

Abstract
The bacterial receptor, Toll-like receptor (TLR) 4 mediates inflammatory responses and have been linked to a broad array of diseases. TLR4 agonists are being explored as potential treatments for cancer and other diseases. We have previously shown that activation of TLR4 by lipopolysaccharide (LPS) leads to down-regulation of drug metabolizing enzymes/transporters (DMETs), and altered pharmacokinetics/pharmacodynamics (PK/PD) of drugs. These changes can increase the risk of drug-drug interactions (DDIs) in patients on multiple medications. Clinically, DDI was observed for combination chemotherapy of paclitaxel (TLR4 ligand) and irinotecan. To determine the role of TLR4 in DDI between paclitaxel and irinotecan in vitro, primary hepatocytes from TLR4-wild-type (WT) and mutant mice were pre-treated with paclitaxel, followed by irinotecan. Gene expression of DMETs was determined. Paclitaxel treatment increased the levels of irinotecan metabolites, SN-38 and SN-38 glucuronide (SN-38G) in TLR4-dependent manner. Paclitaxel-mediated induction of genes involved in irinotecan metabolism such as Cyp3a11 and Ugt1a1 was TLR4-dependent, while induction of the transporter Mrp2 was TLR4-independent. These novel findings demonstrate that paclitaxel can affect irinotecan metabolism by a TLR4-dependent mechanism. This provides a new perspective towards evaluation of marketed drugs according to their potential to exert DDIs in TLR4-dependent manner.
Keywords: Paclitaxel, Irinotecan, Toll-like receptor 4, Drug-drug interactions, Drug metabolizing enzymes
Introduction
Toll-like receptor 4 (TLR4) is a transmembrane receptor that detects components of microbial pathogens and plays a critical role in innate immunity (Marshak-Rothstein, 2006). Emerging genetic data also support the association of TLR4 with several diseases (Bochud et al., 2009; de Oliveira and Silva, 2012). Thus, activating or suppressing TLR4 provides access to a new generation of therapeutics (Manthey et al., 1993; Thoelen et al., 2001; Mullarkey et al., 2003; Ziakas et al., 2013).
Currently, TLR4 agonists are being explored extensively as immunomodulators and vaccine adjuvants for allergic diseases, cancers and infectious diseases (Hawkins et al., 2002; Evans et al., 2003; Lee et al., 2003; Przetak et al., 2003; Stover et al., 2004; Cluff et al., 2005; Krieg, 2006; Kanzler et al., 2007; Casella and Mitchell, 2008). Additionally, medications including opioid agonists (Hutchinson et al., 2010a; Hutchinson et al., 2010b) and widely-used chemotherapy drug, Paclitaxel (Byrd-Leifer et al., 2001; Zimmer et al., 2008) have been reported to activate TLR4 signaling in macrophages. Studies show that TLR4 signaling enhances resistance to paclitaxel therapy in breast and ovarian cancers (Rajput et al., 2013; Wang et al., 2014), and contributes to paclitaxel-induced peripheral neuropathy (Li et al., 2014).
Marked antitumor activity of paclitaxel in ovarian and breast cancer has led to its extensive evaluation as combination therapy with other cytotoxic agents for various metastatic cancers (Bellmunt et al., 2000; Kondagunta et al., 2005; Grau et al., 2009; Li et al., 2011). However, pharmacokinetics (PK) drug-drug interactions (DDIs) pose major clinical problem with such combination therapy. In a Phase-I study, DDI was observed in patients with advanced small non-small cell lung cancer (NSCLC) (Hotta et al., 2004) treated with paclitaxel and the potent anticancer drug, irinotecan. In this study, pre-treatment of paclitaxel, followed by irinotecan, increased the area under curve (AUC) of the toxic metabolite of irinotecan, 7-ethyl-10-hydroxycamptothecin (SN-38) (Kasai et al., 2002).
The plasma level of both paclitaxel and irinotecan is determined by drug metabolism and disposition. Paclitaxel is an auto inducer, and induces drug metabolizing enzymes (DMEs) and transporters involved in its disposition (Nallani et al., 2003). It undergoes metabolism via cytochrome P450 (CYP) 2C8 (Rahman et al., 1994; Dai et al., 2001) and 3A4 (Cresteil et al., 1994; Harris et al., 1994); with biliary excretion via MDR1B (also known as P-glycoproteins [Pgp]) (Monsarrat et al., 1993; Kang et al., 2001). Irinotecan, a topoisomerase I inhibitor, is metabolized by two pathways (Fig. 1) (i) bio activation by carboxylesterases (CES) to form active and toxic metabolite, SN-38 (Satoh et al., 1994; Slatter et al., 1997) which is detoxified to SN-38 glucuronide (SN-38G) by uridine diphosphate glucuronosyltransferases (UGT) 1A1; (ii) oxidation by CYP3A4/5 enzyme (Santos et al., 2000) to form APC (7-ethyl-10[4-N-(5-aminopentanoicacid)-1-piperidino] carbonyloxycamptothecin) and NPC (7-ethyl-10[4-amino-1-piperidino]carbonyloxycamptothecin). NPC is converted to SN-38 by CES. Irinotecan and its metabolites are predominantly eliminated in the bile. Accumulation of SN-38 in the intestine is primarily due to deconjugation of SN-38G to SN-38 by bacterial β-glucuronidase, which accounts for life-threatening diarrhea (Sparreboom et al., 1998; Buajordet et al., 2001).
Figure 1.
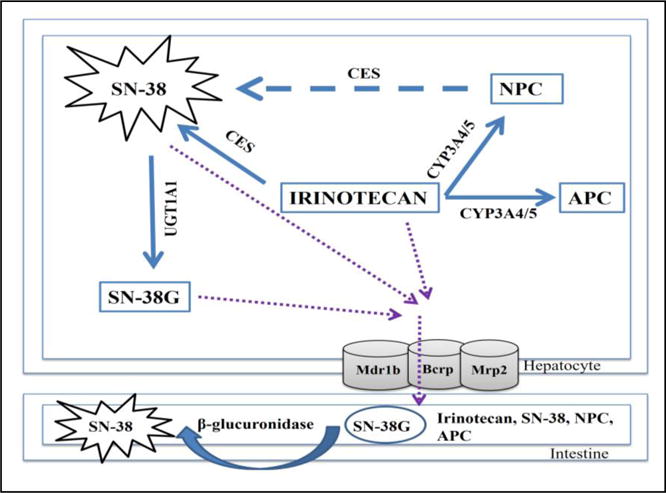
Schematic representation of the metabolic pathway of irinotecan.
We have previously shown that activation of TLR4 by lipopolysaccharide (LPS) downregulate DME/transporters (DMET) genes (Ghose et al., 2009) leading to altered PK/pharmacodynamics (PD) of drugs (Gandhi et al., 2012). We observed that downregulation of DMETs by LPS was associated with reduced expression of the xenobiotic nuclear receptor (NR), pregnane X receptor (PXR) (Ghose et al., 2004a). PXR forms a dimer with the central NR, retinoid X receptor (RXR)-alpha to regulate DMET genes. It is well-established that alterations in DMET expression/activity can cause DDIs (Kohler et al., 2000; Polasek et al., 2011). Since TLR4 is activated by paclitaxel, we hypothesized that paclitaxel will cause TLR4-mediated alterations in DMETs, leading to changes in irinotecan PK. We found that in TLR4-wild type (WT) hepatocytes, paclitaxel increased the levels of SN-38 and SN-38G, while no such induction was observed in TLR4-mutant hepatocytes. Gene expression of Cyp3a11 and Ugt1a1 was induced up to ~ 600 and ~ 4 fold respectively by paclitaxel in TLR4-WT hepatocytes. In the TLR4-mutant hepatocytes Cyp3a11 was induced ~300-fold by paclitaxel, while Ugt1a1 expression was not increased. Thus, our finding proposes a novel mechanism of regulation of DDIs by TLR4 and indicates two possible unwanted effects due to TLR4 involvement in regulation of DMEs, (i) SN-38 levels may be affected in individuals with TLR4 polymorphism, resulting in changes irinotecan efficacy and safety; and (ii) TLR4 agonists pose potential for DDIs when given as combination therapy with other drugs.
