

Abstract
The unique properties of entirely aliphatic TAML activator [FeIII{(Me2CNCOCMe2NCO)2CMe2}-OH2]− (3), namely the increased steric bulk of the ligand and the unmatched resistance to the acid-induced demetalation, enables the generation of high-valent iron derivatives in pure water at any pH. An iron(V)oxo species is readily produced with NaClO at pH values from 2 to 10.6 without any observable intermediate. This is the first reported example of iron(V)oxo formed in pure water. At pH 13, iron(V)oxo is not formed and NaClO oxidizes 3 to an iron(IV)oxo derivative.
In Nature, the highest oxidation states of peroxidases and cytochrome P450, which are by two oxidation equivalents above the iron(III) resting state, are produced in aqueous media.1–3 For a long time, such states were unknown for mimetics of the active sites of these enzymes. The first thoroughly characterized case of the iron(V)oxo unit formed from the iron(III) precursor was reported in 2007 by the example of Generation 1 TAML activator 1.4 Organic nitriles were used as solvents and the compound was made from 1 and m-chloroperoxybenzoic acid (mCPBA) at low temperatures. All examples of iron(V)oxo and related derivatives that followed that report were also made in nonaqueous solvents.5–11 Generating such a species in water remained a huge challenge.12 Using TAMLs 2 of Generation 5, Panda et al. reported the formation of their iron(V)oxo derivatives in a 50% water–acetonitrile mixture13 and the water content was then increased to 90%.14 Water decreases markedly the stability of the iron(V)oxo species. At 90% water, the iron(V)oxo species degrade 10 times faster than in pure acetonitrile.14
Here we show that high-valent iron species, including the iron(V)oxo, of a TAML activator are affordable without any organic cosolvent in pure water at any pH from 2 to 13. The high-valent iron compounds can be effortlessly generated using NaClO. This became possible using the recently introduced TAML activator 315 which, in contrast to TAMLs 1 and 2, does not have an aromatic ring at the head part of the molecule (“beheaded” TAML) (Chart 1).15 We demonstrate in particular that (i) the iron(V)oxo unit is rapidly and quantitatively produced at 13 °C by reacting 3 with NaClO at pH 2 and 10.6; (ii) iron(V)oxo is not formed under basic conditions (pH 13) and the iron(IV)oxo unit is generated instead; (iii) the iron(V)oxo species is converted to iron(IV)oxo by increasing pH from 2 to 13 but (iv) decreasing pH from 13 to 2 induces the unique disproportionation of iron(IV)oxo to form iron-(V)oxo and iron(III) species (Scheme 1).
Chart 1. TAML Activators Referred to (1 and 2) and Explored (3) in This Worka.

aTAML is a registered trademark of Carnegie Mellon University, covering tetra-organic-N macrocyclic ligand complexes.
Scheme 1. pH-Dependent Transformations of 3 in Aqueous Solutions Induced by NaClO at 13 °Ca.

aCharges of TAML species are omitted for clarity.
We have recently reported that at −40 °C in MeCN iron(III) TAML 1 is oxidized into the iron(V)oxo species by NaClO and mCPBA though H2O2 and organic peroxides convert 1 into the diiron(IV)(μ-oxo) dimer.16 The advantages of using NaClO over mCPBA as an oxidant include higher reactivity and unlimited solubility in water. Figure 1 shows the spectra of 3 and the products formed in the presence of NaClO at pH 10.6 and 13. At pH 10.6, the reaction was quantitatively completed within 2 min when 1 equiv of NaClO is added. There are just two absorbing species in solution supported by three isosbestic points at 312, 347, and 384 nm. The inset to Figure 1A confirms the 1:1 reaction stoichiometry implying that the product should be by two oxidation equivalents above the iron(III) resting state (eq 1 in Scheme 1). Similar results were obtained at all pH from 1.0 to 10.6 indicating that the form of hypochlorite (e.g., HClO or ClO−, pKa = 7.5317) is not essential.
Figure 1.
