

WNT antagonism displays marked synergy with taxane chemotherapy and reverses taxane-induced enrichment of cancer stem cells.
Keywords: WNT antagonism, taxane resistance, Cancer stem cells, mitotic catastrophe, Ovarian cancer, pancreatic cancer, Breast Cancer, drug resistance pathways, drug combination strategies, chemotherapy resistance
Abstract
The WNT pathway mediates intercellular signaling that regulates cell fate in both normal development and cancer. It is widely appreciated that the WNT pathway is frequently dysregulated in human cancers through a variety of genetic and epigenetic mechanisms. Targets in the WNT pathway are being extensively pursued for the development of new anticancer therapies, and we have advanced two WNT antagonists for clinical development: vantictumab (anti-FZD) and ipafricept (FZD8-Fc). We examined the antitumor efficacy of these WNT antagonists in combination with various chemotherapies in a large set of patient-derived xenograft models. In responsive models, WNT blockade led to profound synergy with taxanes such as paclitaxel, and the combination activity with taxanes was consistently more effective than with other classes of chemotherapy. Taxane monotherapy increased the frequency of cells with active WNT signaling. This selection of WNT-active chemotherapy-resistant tumorigenic cells was prevented by WNT-antagonizing biologics and required sequential dosing of the WNT antagonist followed by the taxane. The WNT antagonists potentiated paclitaxel-mediated mitotic blockade and promoted widespread mitotic cell death. By blocking WNT/β-catenin signaling before mitotic blockade by paclitaxel, we found that this treatment effectively sensitizes cancer stem cells to taxanes. This combination strategy and treatment regimen has been incorporated into ongoing clinical testing for vantictumab and ipafricept.
INTRODUCTION
The WNT/β-catenin signaling pathway plays an important role in controlling cell fate, self-renewal, and maintenance of normal and cancer stem cells (CSCs) (1, 2). The 19 WNTs are secreted proteins, which are ligands for the Frizzled (FZD) receptors and co-receptors (3). WNT signaling activates diverse responses including cellular proliferation, migration, polarity, and maintenance of stemness. Activation of canonical WNT signaling results in β-catenin stabilization and translocation to the nucleus, where it promotes expression of WNT target genes, such as LEF1 and AXIN2. This activation of the WNT/β-catenin signaling pathway is found in many human cancers, and the therapeutic utility of WNT/β-catenin inhibitors is under investigation in ongoing clinical trials (4). Several agents are being developed to antagonize this pathway including small molecules, such as porcupine inhibitors, and biologics that directly target WNT ligands or receptors.
WNT/β-catenin is involved in multiple aspects of cell cycle progression. WNT signaling plays a major role in cellular proliferation by transcriptional and translational up-regulation of G1 effectors such as cyclin D1 (5). In addition, β-catenin plays a key role in the M phase of the cell cycle, and of particular importance, β-catenin levels have been identified to peak at the G2-M phase in model organisms (6). Several other WNT signaling components are involved in regulating different aspects of mitotic programs, including microtubule dynamics, spindle formation, and centrosome division. β-Catenin, adenomatosis polyposis coli (APC), and axis inhibition protein 2 (AXIN2) have been implicated as direct regulators of mitosis (7). Inhibition of WNT signaling results in mitotic spindle defects (8), and β-catenin was shown to be essential for centrosome separation at the onset of mitotic spindle formation (9). In addition, APC and Dishevelled have been observed to regulate the attachment of the mitotic spindle to the kinetochores and, together with FZD receptors and LRP6 (low-density lipoprotein receptor–related protein 6), modulate spindle orientation (10). Thus, there is abundant evidence that WNT/β-catenin signaling serves multiple functions in governing cell cycle control during the process of mitosis, both at the transcriptional level and in chromosomal dynamics.
We have developed two clinical-stage WNT pathway antagonists: vantictumab (OMP-18R5), which blocks binding of WNT ligands to FZD receptors 1, 2, 5, 7, and 8 and inhibits downstream signaling (11), and ipafricept (OMP-54F28), a fusion protein containing the extracellular ligand-binding domain of FZD8 and a human immunoglobulin G1 (IgG1) Fc domain. Ipafricept blocks WNT signaling by sequestering secreted WNTs. These agents demonstrated antitumor activity in several patient-derived xenograft (PDX) models and were shown to reduce the frequency of tumorigenic cells (11, 12). Both vantictumab and ipafricept have an effect on bone metabolism that has been observed clinically, and a comprehensive strategy has been implemented to mitigate this effect, including patient selection, monitoring, zoledronic acid administration, and modification of the dose and schedule (12, 13).
Chemotherapeutic agents are widely used for cancer treatment and can provide benefit but are rarely curative (14). A few of the major limitations that restrict the usefulness of many cancer chemotherapeutic agents are treatment-related side effects and development of resistance, leading to subsequent recurrence of cancers. Activation of the WNT pathway has been found to be associated with resistance to chemotherapeutic agents (15). We and others have found that chemotherapy is generally ineffective in reducing tumorigenic cell frequency, whereas we have shown that WNT blockade promotes increased sensitivity to chemotherapeutic agents and effectively reduces the tumorigenicity of cancer cells (11).
Here, we examined the antitumor efficacy of WNT antagonists combined with standard-of-care (SOC) chemotherapeutic agents in a diverse panel of PDXs. PDX models have become a useful tool for preclinical drug discovery because these models are considered to retain much of the cellular heterogeneity of the patient’s primary tumors (16) and reflect the diversity of clinical subtypes and patients’ drug responses (17, 18). We discovered that the combination of a WNT antagonist with a taxane effectively inhibits PDX tumor growth and is more effective than that with other classes of chemotherapeutic agents. Taxane chemotherapy as a monotherapy enriched for tumor cells with high WNT/β-catenin pathway activity and tumorigenicity. Sequential dosing of WNT antagonists, followed by taxane treatment, produced superior antitumor efficacy that was coupled with mitotic cell death in tumors. By blocking WNT/β-catenin signaling before mitotic blockade by paclitaxel, we found that this treatment schedule effectively sensitizes cancer cells to taxanes.
RESULTS
WNT antagonists synergize with taxane chemotherapies
We used PDXs as a preclinical drug discovery platform to discover tumor types that are responsive to WNT antagonists using the anti-FZD antibody (OMP-18R5, vantictumab) (11). We also developed a decoy receptor (OMP-54F28, ipafricept) that functions as a WNT antagonist composed of the extracellular domain of FZD8 and the IgG1 Fc domain [(12); see Materials and Methods].
In pancreatic cancer PDX models, our initial drug combinatorial experiments focused on the preexisting SOC agent gemcitabine. We subsequently examined efficacy in combination with the more recently established SOC, nab-paclitaxel in combination with gemcitabine, because this was shown to significantly improve response rate and survival in pancreatic adenocarcinoma patients (19). Studies in pancreatic PDX models indicated that the combination of vantictumab with nab-paclitaxel was more effective than that of vantictumab with gemcitabine (Fig. 1A). Similarly, we found that the addition of nab-paclitaxel to gemcitabine could markedly increase the antitumor activity of ipafricept in pancreatic PDX (Fig. 1B). Thus, the pharmacological activities of these WNT antagonists are similar in regard to synergy with taxanes, and we used both these agents for further studies in pancreatic, breast, and ovarian cancer PDX models.
Fig. 1. WNT antagonists synergize with taxanes.
