

Abstract
Post-inflammatory scarring is the end-result of excessive extracellular matrix (ECM) accumulation and tissue architectural destruction. It represents a failure to effectively remodel ECM and achieve proper reinstitution and healing during chronic relapsing inflammatory processes. Scarring may affect the functionality of any organ, and in the case of inflammatory bowel disease (IBD)-associated fibrosis leads to stricture formation and often surgery to remove the affected bowel. The activated myofibroblast is the final effector cell that overproduces ECM under the influence of various mediators generated by an intense interplay of classic and non-classic immune cells. This review focuses on how proinflammatory mediators from various sources produced in different stages of intestinal inflammation can form profibrotic pathways that eventually lead to tissue scarring through sustained activation of myofibroblasts.
Keywords: Intestinal fibrosis, inflammatory bowel disease, myofibroblast, extracellular matrix, Crohn’s disease, ulcerative colitis
Introduction
Inflammatory bowel diseases (IBDs) comprise ulcerative colitis (UC) and Crohn’s disease (CD) and are characterized by chronic and recurrent inflammation of the intestinal tract [1]. Their pathophysiology is yet to be fully elucidated. Nonetheless, accumulated scientific data suggest that IBDs are caused by aberrant immune responses to the intestinal microflora, triggered by various environmental insults, in the context of a susceptible genetic background of the host [1]. The recurrent nature of IBD is characterized by repeated cycles of tissue inflammation, ulceration and repair that ultimately lead to tissue scarring [2]. Scarring is the end result of fibrosis, a process of excessive accumulation of extracellular matrix (ECM), and especially collagen, combined with the failure to effectively remodel ECM, that ultimately leads to the formation of strictures [2]. The clinical impact of this process is major in CD patients with the “fibrostenosing” phenotype, where symptoms and complications associated with small bowel obstruction predominate over those related to intestinal inflammation. In contrast, intestinal fibrosis and bowel obstruction rarely dominate the clinical picture in UC, as the inflammation is superficial and confined to the colon, which has a larger luminal diameter [3].
Although chronic intestinal inflammation is absolutely required for the development of fibrosis, it plays a minor role once fibrosis is established [4]. This is supported by the fact that anti-inflammatory medication has minimal or no effect on fibrostenotic CD, where surgery is still the only treatment option. Unfortunately, intestinal strictures are likely to recur following surgical treatment [4]. Specifically, almost 30% of patients with CD and 5% of patients with UC will have to undergo surgery for the treatment of intestinal strictures, with the risk of recurrence reaching 50% [5]. A considerable number of CD patients will be subjected to repeated surgeries, and occasionally, following resection of a significant part of the small bowel, will suffer from short bowel syndrome and malnutrition [6]. Therefore, there is an urgent need for the development of drugs that could prevent or even reverse intestinal fibrosis [5].
Under physiological conditions, acute inflammation leads to the activation of mesenchymal cells that increase the production of ECM in an effort to heal the wound. Chronic accumulation of immune cells and the constant release of inflammatory mediators lead to persistent tissue damage and excessive production of ECM [3]. The myofibroblast seems to be the protagonist in this process, being the leading mesenchymal cell in producing ECM components [7,8]. This review focuses on the interactions between immune cells and myofibroblasts that contribute to fibrosis and highlights pathways that could be potentially targeted.
Intestinal stricturing and ECM remodeling failure
Intestinal strictures are developed by thickening of all layers of the intestinal wall. This is mediated via hyperplasia and hypertrophy of smooth muscle cells, leading to hypertrophic and disordered muscularis layers, and increased production and disorganized deposition of ECM [9]. ECM consists of various structural proteins, such as collagen, glycoproteins and proteoglycans. Type I collagen is the major collagen subtype found in normal bowel, followed by type III and type V. Bowel fibrosis is characterized by an increase in the total collagen content, as well as an increase in the relative amounts of types III and V [10].
ECM remodeling is regulated by two different groups of proteins, the matrix metalloproteinases (MMPs) and their inhibitors. MMPs are proteolytic enzymes that cleave various ECM components. Different classes of MMPs cleave different ECM substrates, sometimes acting sequentially—for example in the case of collagen, initially cleaved by collagenases and then by gelatinases. MMPs can be regulated either transcriptionally, by various factors such as cytokines, growth factors, hormones, cell-cell and cell-matrix interactions, or post-transcriptionally and through epigenetics [11]. Among other important MMP regulators are the tissue inhibitors of metalloproteinases (TIMP)-1-4 [9]. It has been suggested that an imbalance between MMPs and TIMPs, due to a decrease in MMP activity and/or an increase in TIMP activity, may lead to ECM accumulation and eventually fibrogenesis [9]. The contribution of the imbalance between MMPs and TIMPs in tissue fibrosis has been demonstrated by studies in mice that lack TIMP-1. Specifically, TIMP-1 depletion in the chronic model of dextran sulfate sodium (DSS)-induced colitis resulted in reduced inflammation and collagen disposition, with elevated expression levels of MMP-9, but not MMP-2 [12].
The theory of MMP/TIMP imbalance contributing to intestinal fibrosis is also supported by descriptive studies in patients with fibrostenotic CD. Intestinal subepithelial myofibroblasts (SEMFs) isolated from fibrotic areas of patients with CD exhibited increased TIMP-1 transcripts and unchanged expression of MMPs compared to healthy controls or UC [13]. Treatment with transforming growth factor (TGF)-β1 on SEMFs isolated from healthy individuals resulted in TIMP-1 induction [13], whereas blockage of TGF-β in normal intestinal biopsies resulted in higher levels of MMP-3 but not TIMP-1 [14]. In addition, higher levels of TIMP-1, along with lower levels of MMP-12 and -3 transcripts were reported in mucosa overlying uninflamed strictured areas, when compared to mucosa lining uninflamed or inflamed non-strictured regions [15]. Resent studies indicate that elevated MMP-9 levels can be found in feces of patients with active UC; this correlates with clinical, endoscopic and histological activity, suggesting MMP-9 as a possible new fecal marker for monitoring disease activity [16]. However, alteration of the balance between MMPs and TIMPs cannot always predict ECM accumulation or degradation. In the case of MMP-7, which cleaves galectin-3, a crucial molecule of epithelial cell migration, the increase in MMP-7, and the subsequent degradation of galectin-3, inhibits re-epithelialization and eventually contributes to fibrosis [17,18]. MMP-10 has been found to be upregulated at the healing edges of ulcers in patients with UC, suggesting a possible role in disease resolution [19], while DSS treatment in mice lacking MMP-10 increased the severity of colitis [20]. Therefore, given the complex interactions between the various MMPs, their substrates and different TIMPs during ECM remodeling, targeting MMP/TIMP balance as a therapeutic strategy to prevent or treat fibrosis may produce highly unpredictable outcomes.
The activated myofibroblast
ECM production depends on the activation state of ECM-producing myofibroblasts. Activation and proliferation of myofibroblasts by inflammatory and profibrotic mediators is a central event in the process of normal wound healing after acute injury (Table 1). Following the resolution of an acute inflammation, the healing process is gradually terminated and the connective tissue gains its initial architecture. In the case of IBD, the ongoing and persistent inflammation leads to a sustained activation and proliferation of ECM-producing myofibroblasts [3].
Table 1.
Factors with a profibrotic effect and their mechanisms of action in myofibroblast biology. Data from preclinical studies and experimental models

Myofibroblasts are characterized by specific cellular markers that grant them an intermediate phenotype between fibroblasts and smooth muscle cells, their contractile shape and their ability to produce great quantities of ECM components when activated [9,21]. There are two main cell types of myofibroblasts in the intestine. The first ones are the intestinal cells of Cajal, which reside in the submucosa and the muscularis propria and provide signals that facilitate gastrointestinal motility [22]. The second type is the SEMFs, which express vimentin and α-smooth muscle actin (α-SMA) (Fig. 1). SEMFs are located within the lamina propria underneath the epithelial cells, forming an anastomosing syncytium that is dense in the crypt region and sparse at the colonic surface or the villi of the small bowel [7]. Gene expression profiles of pericryptal myofibroblasts suggest their participation in the intestinal stem cell niche through the production of bone morphogenetic protein (BMP) antagonists and R-spondins, well-described Wnt agonists, that maintain epithelial stem cell Wnt signaling and therefore self-renewal and expansion [23]. A recent study showed that SEMFs produce angiopoietin-like protein 2, an essential factor for epithelial regeneration that acts on epithelial cells through the BMP/β-catenin signaling pathway [24]. In addition, they secrete growth factors such as TGF-β [25], cyclooxygenase products [26] such as prostaglandin E2 (PGE2) [27], and ECM-related proteins [26], which suggests a primary role in epithelial reinstitution upon injury.
Figure 1.
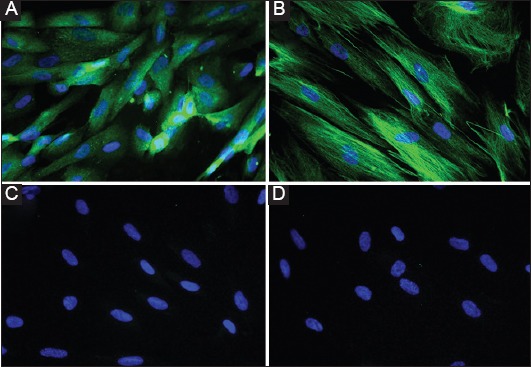
Human intestinal subepithelial myofibroblasts. Immuno-fluorescence staining of subepithelial myofibroblasts isolated from human colonic biopsies, expressing α-smooth muscle actin (A) and vimentin (B), negative for desmin (C), and isotype matched control antibody (D) (unpublished data)
There are accumulating data that incriminate SEMFs in autoimmune inflammation and post-inflammatory fibrosis. Specifically, SEMFs respond to proinflammatory cytokines by upregulating the expression of interleukin (IL)-8, IL-6, monocyte chemotactic protein-1 (MCP-1/CCL-2), granulocyte macrophage colony-stimulating factor and macrophage colony-stimulating factor [28,29]. They also express Toll-like receptor (TLR)-2 and TLR-4, which induce MCP-1, growth-related oncogene-α (GRO-α), IL-6 and IL-8 upon stimulation with lipopolysaccharide [30,31] and TLR-5, which induce IL-6 and IL-8 in response to flagellin [31]. Furthermore, they have been found to constitutively express class II major histocompatibility complex, and possibly act as non-professional antigen-presenting cells to promote CD4+ T cell differentiation [32]. Recently, we have shown that stimulation of human SEMFs with proinflammatory cytokines results in production of tumor necrosis factor (TNF)-like ligand 1A (TL1A), a molecule recently implicated in both T-cell differentiation and intestinal fibrosis [33]. Concerning their possible participation in post-inflammatory intestinal fibrosis, SEMFs have been found to respond to proinflammatory and profibrotic cytokines, such as IL-17A [34] and TGF-β [35], by increasing production of collagen and MMPs. Finally, we have shown that conditioned medium from epithelial cell cultures stimulated with proinflammatory cytokines can enhance SEMF migration, and production of collagen and MMPs, suggesting the existence of a crosstalk between SEMFs and overlying epithelial cells during intestinal inflammation that can generate profibrotic responses [35].