Materials and Methods
Chemicals
Camptothecin (CPT; internal standard (I.S)), Irinotecan hydrochloride (Cat # I1406), Paclitaxel (Cat # T7402) and 4-Nitrophenyl acetate (PNPA; Cat # N8130) were purchased from Sigma-Aldrich, St. Louis, MO. SN-38 and SN-38G were kind gifts from Dr. Ming Hu’s laboratory at the University of Houston, Houston, TX. The sequences of the primers and probes were reported in our previous publications (Ghose et al., 2007; 2008; 2009). Bicinchoninic acid (BCA) assay kit (Cat # 23225) was purchased from Thermo Fisher Scientific Inc. All other reagents for real-time PCR were purchased from Applied Biosystems (Foster City, CA). Liquid chromatography mass spectrometry (LCMS) grade solvents were purchased from VWR international, LLC (Suwanee, GA, USA) for chromatography. Unless specified, all other materials were purchased from Sigma-Aldrich (St Louis, MO, USA.).
Animals
Adult, male, 6–8 week old C3HeB/FeJ (TLR4-wildtype; TLR4-WT) and C3H/HeJ (TLR4-mutant) mice were purchased from Jackson Labs, Bar Harbor, ME. The C3H/HeJ mice are homozygous mutants that have spontaneous mutation in TLR4 gene as a result of a missense mutation (proline → histidine) at codon 712 (Poltorak et al., 1998). TLR4-mutant mice exhibit a defective response to LPS stimulation. They are genetically similar to TLR4-WT (C3HeB/FeJ) mice but display significantly reduced expression of pro-inflammatory genes compared TLR4-WT mice (Ghose et al., 2008). All animals were maintained in a 12 h dark/light cycle and a temperature-and-humidity-controlled environment. The mice had access to rodent chow ad libitum. All the animal care and use protocols were approved by the Institutional Animal Care and Use Committee guidelines. All experiments were performed in triplicate and repeated at least three times.
Primary mouse hepatocyte culture
Primary hepatocytes were isolated from adult male mice according to the two-step perfusion procedure, as described previously (Li and Koda, 2002; Ghose et al., 2011; Shah et al., 2014) with some modifications (Ghose et al., 2016). Cell viability was measured using trypan exclusion method. Cells plated at a density of 250,000 cells/ml in six-well Primaria plates (BD, Franklin Lakes, NJ) were allowed to attach for 4 h in Williams E medium (Invitrogen) containing the following; 10,000 U/ml of Penicillin/streptomycin solution (Invitrogen), 200 mM of L-glutamine, 5 mg of gentamicin, 5 μg/ml Insulin-transferrin-sodium selenite [ITS], 4 ng/ml glucagon and 10% fetal bovine serum [FBS] (Invitrogen). Hepatocyte preparations with more than 85% viability were used in experiments. Cells were maintained for 48 h with a daily change of medium.
Cell treatments
After 48 h in culture, on the day of treatment, primary mouse hepatocytes were incubated with serum-free Williams E treatment media 2 h prior to treatment with drugs. For gene expression studies, cells were incubated with vehicle (< 0.1% DMSO) or paclitaxel for 4 and 24 h. The assay was terminated by addition of 0.25 ml of cold TRIzol reagent to each well. Cells were harvested for RNA isolation for real-time PCR analysis. For DDI studies, cells were pretreated with either vehicle or 20 μM paclitaxel for 24 h to induce DMEs, followed by addition of irinotecan (20 and 50 μM) or SN-38 (5 μM) for additional 48 h. Supernatants were collected at different time points till 48 h. The concentrations of SN-38 and SN-38G were quantified by LCMS/MS. All experiments were performed in triplicate.
Preparation of microsomes
Microsomes were prepared from the hepatocytes as described in our previous publication Briefly, hepatocytes were homogenized using a motorized homogenizer in ice-cold homogenization buffer [50 mM potassium phosphate buffer (pH 7.4), 250 mM sucrose, 1 mM EDTA] and centrifuged at 18 500×g for 15 min at 4°C. The pellet was discarded, and the supernatant was collected and centrifuged again at 85 600×g for 60 min at 4°C to yield the microsome pellets. The microsomal pellet was resuspended in 250 mM sucrose. Protein concentration was determined using a BCA protein assay kit (Pierce, Rockford, IL) using BSA as the standard.
Immunoblotting
Mouse liver nuclear and whole cell extracts containing equal amounts of protein were separated by 10% SDS-polyacrylamide gel electrophoresis and transferred to Trans-blot membranes (Bio-Rad, Hercules, CA). Membranes were blocked for 1 h at room temperature in 5% nonfat dry milk or 5% BSA (for phosphoproteins) dissolved in Tris-buffered saline with Tween-20 (TBST) prior to incubation with primary antibodies. Membranes were subsequently washed and probed with a goat anti-rabbit IgG-AP secondary antibody (1:2000) for 1 h at room temperature. The blots were then washed with TBST and incubated with Tropix CDP Star® Nitro block II™ ECL reagent according to the manufacturer’s instructions (Applied Biosystems, Foster City, CA). Protein bands were analyzed and quantified by FluorChem FC Imaging System (Alpha Innotech, San Leandro, CA).
Real-Time PCR
Total RNA was isolated from mouse liver using TRIzol reagent (Sigma-Aldrich, St Louis, MO, U.S.A) according to the manufacturer’s protocol. cDNA was synthesized using the High Capacity Reverse Transcription Kit from Applied Biosystems. Real-time PCR was performed using an ABI PRISM 7300 Sequence Detection System instrument and software (Applied Biosystems) as described previously (Ghose et al., 2004a). Briefly, each reaction mixture (total of 25 μL) contained 50–100 ng of cDNA, 300 nM forward primer, 300 nM reverse primer, 200 nM fluorogenic probe, and 15 μL of TaqMan Universal PCR Master Mix. The sequences of the primers and probes were reported in our previous publications (Ghose et al., 2007; 2008; 2009). Quantitative expression values were normalized to cyclophilin.
Liquid chromatography mass spectrometry (LC/MS) analysis of SN38 and SN-38G in hepatocyte media
The assay supernatants were collected at 0, 5, 15, 30, 60, 120, 240 and 480 min. Seventy five μL of the supernatant was first acidified with 20 μL of 90% acetonitrile/10% water/6% glacial acetic acid containing 0.5 mg/ml CPT (internal standard) followed by acetonitrile protein precipitation. The mixture was vortexed rigorously for 1 min and centrifuged at 13,000 rpm for 15 min at room temperature. The supernatants were air dried and reconstituted with 50% acetonitrile/50% methanol/1% glacial acetic acid. Quantification of SN-38 and SN-38G was performed using API 5500 Qtrap triple quadrupole mass spectrophotometer (AB Sciex, USA) equipped with a Turbospray™ source as described previously (Mallick et al., 2015; Basu et al., 2016). LCMS/MS data is reported as fold change in metabolite levels in the presence or absence of paclitaxel pretreatment. The rate of SN-38 or SN-38G formation over time is the slope derived from curve fitting to the data using GraphPad Prism 5.2 software.
Carboxylesterase (CES) activity
Whole cell extracts were prepared as described previously (Rahman et al., 1994; Zimmer et al., 2008). Protein concentration was determined by BCA assay according to the manufacturer’s protocol (Pierce Chemical, Rockford, IL). Total CES activity was monitored by a spectrophotometric method described previously (Brzezinski et al., 1994). Briefly, whole cell extracts were incubated with 750 μM PNPA at 37°C. The formation of p-nitrophenolate (PNP) at 405 nm was monitored from 10 to 30 min (Mallick et al., 2015). The activity was linear over this time period. The specific activity is expressed as μmoles PNP formed/min/mg protein.
