

Abstract
Chronic liver disease is accompanied by multiple hematological abnormalities. Iron deficiency anemia is a frequent complication of advanced liver disease. The etiology is multifactorial, mostly due to chronic hemorrhage into the gastrointestinal tract. The diagnosis of iron deficiency anemia is very challenging, as simple laboratory methods, including serum iron, ferritin, transferrin saturation (Tsat), and mean corpuscular volume are affected by the liver disease itself or the cause of the disease, resulting in difficulty in the interpretation of the results. Several new parameters, such as red blood cell ferritin, serum transferrin receptor test and index, and hepcidin, have been studied for their utility in indicating true iron deficiency in combination with chronic liver disease. Once iron deficiency anemia is diagnosed, it should be treated with oral or parenteral iron as well as portal pressure reducing drugs. Blood transfusion is reserved for symptomatic anemia despite iron supplementation.
Keywords: Iron deficiency anemia, homeostasis, hepcidin, ferritin, chronic liver disease, cirrhosis
Introduction
Chronic liver disease (CLD) of any cause is frequently associated with hematological abnormalities. Among these, anemia is a frequent occurrence, seen in about 75% of patients with advanced liver disease. The etiology of anemia, especially in cirrhotic patients, is complex and multifactorial [1].
The liver performs a major role in iron homeostasis. It is the main organ for the production of the iron regulatory hormone hepcidin, expressed in iron excess conditions as well as in cases of inflammation, blocking the absorption of iron from the enterocytes. The role of hepcidin in liver diseases, with or without cirrhosis, is still under investigation, but is probably one of the contributing factors to the anemia of chronic disease present in a variety of liver conditions.
Hemolysis also represents a common cause of anemia in patients with liver disease, especially of alcoholic cause, and is usually attributed to spur-cell anemia. It is related to abnormal cholesterol loading of the red blood cell membrane, which results in spiculated erythrocytes with a short lifespan, called acanthocytes [2,3].
In patients with chronic hepatitis C, the standard of care until recently, namely treatment with pegylated interferon in combination with ribavirin, can also cause significant anemia. Interferon treatment has been associated with bone marrow toxicity, while ribavirin-induced hemolytic anemia is a well-recognized, dose-dependent side effect of the drug [3].
Anemia in alcoholic liver disease is also associated with a direct toxic effect of alcohol on the bone marrow, causing reversible suppression of hematopoiesis and subsequently anemia with impaired platelet production and function. Moreover, the majority of the patients with chronic alcohol abuse also present with malnutrition, resulting in iron, folate, vitamin B12 and B6 deficiency [4].
One of the major, and potentially treatable, causes of anemia in patients with liver cirrhosis is acute or chronic blood loss into the gastrointestinal tract, often resulting in iron deficiency anemia (IDA). The hemorrhage is usually secondary to complications of portal hypertension such as gastroesophageal variceal rupture, gastropathy, gastric antral vascular ectasia (GAVE) or peptic ulcers, more common in patients with cirrhosis [5,6]. The fact that patients with liver disease have also had impaired coagulation is a contributing factor to the tendency of bleeding, as well as the thrombocytopenia due to spleen enlargement.
Iron deficiency (ID), with or without anemia, is associated with many symptoms and complications that have a significant and negative impact on patients. It can increase cardiovascular morbidity and mortality, impair cognition, and decrease quality of life [7]. The impact on quality of life in particular is substantial and can affect patients physically and emotionally, impairing their cognition and their ability to work. ID can also induce a variety of metabolic changes in the liver. In a study with rats, Kamei et al showed that ID can cause impairment of lipid, glucose and nutrient metabolism, as well as induction of cell apoptosis [8,9].
This review focuses on IDA in the group of patients with CLD, as it is a very common finding and can be easily corrected when appropriate. The diagnosis of this condition is not always straightforward, since many of the conventional tests commonly used in the diagnosis of ID are affected by hepatic cell injury [10,11].
Materials and methods
We conducted an extensive English language literature search using PubMed, Medline, and Google to identify peer-reviewed original and review articles. The keywords used were: Iron deficiency anemia diagnosis, hepcidin, iron homeostasis, CLD, and liver cirrhosis. The references of articles were searched thoroughly in order to find relevant articles.
Iron homeostasis and pathogenesis of ID
Iron is a micronutrient of vital importance for human oxygenation, given its critical role in oxygen transport via hemoglobin (Hb) and myoglobin. It also plays an important role in multiple metabolic pathways, such as DNA synthesis and mitochondrial function. The concentration of iron in the body is 2-4 g, and over 80% is contained in the Hb of red blood cells.
The homeostasis of iron is regulated via multiple complicated pathways that balance the iron needs and the absorption of iron in the intestine. There are three major cellular regulators recognized as the “players” in the orchestra of iron homeostasis: The enterocyte, the hepatocyte, and the macrophage.
The absorption of dietary iron occurs in the small intestine, specifically in the duodenum, via the Divalent Metal Transporter-1 (DMT-1). After transfer from the gut lumen to the enterocyte, the iron can be stored as ferritin or transferred into the plasma by a transmembrane transporter named ferroportin (FPN)/SLC40A1. The plasma iron is then conjugated to transferrin, the major iron transporter in the circulation. Impaired synthesis of any of these participants can cause inhibition of the absorption of iron and dysregulation of its homeostasis [12]. However, the iron absorbed from alimentation is not sufficient to meet daily needs. Another important source of iron is provided by the second player: The macrophages that degrade senescent or damaged erythrocytes through erythrophagocytosis in the spleen, liver or bone marrow. Macrophages also have the ability to store iron as ferritin or to release it to circulation via FPN. Hepatocytes represent the third player. The liver constitutes a significant storage site for iron, which it receives through the portal circulation and, in case of increased demand, releases back to the systemic circulation through the only known iron exporter, FPN [13] (Fig. 1).
Figure 1.
