

Abstract
Increased small intestinal permeability has been proposed to be an integral element, along with genetic makeup and environmental triggers, in the pathogenies of chronic inflammatory diseases (CIDs). We identified zonulin as a master regular of intercellular tight junctions linked to the development of several CIDs. We aim to study the role of zonulin-mediated intestinal permeability in the pathogenesis of CIDs. Zonulin transgenic Hp2 mice (Ztm) were subjected to dextran sodium sulfate (DSS) treatment for 7 days, followed by 4–7 days recovery and compared to C57Bl/6 (wild-type) mice. Intestinal permeability was measured in vivo and ex vivo, and weight, histology, and survival were monitored. To mechanistically link zonulin-dependent impairment of small intestinal barrier function with clinical outcome, Ztm were treated with the zonulin inhibitor AT1001 added to drinking water in addition to DSS. We observed increased morbidity (more pronounced weight loss and colitis) and mortality (40–70% compared with 0% in wild type) at 11 days post-DSS treatment in Ztm compared with wild-type mice. Both in vivo and ex vivo measurements showed an increased intestinal permeability at baseline in Ztm compared to wild-type mice, which was exacerbated by DSS treatment and was associated with upregulation of zonulin gene expression (fourfold in the duodenum, sixfold in the jejunum). Treatment with AT1001 prevented the DSS-induced increased intestinal permeability both in vivo and ex vivo without changing zonulin gene expression and completely reverted morbidity and mortality in Ztm. Our data show that zonulin-dependent small intestinal barrier impairment is an early step leading to the break of tolerance with subsequent development of CIDs.
Keywords: zonulin, intestinal permeability, tight junctions, barrier function, inflammation
Introduction
The incidence of chronic inflammatory diseases (CIDs) in Western countries has steadily increased since the mid-20th century.1 This phenomenon has led to the formulation of the hygiene hypothesis,1, 2 which has recently been challenged by evidence that increased hygiene in developing countries did not lead to similar CID epidemics. It has been hypothesized that the development of CIDs is the result of the exposure to environmental triggers in genetically susceptible individuals, but recent epidemiological evidence suggests that, while both genetic predisposition and antigenic triggers are necessary, they are not sufficient to develop CIDs.3 Additional factors instrumental in causing CIDs include an inappropriate immune response, particularly related to the coordination between the innate and adaptive immune responses,4 changes in gut microbiome composition (dysbiosis),5 and impaired mucosal barrier function.6
We identified zonulin as a human protein master regulator of paracellular permeability by reversibly modulating intercellular tight junctions (TJs).7–9 Zonulin release is triggered by specific microbiota10 or exposure to gliadin,11 and it is augmented in CIDs associated with TJs dysfunction.8,12,13 Gliadin causes zonulin release from enterocytes and monocytes through binding to the CXCR3 receptor in a MyD88-dependent manner.11 Zonulin causes TJ disassembly through proteinase-activating receptor 2 (PAR2) transactivation of the epidermal growth factor receptor (EGFR).14,15 It is hypothesized that zonulin causes actin cytoskeleton rearrangement in a protein kinase C (PKC)–dependent mechanism, similar to its prokaryotic analog, zonula occludins toxin (Zot).16 The cytoskeleton rearrangement and subsequent TJ disassembly is caused by displacement of ZO-1 and occludin from the cell junction, secondary to PKCα-dependent phosphorylation of ZO-1 and myosin 1C.17
Zonulin is overexpressed in the intestinal mucosa of celiac disease (CD) patients,14 and serum zonulin levels correlate with increased intestinal permeability (IP).8,9,12,18 Serum zonulin levels have been associated with many CIDs, which affect almost every major system in the body.13 Many of the CIDs associated with elevated serum zonulin levels have unknown etiologies, and the early events leading to disease development are unknown. A zonulin-dependent loss of intestinal barrier function leading to uncontrolled influx of environmental antigens may be an early event in many CIDs.
We have identified zonulin as the precursor of haptoglobin-2 (pre-Hp2).14 Haptoglobin (Hp) is an acute-phase protein that binds hemoglobin (Hb) to prevent oxidative stress caused by free Hb. Human haptoglobin (HP) exists as two common alleles, HP1 and HP2, giving rise to three possible genotypes: HP1-1, HP2-1, and HP2-2. The HP1 allele shares homology with Hp found in animals from early reptiles to other mammals. The Hp2 allele is only found in humans and arose from an uneven crossover event resulting in the duplication of exons 3 and 4 of Hp1. Hp is translated as a pro-protein before enzymatic cleavage into its α and β chains and subsequent mature Hp. Uncleaved, the pro-protein of Hp2 (pre-Hp2) is active as zonulin. Therefore, only patients who have an Hp2 gene can produce zonulin.
The presence of the zonulin (HP2) gene is more frequently encountered in CIDs, including CD,19 inflammatory bowel disease (IBD),20,21 and schizophrenia,22 and homozygous expression of the zonulin gene (HP2-2) correlates with more severe clinical manifestations.19,20 Wild-type (WT) C57Bl/6 mice express only the HP1 allele (> 90% homology with human HP1 allele) and, therefore, are used as a surrogate for the human HP1-1 genotype. To work on a murine model overexpressing zonulin (pre-HP2), we acquired a mouse model in which the native mouse Hp1 allele was substituted with a murine Hp2 allele via targeted insertion to generate mice with the Hp2-2 genotype.23 We have used this zonulin transgenic Hp2 mouse (Ztm) model to establish the role of zonulin-dependent small intestinal barrier dysfunction as an early step in breaching mucosal tolerance with subsequent onset of inflammation in the mouse model of DSS colitis.
In this study, we show that the presence of the zonulin gene under baseline conditions causes increased small intestinal permeability not associated with any pathological phenotype. The addition of DSS as a trigger of inflammation causes increased morbidity and mortality in Ztm secondary to a zonulin-dependent increase in small intestinal permeability. Blocking the zonulin pathway with the zonulin antagonist AT1001 ameliorates the colitis, similar to what was previously reported in the IL-10 knockout model of colitis24 and prevented Ztm mortality.
Methods
Animals
The animal studies in this paper were approved by the Institutional Animal Care and Use Committee at Massachusetts General Hospital (MGH) (2013N000013). C57Bl/6 (WT) mice were obtained from the Jackson Laboratory (Bar Harbor, ME), and a colony was maintained in our facility at Massachusetts General Hospital. Ztm were created as previously described,23 and breeding pairs were generously provided by Andrew Levy. A colony of Ztm were maintained at MGH. WT mice and Ztm were housed in separate cages within the same facility for the duration of the study.
