

Abstract
In the era of antiretroviral therapy (ART), HIV-1 infection is no longer tantamount to early death. Yet the benefits of treatment are available only to those who can access, afford, and tolerate taking daily pills. True cure is challenged by HIV latency, the ability of chromosomally integrated virus to persist within memory CD4+ T cells in a non-replicative state and activate when ART is discontinued. Using a mathematical model of HIV dynamics, we demonstrate that treatment strategies offering modest but continual enhancement of reservoir clearance rates result in faster cure than abrupt, one-time reductions in reservoir size. We frame this concept in terms of compounding interest: small changes in interest rate drastically improve returns over time. On ART, latent cell proliferation rates are orders of magnitude larger than activation and new infection rates. Contingent on subtypes of cells that may make up the reservoir and their respective proliferation rates, our model predicts that coupling clinically available, anti-proliferative therapies with ART could result in functional cure within 2–10 years rather than several decades on ART alone.
Introduction
The most significant accomplishment in HIV medicine is the suppression of viral replication and prevention of AIDS with antiretroviral therapy (ART). However, HIV cure remains elusive due to viral latency, the ability of integrated virus to persist for decades within CD4+ T cells in a latent state. When ART is discontinued, latent cells soon activate, and virus rebounds1, 2. HIV cure strategies aim to eradicate the latent reservoir of infected cells3 but have been unsuccessful except in one notable example4. In addition, substantial technological and financial hurdles preclude the widespread use of many developing cure strategies. The anti-proliferative therapies we propose here are used widely, permitting broad and immediate availability following a proof of efficacy study.
Several recent studies link cellular proliferation (both antigen-driven expansion and homeostatic proliferation) with persistence of the HIV reservoir on long-term ART (>1 year)5–13. Using a mathematical model, we demonstrate that continuous, modest reductions in latent cell proliferation rates would deplete the latent reservoir more rapidly than comparable increases in HIV activation as occurs with latency reversing agents. Further, we find that more rapid reservoir elimination on anti-proliferative therapy occurs with lower pre-treatment reservoir size and higher proportions of rapidly proliferating effector and central memory CD4+ T cells in the reservoir.
Based on analogies to finance, we call this strategy “compound interest cure”. We demonstrate the promise of the compound interest approach by identifying reservoir reduction commensurate with predictions from our model in HIV-infected patients treated with mycophenolate mofetil (MMF) in past studies. We confirm the anti-proliferative effect of MMF on naïve and memory CD4+ T cell subsets via in vitro experiments.
Results
ART decouples latent pool dynamics from ongoing infection
Our model is visualized in Fig. 1 and detailed in the Methods. If ART is perfectly effective, all susceptible cells are protected from new infection, even when cells activate from latency. Thus, the dynamics of the latent cells can be considered separately, decoupled from the dynamics of the other cell types, and the only mechanisms changing the latent cell pool size are cell proliferation, death, and activation (bottom panel, Fig. 1).
Figure 1.

Schematics of models for HIV dynamics on and off ART. The top panel shows all possible transitions in the model (equation (1)). The bottom shaded panel shows the available transitions for the decoupled dynamic equations when ART suppresses the virus. Model parameters are given in Table 1. HIV virus V infects susceptible cells S at rate β reduced by ART of efficacy ε to β ε. The probability of latency given infection is τ. The rate of activation from latently infected cells (L) to actively infected cells (A) is ξ. Cellular proliferation and death are determined by rates α and δ for each compartment. The mechanisms of action of anti-proliferative and latency reversal therapies are to decrease α L and increase ξ, respectively.
However, perfectly effective ART is not strictly necessary to consider the latent pool separately. As previously described14, 15, we define ART “critical efficacy” ε c as the ART efficacy above which there is no set-point viral load, i.e. virus decreases rapidly with time (see Methods). Above the critical efficacy, viral production from activation could cause some new cell infection, but because the probability of latency (τ) is so low, new infection does not affect reservoir size or dynamics meaningfully. Using parameters from Table 1, we find ε c ~ 85%. Because true ART efficacy is generally greater than this efficacy16, we predict little de novo infection in ART-suppressed patients, consistent with the lack of viral evolution following years of ART without re-seeding of the latent reservoir8, 10, 11, 13, 17.
Table 1.
Parameters used in the HIV latency model.
| Parameter | Value | Dimensions | Source | Meaning |
|---|---|---|---|---|
| θ L | −5.2 × 10−4 | day−1 | 1, 26 | net latent clearance rate on ART |
| δ L | 0.0155 | day−1 | calculated* | latent central memory cell death rate |
| α L | 0.015 | day−1 | 49 | latent cell proliferation rate |
| α cm | 0.015 | day−1 | 49 | latent central memory cell Tcm proliferation rate |
| α em | 0.047 | day−1 | 49 | latent effector memory cell Tem proliferation rate |
| α n | 0.002 | day−1 | 49 | latent naïve cell Tn proliferation rate |
| ξ | 5.7 × 10−5 | day−1 | 21 | activation rate |
| α A | 0 | day−1 | 45 | active proliferation rate |
| δ A | 1.0 | day−1 | 50 | active death rate |
| τ | 10−4 | day−1 | 29 | probability of latency given infection |
| α S | 300 | cells/(μL-day) | 51 | susceptible growth rate |
| δ S | 0.2 | day−1 | 51 | susceptible death rate |
| β | 10−4 | μL/(virus-day) | 45, 51 | HIV infectivity off ART |
| β ε | β(1 − ε) | μL/(virus-day) | 14, 15 | HIV infectivity on ART (with efficacy ) |
| π | 103 | virus/(cell-day) | 45, 52 | viral production rate |
| γ | 23 | day−1 | 53 | viral clearance rate |
All cellular rates are for CD4+ T cells. *Death rates for each cell type are calculated using the total clearance as δ i = α i − ξ − θ L with .
