z., Ionic skin. Adv. Mater. 26, 7608–7614 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Kim C.-C., Lee H.-H., Oh K. H., Sun J.-Y., Highly stretchable, transparent ionic touch panel. Science 353, 682–687 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Yang C. H., Chen B., Zhou J., Chen Y. M., Suo Z., Electroluminescence of giant stretchability. Adv. Mater. 28, 4480–4484 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Larson C., Peele B., Li S., Robinson S., Totaro M., Beccai L., Mazzolai B., Shepherd R., Highly stretchable electroluminescent skin for optical signaling and tactile sensing. Science 351, 1071–1074 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Tang J., Li J., Vlassak J. J., Suo Z., Adhesion between highly stretchable materials. Soft Matter 12, 1093–1099 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Rose S., Prevoteau A., Elzière P., Hourdet D., Marcellan A., Leibler L., Nanoparticle solutions as adhesives for gels and biological tissues. Nature 505, 382–385 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Yuk H., Zhang T., Lin S., Parada G. A., Zhao X., Tough bonding of hydrogels to diverse non-porous surfaces. Nat. Mater. 15, 190–196 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yuk H., Zhang T., Parada G. A., Liu X., Zhao X., Skin-inspired hydrogel–elastomer hybrids with robust interfaces and functional microstructures. Nat. Commun. 7, 12028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.I. Gibson, D. Rosen, B. Stucker, Additive Manufacturing Technologies (Springer, 2015). [Google Scholar]
- 18.Zhao X., Multi-scale multi-mechanism design of tough hydrogels: Building dissipation into stretchy networks. Soft Matter 10, 672–687 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gong J. P., Why are double network hydrogels so tough? Soft Matter 6, 2583–2590 (2010). [Google Scholar]
- 20.Wu H., Sariola V., Zhu C., Zhao J., Sitti M., Bettinger C. J., Transfer printing of metallic microstructures on adhesion-promoting hydrogel substrates. Adv. Mater. 27, 3398–3404 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Kusaka I., Suëtaka W., Infrared spectrum of α-cyanoacrylate adhesive in the first monolayer on a bulk aluminum surface. Spectrochim. Acta A 36, 647–648 (1980). [Google Scholar]
- 22.Cao Y., Morrissey T. G., Acome E., Allec S. I., Wong B. M., Keplinger C., Wang C., A transparent, self-healing, highly stretchable ionic conductor. Adv. Mater. 29, 1605099 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Chortos A., Liu J., Bao Z., Pursuing prosthetic electronic skin. Nat. Mater. 15, 937–950 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Kim D.-H., Lu N., Ma R., Kim Y.-S., Kim R.-H., Wang S., Wu J., Won S. M., Tao H., Islam A., Yu K. J., Kim T.-i., Chowdhury R., Ying M., Xu L., Li M., Chung H.-J., Keum H., McCormick M., Liu P., Zhang Y.-W., Omenetto F. G., Huang Y., Coleman T., Rogers J. A., Epidermal electronics. Science 333, 838–843 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Kaltenbrunner M., Sekitani T., Reeder J., Yokota T., Kuribara K., Tokuhara T., Drack M., Schwödiauer R., Graz I., Bauer-Gogonea S., Bauer S., Someya T., An ultra-lightweight design for imperceptible plastic electronics. Nature 499, 458–463 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Bundy K., Schlegel U., Rahn B., Geret V., Perren S., An improved peel test method for measurement of adhesion to biomaterials. J. Mater. Sci. Mater. Med. 11, 517–521 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Gobble K., Stark A., Stagon S. P., Improved bond strength of cyanoacrylate adhesives through nanostructured chromium adhesion layers. Nanoscale Res. Lett. 11, 416 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rivlin R. S., Thomas A. G., Rupture of rubber. I. Characteristic energy for tearing. J. Polym. Sci. 10, 291–318 (1953). [Google Scholar]
- 29.Rosset S., Shea H. R., Small, fast, and tough: Shrinking down integrated elastomer transducers. Appl. Phys. Rev. 3, 031105 (2016). [Google Scholar]
- 30.Shian S., Diebold R. M., Clarke D. R., Tunable lenses using transparent dielectric elastomer actuators. Opt. Express 21, 8669–8676 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Sydney Gladman A., Matsumoto E. A., Nuzzo R. G., Mahadevan L., Lewis J. A., Biomimetic 4D printing. Nat. Mater. 15, 413–418 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Shepherd R. F., Ilievski F., Choi W., Morin S. A., Stokes A. A., Mazzeo A. D., Chen X., Wang M., Whitesides G. M., Multigait soft robot. Proc. Natl. Acad. Sci. U.S.A. 108, 20400–20403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trivedi D., Rahn C. D., Kier W. M., Walker I. D., Soft robotics: Biological inspiration, state of the art, and future research. Appl. Bionics Biomech. 5, 99–117 (2008). [Google Scholar]