DSS colitis
Mice (8–12 weeks) were given 3% (w/v) DSS (molecular weight 36,000–50,000, MP Biomedicals) in their drinking water ad libitum for 7 days, followed by up to 7 days of normal drinking water for recovery. Mice were euthanized on either day 7 following DSS treatment or day 14 following recovery. Body weight and water intake were measured daily. After euthanasia, the intestine was removed en bloc, and colon weight and length were measured. To quantify the extent of mucosal damage, a 0.5-cm segment from the distal colon was fixed in 4% paraformaldehyde, paraffin embedded, sectioned (5 um), and stained with hematoxylin and eosin (H&E). Assessment of microscopic inflammation was performed in a blinded fashion using the following scores. Epithelium: 0, no pathological change; 1, loss of basal 1/3 of crypts; 2, loss of basal 2/3 of crypts; 3, entire crypt loss; and 4 – erosion of epithelium. Inflammatory cell infiltration: 0, rare inflammatory cells in the lamina propria; 1, increased number of inflammatory cells in the lamina propria; 2, confluence of inflammatory cells extending into the submucosa; and 3, transmural extension of the inflammatory infiltrate. Submucosal edema: 0, no pathological change; 1, mild edema; 2, moderate edema; and 3, profound edema. The score for each category was then multiplied by 1 for focal, 2 for patchy, and 3 for diffuse changes. The combined score for each tissue is the sum of the above and ranges from 0–30 units with the following grades of total inflammation: 0, no inflammation; 1–7, minimal inflammation; 8–14, slight inflammation; 15–21, moderate inflammation; and 21–30, severe inflammation.
Intestinal permeability to FITC-dextran
On day 11 following DSS treatment, mice were deprived of food for 2 h and orally gavaged with 0.6 mg/g body weight 4-kDa fluoresceinyl (FITC)-dextran at a concentration of 80 mg/mL 1 h before euthanasia. Blood was collected by retro-orbital eye bleed and centrifuged to collect serum. The fluorescence in the serum was measured by a fluorescent spectrophotometer with 485 nm excitation and 535 nm emission. A standard curve was created diluting FITC-dextran in PBS. The concentration of FITC-dextran in the serum was calculated using the standard curve.
Cascade blue flux
Sections of duodenum, jejunum, and colon were stripped of smooth muscle, mounted in microsnapwell chambers as previous described,10 and incubated in the dark. After a 30-min equilibration period, 200 μg/mL of cascade blue–labeled dextran (10K for small intestine and 3K for colon) was added to the apical chamber. After 3 hours postmounting, supernatant was collected from both apical and basal lateral compartments. Cascade blue translocation was measure with a fluorescent spectrophotometer with 355 nm excitation and 425 nm emission. A standard curve was created diluting cascade blue dextran in PBS. The concentration of cascade blue translocation in the basal lateral compartment was calculated using the standard curve.
Zonulin gene expression
Total RNA was extracted from tissue with TRIzol reagent using the Direct-zol RNA kits from Zymo Research per the manufacturers’ instructions. RNA samples were reverse transcribed to cDNA using the Maxima H Minus First Strand cDNA synthesis kit (Thermo Scientific). Zonulin quantitative polymerase chain reaction (qPCR) was performed with SYBR green in an iCycler96X (Bio-Rad, Hercules, CA) using primers designed over the unique sequence in HP2 caused by the duplication of part of the α chain of HP1. The primers used were: F – GAATGTGAGGCAGATGACAG, R – GTGTTCACCCATTGCTTCTC.
AT1001 treatment
Mice (8–12 weeks) were given 3% DSS plus 1.0 mg/mL AT1001 (Biopolymer Laboratories, University of Maryland, Baltimore, MD) dissolved in their drinking water as we have recently described25 for 7 days, followed by 4 or 7 days of recovery. The drinking water was prepared daily. Body weight and water consumption were measured daily. Mice were euthanized and processed as described above.
Statistical analysis
Data are expressed as means with error bars representing the standard deviation. Statistics were performed as t-test or analysis of variance (ANOVA) using Graph Pad Prism 7.
Results
Phenotypic characterization of the zonulin transgenic Hp2 mice
Male and female Ztm were compared to WT mice to look for possible phenotypic differences. While, at 8 weeks, the mean weight of the female mice was not different between the two groups, the mean weight of the male Ztm was less than the WT mice (Fig S1A). No difference was observed between Ztm and WT mice for either males or females in intestinal histology (Fig 1A), colon length (Fig S1B), or colon weight (Fig. S1C). Although Ztm developed normally and did not show any overt disease, they did show increased in vivo transepithelial permeability to antigen flux, measured by FITC-dextran passage, in the small intestine of both male and female mice (Fig 1B).
Figure 1.
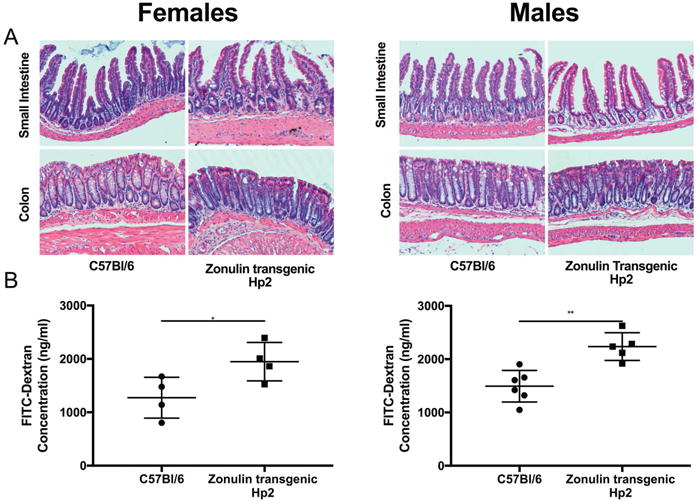
Changes in intestinal histology and barrier function seen in Ztm compared with WT mice. (A) Representative histology pictures of small intestine and colon of male and female Ztm and WT mice. (B) In vivo FITC-dextran passage in small intestine of untreated WT mice and Ztm (4–6 mice per group). *, P < 0.05; **, P < 0.01.
DSS-induced colitis is more severe in zonulin transgenic Hp2 mice
WT mice and Ztm were given 3.0% DSS in their drinking water ad libitum for 7 days, followed by normal drinking water for an additional 7 days, and weighed daily. Both male and female Ztm showed an increased rate of mortality after DSS treatment, with the rate higher in males (~70%) than females (~40%), compared to WT mice (0%) (Fig. 2A and 2B). Since male Ztm had the most severe phenotype following DSS treatment, we continued our experiments exclusively in males.
Figure 2.