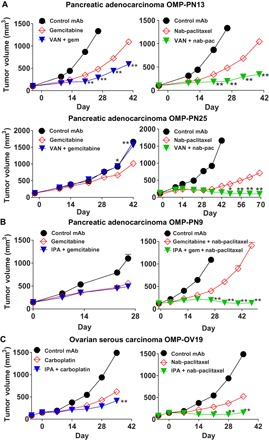
(A) Vantictumab (VAN) has greater tumor growth inhibition when combined with nab-paclitaxel than gemcitabine. OMP-PN13 was treated with vantictumab (25 mg/kg) every 2 weeks and with gemcitabine (gem; 25 mg/kg) or nab-paclitaxel (nab-pac; 30 mg/kg) every week (n = 7 to 9 per group). OMP-PN25 was treated with vantictumab (25 mg/kg) every 2 weeks and with gemcitabine (20 mg/kg) or nab-paclitaxel (15 mg/kg) every week (n = 5 to 10 per group). mAb, monoclonal antibody. (B) Ipafricept (IPA) promotes tumor growth inhibition when combined with weekly gemcitabine and nab-paclitaxel. Left: OMP-PN9 was treated with ipafricept (10 mg/kg) and gemcitabine (50 mg/kg) every week (n = 9 to 10 per group). Right: OMP-PN9 treated with ipafricept (25 mg/kg) every 2 weeks and gemcitabine (5 mg/kg) combined with nab-paclitaxel (10 mg/kg) every week (n = 7 to 8 per group). (C) Ipafricept results in greater tumor growth inhibition in combination with nab-paclitaxel than carboplatin in ovarian cancer. OMP-OV19 was treated with ipafricept (45 mg/kg) every 2 weeks and carboplatin (30 mg/kg) or nab-paclitaxel (7.5 mg/kg) every week (n = 8 per group). *P < 0.01; **P < 0.001 combination versus chemotherapy. Data are means + SEM.
Ovarian cancer is typically treated with either platinum or taxane chemotherapy (20), and we compared the activity of the WNT antagonists with platinum and taxane chemotherapies in this disease setting. In one ovarian cancer model, OMP-OV19, the antitumor activity of WNT blockade was greater when ipafricept was combined with nab-paclitaxel than with carboplatin (Fig. 1C), and similar results were obtained with taxanes (either paclitaxel or nab-paclitaxel) in other pancreatic and ovarian tumors.
The chemotherapeutic agents tested all affect the cell cycle but do so through different mechanisms. Whereas taxanes act on microtubules and block cell division at the M phase, gemcitabine and carboplatin inhibit DNA synthesis and arrest cells at the S phase (21–23). WNT/β-catenin signaling is tightly associated with the cell cycle; it peaks at the G2-M phase and is involved in centrosome function and chromosomal segregation during mitosis (6–9). We thus hypothesized that the combination activity of WNT antagonists with chemotherapeutic agents relied, at least in part, on the ability of WNT antagonism to affect the cell cycle and that nontranscriptional mechanisms of action may also contribute to the enhanced antitumor activity of WNT antagonists with taxanes.
WNT antagonists reduce β-catenin accumulation within mitotic cells
To examine the effects of different chemotherapeutic agents on WNT pathway activation in the PDX tumors, we performed immunohistochemistry (IHC) analyses for β-catenin. The mitotic marker histone H3 phosphorylated at Ser10 (pHH3) was used as a specific marker for the G2-M stage. In the pancreatic tumor OMP-PN13, nab-paclitaxel treatment resulted in both enrichment of cells at the G2-M stage and elevated levels of β-catenin (Fig. 2A). In contrast, gemcitabine reduced the frequency of mitotic cells (Fig. 2A). Immunofluorescence confocal microscopy demonstrated that mitotic tumor cells were often enriched for intracellular β-catenin and that this enrichment was enhanced by nab-paclitaxel (Fig. 2B). The mitotic cells present in the gemcitabine treatment group were not associated with enrichment for β-catenin. Using IHC for dual detection of both β-catenin and pHH3, we quantified that nab-paclitaxel induced a threefold enrichment for β-catenin accumulation within mitotic cells (Fig. 2C). However, treatment with gemcitabine or vantictumab combined with gemcitabine prevented the accumulation of mitotic cells with β-catenin. This response was also observed in OMP-PN25 (fig. S1), where the combination of vantictumab with nab-paclitaxel was more efficacious than with gemcitabine. Thus, paclitaxel enriched for mitotic cells with β-catenin expression, and a reduction of total β-catenin levels was dependent on the combination of vantictumab with gemcitabine and nab-paclitaxel (Fig. 2C).
Fig. 2. Nab-paclitaxel arrests cells at mitosis and promotes mitotic cell accumulation of β-catenin.
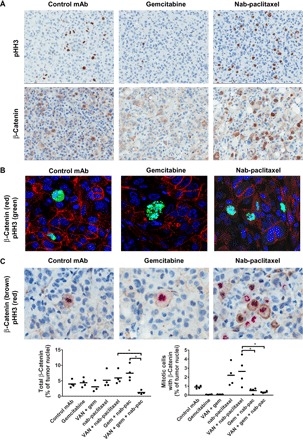
Pancreatic adenocarcinoma OMP-PN13 was treated with gemcitabine and/or nab-paclitaxel every week with and without vantictumab (25 mg/kg) every 2 weeks for 6 weeks. (A) Tumors on day 41 were preserved as formalin-fixed, paraffin-embedded (FFPE), IHC was performed for pHH3 or β-catenin, and bound primary antibody was detected with horseradish peroxidase (HRP)–labeled secondary antibody with hematoxylin counterstain. Positive regions are labeled in brown, and nuclei are labeled in blue. Slides were imaged on an Aperio AT scanner with ×20 magnification. Representative images for control, gemcitabine, and nab-paclitaxel single agents are shown. (B) Nab-paclitaxel enriches for nuclear β-catenin. FFPE sections were costained for pHH3 and β-catenin and then with fluorescently labeled secondary antibodies, and nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Images were taken on an Olympus confocal microscope (magnification, ×100). (C) FFPE sections costained for pHH3 and β-catenin. Quantifications of cells expressing total β-catenin and pHH3-positive mitotic cells with β-catenin expression were performed. Total β-catenin was detected with HRP–3,3′-diaminobenzidine (DAB), and pHH3 was detected with alkaline phosphatase (AP)–Warp Red, with hematoxylin counterstain. Digital scans were performed on an Aperio AT scanner with ×20 magnification and were analyzed with Definiens Tissue Studio. *P < 0.05 combination versus chemotherapy. Data are means with four replicates.
WNT antagonists are active when dosed before taxanes
We carried out dose-response and dose partition experiments to optimize the dose and schedule for the WNT antagonists. Vantictumab produced durable tumor growth inhibition at a minimum dose of 25 mg/kg administered every 3 weeks (fig. S2A). Comparing weekly versus twice per week dosing in a prophylactic tumor model, we also found that ipafricept could be dosed less frequently (fig. S2B). Subsequently, we varied the schedule of dosing for vantictumab and determined, using the same total amount of antibody, that higher doses given less frequently were more efficacious than lower doses given more frequently (fig. S2C). Ipafricept produced significant tumor growth inhibition in combination with chemotherapy at 20 mg/kg every 2 weeks while demonstrating no activity at 10 mg/kg weekly (fig. S2D). Furthermore, we found that dosing the WNT antagonists every 2 or 3 weeks led to less bone turnover compared with a lower dose given weekly and, therefore, a more favorable therapeutic index. These findings led us to generally use a dosing regimen of 25 mg/kg, every 2 or 3 weeks, in subsequent studies.