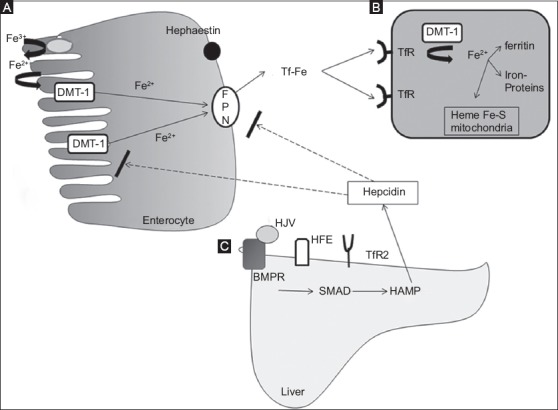
Iron homeostasis. (A) Iron absorption. Fe3+ is reduced to Fe2+ by duodenal cytochrome B (Dcytb) and is transported from enteral lumen into the enterocyte by the divalent metaltrasporter-1 (DMT-1). Iron is transported by the ferroportin (FPN) to the circulation, after oxidization by hephaestin and binding to transferrin (Tf) (B) Cellular iron uptake takes place via the transferrin receptors (TfR) on the membrane of the cell and iron is internalized and released from Tf with the help of DMT-1 (C) The hepcidin is produced mainly in the liver after activation of the BMP/SMAD pathway via the interaction of transferrin with TfR2, protein HFE and the co-receptor hemojuvelin (HJV)
BMPR, bone morphogenetic protein receptor, HAMP, hepcidin antimicrobial protein.
FPN can be considered the “first violin” in this orchestra, since FPN is the only “on demand” iron exporter from enterocytes, macrophages, and hepatocytes. But the major regulator of the iron homeostasis is hepcidin, which could be considered the “conductor” of the iron homeostasis. Hepcidin is a 25-amino-acid peptide synthesized and secreted by hepatocytes, circulates in blood plasma, mostly free or binding weakly to albumin and a2-macroglobulin, and is filtered by the kidney. Hepcidin binds to FPN, responsible for transfer of iron from the intracellular space to the systematic circulation. In that way hepcidin blocks the transfer of iron from enterocytes, hepatocytes and macrophages. Hepcidin also inhibits the transcription of DMT-1 and therefore blocks the absorption of iron from the duodenum. Its expression increases when there is an excess of iron in the body and in cases of inflammation, whilst its expression is suppressed when there is tissue hypoxia, ID or increased erythroblastic activity. In this context, hepcidin reacts as an acute phase protein [14,15] (Figs. 1 and 2).
Figure 2.
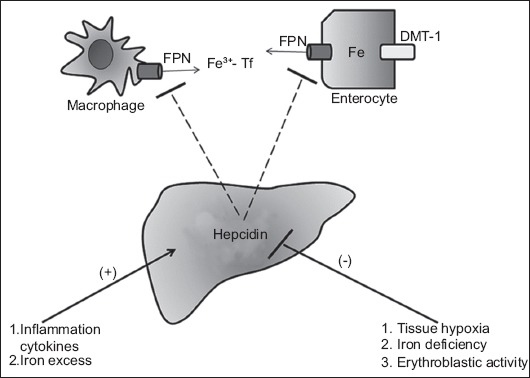
Hepcidin production by the liver. The production of hepcidin causes ferroportin (FPN) internalization, degradation and thus inhibition of iron export to the circulation. Hepcidin also inhibits divalent metal transporter-1 (DMT-1), blocking the absorption of iron in the duodenum. The production of hepcidin is inhibited by hypoxia, iron deficiency and erythroblastic activity, and triggered by inflammation and iron excess Tf, transferrin
Iron homeostasis and liver disease
Since the liver plays a major role in iron balance, it is obvious that liver diseases of different etiology, and especially advanced CLD with portal hypertension, are directly related to abnormalities in iron homeostasis.
Liver and hepcidin
The main regulator of iron homeostasis, hepcidin, is secreted mainly by the liver. Therefore, in CLD, and especially in cases of severe hepatic injury, we expect to find low hepcidin levels. On the other hand, many CLDs (alcoholic and non-alcoholic liver disease, chronic hepatitis C virus [HCV] infection) are associated with a variable grade of iron overload that represents, together with inflammation, a complex and still not clearly known condition for hepcidin synthesis and regulation [16].
A growing number of studies have investigated the levels of hepcidin in liver diseases of different etiology and their clinical significance. A close relationship of hepcidin levels with the grade of hepatic inflammation has been documented by Tsochatzis et al, who found a positive correlation between a decrease in hepcidin levels and the biochemical and histological markers of inflammation, hepatocellular damage and fibrosis in patients with HCV [17].
Several studies have suggested that in HCV infection there is a down-regulation of hepcidin expression that causes iron deposition and therefore liver injury [18,19]. Liver hepcidin, however, appears to be reduced not only in HCV, but also in liver diseases of different etiology. A recent study of patients with autoimmune liver diseases revealed significantly lower serum hepcidin levels and serum hepcidin/ferritin ratio in patients with autoimmune hepatitis, primary biliary cholangitis or primary sclerosing cholangitis, compared with patients with hepatitis B, C or non-alcoholic liver disease; however, there was no comparison with healthy controls [20].
The hepcidin/ferritin ratio was also studied by Tan et al in 332 patients with CLD versus healthy controls and patients with non-liver disease. They showed that hepcidin level was reduced in the CLD group compared with the non-liver disease group but not with healthy controls. When the hepcidin was combined with ferritin, however, the hepcidin/ferritin ratio was found to be reduced significantly in CLD patients compared with the other 2 groups and was also independently associated with progressive fibrosis, indicating that the hepcidin/ferritin ratio could have diagnostic implications [21].
In contrast, Wang et al, in an observational study of 46 patients who had chronic hepatitis B with or without cirrhosis, found that the mean hepcidin level was higher in patients with chronic hepatitis B without cirrhosis and in patients with hepatocellular carcinoma (HCC), compared with the healthy population but not the cirrhosis group, suggesting that iron loading, viral infection and liver inflammation can upregulate hepcidin in non-cirrhotic patients. There is definitely great interest in the complicated mechanisms of hepcidin regulation and further research in the future may demonstrate additional factors that affect its expression in liver diseases of different etiology.
Liver and iron storage
Apart from producing vital proteins for the iron homeostasis, the liver can uptake and store an excess of iron. The most important mechanism for iron uptake is the transferrin/transferrin-receptor system. But even in cases of severe iron overload, when there is no further transferrin capacity for binding, the liver has the ability to take up the non-transferrin bound iron. Iron that enters the hepatocytes is stored and mobilized for systemic metabolic demands. The majority of iron is stored in ferritin, but during severe iron overload storage hemosiderin is also used [22]. Ferritin is classically considered the main marker of iron homeostasis. The hepatocyte is the major site for ferritin synthesis, but also for synthesis of transferrin, which represents the major iron-binding protein. In the case of cirrhosis, an imbalance between iron loss/iron accumulation and ferritin/transferrin synthesis has also been documented.