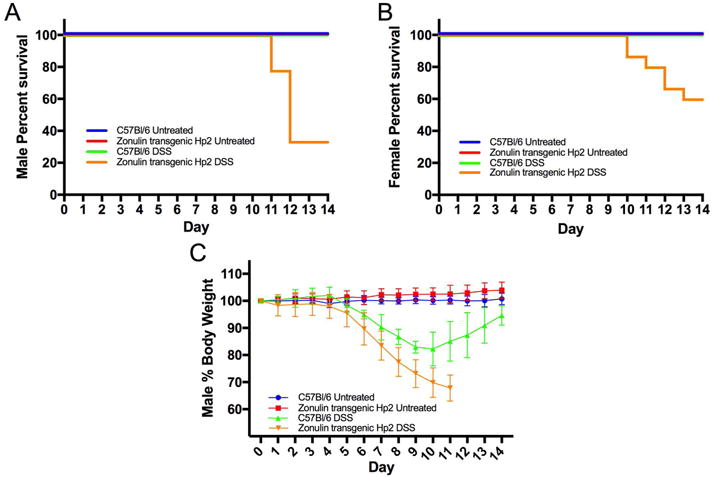
Effects of DSS treatment on survival and body weight. (A & B) Kaplan-Meier survival analysis of untreated and DSS-treated Ztm and WT (A) male (P < 0.001) and (B) female (P < 0.01) mice. (C) Mean body weight of untreated and DSS-treated Ztm and WT mice (Ztm versus WT DSS-treated P < 0.001) (4–14 mice per group).
Owing to the rate of mortality after day 11 in the male Ztm, we shortened the recovery to 4 days and euthanized animals on day 11 after DSS treatment. Ztm treated with DSS showed significant weight loss compared with untreated mice on day 5 (P < 0.01) and WT mice on day 6 (P < 0.01) (Fig. 2C). DSS-treated Ztm lost significantly more weight compared with WT DSS-treated mice on day 7 (P < 0.001) (Fig 2C). Overall, DSS-treated Ztm showed a more significant weight loss compared with WT DSS-treated mice (Fig 2C). WT DSS-treated mice started to recover their lost weight on day 9 or 10, while Ztm did not show any recovery (Fig. 2C). Most Ztm exhibited bloody stools after 5 days of DSS treatment that did not resolve over time, while less than 10% of WT mice had bloody stools that subsided following DSS withdrawal.
Macroscopic and microscopic damage
Colon length and weight of the Ztm treated with DSS were significantly decreased compared with WT animals treated with DSS (Fig 3A and 3B). The duodenum, jejunum, distal ileum, and distal colon of mice were scored for microscopic damage. We saw no histological damage in either the Ztm or the WT animals following DSS treatment in any portion of the small intestine (Fig. 3C). There was significant damage in the colon of both WT and DSS-treated Ztm in the colon (Fig. 3D). The average colonic inflammatory score was more severe in DSS-treated Ztm than in WT mice, but this difference did not reach statistical significance because of the uneven distribution of the score in WT mice. Indeed, only a subgroup of WT mice showed complete histological recovery (Fig. 3D, bottom panel), while others showed persistence of colitis (Fig. 3D, middle panel); all Ztm showed severe colitis with mucosal erosion and absence of crypts (Fig. 3D). Due to the dichotomous results in the WT mice (Fig. 3E), we also analyzed the histological data on a categorical basis, as described in the methods. This analysis revealed that 93% of Ztm showed some degree of inflammation (scores of 8 and above), compared with 45% of WT mice (P < 0.001).
Figure 3.
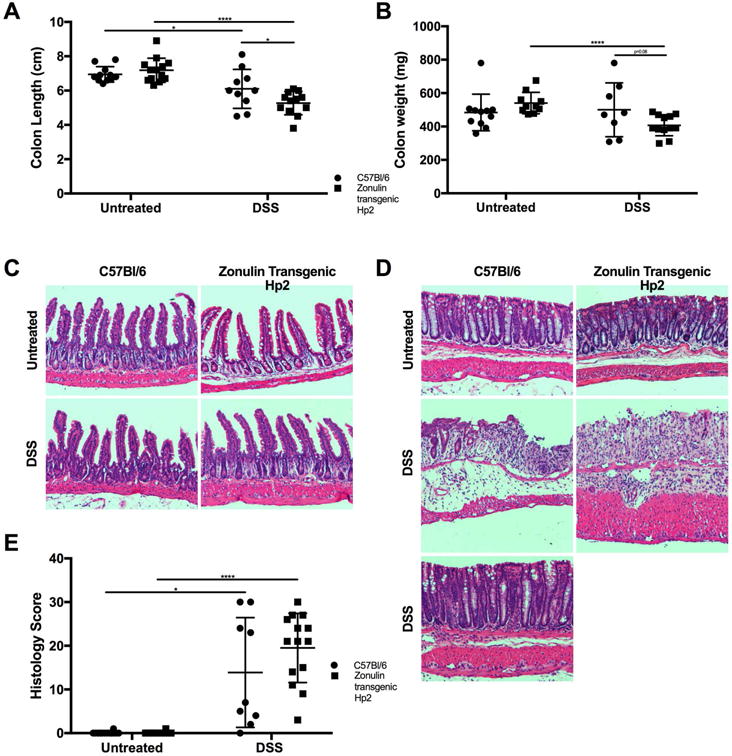
Effect of DSS treatment on the intestine of both Ztm and WT male mice on colon weight, length, and histology. (A) Colon length and (B) colon weight after 11 days ofDSS treatment. (C) Representative histology of small intestine in both untreated and DSS-treated Ztm and WT mice. (D) Representative histology of colon of either untreated or DSS-treated Ztm and WT mice. In WT mice, we observed two distinct groups of animals, with one group showing persistent colitis with occasional mucosal erosion and crypt distortion associated with increased inflammatory infiltrate in the lamina propria (middle panel), while in the other group a complete recovery of the inflammatory process was detected (bottom panel). Conversely, in DSS-treated Ztm, there was a homogeneous and more severe inflammatory process characterized by severe mucosal erosion, severe inflammatory infiltrate in the lamina propria, and loss of the regenerative crypt compartment. (E) Histology scores show dichotomous results in DSS-treated WT mice, while DSS-treated Ztm show more homogeneous severe inflammation (8–12 mice per group). *, P < 0.05; ****, P < 0.0001.
DSS colitis increases small intestinal permeability and zonulin expression in Hp2 mice
Changes in small intestinal permeability were measured both in vivo and ex vivo by FITC-dextran and cascade blue–dextran passage, respectively. In vivo small intestinal permeability test showed that, at baseline, Ztm had significantly higher permeability compared with WT mice (Fig. 4A). Following DSS treatment, small intestinal permeability significantly increased in Ztm but not in WT mice (Fig. 4A). The in vivo permeability changes were confirmed in ex vivo experiments. Ztm at baseline has a trending increased permeability in the duodenum (P = 0.08) (Fig. 4B) but no difference in the jejunum (Fig. 4C) compared with WT. Following DSS treatment, both WT mice and Ztm had increased duodenal permeability compared with untreated mice (Fig. 4B), but the increase was much more pronounced in Ztm than in WT mice. Only the Ztm treated with DSS showed increased permeability in the jejunum compared with untreated animals (Fig. 4C). The increased permeability corresponded with an increase in zonulin gene expression in both the duodenum and jejunum of Ztm (Fig. 4D and 4E). Not surprisingly, on the basis of the histological damage, there was no significant difference in colonic permeability between WT mice and DSS-treated Ztm, but both were significantly increased over untreated mice (Fig. S2). Zonulin mRNA expression was not measured in the colon, as the zonulin receptor is not present in the colon and the zonulin pathway is not active.26 The histological damage in the colon in both WT mice and Ztm following DSS treatment could explain the increased permeability due to a structural damage of the mucosal barrier rather than its functional modulation.