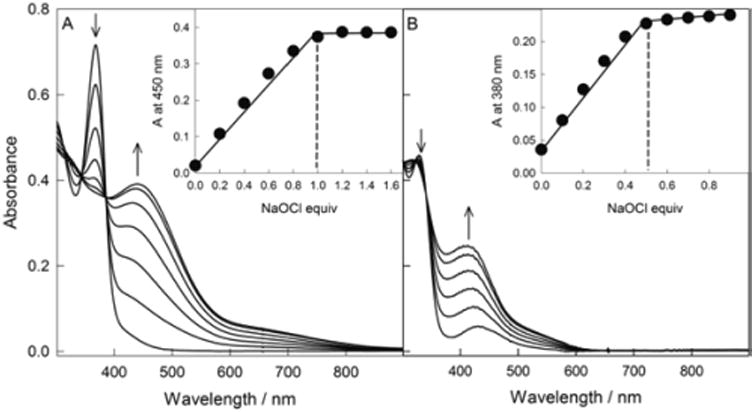
Titration of 3 with NaClO at pH 10.6 (A) and 13 (B). Each spectrum represents the effect of an aliquot of NaClO. The insets show the absorbance changes at the λmax of new species produced. Conditions: [3] = 1 × 10−4 M, 0.01 M phosphate, 13 °C.
The spectrum of the product generated similarly at pH 13 is presented in Figure 1B. The major difference between pH 10.6 and 13 is that the aqua ligand of 3 is now deprotonated (pKa = 11.4).18 e oxidation at pH 13 is very clean as well with the isosbestic points at 302, 324, and 341 nm. The inset to Figure 1B reveals, however, the 2:1 iron to hypochlorite stoichiometry pointing to the iron(IV) product which is by one oxidation equivalent above the iron(III) state. Thus, the oxidation state of iron in the product of oxidation of 3 by NaClO in pure water is rather unexpectedly dictated by pH (eq 2 in Scheme 1).
The identity of the products generated at pH 10.6 and 13 were confirmed by EPR and Mossbauer spectroscopy. The spectroscopy of the S = 3/2 iron(III) complex was reported previously.18 Upon addition of 1 equiv of NaClO to the iron(III) complex in water at pH 10.6, the EPR signal from the iron(III) complex vanished with concomitant generation of the new S = 1/2 signal with g = (2.02, 1.98, 1.84) shown in Figure 2. The reaction was reproduced in 10% glycerol, which generated the same species but with a significantly sharper spectrum. The same sharp EPR spectrum of the S = 1/2 species was also generated in acetonitrile at −40 °C. The g-values are near to those of previously characterized iron(V)oxo complexes of TAML: g = (1.99, 1.97, 1.74)4 and g = (1.98, 1.94, 1.73).19 Spin quantitation indicated that the concentration of the iron(V)oxo complex generated in water or acetonitrile were both in quantitative agreement with the concentration of the initial iron(III) complex.
Figure 2.
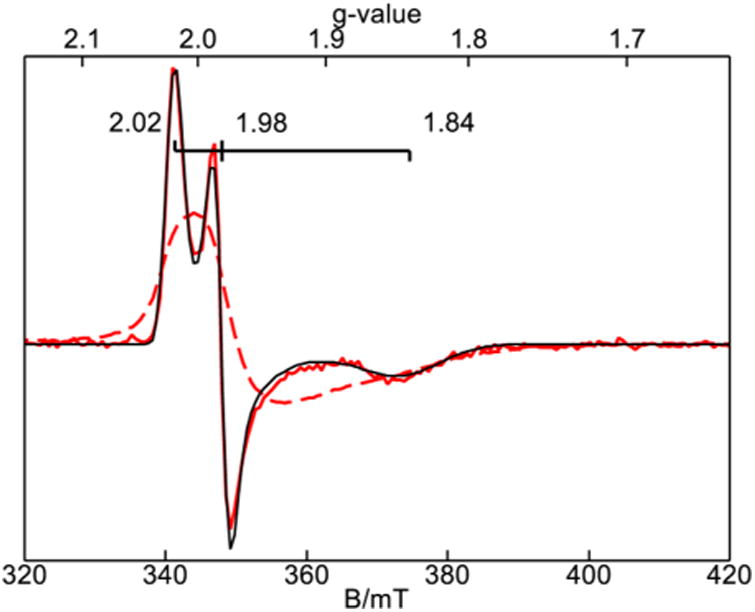
Frozen solution EPR spectra of 3 × 10−4 M 3 after reaction with 1 equiv NaClO at pH 10 with 10% glycerol (red line) and without glycerol (red dashed line). The black line is a S = 1/2 simulation. Temperature, 21 K; microwaves, 20 μW at 9.646 GHz.