There is evidence that cirrhotic patients compared with non-cirrhotics or healthy controls present significantly lower serum transferrin and serum iron levels, but higher levels of serum ferritin. However, a positive association between alanine transaminase and ferritin levels has also been observed, again suggesting the important role of inflammation [23]. Mao et al, in a study of patients with chronic hepatitis B (with and without cirrhosis), found that in cirrhotic chronic hepatitis B patients the levels of serum transferrin were lower, while ferritin and serum iron were higher compared with the iron markers of non-cirrhotic patients and healthy controls [24].
Iron indices and liver disease prognosis
An association between iron indices and the prognosis of liver disease has also been documented. In a recent study, Bruns et al proved the important role of serum transferrin as a prognostic factor of early mortality in patients with decompensated cirrhosis. They analyzed all the common iron parameters of 292 patients with decompensated cirrhosis, 78 of whom had acute liver failure (ACLF), and showed that, although increased ferritin levels were associated with more severe grades of ACLF, there was no significant predictive value in terms of 30-day mortality. In contrast, low serum transferrin and high Tsat were correlated significantly with poor short-term mortality (at 30 days). These results indicate the failure of iron homeostasis in patients with advanced liver disease and suggest possible therapeutic approaches in patients with ACLF [25].
The decreased levels of hepcidin in cirrhotic patients have been found to cause hepatic iron accumulation and may also contribute to the progression of liver fibrosis [26]. Hepatic iron accumulation is known to be associated with a worse prognosis and the development of HCC [27]. A recent cohort study of 237 patients with alcoholic cirrhosis demonstrated that low hepcidin levels could be a negative prognostic factor for survival and risk of HCC development. It is not yet known whether this is the result of hepcidin-associated disease progression, or whether hepcidin represents only a negative prognostic indicator [28]. Kato et al, in a study of 34 patients with HCV infection, showed that phlebotomy and a low-iron diet can reduce the levels of reactive oxygen species and hepatitis severity by improving liver function tests, as well as reducing the risk of developing HCC [29].
Causes of IDA in CLD
The leading cause of ID in advanced CLD is chronic gastrointestinal hemorrhage. Acute hemorrhage is usually seen in variceal rupture or in a bleeding peptic ulcer [30]. Chronic blood loss is mainly due to portal hypertensive gastropathy (PHG) and GAVE. Both of them constitute lesions of the gastric mucosa caused by ectatic vessels in the stomach.
PHG
PHG is very common, with a prevalence that varies from 20-80%, depending on the underlying conditions. Advanced CLD and previous variceal endoscopy treatment, either sclerotherapy or variceal ligation, predisposes to PHG [31-34]. Histologically, it is characterized by vascular dilation in mucosa and submucosa in the body and fundus of the stomach, without significant inflammation. The severity of PHG appears to correlate with the degree of portal hypertension [35,36]. Other influences, such as gastric mucosal blood flow or other local factors, can contribute to its pathogenesis [37].
GAVE
GAVE, also known as “watermelon stomach”, is a rare disorder, but it causes up to 4% of non-variceal gastrointestinal bleeds [38]. It is characterized endoscopically by tortuous ectatic vessels in the longitudinal folds of the antrum and portal hypertension appears not to be necessary for its development. Liver failure probably has a more important role in the pathogenesis of GAVE, as this condition is more commonly found in severe liver disease and resolves after liver transplantation. Other diseases apart from liver failure can also cause “watermelon stomach”, including chronic kidney disease, connective tissue disorders and bone marrow transplantation [34]. The pathogenesis is not yet clear, but one hypothesis is that vasodilator agents that the liver has failed to metabolize, such as prostaglandin E2, may play a role. Other factors that have also been associated with GAVE are mechanical stress and hypomobility of the stomach [39,40].
Peptic ulcer
The incidence of peptic ulcer disease is increased in cirrhosis, but the underlying pathogenetic mechanism is not known [41]. In contrast to the general population, in whom peptic ulcers represent the most common cause of bleeding, the prevalence in cirrhotic patients is not high (ranging from 35-53%) [42]. The role of Helicobacter pylori in the cirrhotic population is unclear.
Coagulation defect
The bleeding risk, and subsequently the risk of developing IDA, is also increased by coagulation defects, usually associated with CLD. There is impaired synthesis of factors II, VII, IX, and X, inhibitors of these factors, thrombocytopenia and impaired platelet function, increased fibrinolysis and disseminated intravascular coagulation. Although usually a “rebalance” in hemostasis can be observed, in some cases a bleeding tendency that facilitates the development of IDA may occur, significantly increasing both morbidity and mortality [43].
Diagnosis of IDA in CLD
When evaluating a patient with CLD IDA everyone should have in mind that the tests commonly used in the general population show a wide variation in these patients, because they are affected directly or indirectly by the hepatic cell damage.
The laboratory methods used in the diagnosis of IDA can be divided into two categories. There are screening measurements (Hb, Tsat, mean corpuscular volume [MCV], mean corpuscular Hb [MCH], red blood cell distribution width [RDW], and reticulocytes) that detect iron-deficient erythropoiesis, and measurements that indicate tissue iron status, either by measuring storage proteins or by direct examination of bone marrow aspiration for ID [44]. The only definitive method still remains bone marrow aspiration, stained with Prussian blue for iron. However, the fact that the procedure is invasive, painful and expensive makes biopsy a tool that is not frequently used, and there is need for less- or even noninvasive tests with a good diagnostic accuracy [11].