Other stromal cells capable of transforming into activated ECM-producing intestinal myofibroblasts include fibroblasts and pericytes [36]. Fibroblasts are potent ECM-producing cells, present in small numbers in almost every tissue. They express specific cellular markers, such as CD90 and vimentin, and they are negative for α-SMA and desmin expression [21]. In case of acute injury or inflammation, fibroblasts are activated, migrate and proliferate at the site of the wound and contribute to ECM production [37]. It has also been reported that upon stimulation with specific growth factors, such as TGF-β1, fibroblasts express α-SMA and differentiate into myofibroblast-like cells [38].
Pericytes are mainly found in capillaries and small blood vessels surrounding endothelial cells [36] and play a major role in angiogenesis, as they express receptors for platelet-derived growth factor (PDGF), endothelin-1 and angiotensin II [39]. Pericytes are also implicated in fibrosis, as they are characterized as α-SMA and desmin positive cells and upon acute or chronic inflammation they differentiate into fibroblast-like cells, producing large quantities of ECM components. In a recent study, sections from inflamed mucosa from patients with long-lasting UC showed an increased population of active pericytes compared to controls [40]. However, their contribution to intestinal fibrosis has not yet been clearly defined [36,41].
Another source of ECM-producing cells is the bone marrow-derived fibrocytes, which originate from multipotent bone marrow mesenchymal stem cells. Under certain circumstances, bone marrow mesenchymal stem cells differentiate and give rise to fibrocytes, which in turn travel through the bloodstream to the affected tissue. Fibrocytes express specific markers of hematopoietic origin, such as CD45, CD34, CD11, CD13, CD14, CD80 and CD86, and they are also positive for collagen I and α-SMA expression [36]. Fibrocytes’ implication in fibrosis has been indicated by a study showing an increased fibrocyte population in inflamed specimens and peripheral blood from patients with CD. In the same study, peripheral blood-isolated fibrocytes expressed increased collagen type I and TNF-α mRNA levels in response to lipopolysaccharide, indicating that fibrocytes may actively contribute to fibrosis upon injury [42].
Additionally, non-mesenchymal cells, such as epithelial and endothelial cells, are able to contribute to fibrosis via epithelial-to-mesenchymal transition (EMT) or endothelial-to-mesenchymal transition [43]. Both are common events in embryogenesis and carcinogenesis, but have also been observed during fibrogenesis. In this process, epithelial or endothelial cells lose their initial markers, rearrange their cytoskeleton, change morphology and acquire a fibroblast-like phenotype and function. Eventually, cells express α-SMA or vimentin and acquire the ability to produce ECM components, such as collagen and fibronectin [44].
From endothelial and epithelial injury to myofibroblast activation
Wound-healing responses are initiated by the activation and proliferation of ECM-producing myofibroblasts in response to acute epithelial and/or endothelial injury. Endothelial injury activates coagulation cascades as a part of the normal wound-healing response. We and others have shown that activated coagulation factors, such as thrombin, activated coagulation factor X and endothelin-1, potentially produced by endothelial cells, can induce myofibroblast differentiation, migration and pro-collagen production (Fig. 2) [45-47]. Interactions of platelet glycoprotein receptors Ia/Ib/V/VI/IX with von Willebrand factor and exposed collagen induce the arrest and activation of adherent platelets [48]. Activated platelets secrete many profibrotic factors, including TGF-β, connective tissue growth factor (CTGF) and PDGF, that act upon precursors of myofibroblasts to drive differentiation to activated myofibroblasts with profibrotic phenotype.
Figure 2.
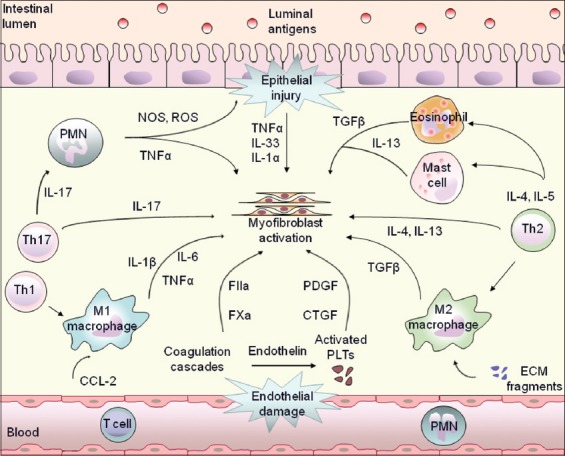
Immune pathways that drive myofibroblast activation. Classic and non-classic immune cells participating in the different stages of tissue inflammation, from initial endothelial or epithelial injury to innate and subsequent adaptive immune activation, drive myofibroblast activation. These include coagulation factors, growth factors (PDGF and CTGF), chemokines (CCL-2), cytokines (TGF-β, IL-13, TNF-α, IL-17) and reactive oxygen (ROS) and nitrogen (NOS) species
TGF-β is a pluripotent cytokine implicated in many cellular and immunological processes. The TGF-β/SMAD pathway is a major “core” pathway for fibrosis that includes IL-13 and CTGF as upstream and downstream mediators, respectively, and can be enhanced or inhibited by various pro- or antifibrotic molecules [49]. Activated TGF-β1 differentiates human fibroblasts to myofibroblasts, via upregulation of α-SMA expression, enhances their proliferation and induces resistance to apoptosis [50]. Acting upon human SEMFs, TGF-β enhances their migratory capacity and increases production of collagen and TIMP-1 [13,35,51]. TGF-β blockade ameliorated fibrosis in animal models of chronic colitis [52], whereas overexpression of TGF-β leads to intestinal stricture formation [53]. In a newly established model for fibrosis using human intestinal organoids, TGF-β treatment resulted in upregulation of fibrosis-related factors, such as collagen type I, fibronectin, α-SMA, actin contractile gene MYLK and fibrogenic transcription factor MLK1 [54]. Despite the undisputable contribution of TGF-β to intestinal fibrosis, the optimal TGF-β inhibition strategy is still under investigation so as to selectively block profibrotic, but not immune-regulatory pathways that are vital to mucosal homeostasis [9].
PDGF is a potent mitogen for myofibroblasts, enhancing chemotaxis and collagen production [55]. However, evidence for a role of PDGF in intestinal fibrosis is still sparse and indirect. Specifically, increased PDGF-A transcripts have been detected in the colonic mucosa in UC. SEMFs isolated from UC patients have been found to express the PDGF receptor β (PDGFRβ), while isolated intestinal SEMFs have been found to respond to PDGF-AB and PDGF-BB by increasing migration and proliferation [51,56]. Kurahashi et al identified a subpopulation of intestinal fibroblasts that express PDGFR and present distinct characteristics [57]. In another study, stimulation of CCD-18Co colonic fibroblasts or colonic epithelial cell line T84 with PDGFA led to an increased proliferation rate. Furthermore, PDGFA activity was found to be regulated by vasopressin, a neurohormone mainly involved in the regulation of water reabsorption in the kidney and large intestine, indicating a possible mechanism in wound repair upon epithelium injury [58]. Therefore, further investigation is warranted of the role for PDGF in intestinal fibrosis.
CTGF is a cysteine-rich peptide, induced by TGF-β, PDGF and basic fibroblast growth factor in fibroblasts, epithelial and endothelial cells, and is crucial for angiogenesis and wound healing [59]. It is a heparin-binding ECM-associated protein that regulates cell adhesion, migration, proliferation, and differentiation through integrin receptors on responding cells that include endothelial cells, platelets and fibroblasts [60]. Being a downstream mediator of the TGF-β pathway, CTGF acts upon stromal cells to promote fibroblast proliferation and production of ECM proteins and MMPs [60,61]. CTGF expression has been found to increase progressively along with the development of intestinal fibrosis in chronic 2,4,6-Trinitrobenzenesulfonic acid (TNBS) colitis, whereas knocking-down Smad3 decreased CTGF production and fibrosis development [62,63]. Increased expression of CTGF has been found in intestinal strictures of CD patients [64-66]. Further studies using in situ hybridization have demonstrated localization of CTGF transcripts primarily in fibroblasts within the submucosal layer [66].
Increased expression of IL-1β and IL-6 has been detected in inflamed tissue from IBD patients, produced mostly by lamina propria mononuclear cells [67] Okuno et al showed that intestinal SEMFs overexpress collagen type I and type IV, as well as MCP-1 and MMP-1, in response to IL-1β stimulation [28]. In a more recent study, IL-1β upregulated the mRNA levels of IL-36γ in human SEMFs [68], a cytokine belonging to the IL-1 family that was recently found to be elevated in active inflamed tissues of patients with IBD [69]. The role of IL-1β in intestinal wound repair is highlighted by an in vitro study where conditioned media from CCD-18Co previously stimulated with IL-1β induced the migration and differentiation of epithelial cell line Caco-2. Furthermore, IL-1β induced the overexpression of Wnt5a in CCD-18Co, an essential molecule of epithelium integrity and regeneration. Conditioned media depleted from Wnt5a failed to promote epithelial migration and differentiation, indicating that IL-1β indirectly induces epithelial migration through its direct action on myofibroblasts’ ability to express Wnt5a [70]. However, increased local and systemic IL-6 levels have been mainly associated with disease activity and severity. SEMFs isolated either from inflamed or non-inflamed tissue of patients with CD expressed elevated levels of IL-6, compared with SEMFs isolated from non-IBD patients [71]. Nonetheless, a link between IL-6 and fibrosis in IBD has not yet been established.