Sustained mild effects on clearance rate deplete the reservoir more rapidly than large, one-time reservoir reductions
The HIV cure strategy most extensively tested in humans is “shock-and-kill” therapy: latency reversing agents activate HIV in latent cells to replicate and express HIV proteins, allowing immune clearance while ART prevents further infection3. Other strategies in development include therapeutic vaccines18, viral delivery of DNA cleavage enzymes19, and transplantation of modified HIV-resistant cells20 informed by the “Berlin patient”4. Some of these therapies manifest as one-time reductions in the number of latent cells. We simulate such instantaneous decreases using equation (4) and cure thresholds described in Methods. Briefly, using ART interruption data, Hill et al. and Pinkevych et al. estimated the number of latently infected cells that would result in ART-free suppression of viremia for one year (Hill 1-yr and Pinkevych 1-yr) versus 30 years, Hill cure (Hc), in 50% of HIV-infected patients21, 22. With the reservoir clearance rate θ L constant and a 100-fold reduction in reservoir size L 0, the Pinkevych 1-yr threshold is immediately satisfied, but the Hill 1-yr and Pinkevych cure still require 15 years of ART. Hill cure requires a 1,000-fold reduction and more than 10 subsequent years of ART (Fig. 2a).
Figure 2.
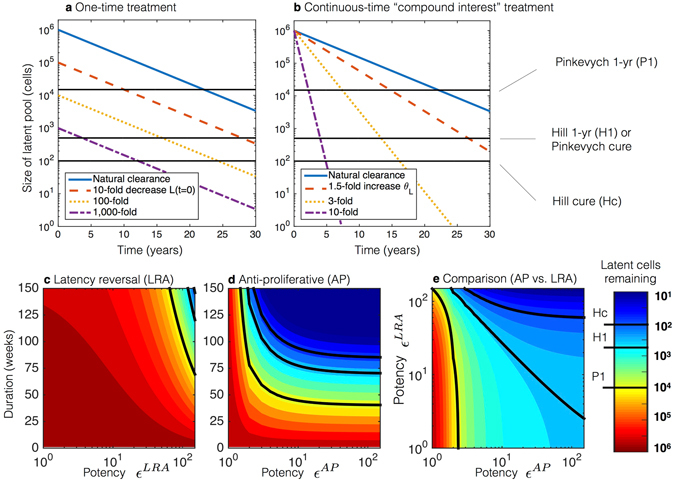
Simulated comparisons of latent reservoir eradication strategies on standard antiretroviral (ART) treatment. Treatment thresholds (discussed in Methods) are shown as solid black lines both in the plots and color bar, which is consistent between panels. (a) One-time therapeutic reductions of the latent pool (L 0). (b) Continuous therapeutic increases in the clearance rate (θ L). Relatively small decreases in the clearance rate θ L produce markedly faster times to cure than much larger decreases in the initial reservoir size. (c–e) Latency reversal agent (LRA) and anti-proliferative (AP) therapies are given continuously for durations of weeks with potencies given in fold increase in activation rate (ε LRA) and fold decrease in proliferation rate (ε AP), respectively. The color bar is consistent between panels, and thresholds of cure are shown as solid black lines both on plots and on the color bar. (c) Latency reversing agent therapy (LRA) administered alone requires years and potencies above 100 to achieve the cure thresholds. (d) Anti-proliferative therapies (AP) administered alone lead to cure thresholds in 1–2 years provided potency is greater than 2–3. (e) LRA and AP therapies are administered concurrently, and the reduction in the latent pool is measured at 70 weeks. Because the proliferation rate is naturally greater than the activation rate, increasing the AP potency has a much stronger effect than increasing the LRA potency.
Continuous-time interventions are more promising. Relatively small changes in θ L in equation (4) lead to significant changes in the time to cure (Fig. 2b). On ART alone, estimated cure occurs at roughly 70 years1. However, just a 3-fold increase in clearance rate achieves Hill cure in fewer than 20 years. A 10-fold sustained increase requires only five years for Hill cure.
Further, when continuous-time therapies are given, outcomes improve more by extending duration than by equivalent increases in potency (Fig. 2c,d demonstrate this given the substantial asymmetry of the contours over their y = x axes). Analogous to the so-called “miracle of compound interest,” increasing the clearance rate for an extended duration produces profound latency reduction.
Smaller reductions in proliferation rate achieve more rapid reservoir depletion than comparable relative increases in activation rate
Latency reversing therapy can be modeled with equation (3) if treatment is assumed to be a continuous-time multiplication of activation. Simulations at various potencies and therapy durations indicate both Hill and Pinkevych cure thresholds require more than a 100-fold multiplication of ξ sustained for two or three years, respectively (Fig. 2c).
The latent cell proliferation rate is considerably larger than the activation rate (, Table 1). Thus, anti-proliferative therapies would clear the reservoir faster than equivalently potent latency reversing strategies. When the reservoir of CD4+ T cells harboring replication-competent HIV is assumed to consist only of central memory cells (Tcm), a 10-fold reduction in α cm leads to Pinkevych 1-yr, Hill 1-yr, Pinkevych cure, and Hill cure in 0.8, 1.6, 1.6, and 1.8 years, respectively (Fig. 2d).
The improvement in cure time (when compared to an equivalent 10-fold increase in net reservoir clearance rate θ L) is possible because decreasing the proliferation rate means the net clearance rate approaches the latent cell death rate δ L. In fact, potency is relatively unimportant beyond reducing the proliferation rate by a factor of ten because the underlying death rate δ L is the bound on clearance rate. The relative impact of anti-proliferative therapy is greater than that of latency reversing therapy when the two therapies are given concurrently for 70 weeks (Fig. 2e).