Full blood count
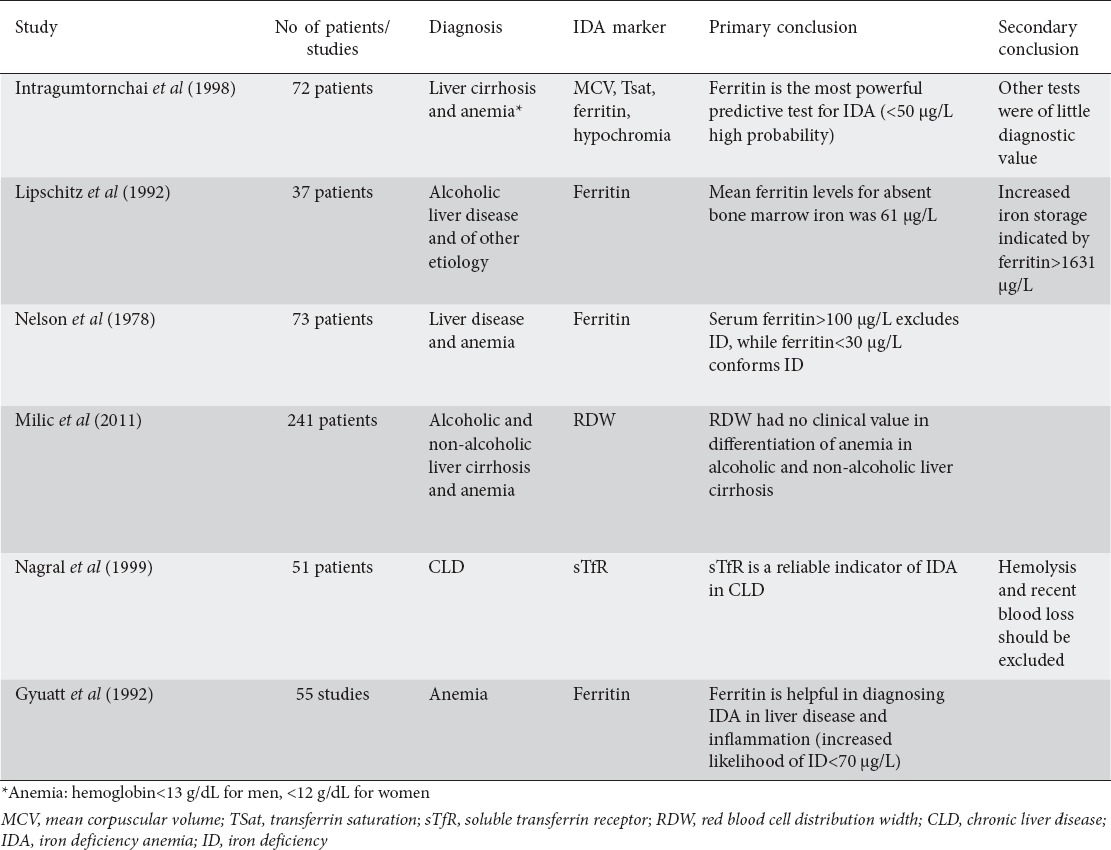
The approach begins with the full blood count (FBC). Hb determination is the most widespread screening method for IDA and the World Health Organization places the lower limit of normal for Hb concentration at 12 g/dL for non-pregnant women, below 11 g/dL in pregnant women and 13 g/dL for men. However, FBC has many limitations when used alone, because of its low specificity and sensitivity [45]. For that reason, in the diagnostic workup of anemia, erythrocyte parameters such as MCV, MCH and RDW are measured and evaluated in combination with Hb. For example hypochromia and microcytosis combined with low ferritin makes the diagnosis of IDA fairly easy, but in patients with cirrhosis MCV is increased because of vitamin deficits and membrane deposition of lipoproteins. In a cross-sectional study of 72 patients with liver cirrhosis Intragumtornchai et al found that 48% of patients with ID, proved by bone marrow aspiration, had an MCV greater than 100 fL [11].
Another parameter of erythrocytes that is often used together with the MCV results is the RDW, which depicts the variation of red blood cell width. Unfortunately, this tool is also not useful in cirrhotic patients, as was shown by a retrospective study of 241 patients with liver cirrhosis and anemia. In this study Milic et al did not manage to prove any clinical value of RDW in the differentiation of anemia, or any statistically significant increase of RDW in relation to the severity of the disease [46].
Likewise, the thrombocytosis that exists in both IDA and anemia of inflammatory conditions, due to the effect of erythropoietin and interleukin-6 on megakaryocytes, is not seen in patients with CLD and portal hypertension because of spleen sequestration.
Iron studies
Tsat
Tsat is another conventional test for the diagnosis of ID. A level of Tsat lower than 16% indicates insufficient supply of iron for erythropoiesis. A systematic review of 127 guidelines for the diagnosis and treatment of IDA showed that a cutoff of <20% was, in most conditions, the level to start treatment [22]. However, Tsat has also some acute-phase reactivity, as transferrin may be elevated in inflammations, which would lower the Tsat. On the other hand, suppressed transferrin synthesis due to malnutrition and chronic disease would raise the Tsat. It should also be noted that there is a degree of diurnal fluctuation in Tsat, which makes the interpretation of the test difficult when comparing results obtained at different times of day [45,47,48]. In a study among cirrhotic patients, a Tsat of <12% had a high diagnostic probability of ID, but the measurement of Tsat alone or in combination with ferritin did not provide any additional diagnostic value over that of ferritin alone [11].
Ferritin
The superiority of ferritin over other measurements in the diagnosis of ID has been proven by several studies. However, the fact that ferritin is an acute phase protein, affected not only by the iron status of the body, but also by acute or chronic inflammation, malignancies and liver disease, makes the interpretation of that test challenging. In the general population, a level of ferritin ≤12 μg/L was used to be suggestive of ID. However, in more recent studies, when these results combined with the bone marrow iron, it was shown that a ferritin cutoff level of ≤12 μg/L had very low sensitivity (25%), whilst the level of 30 μg/L had greater sensitivity (92%) and better predictive value (83%) [49,50]. The level is significantly higher in patients with inflammation or CLD, with the best predictor being ≤100 μg/L [14,51].
The valuable role of ferritin was also demonstrated many years ago by Lipschitz el al, who analyzed the serum ferritin levels (compared with the iron stores in bone marrow) in patients with CLD and showed that the mean ferritin level for absent bone marrow iron was 61 μg/L, while increased iron storage was indicated by ferritin level above 1631 μg/L [52]. Likewise, Nelson et al demonstrated a significant relationship between serum ferritin and bone marrow iron stores in patients with liver disease and other chronic conditions [53].
In a systematic analysis, Guyatt et al found that measurement of serum ferritin was particularly helpful, even in a population with CLD or inflammation, but the test in these patients needs a different interpretation. In their overview of 55 studies, the investigators found that a ferritin level of ≤15 μg/L could be considered a highly specific indicator of ID, while a value ≥100 μg/L ruled out ID. The intermediate values needed further interpretation. They also demonstrated that the traditional cutoff point of 12-20 μg/L is not optimal, as the likelihood of ID drops after the level of 40 μg/L for the general population and 70 μg/L for patients with cirrhosis or an inflammatory condition [54].