Intestinal epithelial necrosis release damage-associated molecular patterns (DAMPs) as part of a sterile inflammatory response, which takes place in parallel with an aberrant response to pathogen-associated molecular patterns (PAMPs) and contributes to the development of chronic autoimmune intestinal inflammation. IL-1α is part of the DAMPs released from damaged intestinal epithelium, and neutralization of IL-1α or its receptor has been found to downgrade experimental intestinal inflammation [72,73]. IL-1α has been found to amplify mucosal and skin inflammation, partly by acting on intestinal subepithelial fibroblasts to induce production of IL-6 and IL-8 [74]. Furthermore, IL-1α upregulates MMP-9 and promotes contractility and CTGF expression on skin fibroblasts and intestinal myofibroblasts [35,75]. Despite the paucity of data regarding intestinal fibrosis, IL-1α is considered a profibrotic cytokine, as IL-1α depletion or deficiency reduced collagen deposition and expression of fibrosis-associated genes in bleomycin-induced lung fibrosis and in models of liver fibrosis [76,77]. IL-33 is another DAMP that is released upon epithelial or endothelial injury and its role in fibrosis has not been adequately studied [78]. Epithelial-derived IL-33 has been found to promote regulatory T cell responses and to antagonize IL-23 in experimental colitis [79]. Intestinal SEMFs and hepatic stellate cells have been identified as important additional sources of IL-33 [80]. IL-33 has been associated with fibrosis in lung and skin animal models, mainly because of its ability to induce alternative (M2) macrophage polarization and strong Th2-associated responses by innate lymphoid and CD4 helper T cells [81-83].
Glycosaminoglycan hyaluronan (HA) is a large linear polymer, composed of repeating disaccharide units of glucuronic acid and N-acetylglucosamine, and it is a component of ECM, implicated in both inflammation and fibrosis. Fragments of HA are considered DAMPs and are recognized by various cell types through receptors, including CD44, TLR2, and TLR4 [84]. HA degradation into fragments seems to be regulated by a recently discovered protein, KIAA1199, and this is supported by a study showing that skin fibroblasts lacking this protein are unable to catalyze HA [85]. In a recent study, KIAA1199 was found to be highly elevated in submucosal regions of inflamed and fibrotic colon sections from patients with CD, with its substrate, HA, being in close proximity. In addition, intestinal fibroblasts isolated from patients with CD expressed high levels of KIAA1199 in ECM, and its expression was specifically regulated by IL-6. Silencing KIAA1199 in intestinal fibroblasts resulted in failure of HA degradation, indicating that intestinal fibroblasts are important players in HA degradation [86].
Immune responses in fibrotic process
Innate immune responses initiate profibrotic cascades
Epithelial injury and pathogen entry release DAMPs and PAMPs, resulting in activation of TLRs, NOD-like receptors, c-type lectin receptors and inflammasomes on resident macrophages that drive the secretion of proinflammatory cytokines and chemokines [87]. Complex chemokine gradients are formed via the cooperation of immune and non-immune cells to recruit innate immune cells. Platelet adhesion to the damaged endothelium and subsequent PDGF production drives MCP-1/CCL-2 production by endothelial cells, mesenchymal cells and monocytes, which in turn induces monocyte migration and infiltration at the site of injury [88]. This initial early phase of tissue injury is characterized by local infiltration by classically activated (M1) macrophages that secrete IL-1, IL-12, IL-23, TNF-α and reactive oxygen and nitrogen species [87]. M1 products, such as TNF-α, participate in myofibroblast activation as part of the normal wound healing response, but also link persistent inflammation to fibrosis (Fig. 2) [87].
Accumulating evidence from animal and in vitro studies suggests that TNF-α acts as a profibrotic molecule. In a rat experimental bowel fibrosis model, treatment with anti-TNF-α resulted in decreased inflammation and fibrosis accompanied by reduced expression of pro-collagen, TGF-β and insulin-like growth factor (IGF) [89]. TNF-α has a direct profibrotic effect on intestinal myofibroblasts via its receptor, TNFR2. TNFR2 signaling through ERK1/2 and STAT3 induced myofibroblast proliferation, over-expression of collagen and TMP-1 and inhibition of MMP-2 activity [90]. Recent studies have shown that TNF-α can enhance myofibroblast migration through the P38 MAPK-regulated cyclooxygenase (COX)-2, heat shock protein 27 and protein kinase D signaling pathways [91]. Indirect actions of TNF-α include upregulation of IL-13Rα2 expression on intestinal macrophages in order to increase TGF-β production in response to IL-13 [92]. Furthermore, TNF-α enhances nuclear factor (NF)-κB translocation that upregulates production of IL-1 and IL-6 by lamina propria mononuclear cells and intestinal epithelium [93]. Studies using a chronic TNBS colitis model have shown that NF-κΒ blockade could ameliorate inflammation-associated intestinal fibrosis [94]. However, despite the undisputable benefits of anti-TNF-α antibodies in the treatment of the inflammatory component of IBDs, there is still not enough evidence that TNF-α neutralization can prevent or even ameliorate intestinal fibrosis [95,96].
At the stage where inflammation resolves or becomes chronic, apoptosis and death with release of extracellular traps (ETosis) of immune cells become central elements of the inflammatory processes [97]. Enhanced phagocytosis shifts macrophages to the alternatively-activated M2 phenotype that release mediators of anti-inflammatory and profibrotic functions, such as IL-4, IL-13, IL-10 and TGF-β (Fig. 2) [87]. Adipose macrophages from active lesions of IBD patients exhibited an alternatively-activated phenotype with increased production of Th2 cytokines, including IL-4 and -13. Furthermore, a direct implication of alternatively activated macrophages in fibrosis has been proposed, through the induction of Arginase-I synthesis, an enzyme necessary for the supply of metabolites used in collagen synthesis [98]. Stimulation of myofibroblasts with IL-4 results in increased collagen production [99], whereas IL-13 regulates their TGF-β expression levels [100]. Increased collagen deposition in CD tissue samples correlated with elevated IL-13 and TIMP-1 expression from mononuclear cells [101].
Adaptive immune responses perpetuate fibrosis through the sustained activation of myofibroblasts
Apart from innate immunity, the aberrant adaptive immune responses that characterize IBD are also implicated in fibrosis. Interferon (IFN)-γ, the hallmark of a Th1 immune response, seen in the early stages of CD [102], has been considered as an antifibrotic cytokine. IFN-γ interferes with the TGF-β signaling pathway, blocking both TGF-β and CTGF expression [103]. IFN-γ knockout mice are more susceptible to liver fibrosis, while treatment with IFN-γ has been beneficial [104]. In experimental renal fibrosis, the selective delivery of IFN-γ to myofibroblasts resulted in amelioration of fibrosis with reduced collagen synthesis and α-SMA production [104]. IFN-γ therapy has been used to treat idiopathic pulmonary fibrosis patients who have low IFN-γ levels, with variable success [105].
In contrast, Th17 immune responses that also characterize CD have been associated with fibrogenesis. Biancheri et al reported that IL-17A was found to be considerably elevated in strictured CD tissues, along with elevated collagen, MMP-3, MMP-12 and TIMP-1 [106]. In the same study, myofibroblasts isolated from intestinal biopsies from either patients with fibrostenotic CD or healthy individuals expressed both receptors for IL-17, and upon stimulation with IL-17A there was an upregulation of MMP-3, -12, TIMP-1 and collagen [106]. Additional studies suggest that IL-17 is implicated in fibrosis by regulating the expression of TGF-β and CTGF in myofibroblasts and upregulating collagen production [107]. In addition, IL-22, also produced by Th17 T cells, seems to play a protective role against liver and lung fibrosis [107,108]. Although secukinumab, a monoclonal antibody against IL-17, has proven ineffective for the treatment of inflammation in active CD, it has not been tested in the prevention or treatment of CD-associated intestinal fibrosis [109].
Atypical Th2 immune responses mostly characterize UC. However, Th2 have also been detected during late stages of experimental and human CD [110,111]. Th2-associated cytokines, mainly IL-4 and IL-13, have been reported to directly stimulate myofibroblast activation (Fig. 2). Specifically, IL-4 has been found to induce fibroblast activation and proliferation and to enhance expression of collagen and fibronectin [112]. IL-4 and IL-13 share a common IL-4Rα receptor and both signal through STAT6 [113]. However, experimental schistosome infection of double IL-4/IL-13 knockout mice has demonstrated that IL-13 is more fibrogenic than IL-4 [114]. In the colon, binding of IL-13 to its receptor induces TGF-β in intestinal macrophages, which in turn results in upregulation of profibrotic factors, such as IGF-1 and early growth response protein (Egr)-1, that stimulate collagen production by intestinal myofibroblasts. Inhibition of either IL-13 or TGF-β reduced levels of IGF-1 and Egr-1, an early transcription factor that transduces the fibrogenic activities of TGF-β, resulting in decreased collagen deposition [92]. Taken together, the above experimental evidence suggests that IL-13 is an important mediator of fibrosis that acts through multiple pathways.