Figure 4.
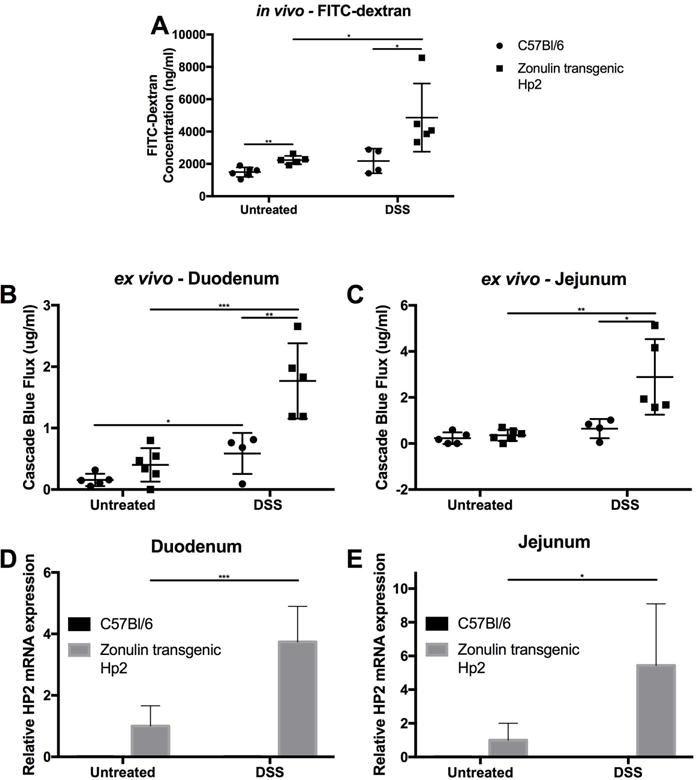
In vivo/ex vivo small intestinal permeability in Ztm and WT mice. (A) In vivo small intestinal permeability was increased in untreated Ztm compared with WT mice. DSS treatment caused further increased permeability in Ztm. Ex vivo measurement of duodenal (B) and jejunal (C) permeability. At baseline, there was a trend of increased duodenal permeability in Ztm (B), while no differences were detected in the jejunum (C). In DSS-treated animals, increased duodenal permeability was detected in both animals, with Ztm showing a more significant increase (C). No changes in jejunal permeability were detected in DSS-treated WT mice, while Ztm showed a significant increase (C). Duodenal and jejunal permeability was significantly higher in DSS-treated Ztm (B & C). (D & E) Relative zonulin mRNA expression in the duodenum (D) and jejunum (E) showed zonulin expression under baseline condition and increased by 4 and 6 fold, respectively, in DSS-treated Ztm (4–6 mice per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Zonulin inhibitor AT1001 ameliorates increased intestinal permeability and colitis
Ztm were treated with AT1001 (a zonulin synthetic peptide antagonist) in addition to DSS to confirm that the increase in small intestinal permeability was zonulin dependent and that increased antigen trafficking in the small intestine is instrumental to the pathogenesis of colitis. The AT1001 treatment completely rescued the DSS-treated Ztm from over 70% mortality to 0% (Fig. 5A). In addition, loss of body weight seen during DSS treatment was recovered in mice treated with DDS + AT1001 (Fig. 5B). These AT1001-dependent clinical rescues paralleled the amelioration of histological damage in the colon induced by AT1001 treatment in Ztm (Fig. 6A and 6B). AT1001 also prevented the decrease in colon weight and length observed after DSS treatment (Fig. 6C and 6D). The abnormal intestinal permeability seen in DSS-treated Ztm was decreased by AT1001 treatment to baseline levels in the small intestine, as established by both in vivo (Fig. 7A) and ex vivo measurements (Fig. 7B and 7C) as well as in the colon (Fig. S3). Even though the small intestinal permeability defect was corrected, the expression of zonulin was still increased in both the duodenum and jejunum (Fig. 7D and 7E), confirming that the rescue effect of AT1001 was related to its antagonistic effect on zonulin, mediating permeating action, rather than on its expression.
Figure 5.
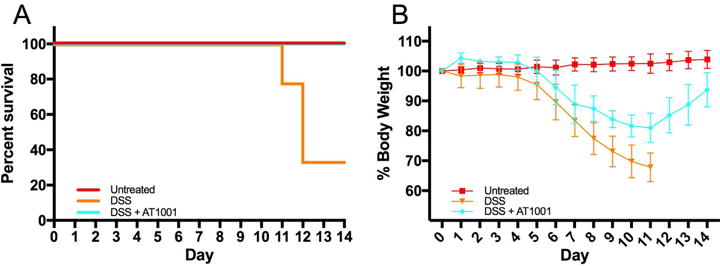
Effects of AT1001 on survival and body weight of DSS-treated Ztm. (A) Kaplan-Meier survival analysis of untreated, DSS-treated, and DSS + AT1001–treated Ztm. DSS-treated animals showed ~70 mortality rate that was completely rescued by AT1001 treatment (P < 0.01). (B) Mean body weight of untreated, DSS-treated, and DSS + AT1001–treated Ztm. DSS + AT1001–treated animals were able to recover the loss of body weight starting at day 11, while DSS-treated animals were not able to recover weight (DSS- versus AT1001- treated P < 0.0001) (4–14 mice per group).
Figure 6.