Heterogeneity in reservoir cell types may necessitate prolonged anti-proliferative therapy
Recent studies indicate that the reservoir is heterogeneous, consisting of CD4+ central memory (Tcm), naïve (Tn), effector memory (Tem), and stem cell-like memory (Tscm) T cells. Further, reservoir cell composition differs dramatically among patients6, 12, 23. This heterogeneity suggests the potential for variable responses to anti-proliferative agents. Proliferation rates of Tcm (once per 66 days) exceed Tn (once every 500 days) but lag behind Tem proliferation rates (once every 21 days, Table 1). In our model Tscm are assumed to proliferate at the same frequency as Tn based on similar properties. We simulate possible reservoir profiles with different percentages of Tn, Tcm, and Tem in Fig. 3a–c. At least 7 years of treatment is needed for Pinkevych functional cure (Hill 1-yr) if slowly proliferating cells (Tn and/or Tscm) comprise more than 20% of the reservoir. In contrast, an increased proportion of Tem has no clinically meaningful impact on time to cure. Slowly proliferating cells are predicted to comprise the entirety of the reservoir within two years of 10-fold anti-proliferative treatment regardless of initial percentage of Tn or Tscm (Fig. 3d,e).
Figure 3.

Simulated comparisons of anti-proliferative therapies on standard antiretroviral therapy (ART) assuming variable reservoir composition. Proliferation and death rates in Table 1. The potency of the therapy is ε AP = 10 (i.e., each cell type i has proliferation rate equal to α i/10 with ). Plausible initial compositions of the reservoir (L i(0)) are taken from experimental measurements6, 12, 23. It is assumed that the HIV activation rate ξ is equivalent across all reservoir subsets. (a–c) Plots of times to therapeutic landmarks on long-term ART and anti-proliferative therapy with heterogeneous reservoir compositions consisting of effector memory (Tem), central memory (Tcm), and naïve plus stem cell-like memory (Tn + Tscm) CD4+ T cells. Tem and Tn + Tscm percentages are shown with the remaining cells representing Tcm. Times to one-year remission and functional cure are extremely sensitive to percentage of Tn + Tscm but not percentage of Tem. (d,e) Continuous 10-fold therapeutic decreases in all proliferation rates (α i) result in Hill 1-yr in (d) 3.5 years assuming Tn + Tscm = 1% and (e) 6 years assuming Tn + Tscm = 10%. The reservoir is predicted to become Tn + Tscm dominant within 2 years under both assumptions, providing an indicator to gauge the success of anti-proliferative therapy in potential experiments.
The uncertainty in the reservoir composition tempers the results in Fig. 2. On the other hand, our model assumes that the HIV activation rate ξ is equivalent across all CD4+ T cell reservoir subsets. It is biologically plausible, though unproven, that latent cell proliferation and activation are linked processes and that therefore HIV rarely or never activates from resting Tn or Tscm. Under this assumption, functional cure might occur once Tem and Tcm reservoirs have been reduced to the Hill cure level, i.e. approximately 1.5 years in Fig. 3d,e.
Initial reservoir size, anti-proliferative potency, and reservoir cell subtypes predict time to cure
Using literature-derived ranges for the parameters of interest, we completed a global sensitivity analysis to examine which factors might impact time to cure in a heterogeneous patient pool developed by Latin Hypercube sampling of a broad parameter space24 (Fig. 4). We correlate variables with time to cure on ART/anti-proliferative combination therapy. Varying the probability of latency given infection (τ) does not change time to cure. Similarly, varying the basic reproductive number on ART (), a measure of ART efficacy, defined as the number of new infected cells generated by one infected cell during ART, does not change time to cure. On the other hand, as the pre-treatment size of the latent pool L 0 increases, the necessary time to cure also increases. Increasing anti-proliferative therapy potency ε AP decreases cure time. Increasing percentages of naïve T cells L n(0)/L 0 in the latent reservoir delay the time to cure while a faster latent decay rate θ L hastens cure. Finally, we simulated the possibility of a diminishing impact of anti-proliferative therapy over time in Fig. 5. The simulation shows that when potency decreases by less than 5% per month, cure thresholds are still achieved within 10 years of ART and anti-proliferative treatment. The fastest waning of potency (20% per month) results in return to the natural clearance rate within the first 2 years of therapy prompting longer times to cure.
Figure 4.
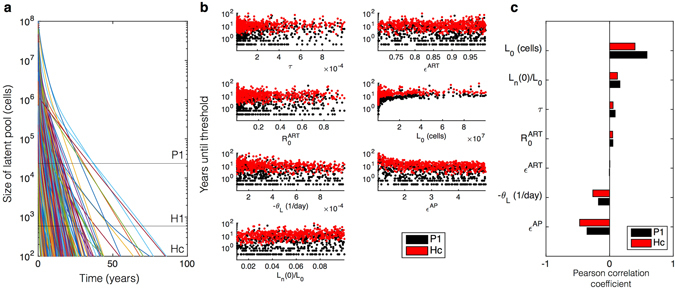
Global sensitivity analysis. We use the ranges of parameters from Supplementary Table S4. (a) 1,000 simulations drawn from Latin Hypercube sample parameter sets where are shown to demonstrate the variability of latent pool dynamics with respect to all combinations of parameter ranges. (b) The time until each cure threshold, Pinkevych 1-yr (P1) and Hill cure (Hc), are calculated as the time when the latent reservoir contains fewer than 20,000 and 200 cells respectively. In some cases cures are achieved within months. In others, cure requires many years. (c) Pearson correlation coefficients indicate the correlations between each variable and time to cure. L 0 is the initial number of latent cells. L n(0)/L 0 is the initial fraction of naïve cells in the latent pool. τ is the probability of latency given infection. is the basic reproductive number on ART. ε ART is the percent decrease in viral infectivity in the presence of ART. θ L is the decay rate of latent cells. ε AP is the fold reduction in proliferation rate.
Figure 5.