Molecules and mechanisms of anti-fibrotic pathways
Anti-fibrotic molecules can be produced by a variety of immune and non-immune cells and play a major role in ECM-remodeling (Table 2). Their relative deficiency has been considered to enhance ECM accumulation and fibrosis. Unfortunately, most of the information on the role of these molecules in fibrosis comes from other tissues. PGE2 can induce myofibroblast dedifferentiation, inhibit proliferation and reduce collagen and ECM production. PGE2 is a bioactive lipid mediator produced by many different cell types, including fibroblasts, and is activated through catalyzation by COX-2 [115]. PGE2 in lung epithelial cells can limit TGF-β-induced myofibroblast differentiation [116,117]. Furthermore, PGE2 signaling through the cAMP-coupled E prostanoid 2 and 4 receptors inhibited fibroblast proliferation and pro-collagen expression [118] and promoted fibroblast apoptosis [119]. PGE2 supplementation was able to rescue murine lungs from bleomycin-induced [120] and colon from TNBS-induced fibrosis [121]. It has been speculated that there is an altered balance of TGF-β and PGE2 in fibrosis, as TGF-β has been found to downregulate COX-2 expression and PGE2 production [122].
Table 2.
Factors with an antifibrotic effect on the implication of myofibroblasts in the fibrotic process. Data from preclinical studies and experimental models
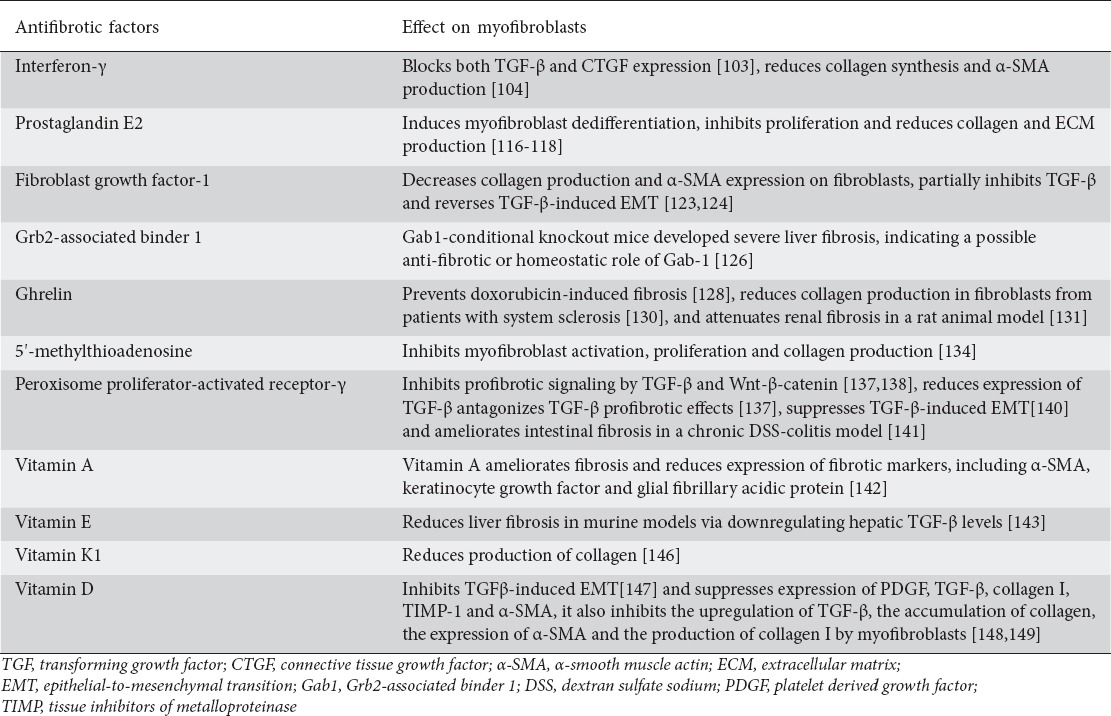
Although fibroblast growth factors (FGFs) are thought to be potent mediators of fibrosis, FGF-1, a member of the FGF family, has been shown to possess anti-fibrotic properties. FGF-1 is able to decrease collagen production and α-SMA expression on fibroblasts, partially inhibit TGF-β and reverse TGF-β-induced EMT [123,124], Grb2-associated binder 1 (Gab1) adaptor protein is a docking protein that interacts with receptor tyrosine kinases, amplifying growth factor signals [125]. In an animal model of liver fibrosis, Kizu et al showed that Gab1-conditional knockout mice developed severe liver fibrosis, indicating a possible antifibrotic or homeostatic role of Gab-1 [126].
Ghrelin is a polypeptide expressed mostly in the stomach and its main role is to increase appetite and enhance food intake [127]. Apart from these well-known actions, ghrelin has been characterized as an anti-fibrotic factor, as it prevents doxorubicin-induced myocardial fibrosis [128] and bleomycin-induced dermal fibrosis [129], reduces collagen production in fibroblasts from patients with system sclerosis [130], and attenuates renal fibrosis in a rat animal model [131]. Another antifibrotic factor is the product of polyamine biosynthesis, the 5’-methylthioadenosine (MTA). MTA is a sulfur-containing adenine nucleoside, present in all mammalian tissues, that has been shown to possess anti-inflammatory properties [132]. Oral administration of MTA reduced colonic inflammation in the DSS colitis model [133], and ameliorated murine liver fibrosis via inhibition of myofibroblast activation, proliferation and collagen production [134].
In addition to extracellular antifibrotic factors that act via interaction with membrane receptors, intracellular mediators can also exhibit antifibrotic effects. Peroxisome proliferator-activated receptor (PPAR)-γ is a ligand-activated nuclear receptor with an important role in fatty acid storage and glucose metabolism. Being expressed in a wide variety of immune and non-immune cells, including macrophages and fibroblasts, it has been shown to decrease inflammatory responses in the intestine and many tissues [135]. PPAR-γ agonists are currently used for the treatment of obesity-associated type 2 diabetes and are currently under investigation in clinical trials for inflammatory diseases, asthma and cancer [136]. PPAR-γ inhibits profibrotic signaling by TGF-β and Wnt-β-catenin [137,138] via distinct tissue or cell-specific pathways [139]. It downregulates TGF-β and actin α1 expression by human intestinal fibroblasts from UC patients, antagonizes TGF-β profibrotic effects, such as collagen and fibronectin production [137], and suppresses TGF-β-induced EMT [140]. A novel PPAR-γ agonist has recently been shown to ameliorate intestinal fibrosis in a chronic DSS colitis model [141].
Finally, vitamins A, E, K1 and D have been reported to exhibit antifibrotic properties. Vitamin A administration in the bile duct ligation model of hepatic injury resulted in amelioration of fibrosis and reduced expression of fibrotic markers, including α-SMA, keratinocyte growth factor and glial fibrillary acidic protein [142]. Vitamin E reduced liver and lung fibrosis in murine models and has been found to improve liver fibrosis in non-alcoholic steatohepatitis patients, possibly via downregulating hepatic TGF-β levels [143-145]. Vitamin K1 reduced production of collagen and ameliorated bile duct ligation-induced liver fibrosis in rats [146]. Finally, vitamin D inhibited TGF-β-induced EMT in human airway epithelial cells [147], and suppressed expression of PDGF, TGF-β, collagen I, TIMP-1 and α-SMA in an animal model of liver fibrosis [148]. Furthermore, it inhibited the upregulation of TGF-β, the accumulation of collagen and the expression of α-SMA and the production of collagen I by myofibroblasts isolated from TNBS-colitis mice [149].
Concluding remarks
Tissue inflammation can initiate multiple pathways, involving a variety of immune and non-immune cells, that result in the activation of myofibroblasts, a central element in ECM remodeling processes. The end result varies from reinstitution, repair and normal tissue healing to excessive collagen accumulation, fibrosis, permanent structural destruction and functional failure. The determinants of tissue fate are far from being clear, because of the multiplicity of cells and the complexity of the pathways involved. This is especially true for intestinal fibrosis, where available information is largely extrapolated from findings in other tissues and in vitro studies. However, although the end result of fibrotic processes is similar in all organs, differential profibrotic pathways have been found to prevail, depending on the tissue or the trigger. Therefore, experimental work focused on post-inflammatory intestinal fibrosis, together with data integration from other organs, are warranted in order to clearly define targets for intervention in the field of IBD.