The previous studies were all carried out with weekly administration of chemotherapy. In some clinical regimens, chemotherapeutic agents are given less frequently. For example, paclitaxel may be administered every 3 weeks to treat platinum-sensitive ovarian cancer or metastatic breast cancer (24, 25). We tested the combination of WNT blockade with intermittent chemotherapeutic regimens to determine the effectiveness of combination treatment in this type of schedule. For these experiments, preestablished xenograft tumors were treated with WNT antagonists every 2 or 3 weeks, and paclitaxel was administered concomitantly, after, or before the biologics. We found that dosing vantictumab 3 days before paclitaxel produced marked efficacy in a breast cancer PDX model (Fig. 3A). Dosing vantictumab on the same day as paclitaxel or paclitaxel before vantictumab was ineffective in blocking tumor progression (Fig. 3A). In ovarian cancer, pretreatment with ipafricept before paclitaxel produced durable tumor growth inhibition that was not evident using simultaneous dosing of both agents or by sequential application of paclitaxel followed by ipafricept (Fig. 3B). This enhancement of efficacy by pretreatment with ipafricept was also observed in an additional ovarian model, OMP-OV19, because sequential application of ipafricept followed by paclitaxel induced tumor regression, and this effect was sustained up to 58 days after treatment was discontinued (Fig. 3C). At the conclusion of the study, three of eight mice had complete tumor regression, and the remaining mice showed evidence of stable disease (fig. S3A). A consistent pattern emerged from our experiments: Sequential dosing of a WNT antagonist before taxane treatment led to enhanced antitumor response relative to simultaneous dosing or dosing in the reverse order. The increased activity of predosing a WNT antagonist before chemotherapy was specific for taxane-containing regimens because an enhanced antitumor response was not observed when a WNT antagonist was given before an S-phase blocker (fig. S3B).
Fig. 3. Ipafricept induces mitotic catastrophe when dosed sequentially before paclitaxel.
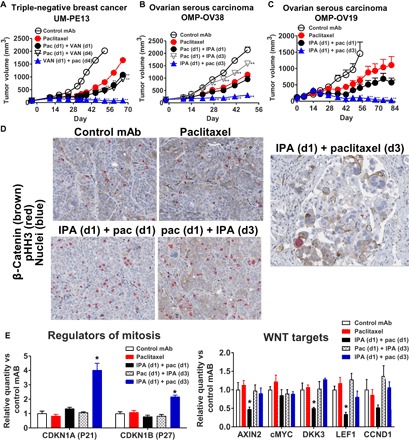
Administration of vantictumab or ipafricept 2 days before paclitaxel is essential for blocking tumor growth. (A) UM-PE13 treated with vantictumab (25 mg/kg) on day 1 or 4 with paclitaxel (pac; 20 mg/kg) given on day 1 or 4 in 3-week cycles (n = 6 to 8 per group). (B) OMP-OV38 treated with ipafricept (25 mg/kg) on day 1 or 3 with paclitaxel (20 mg/kg) on day 1 or 3 in 2-week cycles (n = 9 per group). (C) OMP-OV19 treated with ipafricept (25 mg/kg) on day 1 and paclitaxel (20 mg/kg) on day 1 or 3 in 2-week cycles (n = 8 to 10 per group). *P < 0.01; **P < 0.001 combination versus chemotherapy. Data are means + SEM. (D) Tumors from OMP-OV38 as shown in (B) from study day 51 were processed as FFPE with IHC for pHH3 and β-catenin. β-Catenin was detected with HRP-DAB, and pHH3 was detected with AP–Warp Red, with hematoxylin counterstain (magnification, ×20). (E) Tumors from OMP-OV38 as shown in (B) from study day 51 were processed to RNA, and quantitative real-time polymerase chain reaction (PCR) was performed. Gene expression was normalized to control group using the relative quantification method. Data are means + SEM, four biological replicates per group; *P < 0.05, significant compared to paclitaxel.
WNT antagonists potentiate mitotic cell death
Analysis of tumor samples obtained following multiple rounds of therapy revealed distinct modulations of cell cycle and WNT pathway biomarkers. OMP-OV38 tumors treated by paclitaxel alone, paclitaxel with simultaneous ipafricept, or first with paclitaxel followed by ipafricept resulted in an increase in mitotic cells (Fig. 3D). Colocalization of pHH3 and β-catenin was detected in dividing cells, and mitotic spindles were visible in these cells. Occasionally, giant cells and multinucleated cells were present, but no gross morphological differences were obvious between paclitaxel treatment and control groups (Fig. 3D). Simultaneous treatment reduced β-catenin expression, but there was no evidence of widespread cell death. The sequential administration of ipafricept followed by paclitaxel reduced tumor cell density, and the remaining tumor cells exhibited enlarged cytoplasmic content with enrichment of giant multinucleated cells characteristic of mitotic catastrophe (Fig. 3D). These histological changes were tightly associated with tumor volume reduction; mitotic cell death was not observed without sequential dosing and with the combination of WNT antagonists and S-phase blockers.
Gene expression analyses demonstrated an increased expression of genes associated with negative regulators of cell cycle progression—CDKN1A (P21) and CDKN1B (P27) (Fig. 3E) by sequential application of ipafricept followed by paclitaxel—that was not seen with the other treatment schedules. Simultaneous combination of both agents produced suppression of WNT target genes but did not result in up-regulation of P21 and P27.
Schedule of drug exposure is critical to achieve optimal synergy
In our previous studies in ovarian PDX models, we observed superior activity when WNT antagonists were administered 2 or 3 days before paclitaxel treatment. To identify the optimal timing for WNT blockade in combination with taxanes, paclitaxel was applied on day 2, 3, or 5 relative to the dosing of ipafricept on day 1. Combination activity was observed when paclitaxel was dosed on day 2 or 3 but not on day 5 (Fig. 4A, left). IHC for β-catenin/pHH3 modulation and detection of multinucleated and giant cell formation was consistent with the in vivo effect on tumor volume. The impact on these markers was evident when paclitaxel was applied on day 2 and further enhanced when paclitaxel was applied on day 3. These morphologic changes were not observed when paclitaxel was added on day 5 (Fig. 4A, right).
Fig. 4. Ipafricept blocks selection of WNT active cell types by paclitaxel in ovarian cancer.