Similar results were also demonstrated by a cross-sectional study of 72 patients with CLD and anemia, who underwent bone marrow aspiration for the demonstration of ID. The study proved that serum ferritin remains a very useful tool for the diagnosis of iron restricted anemia, even in patients with liver cirrhosis. The cutoff level in that study, however, was lower than that in Guyatt’s study and the authors recommended iron supplementation in patients with ferritin ≤50 μg/L. Values >400 μg/L were suggestive of decreased probability for the diagnosis. The value interval of 50-400 μg/L had little or no diagnostic value [11].
Although in the past it has been suggested that red blood cell ferritin could be used in patients with CLD as a valuable tool for differentiating anemia, in the same study Guyatt et al proved that red blood cell ferritin was not as good an indicator of ID as ferritin in patients with chronic diseases [54-56].
Serum transferrin receptor (sTfR)
Another alternative iron marker is soluble or sTfR test, elevated in IDA. The underlying theory is based on the fact that the concentration of membrane TfR (mTfR) of erythroblasts in the bone marrow will be increased in a state of ID, and that would seem to correlate with sTfR. In a systematic review in 2009, Koulaouzidis et al showed that sTfR improves the diagnosis of IDA, especially in patients with chronic diseases and gastrointestinal malignancies. Although the sensitivity was high, the specificity remained low, as conditions with increased erythroblastic activity could increase sTfR. The specificity significantly improved when the sTfR-ferritin index was used [44]. Nagral et al, in a study of 51 patients with CLD, showed that the sTfR test is a reliable indicator of ID, provided that hemolysis and recent blood loss have been excluded (sensitivity 91.6%, specificity 84.6%) [57]. Although several studies have proved the superiority of sTfR and the sTfR-ferritin index in chronic disease, it is not used widely in the clinical setting. The cost compared to conventional iron tests is prohibitive and there is also a lack of standardized tests for sTfR [58-61] (Table 1).
Table 1.
Chronic liver disease and diagnosis of iron deficiency anemia
Hepcidin
Despite the fact that hepcidin is a great tool for differentiating ID from other types of anemia, it is currently used only for research and not in everyday clinical practice.
Therapeutic trial
Ultimately, as Goddard et al suggested in their published guidelines in 2011, there is always the option of the therapeutic trial in the diagnosis of ID. They suggested a 3-week trial of oral iron, which may help in the diagnosis. Alternatively, a trial of parenteral iron would demonstrate a significant change in MCH within 7 days in the case of ID [58].
Management of IDA
The management of IDA can be divided into two parts: the first is to identify the underlying pathology, and the second is to treat the cause of anemia, if possible.
Etiologic management
In patients with liver cirrhosis, the target should be the prevention and treatment of the complications of portal hypertension, focused on portal pressure reducing drugs and endoscopic therapy, including ligation or sclerotherapy of varices. In the case of PHG, the use of non-selective β-blockers, particularly propranolol, have been shown to reduce the frequency of bleeding, either as acute event or not [34]. In contrast, in the case of GAVE it has been observed that there is a lack of response to treatments that reduce the portal pressures (β-blockers, transjugular intrahepatic portosystemic shunt), which suggests that portal hypertension does not play a significant role in the pathogenesis of GAVE. The cornerstone of treatment is endoscopic thermoablative techniques, with argon plasma coagulation at the sites of the ectatic lesions. It is usual for several sessions to be necessary in order to control bleeding and to correct the anemia [62].
In cases of ribavirin-induced anemia in patients with HCV infection, it is recommended to reduce the dose or even discontinue therapy. The European Association for the Study of the Liver recommends dose reduction of ribavirin by 200 mg until a stable Hb is achieved and discontinuation of the medication if Hb drops below 8.5 g/dL. In several studies it has been proved that dose reduction of ribavirin does not significantly affect the success of the treatment [63].
Symptomatic management
The second part of the management of ID is to restore the depleted iron stores and therefore to normalize red blood cell indices. The most convenient and effective way to achieve that is the prescription of oral iron (3-10 mg of elemental iron can be absorbed daily), provided the patient is stable. The medication should be used continuously for three months after the correction of the anemia. The absorption can be enhanced with ascorbic acid, although there are no data on its effect in the treatment of IDA. Unfortunately the oral preparations are followed by multiple gastrointestinal side effects, such as epigastric distress, bloating and constipation; although these side effects are not severe, they are frequent and can cause a lack of compliance. Special caution should be observed when these medications are prescribed in cirrhotic patients, because of possible impaired absorption and the risk of hepatic encephalopathy due to constipation. There are no widely used markers in everyday practice to predict the response to treatment. In the future, serum hepcidin levels may possibly be used to identify patients with a good response to oral iron (those with low hepcidin levels) [1,14,58].
For those who are intolerant of or do not respond to oral iron, there is the option of parenteral iron administration, which is more effective and can increase the levels of Hb more quickly than oral iron. The new preparations are safer and the possibility of hypersensitivity reactions has been reduced. An advantage of this route is that in some patients the total dose required can be administered in a single infusion. The dose of iron needed is calculated using this equation:
Total Iron Deficit [mg] = body weight [kg] x (target Hb – actual Hb) [g/dL] x 2.4 + depot iron [mg] [1,58]
Finally, blood transfusion should be reserved for patients who are unstable or remain symptomatic despite iron therapy. Transfusions do not target a normal Hb level, but a safe one, as iron treatment could follow the transfusion [64]. However, there is a growing interest in the impact that iron overload can have on the liver, when it is given either as intravenous iron or with transfusions. Early studies in hemodialysis patients, before the widespread use of erythropoietin, showed that there is extensive hepatic iron deposition, especially in Kupffer cells, in patients receiving intravenous iron, although there was no evidence of liver fibrosis or cirrhosis [65-67]. In cases of concomitant chronic hepatitis B and C infection, known for the increased iron deposition they can cause in the liver, it is unclear whether intravenous iron would accelerate the liver damage. Caramelo et al, in a study of HCV positive liver samples from hemodialysis patients, found that 18 of 33 liver samples had hemosiderosis, but the degree of the liver fibrosis was not associated with the liver iron concentration [68]. There is definitely a need for further research in the field of iron needs in patients with CLD and the possible damage that exogenous iron can cause.