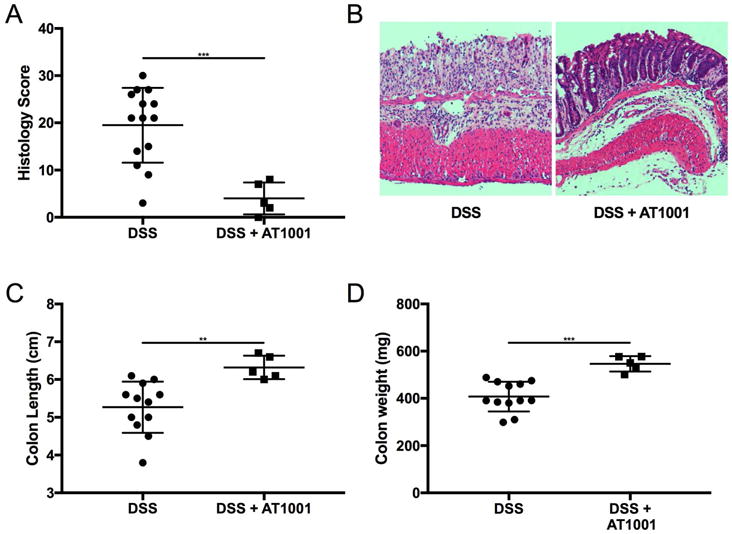
Effects of AT1001 on colon weight, length, and histology in DSS-treated Ztm mice. (A) Histology scores showed a significant decrease in inflammatory score, with DSS + AT1001–treated mice showing scores similar to untreated mice. (B) Representative histology of colon of either DSS-treated or DSS + AT1001–treated Ztm showing complete rescue of the colitis secondary to AT1001 treatment. AT1001 also prevented the decrease in colon length (C) and weight (D) (4–14 mice per group). **, P < 0.01; ***, P < 0.001.
Figure 7.
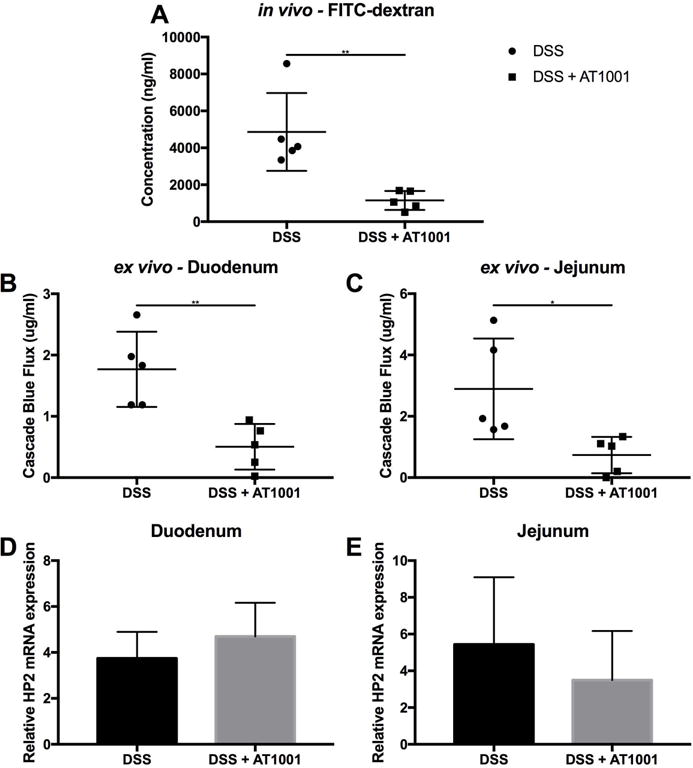
Effect of AT1001 on in vivo and ex vivo small intestinal permeability in DSS-treated Ztm. (A) In vivo measurement of small intestinal permeability by FITC-dextran passage showed that AT1001 completely prevented the increased permeability induced by DSS treatment. (B & C) Ex vivo measurement of small intestinal permeability by cascade blue dextran flux in DSS-treated or DSS + AT1001–treated duodenum (B) and jejunum (C) mounted in the polarized microsnapwell system. AT1001 rescued the increased duodenal and jejunal permeability caused by DSS treatment, paralleling the in vivo findings. (D & E) Relative zonulin mRNA expression in DSS-treated or DSS+AT1001-treated duodenum (D) and jejunum (E). No differences in zonulin gene expression were observed between the two treatment groups, suggesting that the correction of the small intestinal barrier defect by AT1001 is not secondary to changes in zonulin gene expression, but rather to downstream signaling events (4–5 mice per group). *, P < 0.05; **, P < 0.01.
Discussion
Improved hygiene leading to a reduced exposure to microorganisms has been implicated as one possible cause for the epidemic of CIDs, particularly autoimmune diseases, in industrialized countries during the last 3–4 decades.27 Collectively, autoimmune diseases are highly prevalent in the United States, affecting between 14.7 and 23.5 million people—up to 8% of the population.28 The social and financial burdens imposed by these chronic, debilitating diseases include poor quality of life, high healthcare costs, and substantial loss of productivity. For example, in 2002, the average annual medical costs for treating type 1 diabetes (T1D) in the United States were estimated at $6.7 billion.29 In less than a decade, these costs jumped to $14.4 billion.30 Besides genetic makeup and exposure to environmental triggers, a third key element, increased intestinal permeability, which may be influenced by a series of environmental factors, including the composition of the gut microbiota, has been proposed in the pathogenesis of these diseases.31–34 Intestinal permeability, together with luminal antigen sampling by enterocytes and dendritic cells, regulates molecular trafficking between the intestinal lumen and the submucosa, leading to either tolerance or immunity to non-self antigens.35–38 Intercellular TJs tightly regulate paracellular antigen trafficking. TJs are extremely dynamic structures that operate in several key functions of the intestinal epithelium under both physiological and pathological circumstances.6,32 However, despite major progress in our knowledge on the composition and function of the intercellular TJ, the mechanism(s) by which they are regulated is still incompletely understood.
Many in vivo and in vitro models have been used to study the role of TJ components in the development of CIDs. However, current data cannot mechanistically link TJ modulation to onset of CIDs, and the possibility remains that impaired gut barrier function could be the consequence or an epiphenomenon of the inflammatory process rather that its cause.
Using a zonulin transgenic animal model, we have for the first time been able to mechanistically link zonulin-dependent modulation of gut permeability and subsequent enhanced antigen trafficking to the pathogenesis of inflammatory disease. Research in humans has shown that, while the mere presence of two copies of the zonulin gene is not sufficient to develop CIDs, it will increase morbidity and mortality.14,21,22,39–43 The key role of Hp is to be a scavenger for free Hb, preventing tissue oxidative stress secondary to intravascular hemolysis.44 The increased morbidity and mortality associated with the Hp2 genotype has been typically explained by its less efficient Hb scavenging activity compared with Hp1. While this may be true for some disease states, we believe that a more reasonable hypothesis is that zonulin, as the precursor of Hp2, may affect small intestinal antigen trafficking that, associated with a specific genetic background and additional environmental triggering factors, may lead to the onset of CIDs. The data presented here are in line with our hypothesis, suggesting that the presence of the zonulin gene is not sufficient to cause disease (as suggested by normal phenotype of the Ztm under basal conditions) but, together with proinflammatory stimuli, it increases morbidity and mortality through loss of intestinal barrier function and increased antigen trafficking. The experiments using the zonulin inhibitor AT1001 have indeed confirmed that small intestinal antigen trafficking rather than less efficient Hb scavenging activity is responsible for the increased morbidity and mortality in this Ztm model. AT1001, now named larazotide acetate, is currently entering phase III clinical trial as a treatment integrative to the gluten-free diet in CD.45–50 Early phase Ib trials performed in an inpatient setting showed that larazotide acetate was able to decrease intestinal permeability assessed by the lactulose/mannitol double-sugar test and attenuated gastrointestinal and extraintestinal symptoms in CD patients undergoing exposure to gluten.50 However, subsequent phase II gluten-challenge trials performed in outpatient settings failed to confirm larazotide acetate’s antipermeating effect on the gut barrier, despite it still ameliorating symptoms and preventing the increase in tTG antibodies,45,48 casting doubts on its mechanism of action. With this study, in which variables that can affect gut barrier function assessment were tightly controlled, we have confirmed that larazotide acetate functions as a zonulin antagonist capable of correcting the impaired barrier function caused by zonulin upregulation.