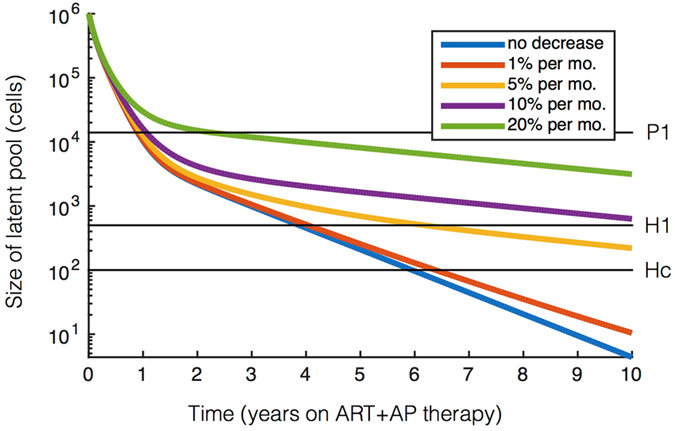
Waning anti-proliferative potency over-time modulates cure. Latent reservoir dynamics on combined ART and anti-proliferative therapy simulated for waning potency of anti-proliferative therapy over time. The latent reservoir size is shown with horizontal black lines corresponding to the cure threshholds used throughout the paper. Cure thresholds are achieved within 10 years if potency decreases by less than 5% per month considering 1% naïve T cells (L n(0)/L 0 = 0.01) and initial anti-proliferative potency ε AP = 5.
Model output is congruent with available clinical data
Chapuis et al. treated eight ART-suppressed, HIV-infected patients with 24 weeks of mycophenolate mofetil, a licensed anti-proliferative agent. As a marker of anti-proliferative effect, the percentages of Ki67+ CD4+ T cells were measured before and after MMF treatment (2 × 500 mg daily) and were found to have decreased on average 2.2-fold. Incorporating that reduction in latent cell proliferation rate ε AP = 2.2 over 24 weeks of treatment, we estimate a 10- to 40-fold reduction in the latent reservoir (see Fig. 2d). Chapuis et al. found a 10- to 100-fold reduction in infectious units per million (IUPM) by quantitative viral outgrowth assay in five patients, comparable to our estimate25. These reductions far exceed natural reservoir clearance rates and are consistent with a therapeutic effect26.
García et al. assessed the effect of MMF (2 × 250 mg daily) on HIV in the context of ART treatment interruption27. Seventeen HIV-infected patients received ART for a year and then were randomized into a control group that remained on ART only and an experimental group that also received MMF for 17 weeks. ART was interrupted in both groups and viral rebound assessed. MMF inhibited CD4+ T cell proliferation (as measured by an in vitro assay) in six of nine MMF recipients (responders). The time to rebound was 1–4 weeks in the control group and 6–12 weeks in the MMF-responder group. Using results from Pinkevych et al., a median time to rebound of seven weeks (see Fig. 5b of ref. 22) corresponds to a 7-fold decrease in the latent reservoir. Using results from Hill et al., the same median time to detection of seven weeks (see Fig. 4 of ref. 21) corresponds to a 50-fold reduction in the latent reservoir. These calculations are congruent with our model’s estimate that 17 weeks of MMF treatment at potency ε AP = 2.2 leads to a 10-fold reduction in the reservoir.
MMF decreases proliferation in CEM cells, CD4+ T cells from HIV positive and negative donors, and all CD4+ T cell subsets
To explain the heterogeneous impact of MMF treatment (three of six in Chapuis et al. did not demonstrate a meaningful reservoir clearance; three of nine patients in García et al. had a weak anti-proliferative response to MMF and no delay in HIV rebound upon ART cessation), we conducted an in vitro study of MMF pharmacodynamics. We titrated the capacity of mycophenolic acid to inhibit spontaneous proliferation of cells from a human T lymphoblastoid cell line (CEM cells)28 and identified a steep Hill slope of −3.7 (Fig. 6a). A Hill slope with absolute value greater than one indicates cooperative binding at the site of drug action and implies a sharp transition from negligible to complete therapeutic effect at a specific drug concentration. These results explain how patients with inadequate MMF dosage could have a limited anti-proliferative effect.
Figure 6.
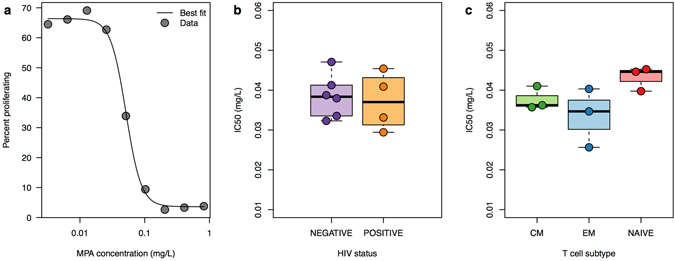
MMF pharmacodynamics. Pure mycophenolic acid (MPA) was added to CEM cells at varying concentrations and proliferation of CEM cells was measured to determine a dose-response curve and Hill slope. CD4+ T cells from stored peripheral blood mononuclear cell samples from 10 participants (4 HIV-infected, 6 HIV-uninfected) were stimulated to proliferate. CD4+ T cells from 3 HIV-negative subjects were sorted into effector memory (EM), central memory (CM), and naïve subsets. Pure MPA was added to these cells at varying concentrations in order to determine IC50s for MPA. (a) Dose-response curve with percentages of CEM cells proliferating at varying doses of MPA. The Hill slope is −3.7. (b) 4 samples from HIV-positive participants and 6 samples from HIV-negative participants had similar IC50s. (c) IC50s were similar among CD4+ effector memory (EM), central memory (CM), and naïve T cell subsets.
We tested the capacity of mycophenolic acid (MPA), the active metabolite of MMF, to inhibit CD4+ T cell proliferation in CD4+ T cells from four HIV-positive and six HIV-negative participants and found similar IC50s (Fig. 6b). Further, CD4+ T cells from three HIV-negative participants were sorted into central-memory, effector-memory, and naïve subsets. Similar proliferation inhibition was observed in all three cell subsets (Fig. 6c). These results suggest a potential for MMF to deplete the HIV reservoir.