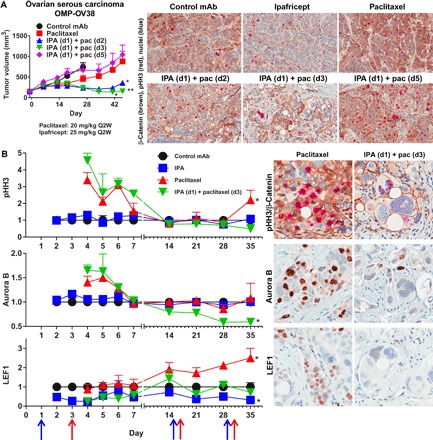
(A) Ipafricept is active when dosed either 1 or 2 days before paclitaxel. Ipafricept dosed 2 days before paclitaxel promotes mitotic catastrophe. Ipafricept (25 mg/kg) was dosed in 2-week cycles on the first day of each cycle. Paclitaxel (20 mg/kg) was dosed every 2 weeks, either on day 2, 3, or 5 of the cycle. Tumors at the end of study were paraffin-embedded and imaged for pHH3 and β-catenin (magnification, ×20). *P < 0.01; **P < 0.001 combination versus chemotherapy. Data are means + SEM; n = 8 to 10 per group. Q2W, once every 2 weeks. (B) Ipafricept dosed 2 days before paclitaxel, in 2-week cycles, prevents the accumulation of drug-resistant, WNT pathway–active ovarian tumor cells. Ipafricept (25 mg/kg) was dosed on days 1, 15, and 29 (blue arrows), and paclitaxel (20 mg/kg) was dosed on days 3, 17, and 31 (red arrows). Time course study: four to five tumors per treatment group per time point; time points were from days 2 to 7, 14, 21, 28, and 35, relative to first ipafricept dose on day 1. IHC performed from FFPE whole-tumor sections for pHH3, Aurora B, and LEF1. Digital scans were performed on an Aperio AT scanner and analyzed with Definiens Tissue Studio. Positive tumor nuclei from the control group of each time point were normalized to “1,” where a fold change of 1 implies no change, and the treatment groups are displayed as fold change as compared to the control from each corresponding time point and are mean + SEM; *P < 0.05 versus control. Representative images are from study day 35 (magnification, ×20).
To further investigate this sequence of events, we conducted a time course study in OMP-OV38 (Fig. 4B). In addition to using β-catenin as a WNT signaling marker, an additional marker for WNT pathway activity was included, the WNT transcriptional target lymphoid enhancer–binding factor 1 (LEF1). Unlike β-catenin, LEF1 is preferentially located in the nucleus, and IHC for LEF1 results in specific nuclear staining that is readily quantifiable. In the time course study, using IHC for LEF1, the mitotic kinase Aurora B, and the mitosis marker pHH3 together with β-catenin, we identified differential expression that was treatment-dependent (Fig. 4B and fig. S4). The mitotic marker pHH3 was up-regulated by paclitaxel and ipafricept followed by paclitaxel (sequential) at the 4-day time point. This up-regulation was transient because levels returned to baseline by day 7 in the paclitaxel-treated tumors and by day 14 in the sequential-treated tumors. The frequency of Aurora B–expressing cells was similarly transiently up-regulated by paclitaxel and sequential treatment. In a previous study, we had also noted that paclitaxel increased the frequency of cells with Aurora B and pHH3 by 24 hours and maintained maximum levels for up to 48 hours (fig. S5). Following the second exposure to sequential treatment, Aurora B expression was reduced to levels below baseline. The sequential treatment therefore prevented the accumulation of Aurora B– and pHH3-positive cells as compared to paclitaxel and, following the second treatment cycle, reduced the frequency below that of baseline and ipafricept alone. The WNT target LEF1 was repressed beginning 1 day following ipafricept exposure, and this down-regulation was maintained optimally for up to 3 days. LEF1 expression was similarly suppressed by the sequential treatment. Tumor cells maintained response to ipafricept, as noted by the suppression of LEF1, and this response was pronounced following each treatment cycle. Paclitaxel did not modulate LEF1 protein levels during the first week. By day 14, paclitaxel treatment selected for cells with LEF1 expression, and upon additional exposure to paclitaxel, this selection for LEF1 positivity continued for the remainder of the study. Sequential treatment prevented this enrichment of WNT pathway–active, LEF1-expressing tumor cells by paclitaxel. On day 21, 6 days after the second dose of ipafricept, a reduced tumor cell density and the presence of an enlarged and multinucleated cell phenotype were evident in the sequential group. In summary, these data indicate that WNT antagonist–mediated inhibition of target expression is an early event and is reversible, whereas sequential combination treatment–induced mitotic catastrophe is an irreversible event. The mitotic blockade was enhanced by repeated rounds of treatment with the WNT antagonist and paclitaxel.
Blocking WNT pathway activation prevents escape from mitotic blockade and reduces CSC frequency
In breast cancer PDX, vantictumab with weekly sequential paclitaxel was more active than with simultaneous dosing (Fig. 5A). Paclitaxel was active as a monotherapy, but these tumors progressed during treatment and this progression was associated with concurrent up-regulation of pHH3 and no change in the frequency of cells expressing cyclin E2, cyclin B1, and P21 (Fig. 5B). Sequential dosing reduced the frequency of cells in mitosis, as determined by pHH3, cyclin E2, and cyclin B1, while increasing P21 expression. The enrichment of P21 was also seen by the simultaneous treatment regimen. Breast tumors exposed to multiple rounds of sequential vantictumab and weekly paclitaxel also displayed the mitotic catastrophe phenotype, as demonstrated in ovarian tumors. With sequential dosing, the membrane localization of β-catenin was pronounced (Fig. 5B).
Fig. 5. Sequential dosing of vantictumab and paclitaxel potentiates mitotic cell death in breast cancer.
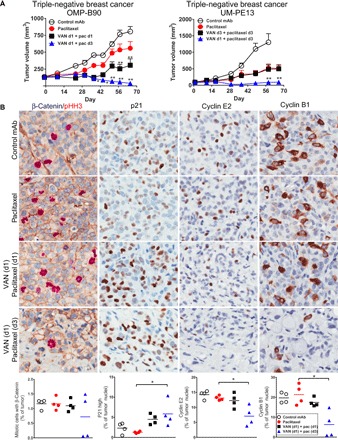
(A) OMP-B90 and UM-PE13 breast tumors respond to sequential vantictumab followed by paclitaxel. For OMP-B90, vantictumab (25 mg/kg) was administered on day 1 in 2-week cycles, for a total of six cycles. Paclitaxel (10 mg/kg) was given on day 1 or 3 in weekly cycles. For UM-PE13, vantictumab (25 mg/kg) was given on day 1 or 3, every other week, whereas paclitaxel (10 mg/kg) was given on day 3 every week. *P < 0.01; **P < 0.001 combination versus chemotherapy. Data are means + SEM; n = 6 to 9 per group. (B) Vantictumab promotes P21 induction and blockade of cyclin expression in OMP-B90. Four tumors from each treatment group were collected on day 5 of the sixth cycle and processed as FFPE. Dual IHC was performed for β-catenin (HRP-DAB; brown) and pHH3 (AP–Warp Red), with hematoxylin counterstain. IHC was performed for P21, cyclin E2, and cyclin B1 (HRP-DAB; brown) with hematoxylin counterstain. Digital scans were performed on an Aperio AT scanner and analyzed with Definiens Tissue Studio. *P < 0.05 combination versus chemotherapy. Data are means with four replicates.
WNT antagonism was determined to reduce tumorigenic cell frequency, as assessed by a functional in vivo study. The limiting dilution assay (LDA) quantifies the frequency of tumorigenic cells following in vivo drug exposure without the selection of tumor cells based on cell surface markers. Live tumor cells depleted of mouse stroma were enumerated, serially diluted, injected into recipient mice, and allowed to grow with no further treatment. In ovarian cancer, ipafricept with sequential paclitaxel reduced the capacity of OMP-OV40 to form tumors in vivo by sevenfold (Fig. 6). In contrast, paclitaxel was found to enrich for tumorigenic cells by fivefold. The remaining live tumor cells following three treatment cycles of the WNT antagonist ipafricept followed sequentially by paclitaxel were significantly depleted of the ability to propagate tumors.