Biography
University of Crete, Heraklion; Democritus University of Thrace, Dragana, Alexandroupolis, Greece
Footnotes
Conflict of Interest: None
References
- 1.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fiocchi C, Lund PK. Themes in fibrosis and gastrointestinal inflammation. Am J Physiol Gastrointest Liver Physiol. 2011;300:G677–G683. doi: 10.1152/ajpgi.00104.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rieder F, Brenmoehl J, Leeb S, Schölmerich J, Rogler G. Wound healing and fibrosis in intestinal disease. Gut. 2007;56:130–139. doi: 10.1136/gut.2006.090456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rieder F, Latella G, Magro F, et al. European Crohn’s and Colitis Organisation Topical Review on Prediction, Diagnosis and Management of Fibrostenosing Crohn’s Disease. J Crohns Colitis. 2016;10:873–885. doi: 10.1093/ecco-jcc/jjw055. [DOI] [PubMed] [Google Scholar]
- 5.Rieder F, de Bruyn JR, Pham BT, et al. Results of the 4th scientific workshop of the ECCO (Group II): markers of intestinal fibrosis in inflammatory bowel disease. J Crohns Colitis. 2014;8:1166–1178. doi: 10.1016/j.crohns.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Keller J, Panter H, Layer P. Management of the short bowel syndrome after extensive small bowel resection. Best Pract Res Clin Gastroenterol. 2004;18:977–992. doi: 10.1016/j.bpg.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 7.Powell DW, Mifflin RC, Valentich JD, Crowe SE, Saada JI, West AB. Myofibroblasts. II. Intestinal subepithelial myofibroblasts. Am J Physiol. 1999;277:C183–C201. doi: 10.1152/ajpcell.1999.277.2.C183. [DOI] [PubMed] [Google Scholar]
- 8.Mifflin RC, Pinchuk IV, Saada JI, Powell DW. Intestinal myofibroblasts: targets for stem cell therapy. Am J Physiol Gastrointest Liver Physiol. 2011;300:G684–G696. doi: 10.1152/ajpgi.00474.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Latella G, Rogler G, Bamias G, et al. Results of the 4th scientific workshop of the ECCO (I): pathophysiology of intestinal fibrosis in IBD. J Crohns Colitis. 2014;8:1147–1165. doi: 10.1016/j.crohns.2014.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Shelley-Fraser G, Borley NR, Warren BF, Shepherd NA. The connective tissue changes of Crohn’s disease. Histopathology. 2012;60:1034–1044. doi: 10.1111/j.1365-2559.2011.03911.x. [DOI] [PubMed] [Google Scholar]
- 11.de Bruyn M, Vandooren J, Ugarte-Berzal E, Arijs I, Vermeire S, Opdenakker G. The molecular biology of matrix metalloproteinases and tissue inhibitors of metalloproteinases in inflammatory bowel diseases. Crit Rev Biochem Mol Biol. 2016;51:295–358. doi: 10.1080/10409238.2016.1199535. [DOI] [PubMed] [Google Scholar]
- 12.Breynaert C, de Bruyn M, Arijs I, et al. Genetic deletion of tissue inhibitor of metalloproteinase-1/TIMP-1 alters inflammation and attenuates fibrosis in dextran sodium sulphate-induced murine models of colitis. J Crohns Colitis. 2016;10:1336–1350. doi: 10.1093/ecco-jcc/jjw101. [DOI] [PubMed] [Google Scholar]
- 13.McKaig BC, McWilliams D, Watson SA, Mahida YR. Expression and regulation of tissue inhibitor of metalloproteinase-1 and matrix metalloproteinases by intestinal myofibroblasts in inflammatory bowel disease. Am J Pathol. 2003;162:1355–1360. doi: 10.1016/S0002-9440(10)63931-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di Sabatino A, Pickard KM, Rampton D, et al. Blockade of transforming growth factor beta upregulates T-box transcription factor T-bet, and increases T helper cell type 1 cytokine and matrix metalloproteinase-3 production in the human gut mucosa. Gut. 2008;57:605–612. doi: 10.1136/gut.2007.130922. [DOI] [PubMed] [Google Scholar]
- 15.Di Sabatino A, Jackson CL, Pickard KM, et al. Transforming growth factor beta signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut. 2009;58:777–789. doi: 10.1136/gut.2008.149096. [DOI] [PubMed] [Google Scholar]
- 16.Farkas K, Saródi Z, Bálint A, et al. The diagnostic value of a new fecal marker, matrix metalloprotease-9, in different types of inflammatory bowel diseases. J Crohns Colitis. 2015;9:231–237. doi: 10.1093/ecco-jcc/jjv005. [DOI] [PubMed] [Google Scholar]
- 17.Cao Z, Said N, Amin S, et al. Galectins-3 and -7, but not galectin-1, play a role in re-epithelialization of wounds. J Biol Chem. 2002;277:42299–42305. doi: 10.1074/jbc.M200981200. [DOI] [PubMed] [Google Scholar]
- 18.Puthenedam M, Wu F, Shetye A, Michaels A, Rhee KJ, Kwon JH. Matrilysin-1 (MMP7) cleaves galectin-3 and inhibits wound healing in intestinal epithelial cells. Inflamm Bowel Dis. 2011;17:260–267. doi: 10.1002/ibd.21443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vaalamo M, Karjalainen-Lindsberg ML, Puolakkainen P, Kere J, Saarialho-Kere U. Distinct expression profiles of stromelysin-2 (MMP-10), collagenase-3 (MMP-13), macrophage metalloelastase (MMP-12), and tissue inhibitor of metalloproteinases-3 (TIMP-3) in intestinal ulcerations. Am J Pathol. 1998;152:1005–1014. [PMC free article] [PubMed] [Google Scholar]
- 20.Koller FL, Dozier EA, Nam KT, et al. Lack of MMP10 exacerbates experimental colitis and promotes development of inflammation-associated colonic dysplasia. Lab Invest. 2012;92:1749–1759. doi: 10.1038/labinvest.2012.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roulis M, Flavell RA. Fibroblasts and myofibroblasts of the intestinal lamina propria in physiology and disease. Differentiation. 2016;92:116–131. doi: 10.1016/j.diff.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 22.Mostafa RM, Moustafa YM, Hamdy H. Interstitial cells of Cajal, the Maestro in health and disease. World J Gastroenterol. 2010;16:3239–3248. doi: 10.3748/wjg.v16.i26.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lei NY, Jabaji Z, Wang J, et al. Intestinal subepithelial myofibroblasts support the growth of intestinal epithelial stem cells. PLoS One. 2014;9:e84651. doi: 10.1371/journal.pone.0084651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Horiguchi H, Endo M, Kawane K, et al. ANGPTL2 expression in the intestinal stem cell niche controls epithelial regeneration and homeostasis. EMBO J. 2017;36:409–424. doi: 10.15252/embj.201695690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKaig BC, Makh SS, Hawkey CJ, Podolsky DK, Mahida YR. Normal human colonic subepithelial myofibroblasts enhance epithelial migration (restitution) via TGF-beta3. Am J Physiol. 1999;276:G1087–G1093. doi: 10.1152/ajpgi.1999.276.5.G1087. [DOI] [PubMed] [Google Scholar]
- 26.Mahida YR, Beltinger J, Makh S, et al. Adult human colonic subepithelial myofibroblasts express extracellular matrix proteins and cyclooxygenase-1 and -2. Am J Physiol. 1997;273:G1341–G1348. doi: 10.1152/ajpgi.1997.273.6.G1341. [DOI] [PubMed] [Google Scholar]
- 27.Roulis M, Nikolaou C, Kotsaki E, et al. Intestinal myofibroblast-specific Tpl2-Cox-2-PGE2 pathway links innate sensing to epithelial homeostasis. Proc Natl Acad Sci U S A. 2014;111:E4658–E4667. doi: 10.1073/pnas.1415762111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okuno T, Andoh A, Bamba S, et al. Interleukin-1beta and tumor necrosis factor-alpha induce chemokine and matrix metalloproteinase gene expression in human colonic subepithelial myofibroblasts. Scand J Gastroenterol. 2002;37:317–324. doi: 10.1080/003655202317284228. [DOI] [PubMed] [Google Scholar]
- 29.Rogler G, Gelbmann CM, Vogl D, et al. Differential activation of cytokine secretion in primary human colonic fibroblast/myofibroblast cultures. Scand J Gastroenterol. 2001;36:389–398. doi: 10.1080/003655201300051216. [DOI] [PubMed] [Google Scholar]
- 30.Otte JM, Rosenberg IM, Podolsky DK. Intestinal myofibroblasts in innate immune responses of the intestine. Gastroenterology. 2003;124:1866–1878. doi: 10.1016/s0016-5085(03)00403-7. [DOI] [PubMed] [Google Scholar]
- 31.Zawahir S, Li G, Banerjee A, Shiu J, Blanchard TG, Okogbule-Wonodi AC. Inflammatory and immune activation in intestinal myofibroblasts is developmentally regulated. J Interferon Cytokine Res. 2015;35:634–640. doi: 10.1089/jir.2014.0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saada JI, Pinchuk IV, Barrera CA, et al. Subepithelial myofibroblasts are novel nonprofessional APCs in the human colonic mucosa. J Immunol. 2006;177:5968–5979. doi: 10.4049/jimmunol.177.9.5968. [DOI] [PubMed] [Google Scholar]
- 33.Bamias G, Filidou E, Goukos D, et al. Crohn’s disease-associated mucosal factors regulate the expression of TNF-like cytokine 1A and its receptors in primary subepithelial intestinal myofibroblasts and intestinal epithelial cells. Transl Res. 2017;180:118–130. doi: 10.1016/j.trsl.2016.08.007. [DOI] [PubMed] [Google Scholar]
- 34.Honzawa Y, Nakase H, Shiokawa M, et al. Involvement of interleukin-17A-induced expression of heat shock protein 47 in intestinal fibrosis in Crohn’s disease. Gut. 2014;63:1902–1912. doi: 10.1136/gutjnl-2013-305632. [DOI] [PubMed] [Google Scholar]
- 35.Drygiannakis I, Valatas V, Sfakianaki O, et al. Proinflammatory cytokines induce crosstalk between colonic epithelial cells and subepithelial myofibroblasts: implication in intestinal fibrosis. J Crohns Colitis. 2013;7:286–300. doi: 10.1016/j.crohns.2012.04.008. [DOI] [PubMed] [Google Scholar]