Discussion
We developed a mathematical model of HIV dynamics to study various cure strategies21, 29. We demonstrate that minor reductions in CD4+ T cell proliferation rates would exhibit powerful reductions in the latent reservoir when therapy duration is extended over time. We call this proposed strategy “compound interest cure” due to the correspondence with financial modeling.
Our results are relevant because the HIV cure strategy most rigorously being tested in humans—latency reversal therapy (“shock-and-kill”)—may not capitalize on the advantages of a compound interest approach. Promising latency reversing agents are typically dosed over short time-frames due to concerns about toxicity. T cell activation does not always lead to induction of HIV replication providing another potential limitation of latency reversing therapy30. Furthermore, even if these agents exert a large relative impact on the activation rate of memory CD4+ T cells, we predict the reduction in the reservoir may be insignificant given that the natural activation rate is orders of magnitude lower than proliferation and death rates. Latency reversal agents are also being considered in conjunction with other interventions such as engineered antibodies and/or T cells. These combined approaches carry additional unknown toxicities and rely on the effectiveness of latency reversal agents. Most challenging of all, these experimental therapies could be prohibitively expensive to implement globally.
The theoretical potential of the anti-proliferative approach is worthy of a clinical trial given the existence of licensed medications that limit T cell proliferation, including MMF. In line with our prediction that duration is more important than potency, these drugs are dosed over months to years for rheumatologic diseases and preventing rejection after solid organ transplant. The most frequent side effects reported are gastrointestinal symptoms and increased risk of infection though the latter risk is obscured by concurrent use of high-dose glucocorticoids31. MMF has been given to several hundred HIV-infected patients suppressed on ART25, 27, 32–40 (reviewed in Supplementary information). In this population, neither opportunistic infections nor adverse events were increased, and CD4+ T cell counts did not decrease significantly during therapy. We hypothesize that whereas MMF decreases proliferation of existing CD4+ T cells, it does not suppress thymic replenishment of these cells. Finally, MMF did not counteract the effects of ART25, 27, and we do not expect viral drug resistance or ongoing viral evolution to occur on anti-proliferative therapy. Despite these reassuring findings, future studies of HIV-infected patients on anti-proliferative agents will require extremely close monitoring for drug toxicity and immunosuppression. In addition, mycophenolic acid has a large Hill coefficient, suggesting a narrow therapeutic range. We suspect that the participants who did not respond to MMF in the clinical studies described above25, 27 required higher drug concentrations.
Our model suggests that slowly proliferating cells in the reservoir could present a barrier to rapid eradication of latently HIV-infected cells. Therefore, anti-proliferative strategies may face a challenge akin to the cancer stem cell paradox, whereby only the rapidly proliferating tumor cells are quickly expunged with chemotherapy. For example, tyrosine kinase inhibitors suppress proliferation of cancer cells in chronic myelogenous leukemia (CML). While many patients achieve “undetectable minimal residual disease,” some patients relapse to pre-therapy levels of disease following therapy cessation—perhaps due to slowly proliferating residual cancer stem cells41. Additional limitations could include insufficient anti-proliferative drug delivery to anatomic sanctuaries, certain cellular subsets that are unaffected by treatment, and cytokine-driven feedback mechanisms that compensate for decreased proliferation by increasing memory CD4+ T cell lifespan. These challenges might be countered by combining anti-proliferative agents with other cure therapies. Avoidance of nucleoside and nucleotide reverse transcriptase inhibitors, which may enhance T cell proliferation, could provide an important adjunctive benefit42, 43.
The anti-proliferative approach is attractive because it is readily testable without the considerable research and development expenditures required for other HIV cure strategies. Anti-proliferative approaches require minimal potency relative to latency reversing agents, and T cell anti-proliferative medications are well studied mainstays of organ rejection prevention. Therefore, we propose trials with anti-proliferative agents as an important next step in the HIV cure agenda.
Methods
Latent reservoir dynamic model
We based our model (schematic in Fig. 1) on previous HIV dynamics models29, 44. We follow the concentrations [cells/μL] of susceptible CD4+ T cells S, latently infected cells L, actively infected cells A, and plasma viral load V [copies/μL] over time. The system of ordinary differential equations (using the over-dot to denote derivative in time)
| 1 |
tracks these state variables. We define α S [cells/μL-day] as the constant growth rate of susceptible cells, δ S [1/day] as the death rate of susceptible cells, and β ε = (1 − ε)β [μL/virus-day] as the therapy-dependent infectivity. We define ε [unitless] as the ART efficacy, ranging from 0 (meaning no therapy) to 1 (meaning perfect therapy). α L and δ L [1/day] are the proliferation and death rates of latent cells, respectively. The death rate of actively infected cells is δ A, and the proliferation rate of activated cells is likely negligible45. τ [unitless] is the probability of latency given infection, and ξ [1/day] is the rate of transition from latent to actively infected cells. The viral production rate is π [virions/cell-day], which describes the aggregate rate of constant viral leakage and burst upon cell death. γ [1/day] is the HIV clearance rate. Parameter values are given in Table 1.
Additional calculations including derivations of equilibrium solutions and stability analysis as well as further discussion of model parameter derivations are presented in the Supplementary information.
The compound interest formula
In the Supplementary information, we determine the critical drug efficacy ε c, the value of ε above which viral load quickly decays. Moreover, when ε > ε c, we can consider the latent cell equation in isolation:
| 2 |
Defining the initial number of latent cells as L 0 gives
| 3 |
Equation (3) implies that the clearance rate of latently infected cells is a function of their proliferation, death, and activation rates. Defining the total clearance rate θ L = α L − δ L − ξ, we see a mathematical correspondence to the principle of continuous compound interest with L 0 as the principal investment and θ L as the interest rate:
| 4 |
Experimental measurements indicate an average latent cell half-life of 44 months (θ L = −5.2 × 10−4 per day)1, 26 and an average latent reservoir size L 0 of one-million cells1. Note that when θ L < 0, the latent reservoir is cleared exponentially. Alternatively, if α L exceeds the sum of ξ and δ L, L grows indefinitely.