Statistical analysis
All data are presented as mean ± S.D from experiments on three different hepatocyte preparations. The data were analyzed by un-paired Student’s t-test and ANOVA using GraphPad Prism 5.2 software (GraphPad Inc., La Jolla, CA) where p < 0.05 was considered to be statistically significant.
Results
PK DDIs between paclitaxel and irinotecan is mediated by TLR4 in vitro
TLR4-WT and mutant hepatocytes were treated with either 20 μM paclitaxel or vehicle (0.1% DMSO) for 24 h followed by incubation with irinotecan or SN-38. In several metabolism studies in hepatocytes, paclitaxel has been used in concentrations ranging from 5 to 50 μM (Kostrubsky et al., 1999). As in our preliminary studies with concentrations ranging from 1 to 20 μM, paclitaxel had significant and consistent effect on Cyp3a11 gene expression at 20 μM (data not shown), therefore, this concentration was used for all studies. The concentration of irinotecan (20 and 50 μM) and SN-38 (5 μM) used are in therapeutic range which is ~10–20 times of their Cmax in blood (Irinotecan: 3392 ± 874 ng/ml and SN38: 56 ± 28.2 ng/ml were achieved at irinotecan dose of 340 mg/kg/i.p) (Pharmacia, 1996). Supernatants were collected at different time points from 0 to 48 h, and analyzed for SN-38 and SN-38G levels by LCMS/MS. To avoid underestimation of DDIs due to loss of cell viability and enzyme stability, incubations were done till 48 h. In TLR4-WT hepatocytes, 24 h paclitaxel pretreatment significantly increased the levels of SN-38 by ~2-fold at time points ranging from 18 to 48 h at both concentrations of irinotecan (Fig. 2). Rate of SN-38 formation increased with paclitaxel (i) ~ 45% with 20 μM irinotecan (0.20 ± 0.03 vs 0.177 ± 0.03 nM/min, p < 0.05) and (ii) ~ 32% with 50 μM irinotecan (15 ± 1.5 vs 12 ± 0.5 nM/min, p < 0.05) as shown in Fig. 2A and B. Interestingly, in TLR4-mutant hepatocytes there was no significant increase (<10%) in rate of SN-38 formation in presence of paclitaxel (i) with 20 μM irinotecan (0.04 ± 0.02 vs 0.03 ± 0.01 nM/min, p > 0.05) and (ii) with 50 μM irinotecan (8.9 ± 8 vs 8.7± 7.55 nM/min, p > 0.05) as shown in Fig. 2A and B.
Figure 2. Effect of paclitaxel on irinotecan metabolism is mediated by TLR4.
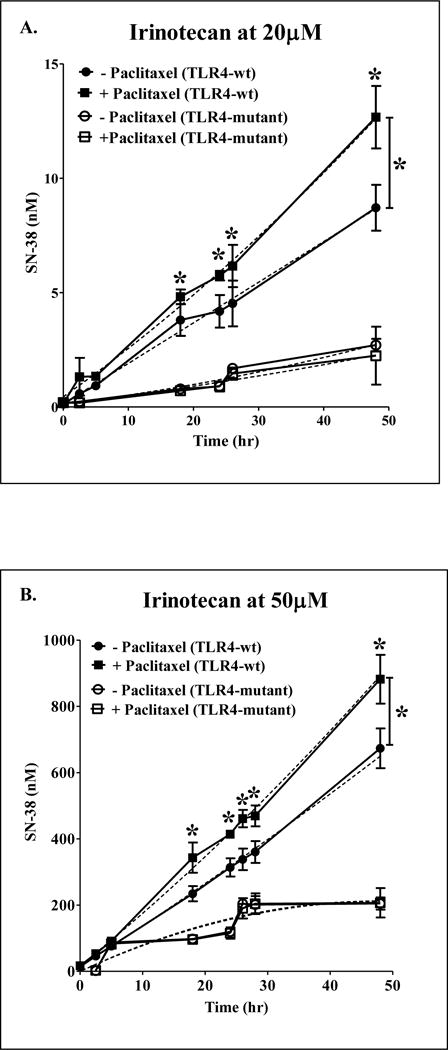
Primary mouse hepatocytes were treated with 0.1% DMSO (vehicle) or 20 μM of paclitaxel for 24 h. After 24 h, the hepatocytes were treated with two concentrations of irinotecan, 20 μM (A); 50 μM (B) for 48 h. Supernatants collected at different time points were analyzed for levels of SN-38 by LCMS/MS. ■ indicates paclitaxel + irinotecan group in TLR4-WT; ● indicates only irinotecan in TLR4-WT; □ indicates paclitaxel + irinotecan group in TLR4-mutant; ○ indicates only irinotecan group in TLR4-mutant. The secondary order polynomial fitting describes the continuous metabolic data for different time points. n = 3 per group. All data are presented as ± S.D. (*) indicate significant difference (p < 0.05) between irinotecan and paclitaxel + irinotecan of TLR4-WT groups with respect to % induction of SN-38 levels by paclitaxel. The experiments were repeated at least thrice.
The TLR4-mediated effect of paclitaxel on SN-38G formation was studied using 5 μM SN-38 as the substrate concentration. This concentration of SN-38 has been previously used by Iyer et al to understand the Ugt1a1 inducibility by phenobarbital in human hepatocytes (Iyer et al., 2002a; 2002b). As shown in Fig. 3, paclitaxel significantly increased the levels of SN-38G by ~100% in TLR4-WT and rate of SN-38G formation was 0.23 ± 0.04 vs 0.094 ± 0.02 nM/min, p < 0.05). In TLR4-mutant hepatocytes, there was no significant increase in rate of SN-38G formation in the presence of paclitaxel (0.053 ± 0.008 vs 0.07 ± 0.012 nM/min, p > 0.05). Thus, our overall data demonstrates the significant role of TLR4 in PK alteration of irinotecan by paclitaxel in vitro.
Figure 3. Effect of paclitaxel on SN38 glucuronidation is mediated by TLR4.
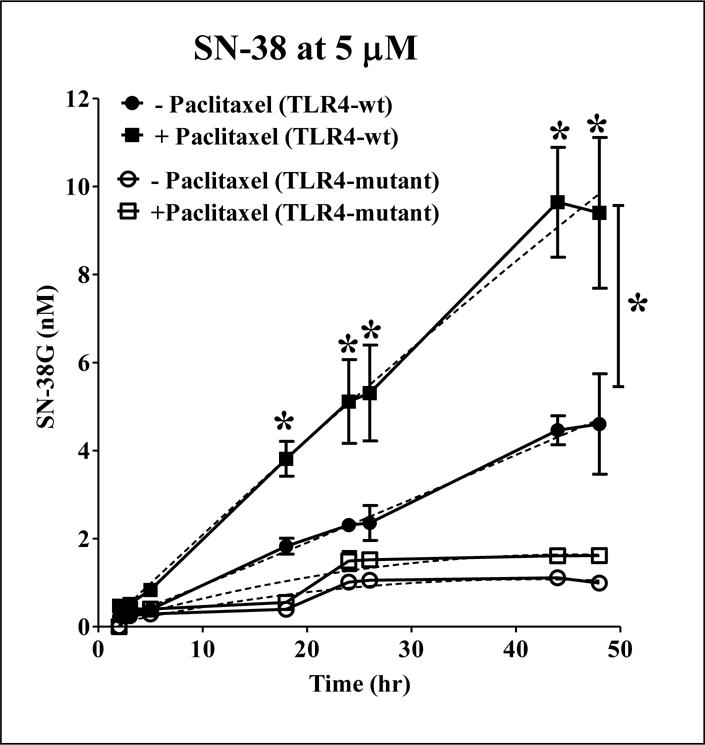
Primary mouse hepatocytes were treated with 0.1% DMSO (vehicle) or 20 μM of paclitaxel for 24 h, followed by treatment with 5 μM SN-38. Supernatants collected at different time points were analyzed for SN-38G levels by LCMS/MS. ■ indicates paclitaxel + irinotecan group in TLR4-WT; ● indicates only irinotecan in TLR4-WT; □ indicates paclitaxel + irinotecan group in TLR4-mutant; ○ indicates only irinotecan group in TLR4-mutant. The secondary order polynomial fitting describes the continuous metabolic data for different time points. n = 3 per group. All data are presented as ± S.D. (*) indicate significant difference (p < 0.05) between irinotecan and paclitaxel + irinotecan of TLR4-WT groups with respect to % induction of SN-38 levels by paclitaxel. The experiments were repeated at least thrice.