- 36.Speca S, Giusti I, Rieder F, Latella G. Cellular and molecular mechanisms of intestinal fibrosis. World J Gastroenterol. 2012;18:3635–3661. doi: 10.3748/wjg.v18.i28.3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rieder F, Fiocchi C. Intestinal fibrosis in IBD—a dynamic, multifactorial process. Nat Rev Gastroenterol Hepatol. 2009;6:228–235. doi: 10.1038/nrgastro.2009.31. [DOI] [PubMed] [Google Scholar]
- 38.Koumas L, Smith TJ, Feldon S, Blumberg N, Phipps RP. Thy-1 expression in human fibroblast subsets defines myofibroblastic or lipofibroblastic phenotypes. Am J Pathol. 2003;163:1291–1300. doi: 10.1016/S0002-9440(10)63488-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Murray IR, West CC, Hardy WR, et al. Natural history of mesenchymal stem cells, from vessel walls to culture vessels. Cell Mol Life Sci. 2014;71:1353–1374. doi: 10.1007/s00018-013-1462-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ippolito C, Colucci R, Segnani C, et al. Fibrotic and vascular remodelling of colonic wall in patients with active ulcerative colitis. J Crohns Colitis. 2016;10:1194–1204. doi: 10.1093/ecco-jcc/jjw076. [DOI] [PubMed] [Google Scholar]
- 41.Kawakami T, Mimura I, Shoji K, Tanaka T, Nangaku M. Hypoxia and fibrosis in chronic kidney disease: crossing at pericytes. Kidney Int Suppl (2011) 2014;4:107–112. doi: 10.1038/kisup.2014.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sazuka S, Katsuno T, Nakagawa T, et al. Fibrocytes are involved in inflammation as well as fibrosis in the pathogenesis of Crohn’s disease. Dig Dis Sci. 2014;59:760–768. doi: 10.1007/s10620-013-2813-8. [DOI] [PubMed] [Google Scholar]
- 43.Flier SN, Tanjore H, Kokkotou EG, Sugimoto H, Zeisberg M, Kalluri R. Identification of epithelial to mesenchymal transition as a novel source of fibroblasts in intestinal fibrosis. J Biol Chem. 2010;285:20202–20212. doi: 10.1074/jbc.M110.102012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–981. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kambas K, Chrysanthopoulou A, Kourtzelis I, et al. Endothelin-1 signaling promotes fibrosis in vitro in a bronchopulmonary dysplasia model by activating the extrinsic coagulation cascade. J Immunol. 2011;186:6568–6575. doi: 10.4049/jimmunol.1003756. [DOI] [PubMed] [Google Scholar]
- 46.Rodriguez-Pascual F, Busnadiego O, Gonzalez-Santamaria J. The profibrotic role of endothelin-1: is the door still open for the treatment of fibrotic diseases? Life Sci. 2014;118:156–164. doi: 10.1016/j.lfs.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 47.Bogatkevich GS, Ludwicka-Bradley A, Silver RM. Dabigatran, a direct thrombin inhibitor, demonstrates antifibrotic effects on lung fibroblasts. Arthritis Rheum. 2009;60:3455–3464. doi: 10.1002/art.24935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J. 2010;74:597–607. doi: 10.1253/circj.cj-09-0982. [DOI] [PubMed] [Google Scholar]
- 49.Latella G, Sferra R, Speca S, Vetuschi A, Gaudio E. Can we prevent, reduce or reverse intestinal fibrosis in IBD? Eur Rev Med Pharmacol Sci. 2013;17:1283–1304. [PubMed] [Google Scholar]
- 50.Sanders YY, Cui Z, Le Saux CJ, et al. SMAD-independent down-regulation of caveolin-1 by TGF-β: effects on proliferation and survival of myofibroblasts. PLoS One. 2015;10:e0116995. doi: 10.1371/journal.pone.0116995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leeb SN, Vogl D, Falk W, Schölmerich J, Rogler G, Gelbmann CM. Regulation of migration of human colonic myofibroblasts. Growth Factors. 2002;20:81–91. doi: 10.1080/08977190290031941. [DOI] [PubMed] [Google Scholar]
- 52.Ma Y, Guan Q, Bai A, et al. Targeting TGF-beta1 by employing a vaccine ameliorates fibrosis in a mouse model of chronic colitis. Inflamm Bowel Dis. 2010;16:1040–1050. doi: 10.1002/ibd.21167. [DOI] [PubMed] [Google Scholar]
- 53.Manresa MC, Tambuwala MM, Radhakrishnan P, et al. Hydroxylases regulate intestinal fibrosis through the suppression of ERK-mediated TGF-β1 signaling. Am J Physiol Gastrointest Liver Physiol. 2016;311:G1076–G1090. doi: 10.1152/ajpgi.00229.2016. [DOI] [PubMed] [Google Scholar]
- 54.Rodansky ES, Johnson LA, Huang S, Spence JR, Higgins PD. Intestinal organoids: a model of intestinal fibrosis for evaluating anti-fibrotic drugs. Exp Mol Pathol. 2015;98:346–351. doi: 10.1016/j.yexmp.2015.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bonner JC. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004;15:255–273. doi: 10.1016/j.cytogfr.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 56.Sponheim J, Pollheimer J, Olsen T, et al. Inflammatory bowel disease-associated interleukin-33 is preferentially expressed in ulceration-associated myofibroblasts. Am J Pathol. 2010;177:2804–2815. doi: 10.2353/ajpath.2010.100378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kurahashi M, Nakano Y, Peri LE, Townsend JB, Ward SM, Sanders KM. A novel population of subepithelial platelet-derived growth factor receptor α-positive cells in the mouse and human colon. Am J Physiol Gastrointest Liver Physiol. 2013;304:G823–G834. doi: 10.1152/ajpgi.00001.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Miró L, Pérez-Bosque A, Maijó M, Naftalin RJ, Moretó M. Vasopressin regulation of epithelial colonic proliferation and permeability is mediated by pericryptal platelet-derived growth factor A. Exp Physiol. 2014;99:1325–1334. doi: 10.1113/expphysiol.2014.080952. [DOI] [PubMed] [Google Scholar]
- 59.Shi-Wen X, Leask A, Abraham D. Regulation and function of connective tissue growth factor/CCN2 in tissue repair, scarring and fibrosis. Cytokine Growth Factor Rev. 2008;19:133–144. doi: 10.1016/j.cytogfr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 60.Chen CC, Chen N, Lau LF. The angiogenic factors Cyr61 and connective tissue growth factor induce adhesive signaling in primary human skin fibroblasts. J Biol Chem. 2001;276:10443–10452. doi: 10.1074/jbc.M008087200. [DOI] [PubMed] [Google Scholar]
- 61.Uchio K, Graham M, Dean NM, Rosenbaum J, Desmoulière A. Down-regulation of connective tissue growth factor and type I collagen mRNA expression by connective tissue growth factor antisense oligonucleotide during experimental liver fibrosis. Wound Repair Regen. 2004;12:60–66. doi: 10.1111/j.1067-1927.2004.012112.x. [DOI] [PubMed] [Google Scholar]
- 62.Zhu MY, Lu YM, Ou YX, Zhang HZ, Chen WX. Dynamic progress of 2,4,6-trinitrobenzene sulfonic acid induced chronic colitis and fibrosis in rat model. J Dig Dis. 2012;13:421–429. doi: 10.1111/j.1751-2980.2012.00607.x. [DOI] [PubMed] [Google Scholar]
- 63.Latella G, Vetuschi A, Sferra R, et al. Smad3 loss confers resistance to the development of trinitrobenzene sulfonic acid-induced colorectal fibrosis. Eur J Clin Invest. 2009;39:145–156. doi: 10.1111/j.1365-2362.2008.02076.x. [DOI] [PubMed] [Google Scholar]
- 64.Li C, Iness A, Yoon J, et al. Noncanonical STAT3 activation regulates excess TGF-β1 and collagen I expression in muscle of stricturing Crohn’s disease. J Immunol. 2015;194:3422–3431. doi: 10.4049/jimmunol.1401779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Günther U, Bateman AC, Beattie RM, Bauer M, MacDonald TT, Kaskas BA. Connective tissue growth factor expression is increased in collagenous colitis and coeliac disease. Histopathology. 2010;57:427–435. doi: 10.1111/j.1365-2559.2010.03652.x. [DOI] [PubMed] [Google Scholar]
- 66.di Mola FF, Di Sebastiano P, Gardini A, et al. Differential expression of connective tissue growth factor in inflammatory bowel disease. Digestion. 2004;69:245–253. doi: 10.1159/000079845. [DOI] [PubMed] [Google Scholar]
- 67.Reinecker HC, Steffen M, Witthoeft T, et al. Enhanced secretion of tumour necrosis factor-alpha, IL-6, and IL-1 beta by isolated lamina propria mononuclear cells from patients with ulcerative colitis and Crohn’s disease. Clin Exp Immunol. 1993;94:174–181. doi: 10.1111/j.1365-2249.1993.tb05997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takahashi K, Nishida A, Shioya M, et al. Interleukin (IL)-1βis a strong inducer of IL-36γexpression in human colonic myofibroblasts. PLoS One. 2015;10:e0138423. doi: 10.1371/journal.pone.0138423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishida A, Hidaka K, Kanda T, et al. Increased expression of interleukin-36, a member of the interleukin-1 cytokine family, in inflammatory bowel disease. Inflamm Bowel Dis. 2016;22:303–314. doi: 10.1097/MIB.0000000000000654. [DOI] [PubMed] [Google Scholar]
- 70.Raymond M, Marchbank T, Moyer MP, Playford RJ, Sanderson IR, Kruidenier L. IL-1βstimulation of CCD-18co myofibroblasts enhances repair of epithelial monolayers through Wnt-5a. Am J Physiol Gastrointest Liver Physiol. 2012;303:G1270–G1278. doi: 10.1152/ajpgi.00458.2011. [DOI] [PubMed] [Google Scholar]
- 71.Catarzi S, Favilli F, Romagnoli C, et al. Oxidative state and IL-6 production in intestinal myofibroblasts of Crohn’s disease patients. Inflamm Bowel Dis. 2011;17:1674–1684. doi: 10.1002/ibd.21552. [DOI] [PubMed] [Google Scholar]
- 72.Bersudsky M, Luski L, Fishman D, et al. Non-redundant properties of IL-1αand IL-1βduring acute colon inflammation in mice. Gut. 2014;63:598–609. doi: 10.1136/gutjnl-2012-303329. [DOI] [PubMed] [Google Scholar]
- 73.Arvanitidis K, Filidou E, Valatas V, Kouroumalis E, Paspaliaris V, Kolios G. The effect of interleukin-1 receptor antagonism in the management of intestinal inflammation;an in vitro and in vivo study. J Crohns Colitis. 2013;7:S14. [Google Scholar]