Concluding remarks
Iron homeostasis is the result of a complex mechanism and may be significantly disturbed in patients with CLD of different etiology and variable stages. ID is often documented, especially in advanced liver disease with portal hypertension, and is usually due to chronic blood loss. However, the concomitant presence of inflammation significantly influences the iron markers, making the diagnosis of ID difficult. The role of iron homeostasis proteins in the pathophysiology of CLD or as markers of prognosis is still controversial and has to be demonstrated in future research.
Biography
University of Athens, Hippokration General Hospital, Athens
Footnotes
Conflict of Interest: None
References
- 1.McHutchison JG, Manns MP, Longo DL. Definition and management of anemia in patients infected with hepatitis C virus. Liver Int. 2006;26:389–398. doi: 10.1111/j.1478-3231.2006.01228.x. [DOI] [PubMed] [Google Scholar]
- 2.Malik P, Bogetti D, Sileri P, et al. Spur cell anemia in alcoholic cirrhosis: Cure by orthotopic liver transplantation and recurrence after liver graft failure. Int Surg. 2002;87:201–204. [PubMed] [Google Scholar]
- 3.Gupte P, Nagral A. Hematological problems and liver disease. Trop Gastroenterol. 2009;30:65–70. [PubMed] [Google Scholar]
- 4.Gonzalez-Casas R, Jones EA, Moreno-Otero R. Spectrum of anemia associated with chronic liver disease. World J Gastroenterol. 2009;15:4653–4658. doi: 10.3748/wjg.15.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ichiyanagui C, Lozano R, Huaman C, Iparraguirre H. Peptic ulcer in patients with cirrhosis. Rev Gastroenterol Peru. 1995;15:15–19. [PubMed] [Google Scholar]
- 6.Luo JC, Leu HB, Hou MC, et al. Cirrhotic patients at increased risk of peptic ulcer bleeding: A nationwide population-based cohort study. Aliment Pharmacol Ther. 2012;36:542–550. doi: 10.1111/j.1365-2036.2012.05225.x. [DOI] [PubMed] [Google Scholar]
- 7.Les I, Doval E, Flavià M, et al. Quality of life in cirrhosis is related to potentially treatable factors. Eur J Gastroenterol Hepatol. 2010;22:221–227. doi: 10.1097/MEG.0b013e3283319975. [DOI] [PubMed] [Google Scholar]
- 8.Kamei A, Watanabe Y, Ishijima T, et al. Dietary iron-deficient anemia induces a variety of metabolic changes and even apoptosis in rat liver: A DNA microarray study. Physiol Genomics. 2010;42:149–156. doi: 10.1152/physiolgenomics.00150.2009. [DOI] [PubMed] [Google Scholar]
- 9.Stein J, Connor S, Virgin G, Ong DE, Pereyra L. Anemia and iron deficiency in gastrointestinal and liver conditions. World J Gastroenterol. 2016;22:7908–7925. doi: 10.3748/wjg.v22.i35.7908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prieto J, Barry M, Sherlock S. Serum ferritin in patients with iron overload and with acute and chronic liver diseases. Gastroenterology. 1975;68:525–533. [PubMed] [Google Scholar]
- 11.Intragumtornchai T, Rojnukkarin P, Swasdikul D, Israsena S. The role of serum ferritin in the diagnosis of iron deficiency anaemia in patients with liver cirrhosis. J Intern Med. 1998;243:233–241. doi: 10.1046/j.1365-2796.1998.00290.x. [DOI] [PubMed] [Google Scholar]
- 12.Anderson ER, Shah YM. Iron homeostasis in the liver. Compr Physiol. 2013;3:315–330. doi: 10.1002/cphy.c120016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fleming RE, Bacon BR. Orchestration of iron homeostasis. N Engl J Med. 2005;352:1741–1744. doi: 10.1056/NEJMp048363. [DOI] [PubMed] [Google Scholar]
- 14.Camaschella C. Iron-deficiency anemia. N Engl J Med. 2015;373:485–486. doi: 10.1056/NEJMc1507104. [DOI] [PubMed] [Google Scholar]
- 15.Davis M, Clarke S. Influence of microRNA on the maintenance of human iron metabolism. Nutrients. 2013;5:2611–2628. doi: 10.3390/nu5072611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Milward E, Johnstone D, Trinder D, Ramm G, Olynyk J. The nexus of iron and inflammation in hepcidin regulation: SMADs, STATs, and ECSIT. Hepatology. 2007;45:253–256. doi: 10.1002/hep.21526. [DOI] [PubMed] [Google Scholar]
- 17.Tsochatzis E, Papatheodoridis GV, Koliaraki V, et al. Serum hepcidin levels are related to the severity of liver histological lesions in chronic hepatitis C. J Viral Hepat. 2010;17:800–806. doi: 10.1111/j.1365-2893.2009.01244.x. [DOI] [PubMed] [Google Scholar]
- 18.Fujita N, Sugimoto R, Takeo M, et al. Hepcidin expression in the liver: Relatively low level in patients with chronic hepatitis C. Mol Med. 2007;13:97–104. doi: 10.2119/2006-00057.Fujita. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miura K, Taura K, Kodama Y, Schnabl B, Brenner DA. Hepatitis C virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology. 2008;48:1420–1429. doi: 10.1002/hep.22486. [DOI] [PubMed] [Google Scholar]
- 20.Lyberopoulou A, Chachami G, Gatselis NK, et al. Low serum hepcidin in patients with autoimmune liver diseases. PLoS One. 2015;10:e0135486. doi: 10.1371/journal.pone.0135486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan TC, Crawford DH, Franklin ME, et al. The serum hepcidin: Ferritin ratio is a potential biomarker for cirrhosis. Liver Int. 2012;32:1391–1399. doi: 10.1111/j.1478-3231.2012.02828.x. [DOI] [PubMed] [Google Scholar]
- 22.Peyrin-Biroulet L, Williet N, Cacoub P. Guidelines on the diagnosis and treatment of iron deficiency across indications: A systematic review. Am J Clin Nutr. 2015;102:1585–1594. doi: 10.3945/ajcn.114.103366. [DOI] [PubMed] [Google Scholar]