Upon addition of 1 equiv of NaClO to the iron(III) complex in water at pH 10.6, the Mössbauer spectrum of the iron(III) complex vanished with concomitant generation of the new S = 1/2 species shown in Figure 3A. The parameters of this new species (see figure caption) were close to those of a previously characterized iron(V)oxo complex of TAML: S = 1/2, δ = −0.42 mm s−1, ΔEq = +4.25 mm s−1, A = (−49, −2, −16) T, η = 0.76.4
Figure 3.
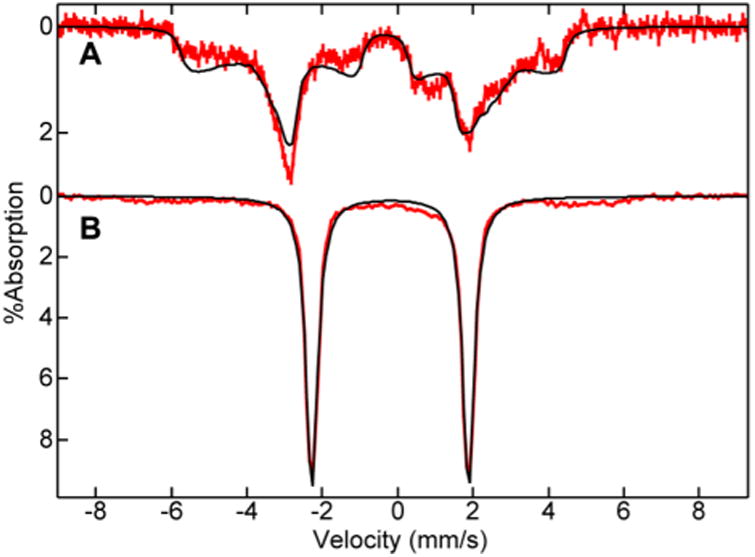
Frozen solution 57Fe-enriched Mössbauer spectra (red lines) of 3 in water and 10% glycerol after reaction with 1 equiv NaClO at (A) pH 10.6 and (B) pH 13. The simulations (black lines) are for (A) S = 1/2, δ = −0.48 mm s−1, AEq = +4.7 mm s−1, A= (−48, +1, 0) T, η = 0.74; and (B) δ = −0.20 mm s−1, ΔEq = 4.15 mm s−1. Spectra recorded at 4.2 K in an applied field of 450 mT parallel to the incident γ beam.
EPR and Mossbauer samples were prepared of the same reactions at pH 13. Upon addition of 1 equiv NaClO, the EPR spectrum of the iron(III) complex vanished with no new species observed. The Mossbauer spectrum of the iron(III) complex also vanished and a new species was observed as shown in Figure 3B. The parameters of this species (see figure caption) are close to that of a previously characterized S = 1 iron(IV)oxo complex of TAML: δ = −0.12 mm s−1, AEq =3.35 mm s−1. The same results were obtained with 0.5 equiv NaClO. Upon addition of 0.25 equiv NaClO to the iron(III) form of 3, spin quantitation indicated that the iron(III) signal decreased by 50%, with no other new EPR signals produced (Figure S1, Supporting Information (SI)). This observation suggested primary formation of 25% iron(V)oxo, which is then rapidly oxidized to an equivalent amount of iron(III) to form 50% iron(IV)oxo, the remaining 50% iron(III) being unreacted.
The conversion of 3 by NaClO into iron(V)oxo occurs without the intermediacy of iron(IV). Such behavior for a TAML activator is new. The oxidations of 1 by NaClO and mCPBA in MeCN at −40 °C and below occur with spectrally detectable diiron(IV)(μ-oxo) species21 because the FeIII→FeIV process is much faster than the FeIV→FeV conversion.16,22 It is worth noting that diiron(IV)(μ-oxo) species are not formed either when 3 was exposed to mCPBA in MeCN at −40 °C. Iron(V)oxo generated under such conditions has UV–vis and EPR spectra identical to those obtained in water. The new reactivity pathway for 3, for which the diiron(IV)(μ-oxo) dimer is not formed, is attributed to steric interactions created by the four adjacent methyl groups. The role of electronic effects cannot be excluded as suggested by the reviewer because extra donating methyl groups may stabilize iron(V)oxo species.
The observations that NaClO converts 3 into different oxidized states depending on pH prompted us to study the fate of iron(V)oxo and iron(IV)oxo species generated at pH 2 and 13 on reverting the solution pH to 13 and 2, respectively. When iron(IV)oxo was generated from 3 by 0.5 equiv NaClO at pH 13 and the solution was acidified to pH 2, the compound underwent rapid disproportionation (within 5 s) to afford equal quantities of iron(III) and iron(V)oxo derivatives (Scheme 1) as shown by the UV–vis (Figure S2, SI) spectroscopy. The bands at 368 and 450 nm were developed that correspond to iron(III) and iron(V)oxo species, respectively.