TLR4-mediated effect of paclitaxel on key enzymes and transporters primarily involved in irinotecan metabolism
Efficacy and toxicity of irinotecan is dependent on its metabolite, SN-38. The DMET(s), UGT1A, CYP3A, and ABC gene families (MRP2 and MDR1B) primarily regulate SN-38 levels in vivo (Fig. 1). Therefore, we studied the effect of paclitaxel on expressions of Cyp3a11, Ugt1a1, Mrp2 and Mdr1b genes by RTPCR and activity of carboxylesterase (CES) by colorimetric assay (Fig. 4). Previous studies have shown that paclitaxel induces CYP3A gene expression and activity in rat and human hepatocytes (Kostrubsky et al., 1999; Synold et al., 2001; Nallani et al., 2003). TLR4 activation by LPS downregulated Cyp3a11 and Ugt1a1 genes (Ghose et al., 2008). Primary hepatocytes from TLR4-WT and mutant mice were treated with 20 μM of paclitaxel for 24 h. As shown in Fig. 4A, in TLR4-WT hepatocytes, paclitaxel significantly induced expression of Cyp3a11 and Ugt1a1 genes by 554 ± 151 and 3.4 ± 0.99 folds respectively, while in TLR4-mutant the induction for Cyp3a11 and Ugt1a1 genes were 228 ± 80 and 1.6 ± 0.55 folds, respectively. Therefore, in TLR4-mutant the induction of DME genes by paclitaxel was significantly attenuated.
Figure 4. Regulation of DME/transporter expression and CES activity in primary hepatocytes by paclitaxel.
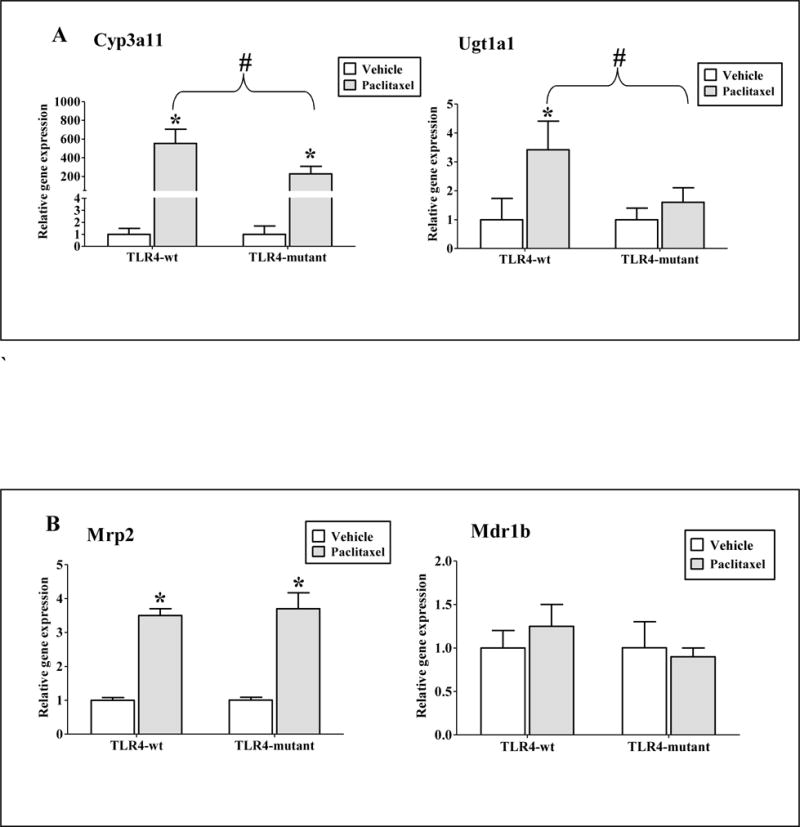
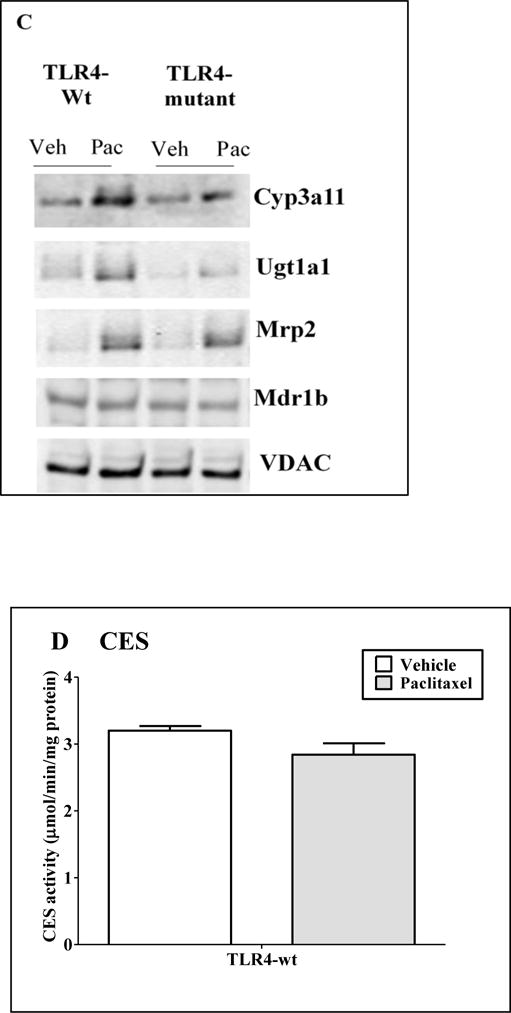
Primary mouse hepatocytes were treated with 0.1% DMSO (vehicle) or 20 μM paclitaxel for 24 h. Gene expression (A) Cyp3a11 and Ugt1a1 (B) Mrp2 and Mdr1b were determined. All data are presented as ± S.D. and standardized for cyclophilin mRNA levels. n = 3 per group. Expression in DMSO group was set to 1, fold change after paclitaxel treatment at 24 h was compared to vehicle. (*) indicates significant difference (p < 0.05) between vehicle and paclitaxel groups; (#) indicates significant differences (p < 0.05) between paclitaxel treated samples of TLR4-wt and mutant groups. Experiments were repeated at least thrice. (C) Representative western blots from n ≥ 3 blots of hepatocytes treated with DMSO (Veh) or paclitaxel (Pac) for 24h. (D) CES activity: Whole cell extracts were prepared and CES activity was determined as described earlier. Activity data are presented as ± S.D. n = 3 per group. Experiments were repeated with three different hepatocyte preparations.