- 23.Drakesmith H, Prentice AM. Hepcidin and the iron-infection axis. Science. 2012;338:768–772. doi: 10.1126/science.1224577. [DOI] [PubMed] [Google Scholar]
- 24.Mao W, Hu Y, Lou Y, Chen Y, Zhang J. Abnormal serum iron markers in chronic hepatitis B virus infection may be because of liver injury. Eur J Gastroenterol Hepatol. 2015;27:130–136. doi: 10.1097/MEG.0000000000000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruns T, Nuraldeen R, Mai M, et al. Low serum transferrin correlates with acute-on-chronic organ failure and indicates short-term mortality in decompensated cirrhosis. Liver Int. 2017;37:232–241. doi: 10.1111/liv.13211. [DOI] [PubMed] [Google Scholar]
- 26.Lunova M, Trautwein C, Strnad P, Nahon P. Reply to: “Hepatic hepcidin expression is decreased in cirrhosis and HCC”. J Hepatol. 2015;62:979–980. doi: 10.1016/j.jhep.2014.11.008. [DOI] [PubMed] [Google Scholar]
- 27.Nahon P, Sutton A, Rufat P, et al. Liver iron, HFE gene mutations, and hepatocellular carcinoma occurrence in patients with cirrhosis. Gastroenterology. 2008;134:102–110. doi: 10.1053/j.gastro.2007.10.038. [DOI] [PubMed] [Google Scholar]
- 28.Nahon P, Nuraldeen R, Rufat P, Sutton A, Trautwein C, Strnad P. In alcoholic cirrhosis, low-serum hepcidin levels associate with poor long-term survival. Liver Int. 2016;36:185–188. doi: 10.1111/liv.13007. [DOI] [PubMed] [Google Scholar]
- 29.Kato J, Kobune M, Nakamura T, et al. Normalization of elevated hepatic 8-hydroxy-2’-deoxyguanosine levels in chronic hepatitis C patients by phlebotomy and low iron diet. Cancer Res. 2001;61:8697–8702. [PubMed] [Google Scholar]
- 30.Abraldes JG, Bosch J. The treatment of acute variceal bleeding. J Clin Gastroenterol. 2007;41(Suppl 3):S312–S317. doi: 10.1097/MCG.0b013e318150d3dd. [DOI] [PubMed] [Google Scholar]
- 31.Mathurin SA, Agüero AP, Dascani NA, et al. Anemia in hospitalized patients with cirrhosis: Prevalence, clinical relevance and predictive factors. Acta Gastroenterol Latinoam. 2009;39:103–111. [PubMed] [Google Scholar]
- 32.Qamar AA, Grace ND, Groszmann RJ, et al. Incidence, prevalence, and clinical significance of abnormal hematologic indices in compensated cirrhosis. Clin Gastroenterol Hepatol. 2009;7:689–695. doi: 10.1016/j.cgh.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Primignani M, Carpinelli L, Preatoni P, et al. Natural history of portal hypertensive gastropathy in patients with liver cirrhosis. The New Italian Endoscopic Club for the study and treatment of esophageal varices (NIEC) Gastroenterology. 2000;119:181–187. doi: 10.1053/gast.2000.8555. [DOI] [PubMed] [Google Scholar]
- 34.Qureshi K, Al-Osaimi AM. Approach to the management of portal hypertensive gastropathy and gastric antral vascular ectasia. Gastroenterol Clin North Am. 2014;43:835–847. doi: 10.1016/j.gtc.2014.08.012. [DOI] [PubMed] [Google Scholar]
- 35.McCormack TT, Sims J, Eyre-Brook I, et al. Gastric lesions in portal hypertension: Inflammatory gastritis or congestive gastropathy? Gut. 1985;26:1226–1232. doi: 10.1136/gut.26.11.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim MY, Choi H, Baik SK, et al. Portal hypertensive gastropathy: Correlation with portal hypertension and prognosis in cirrhosis. Dig Dis Sci. 2010;55:3561–3567. doi: 10.1007/s10620-010-1221-6. [DOI] [PubMed] [Google Scholar]
- 37.Ohta M, Hashizume M, Higashi H, et al. Portal and gastric mucosal hemodynamics in cirrhotic patients with portal-hypertensive gastropathy. Hepatology. 1994;20:1432–1436. doi: 10.1002/hep.1840200609. [DOI] [PubMed] [Google Scholar]
- 38.Selinger CP, Ang YS. Gastric antral vascular ectasia (GAVE): An update on clinical presentation, pathophysiology and treatment. Digestion. 2008;77:131–137. doi: 10.1159/000124339. [DOI] [PubMed] [Google Scholar]
- 39.Quintero E, Pique JM, Bombi JA, et al. Gastric mucosal vascular ectasias causing bleeding in cirrhosis. A distinct entity associated with hypergastrinemia and low serum levels of pepsinogen I. Gastroenterology. 1987;93:1054–1061. doi: 10.1016/0016-5085(87)90569-5. [DOI] [PubMed] [Google Scholar]
- 40.Spahr L, Villeneuve JP, Dufresne MP, et al. Gastric antral vascular ectasia in cirrhotic patients: Absence of relation with portal hypertension. Gut. 1999;44:739–742. doi: 10.1136/gut.44.5.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siringo S, Bolondi L, Piscaglia F, et al. Peptic ulcer in patients with liver cirrhosis: A retrospective endoscopic and clinical study. Ital J Gastroenterol Hepatol. 1997;29:62–68. [PubMed] [Google Scholar]
- 42.Loperfido S, Baldo V, Piovesana E, et al. Changing trends in acute upper-GI bleeding: A population-based study. Gastrointest Endosc. 2009;70:212–224. doi: 10.1016/j.gie.2008.10.051. [DOI] [PubMed] [Google Scholar]
- 43.Violi F, Ferro D. Clotting activation and hyperfibrinolysis in cirrhosis: Implication for bleeding and thrombosis. Semin Thromb Hemost. 2013;39:426–433. doi: 10.1055/s-0033-1334144. [DOI] [PubMed] [Google Scholar]
- 44.Koulaouzidis A, Said E, Cottier R, Saeed AA. Soluble transferrin receptors and iron deficiency, a step beyond ferritin. A systematic review. J Gastrointestin Liver Dis. 2009;18:345–352. [PubMed] [Google Scholar]