That a loss of barrier function is involved in inflammatory bowel diseases (IBDs) has been hypothesized for a long time on the basis of a series of circumstantial evidence but never undisputedly proved. Indeed, the observations that increased gut permeability may precede a flare up of IBD by several months51 and that a subgroup of first-degree relatives of IBD patients showed increased small intestinal permeability without developing overt IBD52–54 and a case report of a healthy first-degree relative of a Crohn’s disease patient who displayed signs of increased intestinal permeability 8 years before her own development of the disesase55 all point to the fact increased small intestinal antigen trafficking is a necessary but not sufficient pathogenic element for the development of IBD. Furthermore, it has been recently reported that in IBD patients there is an overrepresentation of Hp2 compared with controls20 and that zonulin can be mechanistically linked with the onset of IBD (reviewed in Ref. 56). The Ztm model for the first time allowed us to mechanistically prove that the zonulin pathway can indeed be involved in CID pathogenesis, provided that additional factors (e.g., DSS exposure) further jeopardize the small intestinal antigen trafficking by increasing zonulin expression and, therefore, gut permeability.
Since we have shown that both gliadin, the environmental trigger of CD,57,58 and an unbalanced microbiota10 are strong environmental stimuli causing zonulin release, it is possible to hypothesize that zonulin-dependent loss of gut barrier function is involved in several conditions in which dysbiosis and gluten exposure have been identified or suspected as triggers of CIDs. Indeed, several studies have involved zonulin-dependent small intestinal barrier dysfunction in the development of systemic CIDs affecting other organs/districts.24,25 BB diabetic-prone rats have a zonulin-dependent defective small intestine barrier function, which precedes loss of glucose tolerance.25 Administration of AT1001 corrects the small intestinal barrier function and subsequent loss of glucose tolerance. In a murine acute lung injury model, AT1001 or administration of zonulin-neutralizing antibodies was able to reduce the severity of injury.59 AT1001 is also able to increase survival to influenza challenge.60 These animal studies are in line with data generated in humans showing zonulin-dependent increased permeability in several CIDs, including autoimmune diseases, metabolic disorders, and cancer13 and a correlation between intestinal permeability and serum zonulin levels.12,55
In conclusion, the key roadblock to studying the role of gut permeability in the pathogenesis of CIDs has been the lack of a good animal model. The results presented in this manuscript provide evidence that the Ztm model offers a unique opportunity to study early pathogenic events mechanistically linking zonulin-dependent small intestinal permeability regulation and antigen trafficking to the onset of CIDs. This model also offers a unique opportunity to test the efficacy of therapeutic interventions (as we have already demonstrated for AT1001) in ameliorating inflammatory processes secondary to the upregulation of the zonulin pathway and the identification and validation of potential biomarkers for patient-targeted intervention (precision medicine) and disease interception (primary prevention) in genetically predisposed individuals.
Supplementary Material
Figure S1. Body weight and colonic weight and length in both male and female Ztm compared with WT mice.
Figure S2. Ex vivo permeability measurement by cascade blue dextran flux in the colon of either untreated or DSS-treated Ztm and WT mice mounted in the microsnapwell system.
Figure S3. Effect of AT1001 on ex vivo permeability measurement by cascade blue dextran flux in the colon of DSS-treated Ztm mounted in the microsnapwell system.
Acknowledgments
This work was supported by the NIH Grant DK048373 entitled “Zot, Zonulin, and the Pathophysiology of Intestinal Tight Junctions”. C.S. and A.F. designed the experiments, C.S. and J.L. performed experiments, C.S. analyzed data, and C.S. and A.F. wrote the manuscript.
Footnotes
Competing interests
CS and JL have no competing interests. AF is a cofounder and stock holder of Alba Therapeutics.
References
- 1.Bach JF. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med. 2002;347:911–920. doi: 10.1056/NEJMra020100. [DOI] [PubMed] [Google Scholar]
- 2.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Catassi C, Kryszak D, Bhatti B, et al. Natural history of celiac disease autoimmunity in a USA cohort followed since 1974. Ann Med. 2010;42:530–538. doi: 10.3109/07853890.2010.514285. [DOI] [PubMed] [Google Scholar]
- 4.Gasteiger G, Rudensky AY. Interactions between innate and adaptive lymphocytes. Nat Rev Immunol. 2014;14:631–639. doi: 10.1038/nri3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martin VJ, Leonard MM, Fiechtner L, et al. Transitioning From Descriptive to Mechanistic Understanding of the Microbiome: The Need for a Prospective Longitudinal Approach to Predicting Disease. J Pediatr. 2016 doi: 10.1016/j.jpeds.2016.08.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 7.Fasano A. Regulation of intercellular tight junctions by zonula occludens toxin and its eukaryotic analogue zonulin. Ann N Y Acad Sci. 2000;915:214–222. doi: 10.1111/j.1749-6632.2000.tb05244.x. [DOI] [PubMed] [Google Scholar]
- 8.Fasano A, Not T, Wang W, et al. Zonulin, a newly discovered modulator of intestinal permeability, and its expression in coeliac disease. Lancet. 2000;355:1518–1519. doi: 10.1016/S0140-6736(00)02169-3. [DOI] [PubMed] [Google Scholar]
- 9.Wang W, Uzzau S, Goldblum SE, et al. Human zonulin, a potential modulator of intestinal tight junctions. J Cell Sci. 2000;113(Pt 24):4435–4440. doi: 10.1242/jcs.113.24.4435. [DOI] [PubMed] [Google Scholar]
- 10.El Asmar R, Panigrahi P, Bamford P, et al. Host-dependent zonulin secretion causes the impairment of the small intestine barrier function after bacterial exposure. Gastroenterology. 2002;123:1607–1615. doi: 10.1053/gast.2002.36578. [DOI] [PubMed] [Google Scholar]