Biliary excretion of irinotecan, SN-38 and SN-38G involves several ATP-binding cassette transporters, including MDR1B and MRP2 (Fig. 1) (Chu et al., 1998). Paclitaxel significantly induced gene expression of hepatic transporter, Mrp2 to similar extent in both TLR4-WT (3.5 ± 0.2 folds, p < 0.05) and mutant (4.2 ± 0.42 folds, p < 0.050) hepatocytes. Paclitaxel did not induce Mdr1b genes in either TLR4-WT or TLR4-mutant hepatocytes (1.25 ± 0.25 folds, p > 0.05, Fig. 4B). There was no difference in basal RNA levels of these hepatic transporters between both the TLR4-WT and mutant hepatocytes (data not shown). This indicates that TLR4 is likely not involved in regulating the expression levels of hepatic transporters by paclitaxel.
In the present study, we determined the effect of paclitaxel on protein expression of Cyp3a11, Ugt1a1, Mrp2 and Mdr1b in both TLR4-WT and mutant hepatocytes (Fig. 4C). Our protein and gene expression data were in agreement. We observed up-regulation of Cyp3a11 and Ugt1a1 protein expression by paclitaxel in TLR4-depenedent manner, whereas the increased expression of Mrp2 protein was independent of TLR-4. Expression of Mdr1b protein remained unchanged with paclitaxel treatment.
Hepatic CES is the principal non-CYP based Phase-I enzyme responsible for the activation of irinotecan to its active metabolite SN-38, in vivo (Satoh et al., 1994). Therefore, any alteration in CES activity by paclitaxel can contribute to the difference in levels of SN-38. We compared the CES activity using PNPA as a substrate in presence and absence of 20 μM paclitaxel in TLR4-WT hepatocytes, and our results showed that CES activity was not altered by paclitaxel (1.3 ± 0.08 μmol/min/mg protein, p > 0.05, Fig. 4D). These results suggest that TLR4 is involved in upregulation of Cyp3a11 and Ugt1a1 genes by paclitaxel, but not for Mrp2 genes. Interestingly, paclitaxel did not affect Mdr1b gene expression and CES activity.
TLR4 activation in mouse hepatocytes by paclitaxel
Paclitaxel is LPS mimetic, and studies have shown that it activates murine macrophages resulting in the secretion of inflammatory cytokines including tumor necrosis factor (TNF)-α, interleukin (IL) 6 and IL-8 in a TLR4-dependent manner (Byrd-Leifer et al., 2001; Kawasaki et al., 2001a). Likewise, paclitaxel (20 μM, 4 h treatment) induced mRNA levels of the pro-inflammatory cytokines, TNF-α, IL-6 and IL-1β in primary hepatocytes prepared from TLR4-WT mice (Fig. 5A). To evaluate the contribution of TLR4 in paclitaxel-mediated cytokine induction, we measured MRNA levels of these cytokines at 24 h, which is the time-point at which paclitaxel altered DME gene expression. TNF-α and IL-1β mRNA levels were not detected in the TLR4-WT or TLR4-mutant hepatocytes after 24 h of paclitaxel treatment, indicating that induction of these cytokines by paclitaxel was transient, and occurred at early time-points (data not shown). Interestingly, IL-6 mRNA levels were induced ~ 4 folds at 24 h after paclitaxel treatment (Fig. 5B), indicating a long-lasting IL-6 mRNA induction at the time-line corresponding to paclitaxel-mediated changes in DME gene expression. IL-6 mRNA induction by paclitaxel was attenuated in the TLR4-mutant hepatocytes (Fig. 5B). Although these experiments indicate the involvement of TLR4 in cytokine induction by paclitaxel, it is not possible to ascertain the relative contribution of individual cytokines in DME/transporter gene regulation by paclitaxel. This will be addressed in future studies.
Figure 5. Regulation of pro-inflammatory cytokine expression by Paclitaxel.
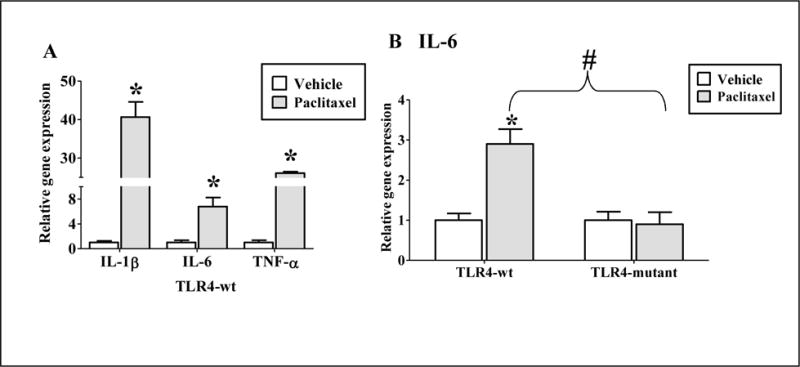
(A) Primary mouse hepatocytes from TLR4-wt mice were treated with 0.1% DMSO (vehicle) or 20 μM of paclitaxel for 4 h. (B) Primary mouse hepatocytes from TLR4-wt and mutant mice were treated with 0.1% DMSO (vehicle) or 20 μM of paclitaxel for 24 h. RNA was isolated from the livers and analyzed by real-time PCR. n = 3 per group. All data are presented as ± S.D. and standardized for cyclophilin mRNA levels. Expression in DMSO group was set to 1, fold change after paclitaxel treatment was compared to vehicle. (*) indicates significant difference (p < 0.05) between vehicle and paclitaxel groups; (#) indicates significant differences (p < 0.05) between paclitaxel treated samples of TLR4-wt and mutant groups. The experiments were repeated at least three times.
Discussion
Our current study demonstrates that the chemotherapy drug, paclitaxel activates TLR4 to alter the levels of irinotecan metabolites, SN-38 and SN-38G in primary mouse hepatocytes. This DDI correlated with TLR4-mediated induction of pro-inflammatory genes by paclitaxel. Furthermore, paclitaxel altered the expression of key enzymes involved in irinotecan metabolism in TLR4-dependent mechanism. Thus, our results demonstrate that TLR4 can be a novel mediator of paclitaxel and irinotecan DDI.
In the present study, paclitaxel pretreatment significantly increased the levels of both SN-38 and SN-38G in primary hepatocytes from TLR4-WT, but not TLR4-mutant mice (Fig. 2 & 3). This can be attributed to the complex metabolic pathways of irinotecan. To a large extent, SN-38 levels are determined by the balance between (i) formation of SN-38 from irinotecan and NPC and (ii) inactivation by glucuronidation (Fig. 1). In our study, paclitaxel induced the expression of Cyp3a11 and Ugt1a1 enzymes in TLR4-WT hepatocytes, but the induction was attenuated in TLR4-mutant hepatocytes; paclitaxel had no effect on CES activity (Fig. 5). Therefore, it is possible that paclitaxel induces Cyp3a11-mediated conversion of irinotecan to NPC, which is then converted to SN-38, resulting in higher SN-38 levels in TLR4-WT hepatocytes, but not the TLR4-mutant hepatocytes (Fig. 3). In our in vivo mice study also, paclitaxel at 40mg/kg/day/i.p induced Cyp3a11 gene (3.8 ± 0.4 fold, p < 0.05) in TLR4-dependent way (data not shown). Therefore, CYP3A will likely be involved in modulating the concentration of irinotecan metabolites by TLR4-dependent mechanism in paclitaxel-treated hepatocytes; while CES may not play a major role. Increased Ugt1a1expression by paclitaxel may lead to higher levels of SN-38G in TLR4-WT hepatocytes. However, further studies with specific inhibitors/activators are required to determine the role of these enzymes in paclitaxel and irinotecan DDI.
In addition to DMEs, impairment of drug transporters, MRP2 and MDR1B genes has been implicated in elevation in plasma levels of irinotecan and its metabolites (de Jong et al., 2007) but due to conflicting results, their role still remains inconclusive (Iyer et al., 2002b; Tagen et al., 2010). In our study, paclitaxel induced Mrp2 expression both in TLR4-WT and mutant hepatocytes but paclitaxel had no effect on Mdr1b expression (Fig. 4B). This result suggests that Mdr1b and Mrp2 transporters will unlikely be involved in the DDI of paclitaxel and irinotecan mediated by TLR4. However, the role of these transporters can only be established by performing further experiments in the future.