- 34.Anderson I. A., Gisby T. A., McKay T. G., O’Brien B. M., Calius E. P., Multi-functional dielectric elastomer artificial muscles for soft and smart machines. J. Appl. Phys. 112, 41101 (2012). [Google Scholar]
- 35.Koh S. J. A., Keplinger C., Li T., Bauer S., Suo Z., Dielectric elastomer generators: How much energy can be converted? IEEE/ASME Trans. Mechatronics 16, 33–41 (2011). [Google Scholar]
- 36.R. D. Kornbluh, R. Pelrine, H. Prahlad, A. Wong-Foy, B. McCoy, S. Kim, J. Eckerle, T. Low, From boots to buoys: Promises and challenges of dielectric elastomer energy harvesting. Proc. SPIE 7976, Electroactive Polymer Actuators and Devices (EAPAD) (2011), pp. 48–66. [Google Scholar]
- 37.Kaltseis R., Keplinger C., Baumgartner R., Kaltenbrunner M., Li T., Mächler P., Schwödiauer R., Suo Z., Bauer S., Method for measuring energy generation and efficiency of dielectric elastomer generators. Appl. Phys. Lett. 99, 162904 (2011). [Google Scholar]
- 38.Kaltenbrunner M., Kettlgruber G., Siket C., Schwödiauer R., Bauer S., Arrays of ultracompliant electrochemical dry gel cells for stretchable electronics. Adv. Mater. 22, 2065–2067 (2010). [DOI] [PubMed] [Google Scholar]
- 39.Gaikwad A. M., Arias A. C., Steingart D. A., Recent progress on printed flexible batteries: Mechanical challenges, printing technologies, and future prospects. Energy Technol. 3, 305–328 (2015). [Google Scholar]
- 40.Gaikwad A. M., Zamarayeva A. M., Rousseau J., Chu H., Derin I., Steingart D. A., Highly stretchable alkaline batteries based on an embedded conductive fabric. Adv. Mater. 24, 5071–5076 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Xu S., Zhang Y., Cho J., Lee J., Huang X., Jia L., Fan J. A., Su Y., Su J., Zhang H., Cheng H., Lu B., Yu C., Chuang C., Kim T.-i., Song T., Shigeta K., Kang S., Dagdeviren C., Petrov I., Braun P. V., Huang Y., Paik U., Rogers J. A., Stretchable batteries with self-similar serpentine interconnects and integrated wireless recharging systems. Nat. Commun. 4, 1543 (2013). [DOI] [PubMed] [Google Scholar]
- 42.Wang L., Zhang Y., Pan J., Peng H., Stretchable lithium-air batteries for wearable electronics. J. Mater. Chem. A 4, 13419–13424 (2016). [Google Scholar]
- 43.Moser R., Kettlgruber G., Siket C. M., Drack M., Graz I. M., Cakmak U., Major Z., Kaltenbrunner M., Bauer S., From playroom to lab: Tough stretchable electronics analyzed with a tabletop tensile tester made from toy-bricks. Adv. Sci. 3, 1500396 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim Y. J., Chun S.-E., Whitacre J., Bettinger C. J., Self-deployable current sources fabricated from edible materials. J. Mater. Chem. B 1, 3781 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Yin L., Huang X., Xu H., Zhang Y., Lam J., Cheng J., Rogers J. A., Materials, designs, and operational characteristics for fully biodegradable primary batteries. Adv. Mater. 26, 3879–3884 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Hwang S.-W., Tao H., Kim D.-H., Cheng H., Song J.-K., Rill E., Brenckle M. A., Panilaitis B., Won S. M., Kim Y.-S., Song Y. M., Yu K. J., Ameen A., Li R., Su Y., Yang M., Kaplan D. L., Zakin M. R., Slepian M. J., Huang Y., Omenetto F. G., Rogers J. A., A physically transient form of silicon electronics. Science 337, 1640–1644 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang S., Murphy R. K. J., Hwang S.-W., Lee S. M., Harburg D. V., Krueger N. A., Shin J., Gamble P., Cheng H., Yu S., Liu Z., McCall J. G., Stephen M., Ying H., Kim J., Park G., Webb R. C., Lee C. H., Chung S., Wie D. S., Gujar A. D., Vemulapalli B., Kim A. H., Lee K.-M., Cheng J., Huang Y., Lee S. H., Braun P. V., Ray W. Z., Rogers J. A., Bioresorbable silicon electronic sensors for the brain. Nature 530, 71–76 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Jeong J.-W., McCall J. G., Shin G., Zhang Y., Al-Hasani R., Kim M., Li S., Sim J. Y., Jang K.-I., Shi Y., Hong D. Y., Liu Y., Schmitz G. P., Xia L., He Z., Gamble P., Ray W. Z., Huang Y., Bruchas M. R., Rogers J. A., Wireless optofluidic systems for programmable in vivo pharmacology and optogenetics. Cell 162, 662–674 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rivnay J., Owens R. M., Malliaras G. G., The rise of organic bioelectronics. Chem. Mater. 26, 679–685 (2014). [Google Scholar]
- 50.P. Mostafalu, S. Amugothu, A. Tamayol, S. Bagherifard, M. Akbari, M. R. Dokmeci, A. Khademhosseini, S. Sonkusale, Smart flexible wound dressing with wireless drug delivery, in 2015 IEEE Biomedical Circuits and Systems Conference (BioCAS) (IEEE, 2015), pp. 1–4; http://ieeexplore.ieee.org/document/7348391/.