Composition of the latent reservoir: modeling T cell subsets
We include heterogeneity in T cell phenotype into the model by splitting the differential equation for the latent cells into three differential equations, one for each subtype L i with . We ignore transitions between phenotype because the composition of the reservoir is reasonably stable over time12. Our extended model is the system
| 5 |
The total number of latent cells is the sum of the subset populations, , and solution is
| 6 |
where θ i = α i − δ i − ξ, and L i(0) are the initial numbers of each subtype.
Simulations assume the same net clearance rate and activation rates among subsets, but different proliferation rates α i and different calculated death rates δ i = α i − θ L − ξ. The initial conditions for each subtype L i(0) are inclusive of several varying measurements in the literature6, 12, 23. We consider Ttm to have the same proliferation rates as Tcm. Similarly, we characterize stem-cell-like memory CD4+ T cells (Tscm) as Tn given their slow turnover rate. Of note, these are conservative estimates that would not favor anti-proliferative therapy. In Fig. 5, we allow the anti-proliferative potency to decrease over time by assuming for each T cell subset in equation 5. Here ϕ is the waning potency rate that ranges from 0–20% per month. We assume the initial potency is a 5-fold decrease ε AP = 5, and we use a 1 million cell reservoir having 1% naïve T cells. We solve the equation for each subset numerically with in Matlab, summing the subset dynamics after solving.
Reservoir reduction targets for cure strategies
We use experimentally derived thresholds to compare potential cure therapies in the framework of our model. Hill et al. employed a stochastic model to estimate that a 2,000-fold reduction in the latent pool would result in HIV suppression off ART for a median of one year. After a 10,000-fold reduction in latent cells, 50% of patients would remain functionally cured (ART-free remission for at least 30 years)21. Pinkevych et al. inferred from analytic treatment interruption data that decreasing the latent reservoir 50–70-fold would lead to HIV remission in 50% of patients for one year22. Using the Pinkevych et al. results, we extrapolate a functional cure threshold as a 2,500-fold decrease in the reservoir size (Supplementary information). Given ongoing debate in the field, we consider all four thresholds—henceforth referred to as Hill 1-yr, Hill cure, Pinkevych 1-yr, and Pinkevych cure.
Sensitivity analysis
To examine the full range of possible outcomes we completed a global sensitivity analysis of the model in which all variables were simultaneously varied in the ranges of Table S5 by logarithmically covering Latin Hypercube sampling24. The simulations were carried out in Matlab using and . We correlated each parameter of interest with the time to reach the Hill and Pinkevych cure thresholds. Calling the time-to-cure T, correlations were calculated with the Pearson correlation coefficient: the covariance of T with each parameter of interest p normalized by both the standard deviation of T and that of p, that is . 1,000 simulations were carried out, keeping only the parameter combinations leading to reservoir decay, i.e. those satisfying .
Mycophenolic acid anti-proliferation assay methods
Blood samples for the MPA in vitro studies were obtained from ART-treated, HIV-infected and healthy, HIV-negative men at the HIV Vaccine Trials Unit Clinic in Seattle, Washington. All procedures were approved by the Institutional Review Boards of the University of Washington and the Fred Hutchinson Cancer Research Center (IRB 1830 and 5567) and were performed in accordance with institutional guidelines and regulations. Written informed consent was obtained from each donor.
Cells were labeled using the CellTrace Violet Cell Proliferation Kit (Invitrogen) by incubation in 40 μM CellTrace Violet in Roswell Park Memorial Institute (RPMI) cell culture media with penicillin/streptomycin and L-glutamine (Gibco) plus 10% fetal bovine serum (Gemini Bio-Products) (R-10 media) for five minutes at room temperature46 followed by washing twice with R-10. Peripheral blood mononuclear cells were stimulated with 1 μg/mL staphylococcal enterotoxin B (SEB; Sigma-Aldrich) and 10 IU/mL IL-2 (Peprotech). Sorted CD4+ T cell subsets (naïve, effector memory, and central memory) were stimulated with Dynabeads Human T-Activator CD3/CD28 beads (Gibco) at a bead to cell ratio of 1:1 with 10 IU/mL IL-2. CEM cells were not stimulated, as they proliferate continuously. Pure mycophenolic acid (Sigma-Aldrich), the active metabolite of MMF, was added at concentrations ranging from 0.01 to 2.56 μM. Cells were cultured in R-10 for 72 h.
After the culture period, cells were washed and stained with Fixable Live/Dead Yellow (Invitrogen), followed by CD45RA FITC, CD4 PE-Cy5, CCR7 BV785 (all BD), and CD3 ECD (Beckman Coulter) at the minimum saturating doses. Cells were then fixed with 1% paraformaldehyde and acquired on a five-laser BD LSRII flow cytometer (355, 405, 488, 535, and 633 nm). Live, single CD4+ T cells were gated into “proliferated” or “not proliferated” on the basis of CellTrace Violet fluorescence.
The IC50s and Hill slope were calculated using the package in (Supplementary information)47, 48.
Electronic supplementary material
Acknowledgements
We thank Keith Jerome for his helpful reading of the manuscript, the VIDD faculty initiative at the Fred Hutchinson Cancer Research Center, and the NIH for grants R01 AI116292 to F.H., 1DP2DE023321-01 to M.P., and U19 AI096111 and UM1 AI12662 to J.T.S. We also thank Claire N. Levy and Fernanda Calienes for assisting in the mycophenolic acid experiments. The following reagent was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: CEM CD4+ T cells from Dr. J. P. Jacobs.