Fig. 6. Sequential ipafricept followed by paclitaxel reduces the paclitaxel resistant CSC population.
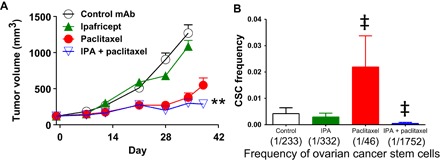
(A) OMP-OV40 was treated with ipafricept (45 mg/kg on days 1, 15, and 29) for three cycles total and/or with paclitaxel (15 mg/kg on days 5, 12, 19, 32, and 39). Thirteen days after the last ipafricept dose and 3 days after the last paclitaxel dose, tumors were processed into single cells for tumorigenicity study. *P < 0.01; **P < 0.001 combination versus chemotherapy. Data are means + SEM; n = 9 per group. (B) For the tumorigenicity study, tumors were collected from four mice per treatment group, as shown in (A), and these four tumors were mixed together to prepare the cells for the limiting dose serial transplantation assay in (B). Tumor cells were injected into recipient mice at two cell doses, 100 and 1000 cells (n = 9 to 10 mice per cell dose). The number of tumors that grew to more than 100 mm3 by day 78 after injection was used to calculate the CSC frequency. Data are means + SEM; ‡P < 0.01, significant to control.
DISCUSSION
WNT pathway activation is associated with cancer cell metastasis, drug resistance, and poor prognosis in several tumor types with unmet medical need for new therapies (26, 27). Taxanes, including paclitaxel, nab-paclitaxel, and docetaxel, are a widely used class of chemotherapy, and agents that could synergize with taxanes have significant potential for improving cancer patient survival.
We have tested our WNT pathway antagonists, vantictumab and ipafricept, in a large set of PDXs. We focused our studies on tumor types such as breast, ovarian, and pancreatic cancers, which lack mutations in WNT pathway intracellular signaling components (for example, APC or β-catenin) that are prevalent in colorectal cancer and other gastrointestinal malignancies. We directly compared the combination activity of either vantictumab or ipafricept with different classes of chemotherapeutic agents, and a clear pattern emerged from the PDX experiments: WNT pathway antagonists were consistently more effective in combination with taxanes than with other classes of chemotherapeutic agents. This effect was observed with both vantictumab and ipafricept and was evident in a variety of tumor types.
A large body of data has accumulated indicating that β-catenin plays an essential role in mitotic spindle formation and proper function during the M phase of the cell cycle (6–10). We observed up-regulation of WNT signaling after paclitaxel treatment, which suggests that taxane treatment selects for cells that are hypersensitive to WNT antagonism and/or enriches for cells that are dependent on high levels of WNT pathway activity as a resistance mechanism. A model to account for our experimental observations is that WNT antagonism enhances the cytotoxic activity of taxanes, by modulation of WNT pathway activity in mitotic cells, but is less effective when combined with S-phase blockers such as gemcitabine or platinum drugs.
Another important experimental observation that emerged from our studies is that sequential dosing of the WNT antagonists before taxane treatment resulted in optimal antitumor activity. In contrast, simultaneous dosing or taxane administration before WNT blockade was substantially less effective. This effect of dose scheduling was observed in numerous PDX models and in different tumor types, indicating the generality of this effect. An interpretation of the requirement for sequential dosing is that the synergistic activity of ipafricept or vantictumab with paclitaxel first requires the modulation of WNT/β-catenin signaling before the mitotic blockade and accumulation of mitotic cells by paclitaxel.
The utilization of PDX models and subsequent histopathology and IHC ex vivo assays were important tools for understanding the biological response to WNT antagonism plus taxane treatment. Using a time course study, we demonstrated that paclitaxel selected for a population of tumor cells that were WNT-active and that the WNT antagonist ipafricept blocked the selection of this paclitaxel-resistant, WNT-active population. We noted a correlation between the onset of tumor regression and distinct morphological changes in tumor cells, notably the formation of enlarged, multinucleated cells. This histology is indicative of a form of cell death known as mitotic catastrophe.
Mitotic catastrophe is an intrinsic cellular response to mitotic failure during or after defective mitosis, culminating in apoptotic, necrotic, or senescence-mediated elimination of mitosis-deficient, genomically unstable cells (28). At high concentrations, paclitaxel can induce mitotic catastrophe by affecting spindle formation and organization (29). Concentrations of paclitaxel in primary breast tumors are more likely to result in chromosome missegregation than mitotic arrest because they seldom achieve the high concentrations used in cell-based assays (30). Consistent with this, we did not find evidence that paclitaxel monotherapy induced widespread mitotic arrest in our PDX models. It has been reported that partial reduction of essential mitotic checkpoint components induced nonlethal mild chromosome missegregation and that these cells were sensitized to low doses of paclitaxel (29). In vitro studies in breast cancer cell lines have reported that mitotic arrest by paclitaxel is associated with induction of the cyclin-dependent kinase inhibitor P21 (31), and small interfering RNA targeting of β-catenin in basal-like breast cancer increases P21 expression (32). In our breast cancer PDX models, induction of P21 and concurrent mitotic catastrophe was dependent on combination treatment and sequential dosing and was not induced by paclitaxel alone. Notably, WNT blockade followed by taxane treatment reduced the frequency of ovarian cancer cells expressing Aurora B, a key regulator of mitotic spindle function. We found that WNT antagonism synergized with paclitaxel, promoting synthetic lethality to the two drug combination. By modulating promitogenic WNT signaling, paclitaxel-mediated mitotic catastrophe was potentiated.
Following exposure to single-agent chemotherapy treatments such as paclitaxel, we have identified an increase in the frequency of cells that form tumors upon serial transplantation. This ability of a cell to reform a tumor upon serial transplantation is a hallmark of tumor-initiating cells or CSCs (33), and this is the method that we have used to quantify the impact of WNT blockade on CSCs. CSCs have been reported to be relatively more resistant to chemotherapy, and we have found these cells to be enriched after chronic exposure to chemotherapy and in tumor cells with acquired chemotherapy resistance (34). In ovarian cancer, we have determined that WNT blockade combined with paclitaxel significantly decreases the CSC frequency, whereas paclitaxel alone enriched for CSCs. By blocking WNT/β-catenin signaling before mitotic blockade by paclitaxel, we found that this treatment effectively sensitizes CSCs to paclitaxel.
In these experiments, we identified taxane treatment as a uniquely effective chemotherapy in combination with WNT pathway inhibition. By inhibiting WNT/β-catenin signaling before mitotic blockade by paclitaxel, we found that this treatment effectively reduced tumor growth and CSC frequency. Combination treatment with taxane-based regimens along with the sequential dosing schedule has been incorporated into the ongoing clinical trials for vantictumab and ipafricept in breast, ovarian, and pancreatic cancers (ClinicalTrials.gov identifiers: NCT01973309, NCT02005315, NCT02092363, and NCT02050178). Early clinical results highlight the promise of this approach (35, 36). Furthermore, we have identified molecular biomarkers that can be used to identify sensitive tumors (35). Vantictumab and ipafricept have the potential to be the first approved agents targeting the WNT pathway for the treatment of cancer.