- 51.Mannsfeld S. C. B., Tee B. C-K., Stoltenberg R. M., Chen C. V. H.-H., Barman S., Muir B. V. O., Sokolov A. N., Reese C., Bao Z., Highly sensitive flexible pressure sensors with microstructured rubber dielectric layers. Nat. Mater. 9, 859–864 (2010). [DOI] [PubMed] [Google Scholar]
- 52.Son D., Lee J., Qiao S., Ghaffari R., Kim J., Lee J. E., Song C., Kim S. J., Lee D. J., Jun S. W., Yang S., Park M., Shin J., Do K., Lee M., Kang K., Hwang C. S., Lu N., Hyeon T., Kim D.-H., Multifunctional wearable devices for diagnosis and therapy of movement disorders. Nat. Nanotechnol. 9, 397–404 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Park S., Kim H., Vosgueritchian M., Cheon S., Kim H., Koo J. H., Kim T. R., Lee S., Schwartz G., Chang H., Bao Z., Stretchable energy-harvesting tactile electronic skin capable of differentiating multiple mechanical stimuli modes. Adv. Mater. 26, 7324–7332 (2014). [DOI] [PubMed] [Google Scholar]
- 54.U.S. Food and Drug Administration (FDA), Premarket Approval Dermabond; www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfpma/pma.cfm?id=p960052.
- 55.Bai Y., Chen B., Xiang F., Zhou J., Wang H., Suo Z., Transparent hydrogel with enhanced water retention capacity by introducing highly hydratable salt. Appl. Phys. Lett. 105, 151903 (2014). [Google Scholar]
- 56.Schausberger S. E., Kalteseis R., Drack M., Cakmak U. D., Major Z., Bauer S., Cost-efficient open source desktop size radial stretching system with force sensor. IEEE Access 3, 556–561 (2015). [Google Scholar]
- 57.J. Crank, The Mathematics of Diffusion (Oxford Univ. Press, 1975). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700053/DC1
Supplementary Materials
Supplementary Methods
fig. S1. Raman spectroscopy of an instantly tough-bonded compound of a 6-μm-thick PET and PHEMA hydrogel using a 1:4 ethyl cyanoacrylate/2,2,4-trimethylpentane dispersion.
fig. S2. Diffusion of bonding agent into PDMS and VHB.
fig. S3. Biaxial stretching of instantly tough-bonded hydrogel.
fig. S4. Uniaxial stretching of physically attached hydrogel versus instantly tough-bonded hydrogels.
fig. S5. Interfacial toughness measurement of hydrogels bonded to various substrates and varying dispersion compositions.
fig. S6. Single-edge notch test of PHEMA and PVA hydrogels.
fig. S7. Model of a human lumbar vertebrae.
fig. S8. Instant tough bonding of PAAm/alginate hydrogel to PMMA and aluminum and instant loading of the bond with more than 1-kg weight.
fig. S9. Reflectance R of PAAm, VHB, and toughly bonded PAAm-VHB sandwich sheets.
fig. S10. Transmission and absorption of PHEMA-PET heterostructures.
fig. S11. Stretchable battery and self-powered stretchable circuit.
fig. S12. Schematic top view and cross section of the stretchable battery in top configuration.
fig. S13. Details for the electronic circuit board of the hydrogel electronic unit.
fig. S14. Illustrating the stretchability of the complex electronic system.
fig. S15. Illustrating the smartphone control of our hydrogel electronic patch.
fig. S16. Interfacial toughness and cell viability studies.
fig. S17. Heat-activated diffusion of a model drug.
video S1. Instant healing of conductive hydrogels.
video S2. Bulk rupture of a tough PAAm/alginate hydrogel undergoing 90° peeling.
video S3. Assembly of a 3D-printed human lumbar vertebrae model.
video S4. Instant tough bonding and instant loading of tough PAAm/alginate hydrogel.
video S5. Visualization of the voltage-controlled focal length change in a soft hydrogel lens.
video S6. Hydrogel electronic skin.