- 74.Scarpa M, Kessler S, Sadler T, et al. The epithelial danger signal IL-1αis a potent activator of fibroblasts and reactivator of intestinal inflammation. Am J Pathol. 2015;185:1624–1637. doi: 10.1016/j.ajpath.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aden N, Nuttall A, Shiwen X, et al. Epithelial cells promote fibroblast activation via IL-1alpha in systemic sclerosis. J Invest Dermatol. 2010;130:2191–2200. doi: 10.1038/jid.2010.120. [DOI] [PubMed] [Google Scholar]
- 76.Suwara MI, Green NJ, Borthwick LA, et al. IL-1αreleased from damaged epithelial cells is sufficient and essential to trigger inflammatory responses in human lung fibroblasts. Mucosal Immunol. 2014;7:684–693. doi: 10.1038/mi.2013.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kamari Y, Shaish A, Vax E, et al. Lack of interleukin-1αor interleukin-1βinhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J Hepatol. 2011;55:1086–1094. doi: 10.1016/j.jhep.2011.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boyapati RK, Rossi AG, Satsangi J, Ho GT. Gut mucosal DAMPs in IBD: from mechanisms to therapeutic implications. Mucosal Immunol. 2016;9:567–582. doi: 10.1038/mi.2016.14. [DOI] [PubMed] [Google Scholar]
- 79.Schiering C, Krausgruber T, Chomka A, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature. 2014;513:564–568. doi: 10.1038/nature13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lopetuso LR, Scaldaferri F, Pizarro TT. Emerging role of the interleukin (IL)-33/ST2 axis in gut mucosal wound healing and fibrosis. Fibrogenesis Tissue Repair. 2012;5:18. doi: 10.1186/1755-1536-5-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Miller AM. Role of IL-33 in inflammation and disease. J Inflamm (Lond) 2011;8:22. doi: 10.1186/1476-9255-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li D, Guabiraba R, Besnard AG, et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. 2014;134:1422–1432. doi: 10.1016/j.jaci.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rankin AL, Mumm JB, Murphy E, et al. IL-33 induces IL-13-dependent cutaneous fibrosis. J Immunol. 2010;184:1526–1535. doi: 10.4049/jimmunol.0903306. [DOI] [PubMed] [Google Scholar]
- 84.de la Motte CA. Hyaluronan in intestinal homeostasis and inflammation: implications for fibrosis. Am J Physiol Gastrointest Liver Physiol. 2011;301:G945–G949. doi: 10.1152/ajpgi.00063.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yoshida H, Nagaoka A, Kusaka-Kikushima A, et al. KIAA1199, a deafness gene of unknown function, is a new hyaluronan binding protein involved in hyaluronan depolymerization. Proc Natl Acad Sci U S A. 2013;110:5612–5617. doi: 10.1073/pnas.1215432110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soroosh A, Albeiroti S, West GA, Willard B, Fiocchi C, de la Motte CA. Crohn’s disease fibroblasts overproduce the novel protein KIAA1199 to create proinflammatory hyaluronan fragments. Cell Mol Gastroenterol Hepatol. 2016;2:358–368. doi: 10.1016/j.jcmgh.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lech M, Anders HJ. Macrophages and fibrosis: How resident and infiltrating mononuclear phagocytes orchestrate all phases of tissue injury and repair. Biochim Biophys Acta. 2013;1832:989–997. doi: 10.1016/j.bbadis.2012.12.001. [DOI] [PubMed] [Google Scholar]
- 88.Sierra-Filardi E, Nieto C, Domínguez-Soto A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol. 2014;192:3858–3867. doi: 10.4049/jimmunol.1302821. [DOI] [PubMed] [Google Scholar]
- 89.Adler J, Rahal K, Swanson SD, et al. Anti-tumor necrosis factor αprevents bowel fibrosis assessed by messenger RNA, histology, and magnetization transfer MRI in rats with Crohn’s disease. Inflamm Bowel Dis. 2013;19:683–690. doi: 10.1097/MIB.0b013e3182802c32. [DOI] [PubMed] [Google Scholar]
- 90.Theiss AL, Simmons JG, Jobin C, Lund PK. Tumor necrosis factor (TNF) alpha increases collagen accumulation and proliferation in intestinal myofibroblasts via TNF receptor 2. J Biol Chem. 2005;280:36099–36109. doi: 10.1074/jbc.M505291200. [DOI] [PubMed] [Google Scholar]
- 91.Saini S, Liu T, Yoo J. TNF-αstimulates colonic myofibroblast migration via COX-2 and Hs 27. J Surg Res. 2016;204:145–152. doi: 10.1016/j.jss.2016.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 93.Neurath MF, Fuss I, Schürmann G, et al. Cytokine gene transcription by NF-kappa B family members in patients with inflammatory bowel disease. Ann N Y Acad Sci. 1998;859:149–159. doi: 10.1111/j.1749-6632.1998.tb11119.x. [DOI] [PubMed] [Google Scholar]
- 94.Lawrance IC, Wu F, Leite AZ, et al. A murine model of chronic inflammation-induced intestinal fibrosis down-regulated by antisense NF-kappa B. Gastroenterology. 2003;125:1750–1761. doi: 10.1053/j.gastro.2003.08.027. [DOI] [PubMed] [Google Scholar]
- 95.Billiet T, Rutgeerts P, Ferrante M, Van Assche G, Vermeire S. Targeting TNF-αfor the treatment of inflammatory bowel disease. Expert Opin Biol Ther. 2014;14:75–101. doi: 10.1517/14712598.2014.858695. [DOI] [PubMed] [Google Scholar]
- 96.Van Assche G, Vermeire S, Rutgeerts P. The potential for disease modification in Crohn’s disease. Nat Rev Gastroenterol Hepatol. 2010;7:79–85. doi: 10.1038/nrgastro.2009.220. [DOI] [PubMed] [Google Scholar]
- 97.Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011;18:581–588. doi: 10.1038/cdd.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jung SH, Saxena A, Kaur K, et al. The role of adipose tissue-associated macrophages and T lymphocytes in the pathogenesis of inflammatory bowel disease. Cytokine. 2013;61:459–468. doi: 10.1016/j.cyto.2012.11.021. [DOI] [PubMed] [Google Scholar]
- 99.Aoudjehane L, Pissaia A, Jr, Scatton O, et al. Interleukin-4 induces the activation and collagen production of cultured human intrahepatic fibroblasts via the STAT-6 pathway. Lab Invest. 2008;88:973–985. doi: 10.1038/labinvest.2008.61. [DOI] [PubMed] [Google Scholar]
- 100.Chiaramonte MG, Donaldson DD, Cheever AW, Wynn TA. An IL-13 inhibitor blocks the development of hepatic fibrosis during a T-helper type 2-dominated inflammatory response. J Clin Invest. 1999;104:777–785. doi: 10.1172/JCI7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bailey JR, Bland PW, Tarlton JF, et al. IL-13 promotes collagen accumulation in Crohn’s disease fibrosis by down-regulation of fibroblast MMP synthesis: a role for innate lymphoid cells? PLoS One. 2012;7:e52332. doi: 10.1371/journal.pone.0052332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Brand S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut. 2009;58:1152–1167. doi: 10.1136/gut.2008.163667. [DOI] [PubMed] [Google Scholar]
- 103.Higashi K, Inagaki Y, Fujimori K, Nakao A, Kaneko H, Nakatsuka I. Interferon-gamma interferes with transforming growth factor-beta signaling through direct interaction of YB-1 with Smad3. J Biol Chem. 2003;278:43470–43479. doi: 10.1074/jbc.M302339200. [DOI] [PubMed] [Google Scholar]
- 104.Inagaki Y, Nemoto T, Kushida M, et al. Interferon alfa down-regulates collagen gene transcription and suppresses experimental hepatic fibrosis in mice. Hepatology. 2003;38:890–899. doi: 10.1053/jhep.2003.50408. [DOI] [PubMed] [Google Scholar]
- 105.Raghu G, Brown KK, Bradford WZ, et al. Idiopathic Pulmonary Fibrosis Study Group. A placebo-controlled trial of interferon gamma-1b in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2004;350:125–133. doi: 10.1056/NEJMoa030511. [DOI] [PubMed] [Google Scholar]
- 106.Biancheri P, Pender SL, Ammoscato F, et al. The role of interleukin 17 in Crohn’s disease-associated intestinal fibrosis. Fibrogenesis Tissue Repair. 2013;6:13. doi: 10.1186/1755-1536-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Meng F, Wang K, Aoyama T, et al. Interleukin-17 signaling in inflammatory, Kupffer cells, and hepatic stellate cells exacerbates liver fibrosis in mice. Gastroenterology. 2012;143:765–776.e1-3. doi: 10.1053/j.gastro.2012.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Simonian PL, Wehrmann F, Roark CL, Born WK, ‘O’Brien RL, Fontenot AP. γδT cells protect against lung fibrosis via IL-22. J Exp Med. 2010;207:2239–2253. doi: 10.1084/jem.20100061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hueber W, Sands BE, Lewitzky S, et al. Secukinumab in Crohn’s Disease Study Group. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut. 2012;61:1693–1700. doi: 10.1136/gutjnl-2011-301668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Valatas V, Bamias G, Kolios G. Experimental colitis models: Insights into the pathogenesis of inflammatory bowel disease and translational issues. Eur J Pharmacol. 2015;759:253–264. doi: 10.1016/j.ejphar.2015.03.017. [DOI] [PubMed] [Google Scholar]
- 111.Kugathasan S, Saubermann LJ, Smith L, et al. Mucosal T-cell immunoregulation varies in early and late inflammatory bowel disease. Gut. 2007;56:1696–1705. doi: 10.1136/gut.2006.116467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Postlethwaite AE, Holness MA, Katai H, Raghow R. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4. J Clin Invest. 1992;90:1479–1485. doi: 10.1172/JCI116015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Aversa G, Punnonen J, Cocks BG, et al. An interleukin 4 (IL-4) mutant protein inhibits both IL-4 or IL-13-induced human immunoglobulin G4 (IgG4) and IgE synthesis and B cell proliferation: support for a common component shared by IL-4 and IL-13 receptors. J Exp Med. 1993;178:2213–2218. doi: 10.1084/jem.178.6.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fallon PG, Richardson EJ, McKenzie GJ, McKenzie AN. Schistosome infection of transgenic mice defines distinct and contrasting pathogenic roles for IL-4 and IL-13: IL-13 is a profibrotic agent. J Immunol. 2000;164:2585–2591. doi: 10.4049/jimmunol.164.5.2585. [DOI] [PubMed] [Google Scholar]