MATERIALS AND METHODS
Research objectives
The objectives of this research were to optimize the combination treatment strategies for vantictumab and ipafricept in various cancer indications. A prespecified hypothesis was that taxane chemotherapies were a highly effective combination partner with WNT antagonists. Early experimental observations suggested that taxane combinations were highly effective. In these studies, we explored combination activity with various chemotherapies. Once we confirmed that combination with taxanes was superior to combination with other chemotherapies, we sought to investigate the optimal dosing strategy and mechanism of action of this drug combination and the resulting effects of these treatment regimens on WNT pathway activity, cell fate, and tumorigenicity.
Ethics statement
The animals used in this study were housed in a U.S. Department of Agriculture–registered facility in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals. The study followed the guidelines set by OncoMed Pharmaceuticals’ Institutional Animal Care and Use Committee under the animal use protocols OMP3 and OMP6. Additional accreditation to this facility was provided by the Association for Assessment and Accreditation of Laboratory Animal Care.
Study design
PDX tumor models were used in these studies. These models were established in immunocompromised mice, and tumors were never propagated in vitro. In these in vivo models, we designed the studies to test the various agents with 10 mice per treatment group (one xenograft per mouse). Mice were randomized on the basis of tumor volume. Each study contained a group treated with a negative control antibody consisting of a murine monoclonal IgG1 antibody [1B7.11, ATCC (American Type Culture Collection) TIB-191]. Because of inherent variability in take rate and individual xenograft growth, we occasionally reduced the group size to as few as seven mice per treatment group, with equal distribution of mice per treatment group. During the study, we did observe variability in treatment response, but no outliers were removed from the analysis.
PDX models
NOD.CB17-Prkdcscid [nonobese diabetic/severe combined immunodeficient (NOD/SCID)] mice were purchased from Envigo or Charles River Laboratories, maintained under specific pathogen–free conditions, and provided with sterile food and water ad libitum. The mice were allowed to acclimate for several days before the PDX studies. PDX models were established at OncoMed Pharmaceuticals Inc. and are listed in table S1.
Preparation of PDX tumor suspensions
Freshly dissociated single-cell suspensions or cryogenically preserved suspensions were used for xenograft implantations. To prepare the suspension, xenograft tumors were minced with a sterile razor blade and digested in collagenase III (300 U; Worthington) in M199 medium (Life Technologies, catalog no. 12350-039) with deoxyribonuclease I (DNase I) (200 U; Worthington) and incubated for 0.5 to 2 hours at 37°C for enzymatic dissociation. These were triturated with a 10-ml serological pipette every 15 to 30 min and then inactivated with PDX buffer [Hank’s balanced salt solution supplemented with 2% heat-inactivated newborn calf serum and 20 mM Hepes (Life Technologies, catalog no. 14175-095, 26010-074, and 15630-080)] and filtered (40-μm mesh) to remove aggregates and undigested tissues. Cells were centrifuged at 150g for 5 min, cleared, and resuspended in 3 ml of ACK buffer (0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM Na2EDTA in water) on ice for 2 min. Then, five volumes of PDX buffer were added to neutralize. These were then centrifuged, cleared, and resuspended in PDX buffer. Injection volumes were 100 μl of an equal volume of PDX buffer with Matrigel (Corning Matrigel Basement Membrane Matrix, LDEV-Free; catalog no. 354234). Subcutaneous injections were made into the left flank region of NOD/SCID mice with a 25-gauge (5/8-inch) needle. Treatments were initiated after randomization with inclusion of tumors at approximately 75 to 125 mm3. Subcutaneous tumor growth was measured with an electronic caliper, and volumes were calculated as (L × W × W)/2. Both antibodies and chemotherapeutic agents were administered by intraperitoneal injection.
Chemotherapeutic agents
Chemotherapies used in this study were nab-paclitaxel [ABRAXANE, National Drug Code (NDC) 68817-134], gemcitabine (GEMZAR, NDC 0002-7501), paclitaxel (NDC 00703-4766), and carboplatin (CARBOplatin; NDC 0703-4244).
In vivo LDA
Single-cell suspensions in PDX buffer from control and treated tumors were incubated with anti-mouse antibodies (CD45-biotin, SF1-1.1, 1:100; H-2Kd–biotin, 1:50, 30-F11; BioLegend) on ice for 30 min followed by addition of streptavidin-labeled magnetic beads (MagnaBind, Thermo Fisher Scientific). Mouse cells were captured with a magnet, whereas the human cells in the suspension were collected, counted, and diluted to 1000 and then 100 cells/80 μl. To a nontreated 96-well plate, 80 μl of cell suspension was added to individual wells, and then, an equal volume of Matrigel was added and the entire volume was injected subcutaneously into the flank of NOD/SCID mice. Tumor formation was monitored for up to 3 months. CSC frequency was determined with L-CALC v.1 (STEMCELL Technologies).
OMP-54F28 (ipafricept)
OMP-54F28 is a fusion protein composed of the N-terminal, ligand-binding domain (also known as the Fri domain or cysteine-rich domain) of ~120 amino acids of human FZD8 linked to a human IgG1 Fc domain. The protein was expressed in Chinese hamster ovary cells and purified by protein A affinity chromatography.
Gene expression
Tumor RNAs were isolated using the RNeasy Fibrous Tissue Mini kit (Qiagen) with DNase I treatment, and 500 ng of total RNA was reverse-transcribed into complementary DNA. Quantitative real-time PCR was performed in an ABI 7900HT and analyzed using SDSv2.3 (Applied Biosystems) using the comparative Ct method. All gene expression assays were obtained from Applied Biosystems.
Molecular pathology
IHC assays were developed and optimized for pHH3 (Ser10; 9701, Cell Signaling), LEF1 (2230, Cell Signaling), P21 (2947, Cell Signaling), cyclin E2 (ab40890, Abcam), cyclin D1 (ab40754, Abcam), Aurora B (ab45145, Abcam), and β-catenin (610154, BD Biosciences). Formalin-fixed, paraffin-embedded tissues were sectioned at 4 μm and mounted on coated glass slides. Tissue sections were subjected to antigen retrieval and then stained for single or dual IHC on a Ventana BenchMark ULTRA instrument using Ventana UltraMap reagents or manually using DAKO EnVision+ HRP reagents and standard IHC techniques. For dual IHC of pHH3 and β-catenin, we visualized pHH3 with Warp Red chromogen following MACH 2 AP-Polymer (Biocare Medical). Sections were then counterstained with hematoxylin and were coverslipped. Slides were scanned using an Aperio AT scanner (Leica) and then analyzed using Definiens Tissue Studio image analysis software. Positively stained cells (single and/or dual) within tumors were identified and counted. These analyses included the entirety of each tumor piece sectioned, after exclusion of nontumor cells (that is, tumor capsule) and uninterpretable regions such as necrosis or folds. For immunofluorescence detection, primary antibodies, as listed above, were detected by Alexa Fluor–conjugated Fab fragments (2 μg/ml in phosphate-buffered saline), preserved with ProLong Gold with DAPI (both by Molecular Probes), and imaged with an Olympus FV10i confocal microscope.
Statistical analysis
Data for the PDX tumor measurements are means + SEM, and differences between groups were quantified with GraphPad Prism 5 using repeated-measures analysis of variance (ANOVA) with multiple planned comparisons. Data for IHC are means with replicates or means with SE and analyzed with one-way ANOVA with Bonferroni’s multiple comparison test.