- 45.Wish JB. Assessing iron status: Beyond serum ferritin and transferrin saturation. Clin J Am Soc Nephrol. 2006;1(Suppl 1):S4–S8. doi: 10.2215/CJN.01490506. [DOI] [PubMed] [Google Scholar]
- 46.Milić S, Mikolasević I, Radić M, Hauser G, Stimac D. Clinical utility of red cell distribution width in alcoholic and non-alcoholic liver cirrhosis. Coll Antropol. 2011;35(Suppl 2):335–338. [PubMed] [Google Scholar]
- 47.Kalantar-Zadeh K, Rodriguez RA, Humphreys MH. Association between serum ferritin and measures of inflammation, nutrition and iron in haemodialysis patients. Nephrol Dial Transplant. 2004;19:141–149. doi: 10.1093/ndt/gfg493. [DOI] [PubMed] [Google Scholar]
- 48.Fishbane S, Maesaka JK. Iron management in end-stage renal disease. Am J Kidney Dis. 1997;29:319–333. doi: 10.1016/s0272-6386(97)90192-x. [DOI] [PubMed] [Google Scholar]
- 49.Mast AE, Blinder MA, Gronowski AM, Chumley C, Scott MG. Clinical utility of the soluble transferrin receptor and comparison with serum ferritin in several populations. Clin Chem. 1998;44:45–51. [PubMed] [Google Scholar]
- 50.Goodnough LT, Nemeth E, Ganz T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood. 2010;116:4754–4761. doi: 10.1182/blood-2010-05-286260. [DOI] [PubMed] [Google Scholar]
- 51.Weiss G, Goodnough LT. Anemia of chronic disease. N Engl J Med. 2005;352:1011–1023. doi: 10.1056/NEJMra041809. [DOI] [PubMed] [Google Scholar]
- 52.Lipschitz DA, Cook JD, Finch CA. A clinical evaluation of serum ferritin as an index of iron stores 1974. Nutrition. 1992;8:443–447. [PubMed] [Google Scholar]
- 53.Nelson R, Chawla M, Connolly P, LaPorte J. Ferritin as an index of bone marrow iron stores. South Med J. 1978;71:1482–1484. doi: 10.1097/00007611-197812000-00011. [DOI] [PubMed] [Google Scholar]
- 54.Guyatt GH, Oxman AD, Ali M, Willan A, McIlroy W, Patterson C. Laboratory diagnosis of iron-deficiency anemia: An overview. J Gen Intern Med. 1992;7:145–153. doi: 10.1007/BF02598003. [DOI] [PubMed] [Google Scholar]
- 55.Brown RD, Benfatto J, Gibson J, Kronenberg H. Red cell ferritin and iron stores in patients with chronic disease. Eur J Haematol. 1988;40:136–141. doi: 10.1111/j.1600-0609.1988.tb00810.x. [DOI] [PubMed] [Google Scholar]
- 56.Isa L, Jean G, Silvani A, Arosio P, Taccagni GL. Evaluation of iron stores in patients with alcoholic liver disease: Role of red cell ferritin. Acta Haematol. 1988;80:85–88. doi: 10.1159/000205608. [DOI] [PubMed] [Google Scholar]
- 57.Nagral A, Mehta AB, Gomes AT, et al. Serum soluble transferrin receptor in the diagnosis of iron deficiency in chronic liver disease. Clin Lab Haematol. 1999;21:93–97. doi: 10.1046/j.1365-2257.1999.00202.x. [DOI] [PubMed] [Google Scholar]
- 58.Goddard AF, James MW, McIntyre AS, Scott BB British Society of Gastroenterology. Guidelines for the management of iron deficiency anaemia. Gut. 2011;60:1309–1316. doi: 10.1136/gut.2010.228874. [DOI] [PubMed] [Google Scholar]
- 59.Nadeem S, Shah S, Iqbal T, Iqbal Z, Hanif E. Serum transferrin receptor, serum ferritin and serum transferrin receptor-ferritin index in adults with iron deficiency anaemia. J Ayub Med Coll Abbottabad. 2011;23:44–46. [PubMed] [Google Scholar]
- 60.Skikne BS, Punnonen K, Caldron PH, et al. Improved differential diagnosis of anemia of chronic disease and iron deficiency anemia: A prospective multicenter evaluation of soluble transferrin receptor and the sTfR/log ferritin index. Am J Hematol. 2011;86:923–927. doi: 10.1002/ajh.22108. [DOI] [PubMed] [Google Scholar]
- 61.Nairz M, Theurl I, Wolf D, Weiss G. Iron deficiency or anemia of inflammation?: Differential diagnosis and mechanisms of anemia of inflammation. Wien Med Wochenschr. 2016;166:411–423. doi: 10.1007/s10354-016-0505-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fuccio L, Zagari RM, Serrani M, et al. Endoscopic argon plasma coagulation for the treatment of gastric antral vascular ectasia-related bleeding in patients with liver cirrhosis. Digestion. 2009;79:143–150. doi: 10.1159/000210087. [DOI] [PubMed] [Google Scholar]
- 63.European Association for Study of Liver. EASL Recommendations on Treatment of Hepatitis C 2015. J Hepatol. 2015;63:199–236. doi: 10.1016/j.jhep.2015.03.025. [DOI] [PubMed] [Google Scholar]
- 64.Fatourou EM, Tsochatzis EA. Transfusion strategies in patients with cirrhosis: less is more. Liver Int. 2016;36:503–504. doi: 10.1111/liv.13056. [DOI] [PubMed] [Google Scholar]
- 65.Fleming LW, Hopwood D, Shepherd AN, Stewart WK. Hepatic iron in dialysed patients given intravenous iron dextran. J Clin Pathol. 1990;43:119–124. doi: 10.1136/jcp.43.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gokal R, Millard PR, Weatherall DJ, Callender ST, Ledingham JG, Oliver DO. Iron metabolism in haemodialysis patients. A study of the management of iron therapy and overload. Q J Med. 1979;48:369–391. [PubMed] [Google Scholar]
- 67.Eschbach JW, Adamson JW. Iron overload in renal failure patients: Changes since the introduction of erythropoietin therapy. Kidney Int Suppl. 1999;69:S35–S43. doi: 10.1046/j.1523-1755.1999.055suppl.69035.x. [DOI] [PubMed] [Google Scholar]
- 68.Caramelo C, Ortiz A, Aguilera B, et al. Liver disease patterns in hemodialysis patients with antibodies to hepatitis C virus. Am J Kidney Dis. 1993;22:822–828. doi: 10.1016/s0272-6386(12)70341-4. [DOI] [PubMed] [Google Scholar]