Cyp3a11 and Ugt1a1 are target genes of the xenobiotic nuclear receptor, PXR. In our previous studies, TLR4 mediated down-regulation of DMETs by LPS was associated with reduced expression of PXR gene (Ghose et al., 2008). Interestingly, in our current study, paclitaxel, a known activator of both PXR and TLR4, induced PXR target genes, Cyp3a11 and Ugt1a1 in TLR4-WT but this induction was significantly attenuated in TLR4-mutant hepatocytes. We therefore propose possible TLR4-PXR crosstalk in primary hepatocytes. Disparate observations have been reported on relationship between PXR and TLR4. Venkatesh et al observed an inverse relationship between PXR and TLR4 mRNA expression in Caco-2 cells (Venkatesh et al., 2014), whereas in another study, PXR agonist rifaximin reduced toxicity of Clostridium difficile Toxin A in Caco-2 by down-regulating TLR4 pathway (Esposito et al., 2016). Role of PXR activation in suppression of NF-κB target genes, including IL-1β and TNF-α has been implicated (Mencarelli et al., 2010; Cheng et al., 2012). More recent studies have shown that PXR activation inhibited LPS-induced NF-κB activation and TNF-α secretion from macrophages (Wallace et al., 2010). Studies indicate role of pro-inflammatory cytokines in downregulation of DMET genes during inflammation (Aitken et al., 2006) through TLR activation (Ghose et al., 2004b; 2008; 2009). Taken together, our results indicate possible involvement of two simultaneous pathways, one being the anti-inflammatory effect of PXR along with inflammatory response to TLR4 activation by paclitaxel.
In the liver, TLR4 is expressed by both the hepatocytes and non-parenchymal cells (NPCs), including liver sinusoidal endothelial cells (LSECs) and Kupffer cells (KCs) (Broering et al., 2008; Mollen et al., 2008). Paclitaxel has been reported to activate TLR4 on murine macrophages to trigger inflammatory responses (Ding et al., 1990; Kawasaki et al., 2000; Byrd-Leifer et al., 2001; Kawasaki et al., 2001b) but an effect on hepatocyte TLR4 is unknown. Our findings demonstrate that paclitaxel activates TLR4 on hepatocytes to alter the expression of DMET genes.
Conclusion
In conclusion, this is the first of its kind study to demonstrate the role of TLR4, the innate immune system in DDI. Paclitaxel significantly increased levels of SN-38 and induced gene and protein expression of Cyp3a11 and Ugt1a1 enzymes in TLR4-dependent manner. This suggests that alteration in drug metabolism and PK of co-administered drug with paclitaxel based chemotherapy may depend on TLR4 expression levels. Further investigation is required to determine the contribution of specific DMEs/transporters in TLR4-dependent DDI between paclitaxel and irinotecan.
These studies provide the foundation for future research to understand the implication of xenobiotic-mediated activation of immune modulators on drug disposition. As TLR agonists are currently being explored most extensively as immunomodulators and vaccine adjuvants (Hawkins et al., 2002; Przetak et al., 2003; Stover et al., 2004; Cluff et al., 2005; Casella and Mitchell, 2008), our finding signifies that attention should be given to the potential of TLR4 agonists to alter DMETs and risk of DDIs during development. Given that ~16% TLR4 polymorphism been reported across different populations (Ferwerda et al., 2007); our results direct towards the possible influence of TLR4 activation on drug metabolism, leading to differences in treatment outcomes and safety.
Highlights.
New mechanism of paclitaxel and Irinotecan drug-drug interaction is proposed.
Paclitaxel activates Toll-like receptor 4 to alter drug metabolizing enzymes/transporters in hepatocytes.
Activation of Toll-like receptor 4 by paclitaxel alters irinotecan pharmacokinetics in hepatocytes.
Potential role of Toll-like receptors as novel modulators of drug-drug interactions.
Acknowledgments
This work was supported in part by the grant 1R21DA035751-01A1 from the National Institutes of Health to BM and RG, and grants R01HL112516, R01ES 009132 and 01ES-019689, and 1R01HL129794 to BM.
Abbreviations
- PXR
pregnane X receptor
- LPS
lipopolysaccharide
- TLR4
Toll-like receptor 4
- DMET
drug metabolizing enzymes and transporters
- CES
carboxyesterases
- CYP
cytochrome P450
- SN-38
(7-ethyl-10-hydroxycamptothecin
- SN-38G
SN-38 glucuronide
- UGT1A1
uridine diphosphate glucuronosyltransferases 1A1
- APC
(7-ethyl-10[4-N-(5-aminopentanoicacid)-1-piperidino] carbonyloxycamptothecin)
- NPC
(7-ethyl-10[4-amino-1 piperidino]carbonyloxycamptothecin)
- DDIs
drug-drug interactions
- MRP
Multidrug resistance-associated protein
- Pgp
P-glycoprotein
- TNF
tumor necrosis factor
- MDR
Multidrug resistance
- IL
Interleukin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authorship Contributions
Participated in research design: Ghose and Mallick
Conducted experiments: Mallick
Contributed new reagents and analytic tools: Ghose, Moorthy
Performed data analysis: Mallick, Basu and Ghose
Wrote or contributed to the writing of the manuscript: Mallick, Moorthy and Ghose
References
- Aitken, et al. Regulation of drug-metabolizing enzymes and transporters in inflammation. Annu Rev Pharmacol Toxicol. 2006;46(2006):123–149. doi: 10.1146/annurev.pharmtox.46.120604.141059. [DOI] [PubMed] [Google Scholar]
- Basu, et al. Development and validation of an UPLC-MS/MS method for the quantification of irinotecan, SN-38 and SN-38 glucuronide in plasma, urine, feces, liver and kidney: Application to a pharmacokinetic study of irinotecan in rats. J Chromatogr B Analyt Technol Biomed Life Sci. 2016;1015–1016(2016):34–41. doi: 10.1016/j.jchromb.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellmunt, et al. Gemcitabine/paclitaxel-based three-drug regimens in advanced urothelial cancer. Eur J Cancer. 2000;36(Suppl 2):17–25. doi: 10.1016/s0959-8049(00)00081-2. 2000. [DOI] [PubMed] [Google Scholar]
- Bochud, et al. Polymorphisms in Toll-like receptor 4 (TLR4) are associated with protection against leprosy. Eur J Clin Microbiol Infect Dis. 2009;28(2009):1055–1065. doi: 10.1007/s10096-009-0746-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broering, et al. Toll-like receptor-stimulated non-parenchymal liver cells can regulate hepatitis C virus replication. J Hepatol. 2008;48(2008):914–922. doi: 10.1016/j.jhep.2008.01.028. [DOI] [PubMed] [Google Scholar]
- Brzezinski, et al. Purification and characterization of a human liver cocaine carboxylesterase that catalyzes the production of benzoylecgonine and the formation of cocaethylene from alcohol and cocaine. Biochem Pharmacol. 1994;48(1994):1747–1755. doi: 10.1016/0006-2952(94)90461-8. [DOI] [PubMed] [Google Scholar]
- Buajordet, et al. Fatal adverse drug events: the paradox of drug treatment. J Intern Med. 2001;250(2001):327–341. doi: 10.1046/j.1365-2796.2001.00892.x. [DOI] [PubMed] [Google Scholar]
- Byrd-Leifer, et al. The role of MyD88 and TLR4 in the LPS-mimetic activity of Taxol. Eur J Immunol. 2001;31(2001):2448–2457. doi: 10.1002/1521-4141(200108)31:8<2448::aid-immu2448>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Casella, Mitchell Putting endotoxin to work for us: monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol Life Sci. 2008;65(2008):3231–3240. doi: 10.1007/s00018-008-8228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, et al. Pregnane X receptor as a target for treatment of inflammatory bowel disorders. Trends Pharmacol Sci. 2012;33(2012):323–330. doi: 10.1016/j.tips.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, et al. Biliary excretion mechanism of CPT-11 and its metabolites in humans: involvement of primary active transporters. Cancer Res. 1998;58(1998):5137–5143. [PubMed] [Google Scholar]