Supplementary Material
Acknowledgments
We thank L. Donigan and S. Satyal for important contributions at the early stage of the Wnt antagonist programs and K. Pickell for the expert oversight of the Animal Research Facility at OncoMed. We also thank the Process Development group at OncoMed for the supply of vantictumab and ipafricept for these studies and D. Pardi for reviewing the manuscript. Funding: This research was funded by OncoMed Pharmaceuticals. Author contributions: M.M.F. designed and performed the PDX efficacy studies and IHC and LDA studies, analyzed the data, and wrote the manuscript. B.C. performed and analyzed the IHC studies and characterized the PDX bank. V.P.Y., F.C., C.L.M., J.C., and X.S. performed and analyzed the PDX efficacy studies. C.C., J.L., A.M.K., and A.G. conceived and designed the WNT antagonist. J.W.E. and G.O. developed and characterized the PDX bank. R.T. and C.-Y.C. performed and analyzed the IHC and biochemistry studies. W.-C.Y. led the PDX efficacy studies conception, design and management, gene expression analysis, and manuscript writing. T.H. conceived and designed the PDX studies, wrote the manuscript, and supervised the study. Competing interests: All authors of this paper are current or former employees and stockholders of OncoMed Pharmaceuticals. J.L. is Executive Vice President, A.G. and T.H. are Senior Vice Presidents, and A.M.K. is Vice President of OncoMed Pharmaceuticals. W.-C.Y. was also a paid consultant for OncoMed Pharmaceuticals. A.G. and W.-C.Y. are authors on pending patent applications related to this work filed in the United States, Australia, Canada, China, Europe, Japan, Mexico, New Zealand, and South Africa based on international application no. PCT/US2015/047102 (filed 27 August 2015) owned by OncoMed Pharmaceuticals. All other authors declare that they have no competing interest. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700090/DC1
fig. S1. Nab-paclitaxel arrests cells at mitosis and synergizes with WNT antagonists.
fig. S2. Higher doses of WNT antagonists administered infrequently are more active than corresponding dosages administered weekly.
fig. S3. Ipafricept induces mitotic catastrophe when dosed sequentially before paclitaxel.
fig. S4. Ipafricept blocks selection of WNT-active cell types by paclitaxel in ovarian cancer.
fig. S5. Paclitaxel up-regulates Aurora B and pHH3 by 24 hours up to 72 hours.
table S1. PDX tumors.
REFERENCES AND NOTES
- 1.Reya T., Clevers H., Wnt signalling in stem cells and cancer. Nature 434, 843–850 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Clevers H., Loh K. M., Nusse R., An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 346, 1248012 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Saito-Diaz K., Chen T. W., Wang X., Thorne C. A., Wallace H. A., Page-McCaw A., Lee E., The way Wnt works: Components and mechanism. Growth Factors 1, 1–31 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Madan B., Virshup D. M., Targeting Wnts at the source—New mechanisms, new biomarkers, new drugs. Mol. Cancer Ther. 14, 1087–1094 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Testu O., McCormick F., β-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398, 422–426 (1999). [DOI] [PubMed] [Google Scholar]
- 6.Davidson G., Shen J., Huang Y.-L., Su Y., Karaulanov E., Bartscherer K., Hassler C., Stannek P., Boutros M., Niehrs C., Cell cycle control of Wnt receptor activation. Dev. Cell 17, 788–799 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Niehrs C., Acebron S. P., Mitotic and mitogenic Wnt signalling. EMBO J. 31, 2705–2713 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stolz A., Neufeld K., Ertych N., Bastians H., Wnt mediated protein stabilization ensures proper mitotic microtubule assembly and chromosome segregation. EMBO Rep. 26, 490–499 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mbom B. C., Siemers K. A., Ostrowski M. A., Nelson W. J., Barth A. I. M., Nek2 phosphorylates and stabilizes β-catenin at mitotic centrosomes downstream of Plk1. Mol. Biol. Cell 25, 977–991 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kikuchi K., Niikura Y., Kitagawa K., Kikuchi A., Dishevelled, a Wnt signaling component, is involved in mitotic progression in cooperation with Plk1. EMBO J. 29, 347–3483 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gurney A., Axelrod F., Bond C. J., Cain J., Chartier C., Donigan L., Fischer M., Chaudhari A., Ji M., Kapoun A. M., Lam A., Lazetic S., Ma S., Mitra S., Park I.-K., Pickell K., Sato A., Satyal S., Stroud M., Tran H., Yen W.-C., Lewicki J., Hoey T., Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc. Natl. Acad. Sci. U.S.A. 109, 11717–11722 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D. C. Smith, M. Gordon, W. Messersmith, R. Chugh, D. Mendelson, J. Dupont, R. Stagg, W.-C. Yen, A. M. Kapoun, L. Xu, R. K. Brachmann, A. Jimeno, A first-in-human Phase 1 study of anti-cancer stem cell (CSC) agent OMP-54F28 (FZD8-Fc) targeting the Wnt pathway in advanced solid tumors, paper presented at the AACR-NCI-EORTC Molecular Targets and Cancer Therapeutics Meeting, Boston, MA, 21 October 2013.
- 13.D. C. Smith, L. Rosen, R. Chugh, J. Goldman, L. Xu, A. M. Kapoun, R. Brachmann, J. Dupont, R. Stagg, T. Tolcher, K. Papadopoulos, First-in-human evaluation of the human monoclonal antibody vantictumab (OMP-18R5; anti-Frizzled) targeting the Wnt pathway in a Phase 1 study for patients with solid tumors, paper presented at the 2013 ASCO Annual Meeting, Chicago, IL, 3 June 2013.