The unavailability of iron(V)oxo at pH 13 was confirmed on basification of a sample of the iron(V)oxo complex generated at pH 10.6. After oxidation of 1 × 10−4 M 3 with 1 equiv NaClO, another aliquot of 3 was added. No comproportionation was observed. When the pH was adjusted to 13 with concentrated NaOH, the spectrum of iron(V)oxo changed instantaneously to that of iron(IV)oxo (98% yield) suggested by the shift of the absorbance maximum from 450 to 410 nm (Figure S3, SI). This observation indicates that the fast comproportionation to form two iron(IV)oxo species, similar to what happens with 1 during the formation of the iron(IV)(μ-oxo) dimer, can be launched by a slight basification of the aqueous solution. This is presumably due to the deprotonation of the axial water of 3 (pKa = 11.4) which makes the hydroxo species a more powerful reducing agent compared to the corresponding aqua species (Scheme 1).
Iron(IV) forms of 3 seem to be stable at pH above 10.6 (iron(IV)oxo species). This suggests that unstable iron(IV) species may form at intermediate pH. Further proof for the instability of FeIV at intermediate pH (2.0−10.6) was obtained adding 1 equiv of K4Fe(CN)6 to iron(V)oxo at pH 10.6. No formation of iron(IV)oxo species was detected. The resulting UV–vis spectrum indicates the presence of a 1:1 mixture of iron(V)oxo and iron(III) (Figure S4, SI). This suggests that, if formed, iron(IV)oxo should rapidly disproportionate. Accordingly, the second equivalent of K4Fe(CN)6 converts all iron(V)oxo into iron(III).
In conclusion, the steric bulk and unparalleled acid resistance18 of TAML activator 3 allows for the chemical generation of high-valent iron complexes in pure water. The pH range of 2.0–10.6 is wide open for the clean and quantitative generation of iron(V)oxo derivative of iron(III) TAML activator 3, which is sufficiently stable at 13 °C for investigating its properties and reactivity. Under more basic conditions, e.g., at pH 13, the iron(V)oxo species is likely a transient species and the iron(IV)oxo derivative is formed instead. Acidification of the latter induces the unprecedented disproportionation of iron(IV)oxo to a mixture of iron(III) and iron(V)oxo species. There are ten methyl groups at the periphery of 3, which are directed above and below the plane of four amide nitrogens. Hypothetically, they create a protective hydrophobic environment around axial water in its resting state and the oxo ligand in the iron(V)oxo form, which allows the formation of surprisingly stable, highly oxidized species in pure water. Perhaps the methyl groups play a role similar to a protein core in peroxidases and cytochrome P450 enzymes.
Supplementary Material
Experimental details; UV–vis spectral changes arising from (1) generating the iron(IV)oxo and adding acid, (2) generating the iron(V)oxo at pH 10.6 and adding aliquots of K4Fe(CN)6, and (3) generating the iron(V)-oxo and adding base; also included are EPR spectra of (1) spin quantitation of remaining iron(III) after addition of 0.25 equiv NaClO at pH 13 (PDF)
Acknowledgments
T.J.C. thanks the Heinz Endowments for support. M.R.M thanks the R. K. Mellon Foundation for support through a Presidential Fellowship. M.P.H. acknowledges the National Institutes of Health GM49970 for funding and the National Science Foundation CHE1126268 for purchase of the EPR spectrometer.
Footnotes
Supporting Information: The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b09572.
Author Contributions: The paper was written through contributions of all authors.
Notes: The authors declare no competing financial interest.