Author Contributions
F.H. and J.T.S. posed the initial question to model the effect of anti-proliferation on HIV latency; D.B.R., E.R.D., and J.T.S. developed the computational model; S.M.H. and F.H. devised, and S.M.H. performed, the in vitro mycophenolic acid experiments; D.B.R. and E.R.D. performed calculations and produced figures; E.R.D., M.P., F.H., J.T.S. performed literature review for parameter values. D.B.R., E.R.D., M.P., F.H., S.M.H., and J.T.S. wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Daniel B. Reeves and Elizabeth R. Duke contributed equally to this work.
Florian Hladik and Joshua T. Schiffer jointly supervised this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04160-3
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Florian Hladik, Email: fhladik@fredhutch.org.
Joshua T. Schiffer, Email: jschiffe@fredhutch.org
References
- 1.Siliciano JD, et al. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med. 2003;9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 2.Finzi D, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 3.Martin A, Siliciano R. Progress toward HIV eradication: Case reports, current efforts, and the challenges associated with cure. Annu Rev Med. 2016;67:011514–023043. doi: 10.1146/annurev-med-011514-023043. [DOI] [PubMed] [Google Scholar]
- 4.Hütter G, et al. Long-term control of HIV by CCR5Δ32 stem-cell transplantation. N Engl J Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 5.Bull ME, et al. Monotypic human immunodeficiency virus type 1 genotypes across the uterine cervix and in blood suggest proliferation of cells with provirus. J Virol. 2009;83:6020–6028. doi: 10.1128/JVI.02664-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jaafoura S, et al. Progressive contraction of the latent HIV reservoir around a core of less-differentiated CD4+ memory T cells. Nat Comm. 2014;5:5407. doi: 10.1038/ncomms6407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Palmer S, et al. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc Natl Acad Sci USA. 2008;105:3879–3884. doi: 10.1073/pnas.0800050105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Von Stockenstrom S, et al. Longitudinal genetic characterization reveals that cell proliferation maintains a persistent HIV-1 DNA pool during effective HIV therapy. J Infect Dis. 2015;1:596–607. doi: 10.1093/infdis/jiv092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wagner T, et al. An increasing proportion of monotypic HIV-1 DNA sequences during antiretroviral treatment suggests proliferation of HIV-infected cells. J Virol. 2013;87:1770–1778. doi: 10.1128/JVI.01985-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wagner TA, et al. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 2014;345:570–573. doi: 10.1126/science.1256304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maldarelli F, et al. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014;345:179–83. doi: 10.1126/science.1254194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chomont N, et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med. 2009;15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bui JK, et al. Proviruses with identical sequences comprise a large fraction of the replication-competent HIV reservoir. PLoS Path. 2017;13:e1006283. doi: 10.1371/journal.ppat.1006283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonhoeffer S, Coffin JM, Nowak MA. Human immunodeficiency virus drug therapy and virus load. J Virol. 1997;71:3275–3278. doi: 10.1128/jvi.71.4.3275-3278.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Callaway D, Perelson A. HIV-1 infection and low steady state viral loads. Bull Math Biol. 2002;64:29–64. doi: 10.1006/bulm.2001.0266. [DOI] [PubMed] [Google Scholar]
- 16.Shen L, et al. A critical subset model provides a conceptual basis for the high antiviral activity of major HIV drugs. Sci Transl Med. 2011;3:91ra63. doi: 10.1126/scitranslmed.3002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brodin J, et al. Establishment and stability of the latent HIV-1 DNA reservoir. eLife. 2016;5:e18889. doi: 10.7554/eLife.18889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fuller DH, et al. Therapeutic DNA vaccine induces broad T cell responses in the gut and sustained protection from viral rebound and AIDS in SIV-infected rhesus macaques. PLoS One. 2012;7:e33715. doi: 10.1371/journal.pone.0033715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aubert M, et al. Successful targeting and disruption of an integrated reporter lentivirus using the engineered homing endonuclease Y2 I-AniI. PLoS One. 2011;6:e16825. doi: 10.1371/journal.pone.0016825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peterson C, Younan P, Jerome K, Kiem H-P. Combinatorial anti-HIV gene therapy: using a multipronged approach to reach beyond HAART. Gene Ther. 2013;20:695–702. doi: 10.1038/gt.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hill A, Rosenbloom D, Fu F, Nowak M, Siliciano R. Predicting the outcomes of treatment to eradicate the latent reservoir for HIV-1. Proc Natl Acad Sci USA. 2014;111:15597. doi: 10.1073/pnas.1406663111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinkevych M, et al. HIV reactivation from latency after treatment interruption occurs on average every 5–8 days? Implications for HIV remission. PLoS Pathog. 2015;11:e1005000. doi: 10.1371/journal.ppat.1005000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buzon MJ, et al. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med. 2014;20:139–142. doi: 10.1038/nm.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marino S, Hogue I, Ray C, Kirschner D. A methodology for performing global uncertainty and sensitivity analysis in systems biology. J Theor Biol. 2008;254:178–196. doi: 10.1016/j.jtbi.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chapuis AG, et al. Effects of mycophenolic acid on human immunodeficiency virus infection in vitro and in vivo. Nat Med. 2000;6:762–768. doi: 10.1038/77489. [DOI] [PubMed] [Google Scholar]