- 11.Lammers KM, Lu R, Brownley J, et al. Gliadin induces an increase in intestinal permeability and zonulin release by binding to the chemokine receptor CXCR3. Gastroenterology. 2008;135:194–204 e193. doi: 10.1053/j.gastro.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sapone A, de Magistris L, Pietzak M, et al. Zonulin upregulation is associated with increased gut permeability in subjects with type 1 diabetes and their relatives. Diabetes. 2006;55:1443–1449. doi: 10.2337/db05-1593. [DOI] [PubMed] [Google Scholar]
- 13.Sturgeon C, Fasano A. Zonulin, a regulator of epithelial and endothelial barrier functions, and its involvement in chronic inflammatory diseases. Tissue Barriers. 2016;4:e1251384. doi: 10.1080/21688370.2016.1251384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tripathi A, Lammers KM, Goldblum S, et al. Identification of human zonulin, a physiological modulator of tight junctions, as prehaptoglobin-2. Proc Natl Acad Sci U S A. 2009;106:16799–16804. doi: 10.1073/pnas.0906773106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fasano A. Zonulin and its regulation of intestinal barrier function: the biological door to inflammation, autoimmunity, and cancer. Physiol Rev. 2011;91:151–175. doi: 10.1152/physrev.00003.2008. [DOI] [PubMed] [Google Scholar]
- 16.Fasano A, Fiorentini C, Donelli G, et al. Zonula occludens toxin modulates tight junctions through protein kinase C-dependent actin reorganization, in vitro. J Clin Invest. 1995;96:710–720. doi: 10.1172/JCI118114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goldblum SE, Rai U, Tripathi A, et al. The active Zot domain (aa 288–293) increases ZO-1 and myosin 1C serine/threonine phosphorylation, alters interaction between ZO-1 and its binding partners, and induces tight junction disassembly through proteinase activated receptor 2 activation. FASEB J. 2011;25:144–158. doi: 10.1096/fj.10-158972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smecuol E, Sugai E, Niveloni S, et al. Permeability, zonulin production, and enteropathy in dermatitis herpetiformis. Clin Gastroenterol Hepatol. 2005;3:335–341. doi: 10.1016/s1542-3565(04)00778-5. [DOI] [PubMed] [Google Scholar]
- 19.Papp M, Foldi I, Nemes E, et al. Haptoglobin polymorphism: a novel genetic risk factor for celiac disease development and its clinical manifestations. Clinical Chemistry. 2008;54:697–704. doi: 10.1373/clinchem.2007.098780. [DOI] [PubMed] [Google Scholar]
- 20.Marquez L, Shen C, Cleynen I, et al. Effects of haptoglobin polymorphisms and deficiency on susceptibility to inflammatory bowel disease and on severity of murine colitis. Gut. 2012;61:528–534. doi: 10.1136/gut.2011.240978. [DOI] [PubMed] [Google Scholar]
- 21.Papp M, Lakatos PL, Palatka K, et al. Haptoglobin polymorphisms are associated with Crohn’s disease, disease behavior, and extraintestinal manifestations in Hungarian patients. Dig Dis Sci. 2007;52:1279–1284. doi: 10.1007/s10620-006-9615-1. [DOI] [PubMed] [Google Scholar]
- 22.Maes M, Delanghe J, Bocchio Chiavetto L, et al. Haptoglobin polymorphism and schizophrenia: genetic variation on chromosome 16. Psychiatry Res. 2001;104:1–9. doi: 10.1016/s0165-1781(01)00298-0. [DOI] [PubMed] [Google Scholar]
- 23.Levy AP, Levy JE, Kalet-Litman S, et al. Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2007;27:134–140. doi: 10.1161/01.ATV.0000251020.24399.a2. [DOI] [PubMed] [Google Scholar]
- 24.Arrieta MC, Madsen K, Doyle J, et al. Reducing small intestinal permeability attenuates colitis in the IL10 gene-deficient mouse. Gut. 2009;58:41–48. doi: 10.1136/gut.2008.150888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Watts T, Berti I, Sapone A, et al. Role of the intestinal tight junction modulator zonulin in the pathogenesis of type I diabetes in BB diabetic-prone rats. Proc Natl Acad Sci U S A. 2005;102:2916–2921. doi: 10.1073/pnas.0500178102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fasano A, Uzzau S, Fiore C, et al. The enterotoxic effect of zonula occludens toxin on rabbit small intestine involves the paracellular pathway. Gastroenterology. 1997;112:839–846. doi: 10.1053/gast.1997.v112.pm9041245. [DOI] [PubMed] [Google Scholar]
- 27.Okada H, Kuhn C, Feillet H, et al. The ‘hygiene hypothesis’ for autoimmune and allergic diseases: an update. Clin Exp Immunol. 2010;160:1–9. doi: 10.1111/j.1365-2249.2010.04139.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Progress in Autoimmune Diseases Research. Report to Congress. National Institutes of Health, The Autoimmune Diseases Coordinating Committee; Mar, 2005. [Google Scholar]
- 29.National Institues of Health Autoimmune Diseases Research Plan. 2002 [Google Scholar]
- 30.Tao B, Pietropaolo M, Atkinson M, et al. Estimating the cost of type 1 diabetes in the U.S.: a propensity score matching method. PLoS One. 2010;5:e11501. doi: 10.1371/journal.pone.0011501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arrieta MC, Bistritz L, Meddings JB. Alterations in intestinal permeability. Gut. 2006;55:1512–1520. doi: 10.1136/gut.2005.085373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fasano A, Shea-Donohue T. Mechanisms of disease: the role of intestinal barrier function in the pathogenesis of gastrointestinal autoimmune diseases. Nat Clin Pract Gastroenterol Hepatol. 2005;2:416–422. doi: 10.1038/ncpgasthep0259. [DOI] [PubMed] [Google Scholar]
- 33.Monsuur AJ, de Bakker PI, Alizadeh BZ, et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat Genet. 2005;37:1341–1344. doi: 10.1038/ng1680. [DOI] [PubMed] [Google Scholar]
- 34.Wapenaar MC, Monsuur AJ, van Bodegraven AA, et al. Associations with tight junction genes PARD3 and MAGI2 in Dutch patients point to a common barrier defect for coeliac disease and ulcerative colitis. Gut. 2008;57:463–467. doi: 10.1136/gut.2007.133132. [DOI] [PubMed] [Google Scholar]
- 35.Chieppa M, Rescigno M, Huang AY, et al. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med. 2006;203:2841–2852. doi: 10.1084/jem.20061884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mowat AM, Millington OR, Chirdo FG. Anatomical and cellular basis of immunity and tolerance in the intestine. J Pediatr Gastroenterol Nutr. 2004;39(Suppl 3):S723–724. doi: 10.1097/00005176-200406003-00003. [DOI] [PubMed] [Google Scholar]