- Cluff, et al. Synthetic toll-like receptor 4 agonists stimulate innate resistance to infectious challenge. Infect Immun. 2005;73(2005):3044–3052. doi: 10.1128/IAI.73.5.3044-3052.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cresteil, et al. Taxol metabolism by human liver microsomes: identification of cytochrome P450 isozymes involved in its biotransformation. Cancer Res. 1994;54(1994):386–392. [PubMed] [Google Scholar]
- Dai, et al. Polymorphisms in human CYP2C8 decrease metabolism of the anticancer drug paclitaxel and arachidonic acid. Pharmacogenetics. 2001;11(2001):597–607. doi: 10.1097/00008571-200110000-00006. [DOI] [PubMed] [Google Scholar]
- de Jong, et al. Irinotecan-induced diarrhea: functional significance of the polymorphic ABCC2 transporter protein. Clin Pharmacol Ther. 2007;81(2007):42–49. doi: 10.1038/sj.clpt.6100019. [DOI] [PubMed] [Google Scholar]
- de Oliveira, Silva Polymorphisms of the TLR2 and TLR4 genes are associated with risk of gastric cancer in a Brazilian population. World J Gastroenterol. 2012;18(2012):1235–1242. doi: 10.3748/wjg.v18.i11.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding, et al. Shared actions of endotoxin and taxol on TNF receptors and TNF release. Science. 1990;248(1990):370–372. doi: 10.1126/science.1970196. [DOI] [PubMed] [Google Scholar]
- Esposito, et al. Rifaximin Improves Clostridium difficile Toxin A-Induced Toxicity in Caco-2 Cells by the PXR-Dependent TLR4/MyD88/NF-kappaB Pathway. Front Pharmacol. 2016;7(2016):120. doi: 10.3389/fphar.2016.00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans, et al. Enhancement of antigen-specific immunity via the TLR4 ligands MPL adjuvant and Ribi.529. Expert Rev Vaccines. 2003;2(2003):219–229. doi: 10.1586/14760584.2.2.219. [DOI] [PubMed] [Google Scholar]
- Ferwerda, et al. TLR4 polymorphisms, infectious diseases, and evolutionary pressure during migration of modern humans. Proc Natl Acad Sci U S A. 2007;104(2007):16645–16650. doi: 10.1073/pnas.0704828104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi, et al. CYP3A-dependent drug metabolism is reduced in bacterial inflammation in mice. Br J Pharmacol. 2012;166(2012):2176–2187. doi: 10.1111/j.1476-5381.2012.01933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose, et al. Regulation of gene expression of hepatic drug metabolizing enzymes and transporters by the Toll-like receptor 2 ligand, lipoteichoic acid. Arch Biochem Biophys. 2009;481(2009):123–130. doi: 10.1016/j.abb.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose, et al. Differential role of Toll-interleukin 1 receptor domain-containing adaptor protein in Toll-like receptor 2-mediated regulation of gene expression of hepatic cytokines and drug-metabolizing enzymes. Drug Metab Dispos. 2011;39(2011):874–881. doi: 10.1124/dmd.110.037382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose, et al. In Vitro Approaches to Study Regulation of Hepatic Cytochrome P450(CYP) 3A Expression by Paclitaxel and Rifampicin. Methods Mol Biol. 2016;1395(2016):55–68. doi: 10.1007/978-1-4939-3347-1_4. [DOI] [PubMed] [Google Scholar]
- Ghose, et al. Rosiglitazone attenuates suppression of RXRalpha-dependent gene expression in inflamed liver. J Hepatol. 2007;46(2007):115–123. doi: 10.1016/j.jhep.2006.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose, et al. Regulation of hepatic drug-metabolizing enzyme genes by Toll-like receptor 4 signaling is independent of Toll-interleukin 1 receptor domain-containing adaptor protein. Drug Metab Dispos. 2008;36(2008):95–101. doi: 10.1124/dmd.107.018051. [DOI] [PubMed] [Google Scholar]
- Ghose, et al. Endotoxin leads to rapid subcellular re-localization of hepatic RXRalpha: A novel mechanism for reduced hepatic gene expression in inflammation. Nuclear receptor. 2004a;2(2004a):4. doi: 10.1186/1478-1336-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghose, et al. Endotoxin leads to rapid subcellular re-localization of hepatic RXRalpha: A novel mechanism for reduced hepatic gene expression in inflammation. Nuclear receptor. 2004b:4. doi: 10.1186/1478-1336-2-4. ed^eds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grau, et al. Weekly paclitaxel for platin-resistant stage IV head and neck cancer patients. Acta Otolaryngol. 2009;129(2009):1294–1299. doi: 10.3109/00016480802590451. [DOI] [PubMed] [Google Scholar]
- Harris, et al. Metabolism of taxol by human hepatic microsomes and liver slices: participation of cytochrome P450 3A4 and an unknown P450 enzyme. Cancer Res. 1994;54(1994):4026–4035. [PubMed] [Google Scholar]
- Hawkins, et al. A novel class of endotoxin receptor agonists with simplified structure, toll-like receptor 4-dependent immunostimulatory action, and adjuvant activity. J Pharmacol Exp Ther. 2002;300(2002):655–661. doi: 10.1124/jpet.300.2.655. [DOI] [PubMed] [Google Scholar]
- Hotta, et al. A phase I study and pharmacokinetics of irinotecan (CPT-11) and paclitaxel in patients with advanced non-small cell lung cancer. Lung Cancer. 2004;45(2004):77–84. doi: 10.1016/j.lungcan.2004.01.001. [DOI] [PubMed] [Google Scholar]
- Hutchinson, et al. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience. 2010a;167(2010a):880–893. doi: 10.1016/j.neuroscience.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson, et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun. 2010b;24(2010b):83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, et al. UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002a;2(2002a):43–47. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- Iyer, et al. Biliary transport of irinotecan and metabolites in normal and P-glycoprotein-deficient mice. Cancer Chemother Pharmacol. 2002b;49(2002b):336–341. doi: 10.1007/s00280-001-0420-4. [DOI] [PubMed] [Google Scholar]
- Kang, et al. The P-glycoprotein antagonist PSC 833 increases the plasma concentrations of 6alpha-hydroxypaclitaxel, a major metabolite of paclitaxel. Clin Cancer Res. 2001;7(2001):1610–1617. [PubMed] [Google Scholar]
- Kanzler, et al. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13(2007):552–559. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- Kasai, et al. Phase I and pharmacokinetic study of paclitaxel and irinotecan for patients with advanced non-small cell lung cancer. Eur J Cancer. 2002;38(2002):1871–1878. doi: 10.1016/s0959-8049(02)00231-9. [DOI] [PubMed] [Google Scholar]
- Kawasaki, et al. Mouse toll-like receptor 4.MD-2 complex mediates lipopolysaccharide-mimetic signal transduction by Taxol. J Biol Chem. 2000;275(2000):2251–2254. doi: 10.1074/jbc.275.4.2251. [DOI] [PubMed] [Google Scholar]
- Kawasaki, et al. Involvement of TLR4/MD-2 complex in species-specific lipopolysaccharide-mimetic signal transduction by Taxol. J Endotoxin Res. 2001a;7(2001a):232–236. [PubMed] [Google Scholar]
- Kawasaki, et al. Cutting edge: Gln22 of mouse MD-2 is essential for species-specific lipopolysaccharide mimetic action of taxol. J Immunol. 2001b;166(2001b):11–14. doi: 10.4049/jimmunol.166.1.11. [DOI] [PubMed] [Google Scholar]