- 14.Weeks J. C., Catalano P. J., Cronin A., Finkelman M. D., Mack J. W., Keating N. L., Schrag D., Patients’ expectations about effects of chemotherapy for advanced cancer. N. Engl. J. Med. 367, 1616–1625 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohammed M. K., Shao C., Wang J., Wei Q., Wang X., Collier Z., Tang S., Liu H., Zhang F., Huang J., Guo D., Lu M., Liu F., Liu J., Ma C., Shi L. L., Athiviraham A., He T. C., Lee M. J., Wnt/β-catenin signaling plays an ever-expanding role in stem cell self-renewal, tumorigenesis and cancer chemoresistance. Genes Dis. 3, 11–40 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ding L., Ellis M. J., Li S., Larson D. E., Chen K., Wallis J. W., Harris C. C., McLellan M. D., Fulton R. S., Fulton L. L., Abbott R. M., Hoog J., Dooling D. J., Koboldt D. C., Schmidt H., Kalicki J., Zhang Q., Chen L., Lin L., Wendl M. C., McMichael J. F., Magrini V. J., Cook L., McGrath S. D., Vickery T. L., Appelbaum E., Deschryver K., Davies S., Guintoli T., Lin L., Crowder R., Tao Y., Snider J. E., Smith S. M., Dukes A. F., Sanderson G. E., Pohl C. S., Delehaunty K. D., Fronick C. C., Pape K. A., Reed J. S., Robinson J. S., Hodges J. S., Schierding W., Dees N. D., Shen D., Locke D. P., Wiechert M. E., Eldred J. M., Peck J. B., Oberkfell B. J., Lolofie J. T., Du F., Hawkins A. E., O’Laughlin M. D., Bernard K. E., Cunningham M., Elliott G., Mason M. D., Thompson D. M. Jr., Ivanovich J. L., Goodfellow P. J., Perou C. M., Weinstock G. M., Aft R., Watson M., Ley T. J., Wilson R. K., Mardis E. R., Genome remodeling in basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Garrido-Laguna I., Uson M., Rajeshkumar N. V., Choon Tan A., de Oliveira E., Karikari C., Villaroel M. C., Salomon A., Taylor G., Sharma R., Hruban R. H., Maitra A., Laheru D., Rubio-Viqueira B., Jimeno A., Hidalgo M., Tumor engraftment in nude mice and enrichment in stroma-related gene pathways predict poor survival and resistance to gemcitabine in patients with pancreatic cancer. Clin. Cancer Res. 17, 5793–5800 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weroha S. J., Becker M. A., Enderica-Gonzalez S., Harrington S. C., Oberg A. L., Maurer M. J., Perkins S. E., AlHilli M., Butler K. A., McKinstry S., Fink S., Jenkins R. B., Hou X., Kalli K. R., Goodman K. M., Sarkaria J. N., Karlan B. Y., Kumar A., Kaufmann S. H., Hartmann L. C., Haluska P., Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin. Cancer Res. 20, 1288–1297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Von Hoff D. D., Ervin T., Arena F. P., Chiorean E. G., Infante J., Moore M., Seay T., Tjulandin S. A., Ma W. W., Saleh M. N., Harris M., Reni M., Dowden S., Laheru D., Bahary N., Ramanathan R. K., Tabernero J., Hidalgo M., Goldstein D., Van Cutsem E., Wei X., Iglesias J., Renschler M. F., Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pinato D. J., Graham J., Gabra H., Sharma R., Evolving concepts in the management of drug resistant ovarian cancer: Dose dense chemotherapy and the reversal of clinical platinum resistance. Cancer Treat. Rev. 39, 153–160 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Horwitz S. B., Taxol (paclitaxel): Mechanisms of action. Ann. Oncol. 5, (suppl. 6), S3–S6 (1994). [PubMed] [Google Scholar]
- 22.Pauwels B., Korst A. E. C., Pattyn G. G. O., Lambrechts H. A. J., Van Bockstaele D. R., Vermeulen K., Lenjou M., de Pooter C. M. J., Vermorken J. B., Lardon F., Cell cycle effect for gemcitabine and its role in the radiosensitizing mechanism in vitro. Int. J. Radiat. Oncol. Biol. Phys. 57, 1075–1083 (2003). [DOI] [PubMed] [Google Scholar]
- 23.Cruet-Hennequart S., Villalan S., Kaczmarczyk A., O’Meara E., Sokol A. M., Carty M. P., Characterization of the effects of cisplatin and carboplatin on cell cycle progression and DNA damage response activation in DNA polymerase eta-deficient human cells. Cell Cycle 8, 3039–3050 (2009). [PubMed] [Google Scholar]
- 24.Rosenberg P., Andersson H., Boman K., Ridderheim M., Sorbe B., Puistola U., Parö G., Randomized trial of single agent paclitaxel given weekly versus every three weeks and with peroral versus intravenous steroid premedication in patients with ovarian cancer previously treated with platinum. Acta Oncol. 41, 418–424 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Lichtman S. M., Hurria A., Cirrincione C. T., Seidman A. D., Winer E., Hudis C., Cohen H. J., Muss H. B. Cancer and Leukemia Group B , Paclitaxel efficacy and toxicity in older women with metastatic breast cancer: Combined analysis of CALGB 9342 and 9840. Ann. Oncol. 23, 632–638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhan T., Rindorff N., Boutros M., Wnt signaling in cancer. Oncogene 36, 1461–1473 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Scheel C., Eaton E. N., Li S. H.-J., Chaffer C. L., Reinhardt F., Kah K.-J., Bell G., Guo W., Rubin J., Richardson A. L., Weinberg R. A., Paracrine and autocrine signals induce and maintain mesenchymal and stem cell states in the breast. Cell 145, 926–940 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vitale I., Galluzi L., Casted M., Kroemer G., Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 12, 385–392 (2011). [DOI] [PubMed] [Google Scholar]
- 29.Janssen A., Kops G. J. P. L., Medema R. H., Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc. Natl. Acad. Sci. U.S.A. 106, 19108–19113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zasadil L. M., Andersen K. A., Yeum D., Rocque G. B., Wilke L. G., Tevaarwerk A. J., Raines R. T., Burkard M. E., Weaver B. A., Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Transl. Med. 6, 229ra43 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi Y. H., Yoo Y. H., Taxol-induced growth arrest and apoptosis is associated with the upregulation of the Cdk inhibitor, p21WAF1/CIP1, in human breast cancer cells. Oncol. Rep. 28, 2163–2169 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Xu J., Chen Y., Huo D., Khramtsov A., Khramtsova G., Zhang C., Goss K. H., Olopade O. I., β-catenin regulates c-Myc and CDKN1A expression in breast cancer cells. Mol. Carcinog. 55, 431–439 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clarke M. F., Dick J. E., Dirks P. B., Eaves C. J., Jamieson C. H. M., Jones D. L., Visvader J., Weissman I. L., Wahl G. M., Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 66, 9339–9344 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Yen W.-C., Fischer M. M., Hynes M., Wu J., Kim E., Beviglia L., Yeung V. P., Song X., Kapoun A. M., Lewicki J., Gurney A., Simeone D. M., Hoey T., Anti-DLL4 has broad spectrum activity in pancreatic cancer dependent on targeting DLL4-Notch signaling in both tumor and vasculature cells. Clin. Cancer Res. 18, 5374–5386 (2012). [DOI] [PubMed] [Google Scholar]
- 35.M. Mita, C. Becerra, D. Richards, A. Mita, E. Shagisultanova, C. Osborne, J. O’Shaughnessy, C. Zhang, R. Henner, A. M. Kapoun, L. Xu, J. Dupont, S. Uttamsingh, R. K. Brachmann, A. Farooki, J. R. Diamond, Phase 1b study of Wnt inhibitor vantictumab (VAN, human monoclonal antibody) with paclitaxel and (P) in patients (pts) with 1st- to 3rd-line metastatic HER2-negative breast cancer (BC), paper presented at the 2016 ASCO Annual Meeting, Chicago, IL, 5 June 2016.
- 36.R. E. O’Cearbhaill, S. McMeekin, G. Mantia-Smaldone, C. Gunderson, M. Morgan, R. Burger, P. Sabbatini, F. Cattaruzza, M. Fischer, A. M. Kapoun, L. Xu, J. Dupont, R. K. Brachmann, S. Uttamsingh, A. Farooki, K. Moore, Phase 1b of Wnt inhibitor ipafricept (IPA, decoy receptor for Wnt ligands) with carboplatin (C) and paclitaxel (P) in patients (pts) with recurrent platinum-sensitive ovarian cancer (OC), paper presented at the 2016 ASCO Annual Meeting, Chicago, IL, 5 June 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/3/6/e1700090/DC1
fig. S1. Nab-paclitaxel arrests cells at mitosis and synergizes with WNT antagonists.
fig. S2. Higher doses of WNT antagonists administered infrequently are more active than corresponding dosages administered weekly.
fig. S3. Ipafricept induces mitotic catastrophe when dosed sequentially before paclitaxel.
fig. S4. Ipafricept blocks selection of WNT-active cell types by paclitaxel in ovarian cancer.
fig. S5. Paclitaxel up-regulates Aurora B and pHH3 by 24 hours up to 72 hours.
table S1. PDX tumors.