- 37.Rescigno M, Lopatin U, Chieppa M. Interactions among dendritic cells, macrophages, and epithelial cells in the gut: implications for immune tolerance. Curr Opin Immunol. 2008;20:669–675. doi: 10.1016/j.coi.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 38.Brandtzaeg P. Homeostatic impact of indigenous microbiota and secretory immunity. Benef Microbes. 2010;1:211–227. doi: 10.3920/BM2010.0009. [DOI] [PubMed] [Google Scholar]
- 39.Gloria-Bottini F, Lucarelli P, Saccucci P, et al. Genetic polymorphism and idiopathic generalized epilepsy. Evidence of interaction between haptoglobin and ACP1 systems. Neuropediatrics. 2008;39:357–358. doi: 10.1055/s-0029-1202834. [DOI] [PubMed] [Google Scholar]
- 40.Nakhoul FM, Zoabi R, Kanter Y, et al. Haptoglobin phenotype and diabetic nephropathy. Diabetologia. 2001;44:602–604. doi: 10.1007/s001250051666. [DOI] [PubMed] [Google Scholar]
- 41.Nakhoul FM, Marsh S, Hochberg I, et al. Haptoglobin genotype as a risk factor for diabetic retinopathy. JAMA. 2000;284:1244–1245. doi: 10.1001/jama.284.10.1244-a. [DOI] [PubMed] [Google Scholar]
- 42.Calderoni DR, Andrade Tdos S, Grotto HZ. Haptoglobin phenotype appears to affect the pathogenesis of American trypanosomiasis. Ann Trop Med Parasitol. 2006;100:213–221. doi: 10.1179/136485906X86356. [DOI] [PubMed] [Google Scholar]
- 43.Jorge SE, Abreu CF, Guariento ME, et al. Haptoglobin genotypes in Chagas’ disease. Clin Biochem. 2010;43:314–316. doi: 10.1016/j.clinbiochem.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 44.Lim YK, Jenner A, Ali AB, et al. Haptoglobin reduces renal oxidative DNA and tissue damage during phenylhydrazine-induced hemolysis. Kidney Int. 2000;58:1033–1044. doi: 10.1046/j.1523-1755.2000.00261.x. [DOI] [PubMed] [Google Scholar]
- 45.Kelly CP, Green PH, Murray JA, et al. Larazotide acetate in patients with coeliac disease undergoing a gluten challenge: a randomised placebo-controlled study. Aliment Pharmacol Ther. 2013;37:252–262. doi: 10.1111/apt.12147. [DOI] [PubMed] [Google Scholar]
- 46.Khaleghi S, Ju JM, Lamba A, et al. The potential utility of tight junction regulation in celiac disease: focus on larazotide acetate. Therap Adv Gastroenterol. 2016;9:37–49. doi: 10.1177/1756283X15616576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurada S, Yadav A, Leffler DA. Current and novel therapeutic strategies in celiac disease. Expert Rev Clin Pharmacol. 2016;9:1211–1223. doi: 10.1080/17512433.2016.1200463. [DOI] [PubMed] [Google Scholar]
- 48.Leffler DA, Kelly CP, Abdallah HZ, et al. A randomized, double-blind study of larazotide acetate to prevent the activation of celiac disease during gluten challenge. Am J Gastroenterol. 2012;107:1554–1562. doi: 10.1038/ajg.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leffler DA, Kelly CP, Green PH, et al. Larazotide acetate for persistent symptoms of celiac disease despite a gluten-free diet: a randomized controlled trial. Gastroenterology. 2015;148:1311–1319 e1316. doi: 10.1053/j.gastro.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paterson BM, Lammers KM, Arrieta MC, et al. The safety, tolerance, pharmacokinetic and pharmacodynamic effects of single doses of AT-1001 in coeliac disease subjects: a proof of concept study. Aliment Pharmacol Ther. 2007;26:757–766. doi: 10.1111/j.1365-2036.2007.03413.x. [DOI] [PubMed] [Google Scholar]
- 51.Wyatt J, Vogelsang H, Hubl W, et al. Intestinal permeability and the prediction of relapse in Crohn’s disease. Lancet. 1993;341:1437–1439. doi: 10.1016/0140-6736(93)90882-h. [DOI] [PubMed] [Google Scholar]
- 52.Buhner S, Buning C, Genschel J, et al. Genetic basis for increased intestinal permeability in families with Crohn’s disease: role of CARD15 3020insC mutation? Gut. 2006;55:342–347. doi: 10.1136/gut.2005.065557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peeters M, Geypens B, Claus D, et al. Clustering of increased small intestinal permeability in families with Crohn’s disease. Gastroenterology. 1997;113:802–807. doi: 10.1016/s0016-5085(97)70174-4. [DOI] [PubMed] [Google Scholar]
- 54.Teahon K, Smethurst P, Levi AJ, et al. Intestinal permeability in patients with Crohn’s disease and their first degree relatives. Gut. 1992;33:320–323. doi: 10.1136/gut.33.3.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Irvine EJ, Marshall JK. Increased intestinal permeability precedes the onset of Crohn’s disease in a subject with familial risk. Gastroenterology. 2000;119:1740–1744. doi: 10.1053/gast.2000.20231. [DOI] [PubMed] [Google Scholar]
- 56.Vanuytsel T, Vermeire S, Cleynen I. The role of Haptoglobin and its related protein, Zonulin, in inflammatory bowel disease. Tissue Barriers. 2013;1:e27321. doi: 10.4161/tisb.27321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Clemente MG, De Virgiliis S, Kang JS, et al. Early effects of gliadin on enterocyte intracellular signalling involved in intestinal barrier function. Gut. 2003;52:218–223. doi: 10.1136/gut.52.2.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Drago S, El Asmar R, Di Pierro M, et al. Gliadin, zonulin and gut permeability: Effects on celiac and non-celiac intestinal mucosa and intestinal cell lines. Scand J Gastroenterol. 2006;41:408–419. doi: 10.1080/00365520500235334. [DOI] [PubMed] [Google Scholar]
- 59.Rittirsch D, Flierl MA, Nadeau BA, et al. Zonulin as prehaptoglobin2 regulates lung permeability and activates the complement system. Am J Physiol Lung Cell Mol Physiol. 2013;304:L863–872. doi: 10.1152/ajplung.00196.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shirey KA, Lai W, Patel MC, et al. Novel strategies for targeting innate immune responses to influenza. Mucosal Immunol. 2016;9:1173–1182. doi: 10.1038/mi.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Body weight and colonic weight and length in both male and female Ztm compared with WT mice.
Figure S2. Ex vivo permeability measurement by cascade blue dextran flux in the colon of either untreated or DSS-treated Ztm and WT mice mounted in the microsnapwell system.
Figure S3. Effect of AT1001 on ex vivo permeability measurement by cascade blue dextran flux in the colon of DSS-treated Ztm mounted in the microsnapwell system.