- Kohler, et al. Drug-drug interactions in medical patients: effects of in-hospital treatment and relation to multiple drug use. Int J Clin Pharmacol Ther. 2000;38(2000):504–513. doi: 10.5414/cpp38504. [DOI] [PubMed] [Google Scholar]
- Kondagunta, et al. Combination of paclitaxel, ifosfamide, and cisplatin is an effective second-line therapy for patients with relapsed testicular germ cell tumors. J Clin Oncol. 2005;23(2005):6549–6555. doi: 10.1200/JCO.2005.19.638. [DOI] [PubMed] [Google Scholar]
- Kostrubsky, et al. The use of human hepatocyte cultures to study the induction of cytochrome P-450. Drug Metab Dispos. 1999;27(1999):887–894. [PubMed] [Google Scholar]
- Krieg Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5(2006):471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- Lee, et al. Molecular basis for the immunostimulatory activity of guanine nucleoside analogs: activation of Toll-like receptor 7. Proc Natl Acad Sci U S A. 2003;100(2003):6646–6651. doi: 10.1073/pnas.0631696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Koda Stability of irinotecan hydrochloride in aqueous solutions. Am J Health Syst Pharm. 2002;59(2002):539–544. doi: 10.1093/ajhp/59.6.539. [DOI] [PubMed] [Google Scholar]
- Li, et al. Paclitaxel based vs oxaliplatin based regimens for advanced gastric cancer. World J Gastroenterol. 2011;17(2011):1082–1087. doi: 10.3748/wjg.v17.i8.1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, et al. Toll-like receptor 4 signaling contributes to Paclitaxel-induced peripheral neuropathy. J Pain. 2014;15(2014):712–725. doi: 10.1016/j.jpain.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick, et al. Impact of obesity on accumulation of the toxic irinotecan metabolite, SN-38, in mice. Life Sci. 2015;139(2015):132–138. doi: 10.1016/j.lfs.2015.08.017. [DOI] [PubMed] [Google Scholar]
- Manthey, et al. Lipopolysaccharide antagonists block taxol-induced signaling in murine macrophages. J Exp Med. 1993;178(1993):695–702. doi: 10.1084/jem.178.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshak-Rothstein Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. 2006;6(2006):823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencarelli, et al. Pregnane-X-receptor mediates the anti-inflammatory activities of rifaximin on detoxification pathways in intestinal epithelial cells. Biochem Pharmacol. 2010;80(2010):1700–1707. doi: 10.1016/j.bcp.2010.08.022. [DOI] [PubMed] [Google Scholar]
- Mollen, et al. Systemic inflammation and end organ damage following trauma involves functional TLR4 signaling in both bone marrow-derived cells and parenchymal cells. J Leukoc Biol. 2008;83(2008):80–88. doi: 10.1189/jlb.0407201. [DOI] [PubMed] [Google Scholar]
- Monsarrat, et al. Hepatic metabolism and biliary excretion of Taxol in rats and humans. J Natl Cancer Inst Monogr. 1993;(1993):39–46. [PubMed] [Google Scholar]
- Mullarkey, et al. Inhibition of endotoxin response by e5564, a novel Toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther. 2003;304(2003):1093–1102. doi: 10.1124/jpet.102.044487. [DOI] [PubMed] [Google Scholar]
- Nallani, et al. Induction of cytochrome P450 3A by paclitaxel in mice: pivotal role of the nuclear xenobiotic receptor, pregnane X receptor. Drug Metab Dispos. 2003;31(2003):681–684. doi: 10.1124/dmd.31.5.681. [DOI] [PubMed] [Google Scholar]
- Pharmacia (1996) CAMPTOSAR- irinotecan hydrochloride injection, solution in: ed^eds), pp.
- Polasek, et al. Perpetrators of pharmacokinetic drug-drug interactions arising from altered cytochrome P450 activity: a criteria-based assessment. Br J Clin Pharmacol. 2011;71(2011):727–736. doi: 10.1111/j.1365-2125.2011.03903.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poltorak, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(1998):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- Przetak, et al. Novel synthetic LPS receptor agonists boost systemic and mucosal antibody responses in mice. Vaccine. 2003;21(2003):961–970. doi: 10.1016/s0264-410x(02)00737-5. [DOI] [PubMed] [Google Scholar]
- Rahman, et al. Selective biotransformation of taxol to 6 alpha-hydroxytaxol by human cytochrome P450 2C8. Cancer Res. 1994;54(1994):5543–5546. [PubMed] [Google Scholar]
- Rajput, et al. TLR4 is a novel determinant of the response to paclitaxel in breast cancer. Mol Cancer Ther. 2013;12(2013):1676–1687. doi: 10.1158/1535-7163.MCT-12-1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos, et al. Metabolism of irinotecan (CPT-11) by CYP3A4 and CYP3A5 in humans. Clin Cancer Res. 2000;6(2000):2012–2020. [PubMed] [Google Scholar]
- Satoh, et al. Metabolic activation of CPT-11, 7-ethyl-10-[4-(1-piperidino)-1-piperidino]carbonyloxycamptothecin, a novel antitumor agent, by carboxylesterase. Biol Pharm Bull. 1994;17(1994):662–664. doi: 10.1248/bpb.17.662. [DOI] [PubMed] [Google Scholar]
- Shah, et al. Role of constitutive androstane receptor in Toll-like receptor-mediated regulation of gene expression of hepatic drug-metabolizing enzymes and transporters. Drug Metab Dispos. 2014;42(2014):172–181. doi: 10.1124/dmd.113.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatter, et al. Bioactivation of the anticancer agent CPT-11 to SN-38 by human hepatic microsomal carboxylesterases and the in vitro assessment of potential drug interactions. Drug Metab Dispos. 1997;25(1997):1157–1164. [PubMed] [Google Scholar]
- Sparreboom, et al. Irinotecan (CPT-11) metabolism and disposition in cancer patients. Clin Cancer Res. 1998;4(1998):2747–2754. [PubMed] [Google Scholar]
- Stover, et al. Structure-activity relationship of synthetic toll-like receptor 4 agonists. J Biol Chem. 2004;279(2004):4440–4449. doi: 10.1074/jbc.M310760200. [DOI] [PubMed] [Google Scholar]
- Synold, et al. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med. 2001;7(2001):584–590. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- Tagen, et al. P-glycoprotein, but not multidrug resistance protein 4, plays a role in the systemic clearance of irinotecan and SN-38 in mice. Drug Metab Lett. 2010;4(2010):195–201. doi: 10.2174/187231210792928251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoelen, et al. A prophylactic hepatitis B vaccine with a novel adjuvant system. Vaccine. 2001:2400–2403. doi: 10.1016/s0264-410x(00)00462-x. ed^eds. [DOI] [PubMed] [Google Scholar]
- Venkatesh, et al. Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity. 2014;41(2014):296–310. doi: 10.1016/j.immuni.2014.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, et al. The PXR is a drug target for chronic inflammatory liver disease. J Steroid Biochem Mol Biol. 2010;120(2010):137–148. doi: 10.1016/j.jsbmb.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, et al. TLR4 induces tumor growth and inhibits paclitaxel activity in MyD88-positive human ovarian carcinoma in vitro. Oncol Lett. 2014;7(2014):871–877. doi: 10.3892/ol.2013.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziakas, et al. The role of TLR4 896 A>G and 1196 C>T in susceptibility to infections: a review and meta-analysis of genetic association studies. PLoS One. 2013:e81047. doi: 10.1371/journal.pone.0081047. ed^eds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer, et al. Paclitaxel binding to human and murine MD-2. J Biol Chem. 2008;283(2008):27916–27926. doi: 10.1074/jbc.M802826200. [DOI] [PMC free article] [PubMed] [Google Scholar]