- 115.Dayer JM, Beutler B, Cerami A. Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J Exp Med. 1985;162:2163–2168. doi: 10.1084/jem.162.6.2163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thomas PE, Peters-Golden M, White ES, Thannickal VJ, Moore BB. PGE(2) inhibition of TGF-beta1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am J Physiol Lung Cell Mol Physiol. 2007;293:L417–L428. doi: 10.1152/ajplung.00489.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Epa AP, Thatcher TH, Pollock SJ, et al. Normal Human Lung Epithelial Cells Inhibit Transforming Growth Factor-βInduced Myofibroblast Differentiation via Prostaglandin E2. PLoS One. 2015;10:e0135266. doi: 10.1371/journal.pone.0135266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang SK, Wettlaufer SH, Chung J, Peters-Golden M. Prostaglandin E2 inhibits specific lung fibroblast functions via selective actions of PKA and Epac-1. Am J Respir Cell Mol Biol. 2008;39:482–489. doi: 10.1165/rcmb.2008-0080OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang SK, White ES, Wettlaufer SH, et al. Prostaglandin E(2) induces fibroblast apoptosis by modulating multiple survival pathways. FASEB J. 2009;23:4317–4326. doi: 10.1096/fj.08-128801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dackor RT, Cheng J, Voltz JW, et al. Prostaglandin E2 protects murine lungs from bleomycin-induced pulmonary fibrosis and lung dysfunction. Am J Physiol Lung Cell Mol Physiol. 2011;301:L645–L655. doi: 10.1152/ajplung.00176.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baird AC, Lloyd F, Lawrance IC. Prostaglandin E2 and polyenylphosphatidylcholine protect against intestinal fibrosis and regulate myofibroblast function. Dig Dis Sci. 2015;60:1603–1616. doi: 10.1007/s10620-015-3552-9. [DOI] [PubMed] [Google Scholar]
- 122.Takai E, Tsukimoto M, Kojima S. TGF-β1 downregulates COX-2 expression leading to decrease of PGE2 production in human lung cancer A549 cells, which is involved in fibrotic response to TGF-β1. PLoS One. 2013;8:e76346. doi: 10.1371/journal.pone.0076346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ramos C, Montaño M, Becerril C, et al. Acidic fibroblast growth factor decreases alpha-smooth muscle actin expression and induces apoptosis in human normal lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2006;291:L871–L879. doi: 10.1152/ajplung.00019.2006. [DOI] [PubMed] [Google Scholar]
- 124.Ramos C, Becerril C, Montaño M, et al. FGF-1 reverts epithelial-mesenchymal transition induced by TGF-{beta}1 through MAPK/ERK kinase pathway. Am J Physiol Lung Cell Mol Physiol. 2010;299:L222–L231. doi: 10.1152/ajplung.00070.2010. [DOI] [PubMed] [Google Scholar]
- 125.Aasrum M, Odegård J, Sandnes D, Christoffersen T. The involvement of the docking protein Gab1 in mitogenic signalling induced by EGF and HGF in rat hepatocytes. Biochim Biophys Acta. 2013;1833:3286–3294. doi: 10.1016/j.bbamcr.2013.10.004. [DOI] [PubMed] [Google Scholar]
- 126.Kizu T, Yoshida Y, Furuta K, et al. Loss of Gab1 adaptor protein in hepatocytes aggravates experimental liver fibrosis in mice. Am J Physiol Gastrointest Liver Physiol. 2015;308:G613–G624. doi: 10.1152/ajpgi.00289.2014. [DOI] [PubMed] [Google Scholar]
- 127.Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999;402:656–660. doi: 10.1038/45230. [DOI] [PubMed] [Google Scholar]
- 128.Pei XM, Yung BY, Yip SP, Ying M, Benzie IF, Siu PM. Desacyl ghrelin prevents doxorubicin-induced myocardial fibrosis and apoptosis via the GHSR-independent pathway. Am J Physiol Endocrinol Metab. 2014;306:E311–E323. doi: 10.1152/ajpendo.00123.2013. [DOI] [PubMed] [Google Scholar]
- 129.Koca SS, Ozgen M, Sarikaya M, Dagli F, Ustundag B, Isik A. Ghrelin prevents the development of dermal fibrosis in bleomycin-induced scleroderma. Clin Exp Dermatol. 2014;39:176–181. doi: 10.1111/ced.12195. [DOI] [PubMed] [Google Scholar]
- 130.Ota Y, Kawaguchi Y, Takagi K, et al. Ghrelin attenuates collagen production in lesional fibroblasts from patients with systemic sclerosis. Clin Immunol. 2013;147:71–78. doi: 10.1016/j.clim.2013.03.001. [DOI] [PubMed] [Google Scholar]
- 131.Sun GX, Ding R, Li M, et al. Ghrelin attenuates renal fibrosis and inflammation of obstructive nephropathy. J Urol. 2015;193:2107–2115. doi: 10.1016/j.juro.2014.11.098. [DOI] [PubMed] [Google Scholar]
- 132.Avila MA, García-Trevijano ER, Lu SC, Corrales FJ, Mato JM. Methylthioadenosine. Int J Biochem Cell Biol. 2004;36:2125–2130. doi: 10.1016/j.biocel.2003.11.016. [DOI] [PubMed] [Google Scholar]
- 133.Benight NM, Stoll B, Marini JC, Burrin DG. Preventative oral methylthioadenosine is anti-inflammatory and reduces DSS-induced colitis in mice. Am J Physiol Gastrointest Liver Physiol. 2012;303:G71–G82. doi: 10.1152/ajpgi.00549.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Latasa MU, Gil-Puig C, Fernández-Barrena MG, et al. Oral methylthioadenosine administration attenuates fibrosis and chronic liver disease progression in Mdr2-/- mice. PLoS One. 2010;5:e15690. doi: 10.1371/journal.pone.0015690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Speca S, Dubuquoy L, Desreumaux P. Peroxisome proliferator-activated receptor gamma in the colon: inflammation and innate antimicrobial immunity. J Clin Gastroenterol. 2014;48(Suppl 1):S23–S27. doi: 10.1097/MCG.0000000000000253. [DOI] [PubMed] [Google Scholar]
- 136.Wei J, Bhattacharyya S, Varga J. Peroxisome proliferator-activated receptor γ: innate protection from excessive fibrogenesis and potential therapeutic target in systemic sclerosis. Curr Opin Rheumatol. 2010;22:671–676. doi: 10.1097/BOR.0b013e32833de1a7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Ghosh AK, Bhattacharyya S, Lakos G, Chen SJ, Mori Y, Varga J. Disruption of transforming growth factor beta signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor gamma. Arthritis Rheum. 2004;50:1305–1318. doi: 10.1002/art.20104. [DOI] [PubMed] [Google Scholar]
- 138.Lu D, Carson DA. Repression of beta-catenin signaling by PPAR gamma ligands. Eur J Pharmacol. 2010;636:198–202. doi: 10.1016/j.ejphar.2010.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhao C, Chen W, Yang L, Chen L, Stimpson SA, Diehl AM. PPARgamma agonists prevent TGFbeta1/Smad3-signaling in human hepatic stellate cells. Biochem Biophys Res Commun. 2006;350:385–391. doi: 10.1016/j.bbrc.2006.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tan X, Dagher H, Hutton CA, Bourke JE. Effects of PPAR gamma ligands on TGF-beta1-induced epithelial-mesenchymal transition in alveolar epithelial cells. Respir Res. 2010;11:21. doi: 10.1186/1465-9921-11-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Speca S, Rousseaux C, Dubuquoy C, et al. Novel PPARγmodulator GED-0507-34 Levo ameliorates inflammation-driven intestinal fibrosis. Inflamm Bowel Dis. 2016;22:279–292. doi: 10.1097/MIB.0000000000000618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Murakami K, Kaji T, Shimono R, et al. Therapeutic effects of vitamin A on experimental cholestatic rats with hepatic fibrosis. Pediatr Surg Int. 2011;27:863–870. doi: 10.1007/s00383-011-2853-0. [DOI] [PubMed] [Google Scholar]
- 143.Parola M, Muraca R, Dianzani I, et al. Vitamin E dietary supplementation inhibits transforming growth factor beta 1 gene expression in the rat liver. FEBS Lett. 1992;308:267–270. doi: 10.1016/0014-5793(92)81290-3. [DOI] [PubMed] [Google Scholar]
- 144.Nakamura H, Sato S, Takahashi K. Effects of vitamin E deficiency on bleomycin-induced pulmonary fibrosis in the hamster. Lung. 1988;166:161–176. doi: 10.1007/BF02714044. [DOI] [PubMed] [Google Scholar]
- 145.Harrison SA, Torgerson S, Hayashi P, Ward J, Schenker S. Vitamin E and vitamin C treatment improves fibrosis in patients with nonalcoholic steatohepatitis. Am J Gastroenterol. 2003;98:2485–2490. doi: 10.1111/j.1572-0241.2003.08699.x. [DOI] [PubMed] [Google Scholar]
- 146.Jiao K, Sun Q, Chen B, Li S, Lu J. Vitamin K1 attenuates bile duct ligation-induced liver fibrosis in rats. Scand J Gastroenterol. 2014;49:715–721. doi: 10.3109/00365521.2014.899618. [DOI] [PubMed] [Google Scholar]
- 147.Fischer KD, Agrawal DK. Vitamin D regulating TGF-βinduced epithelial-mesenchymal transition. Respir Res. 2014;15:146. doi: 10.1186/s12931-014-0146-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Abramovitch S, Sharvit E, Weisman Y, et al. Vitamin D inhibits development of liver fibrosis in an animal model but cannot ameliorate established cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2015;308:G112–G120. doi: 10.1152/ajpgi.00132.2013. [DOI] [PubMed] [Google Scholar]
- 149.Tao Q, Wang B, Zheng Y, Jiang X, Pan Z, Ren J. Vitamin D prevents the intestinal fibrosis via induction of vitamin D receptor and inhibition of transforming growth factor-beta1/Smad3 pathway. Dig Dis Sci. 2015;60:868–875. doi: 10.1007/s10620-014-3398-6. [DOI] [PubMed] [Google Scholar]