References
- 1.Dunford HB. Heme Peroxidases. Wiley-VCH; Weinheim: 1999. [Google Scholar]
- 2.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 3.Ortiz de Montelano PR, editor. Cytochrome P450 Structure, Mechanism, and Biochemistry. 3rd. Springer; New York: 2005. [Google Scholar]
- 4.Tiago de Oliveira F, Chanda A, Banerjee D, Shan X, Mondal S, Que J, Jr, Bominaar EL, Munck E, Collins T. J Science. 2007;315:835–838. doi: 10.1126/science.1133417. [DOI] [PubMed] [Google Scholar]
- 5.Lyakin OY, Bryliakov KP, Britovsek GJP, Talsi EP. J Am Chem Soc. 2009;131:10798–10799. doi: 10.1021/ja902659c. [DOI] [PubMed] [Google Scholar]
- 6.Pan Z, Wang Q, Sheng X, Horner JH, Newcomb M. J Am Chem Soc. 2009;131:2621–2628. doi: 10.1021/ja807847q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Heuvelen KM, Fiedler AT, Shan X, De Hont RF, Meier KK, Bominaar EL, Munck E, Que L., Jr Proc Natl Acad Sci U S A. 2012;109:11933–11938. doi: 10.1073/pnas.1206457109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwon E, Cho KB, Hong S, Nam W. Chem Commun. 2014;50:5572–5575. doi: 10.1039/c4cc01058b. [DOI] [PubMed] [Google Scholar]
- 9.Tse CW, Chow TWS, Guo Z, Lee HK, Huang JS, Che CM. Angew Chem, Int Ed. 2014;53:798–803. doi: 10.1002/anie.201305153. [DOI] [PubMed] [Google Scholar]
- 10.Ren Q, Guo Y, Mills MR, Ryabov AD, Collins TJ. Eur J Inorg Chem. 2015;2015:1445–1452. [Google Scholar]
- 11.Serrano-Plana J, Oloo WN, Acosta-Rueda L, Meier KK, Verdejo B, Garcia-Espana E, Basallote MG, Munck E, Que L, Company A, Costas M. J Am Chem Soc. 2015;137:15833–15842. doi: 10.1021/jacs.5b09904. [DOI] [PubMed] [Google Scholar]
- 12.Ryabov AD. Adv Inorg Chem. 2013;65:118–163. [Google Scholar]
- 13.Panda C, Debgupta J, Diaz Diaz D, Singh KK, Sen Gupta S, Dhar BB. J Am Chem Soc. 2014;136:12273–12282. doi: 10.1021/ja503753k. [DOI] [PubMed] [Google Scholar]
- 14.Singh KK, Tiwari Mk, Ghosh M, Panda C, Weitz A, Hendrich MP, Dhar BB, Vanka K, Sen Gupta S. Inorg Chem. 2015;54:1535–1542. doi: 10.1021/ic502535f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeNardo MA, Mills MR, Ryabov AD, Collins TJ. J Am Chem Soc. 2016;138:2933–2936. doi: 10.1021/jacs.5b13087. [DOI] [PubMed] [Google Scholar]
- 16.Mills MR, Burton AE, Mori DI, Ryabov AD, Collins TJ. J Coord Chem. 2015;68:3046–3057. [Google Scholar]
- 17.Smith RM, Martell AE. Critical Stability Constants. Vol. 4. Springer; New York: 1976. [Google Scholar]
- 18.Mills MR, Weitz AC, Zhang DZ, Hendrich MP, Ryabov AD, Collins T. J Inorg Chem. 2016 doi: 10.1021/acs.inorgchem.6b01988. under revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghosh M, Singh KK, Panda C, Weitz A, Hendrich MP, Collins TJ, Dhar BB, Sen Gupta S. J Am Chem Soc. 2014;136:9524–9527. doi: 10.1021/ja412537m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chanda A, Shan X, Chakrabarti M, Ellis W, Popescu D, Tiago de Oliveira F, Wang D, Que L, Jr, Collins TJ, Munck E, Bominaar EL. Inorg Chem. 2008;47:3669–3678. doi: 10.1021/ic7022902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ghosh A, Tiago de Oliveira F, Yano T, Nishioka T, Beach ES, Kinoshita I, Munck E, Ryabov AD, Horwitz CP, Collins TJ. J Am Chem Soc. 2005;127:2505–2513. doi: 10.1021/ja0460458. [DOI] [PubMed] [Google Scholar]
- 22.Kundu S, Van Kirk Thompson J, Ryabov AD, Collins TJ. J Am Chem Soc. 2011;133:18546–18549. doi: 10.1021/ja208007w. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental details; UV–vis spectral changes arising from (1) generating the iron(IV)oxo and adding acid, (2) generating the iron(V)oxo at pH 10.6 and adding aliquots of K4Fe(CN)6, and (3) generating the iron(V)-oxo and adding base; also included are EPR spectra of (1) spin quantitation of remaining iron(III) after addition of 0.25 equiv NaClO at pH 13 (PDF)