- 26.Crooks AM, et al. Precise quantitation of the latent HIV-1 reservoir: implications for eradication strategies. J Infect Dis. 2015;212:1361–1365. doi: 10.1093/infdis/jiv218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garca F, et al. Effect of mycophenolate mofetil on immune response and plasma and lymphatic tissue viral load during and after interruption of highly active antiretroviral therapy for patients with chronic HIV infection: a randomized pilot study. J Acquir Immune Defic Syndr. 2004;36:823–830. doi: 10.1097/00126334-200407010-00009. [DOI] [PubMed] [Google Scholar]
- 28.Foley GE, et al. Continuous culture of human lymphoblasts from peripheral blood of a child with acute leukemia. Cancer. 1965;18:522–529. doi: 10.1002/1097-0142(196504)18:4<522::AID-CNCR2820180418>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 29.Conway J, Perelson A. Residual Viremia in Treated HIV+ Individuals. PLoS Comput Biol. 2016;12:e1004677. doi: 10.1371/journal.pcbi.1004677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ho Y-C, et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013;155:540–551. doi: 10.1016/j.cell.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mok C. Mycophenolate mofetil for lupus nephritis: an update. Expert Rev Clin Immunol. 2015;11:1353–1364. doi: 10.1586/1744666X.2015.1087314. [DOI] [PubMed] [Google Scholar]
- 32.Müller E, Barday Z, Mendelson M, Kahn D. HIV-positive–to–HIV-positive kidney transplantation—Results at 3 to 5 years. N Engl J Med. 2015;372:613–620. doi: 10.1056/NEJMoa1408896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stock PG, et al. Outcomes of kidney transplantation in HIV-infected recipients. N Engl J Med. 2010;363:2004–2014. doi: 10.1056/NEJMoa1001197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaur R, et al. A placebo-controlled pilot study of intensification of antiretroviral therapy with mycophenolate mofetil. AIDS Res Ther. 2006;3:16. doi: 10.1186/1742-6405-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vrisekoop N, et al. Short communication: no detrimental immunological effects of mycophenolate mofetil and HAART in treatment-naive acute and chronic HIV-1-infected patients. AIDS Res Hum Retrovir. 2005;21:991–996. doi: 10.1089/aid.2005.21.991. [DOI] [PubMed] [Google Scholar]
- 36.Sankatsing SU, et al. Highly active antiretroviral therapy with or without mycophenolate mofetil in treatment-naive HIV-1 patients. AIDS. 2004;18:1925–1931. doi: 10.1097/00002030-200409240-00008. [DOI] [PubMed] [Google Scholar]
- 37.Press N, et al. Case series assessing the safety of mycophenolate as part of multidrug rescue treatment regimens. HIV Clin Trials. 2002;3:17–20. doi: 10.1310/B6T0-N98J-1J3M-EQPK. [DOI] [PubMed] [Google Scholar]
- 38.Margolis DM, et al. The addition of mycophenolate mofetil to antiretroviral therapy including abacavir is associated with depletion of intracellular deoxyguanosine triphosphate and a decrease in plasma HIV-1 RNA. J Acquir Immune Defic Syndr. 2002;31:45–49. doi: 10.1097/00126334-200209010-00006. [DOI] [PubMed] [Google Scholar]
- 39.Millan O, et al. Pharmacokinetics and pharmacodynamics of low dose mycophenolate mofetil in HIV-infected patients treated with abacavir, efavirenz and nelfinavir. Clin Pharmacokinet. 2005;44:525–538. doi: 10.2165/00003088-200544050-00006. [DOI] [PubMed] [Google Scholar]
- 40.Jurriaans S, et al. HIV-1 seroreversion in an HIV-1-seropositive patient treated during acute infection with highly active antiretroviral therapy and mycophenolate mofetil. AIDS. 2004;18:1607–1608. doi: 10.1097/01.aids.0000131367.05823.ce. [DOI] [PubMed] [Google Scholar]
- 41.Ross DM, et al. Safety and efficacy of imatinib cessation for CML patients with stable undetectable minimal residual disease: results from the TWISTER study. Blood. 2013;122:515–522. doi: 10.1182/blood-2013-02-483750. [DOI] [PubMed] [Google Scholar]
- 42.Hladik F. A new perspective on HIV cure. F1000Res. 2014;4:77. doi: 10.12688/f1000research.4529.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hladik F, et al. Mucosal effects of tenofovir 1% gel. eLife. 2015;4:e04525. doi: 10.7554/eLife.04525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rong L, Perelson A. Modeling latently infected cell activation: Viral and latent reservoir persistence, and viral blips in HIV-infected patients on potent therapy. PLoS Comput Biol. 2009;5:e1000533. doi: 10.1371/journal.pcbi.1000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perelson AS, Kirschner DE, De Boer R. Dynamics of HIV infection of CD4+ T cells. Math Biosci. 1993;114:81–125. doi: 10.1016/0025-5564(93)90043-A. [DOI] [PubMed] [Google Scholar]
- 46.Quah BJ, Parish CR. New and improved methods for measuring lymphocyte proliferation emphin vitro and in vivo using CFSE-like fluorescent dyes. J Immunol Methods. 2012;379:1–14. doi: 10.1016/j.jim.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 47.Ritz, C., Baty, F., Streibig, J. C. & Gerhard, D. Dose-response analysis using R. PLoS One10, e0146021, journal.pone.0146021 (2015). [DOI] [PMC free article] [PubMed]
- 48.R Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria (2016).
- 49.Macallan DC, et al. Rapid turnover of effector-memory CD4+ T cells in healthy humans. J Exp Med. 2004;200:255–260. doi: 10.1084/jem.20040341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Markowitz M, et al. A novel antiviral intervention results in more accurate assessment of HIV-1 replication dynamics and T-cell decay in vivo. J Virol. 2003;77:5037–5038. doi: 10.1128/JVI.77.8.5037-5038.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luo R, Piovoso M, Martinez-Picado J, Zurakowski R. HIV model parameter estimates from interruption trial data including drug efficacy and reservoir dynamics. PLoS One. 2012;7:e40198. doi: 10.1371/journal.pone.0040198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hockett RD, et al. Constant mean viral copy number per infected cell in tissues regardless of high, low, or undetectable plasma HIV RNA. J Exp Med. 1999;189:1545–1554. doi: 10.1084/jem.189.10.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramratnam B, et al. Rapid production and clearance of HIV-1 and hepatitis C virus assessed by large volume plasma apheresis. Lancet. 1999;354:1782–1785. doi: 10.1016/S0140-6736(99)02035-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.