

Abstract
Metformin is the most commonly used drug for type 2 diabetes and has potential benefit in treating and preventing cancer. Previous studies indicated that membrane proteins can affect the antineoplastic effects of metformin and may be crucial in the field of cancer research. However, the antineoplastic effects of metformin and its mechanism in gallbladder cancer (GBC) remain largely unknown. In this study, the effects of metformin on GBC cell proliferation and viability were evaluated using the Cell Counting Kit‐8 (CCK‐8) assay and an apoptosis assay. Western blotting was performed to investigate related signaling pathways. Of note, inhibition, knockdown and upregulation of the membrane protein Chloride intracellular channel 1 (CLIC1) can affect GBC resistance in the presence of metformin. Our data demonstrated that metformin apparently inhibits the proliferation and viability of GBC cells. Metformin promoted cell apoptosis and increased the number of early apoptotic cells. We found that metformin can exert growth‐suppressive effects on these cell lines via inhibition of p‐Akt activity and the Bcl‐2 family. Notably, either dysfunction or downregulation of CLIC1 can partially decrease the antineoplastic effects of metformin while upregulation of CLIC1 can increase drug sensitivity. Our findings provide experimental evidence for using metformin as an antitumor treatment for gallbladder carcinoma.
Keywords: apoptosis, chloride intracellular channel 1, gallbladder carcinoma, metformin, proliferation
Although generally considered rare, gallbladder carcinoma is one of the most common malignancies of the biliary tract, represents 80–95% of biliary tract cancers worldwide, and ranks sixth among all gastrointestinal malignancies.1, 2, 3, 4 Even though gallbladder carcinoma is rare in developed countries, it is a highly fatal disease with a low survival rate because it is often diagnosed at the advanced stage.5, 6 The overall mean survival rate of patients with gallbladder carcinoma is 6 months, with a 5‐year survival rate of approximately 5%. From an epidemiological perspective, mortality due to gallbladder carcinoma closely follows its incidence.6, 7 Currently, complete surgical resection is the only potentially curative therapy for gallbladder carcinoma; however, most patients suffer recurrence after surgical treatment. For patients with unresectable and recurrent disease, either chemotherapy or radiotherapy are the available options; unfortunately, the treatment outcomes are unsatisfactory. Therefore, identifying novel effective therapeutic drugs is necessary to combat this deadly disease.
Metformin is the most commonly used drug for the treatment of type 2 diabetes. It also exhibits potential benefits in cancer treatment and prevention.8, 9 After epidemiological studies indicated a lower prevalence of cancer among type 2 diabetic patients treated with metformin, subsequent reports suggested that metformin can slow cancer cell growth and protect against multiple cancers.10, 11 The antineoplastic mechanisms of metformin vary among different kinds of carcinomas.12, 13, 14, 15, 16 However, the effects of metformin on gallbladder cancer cells and the potential mechanism involved have not been reported.
Previous studies have indicated that some membrane proteins can affect the antineoplastic effects of metformin.17, 18 Chloride intracellular channel 1 (CLIC1), a member of the p64 family, acts as a plasma membrane chloride ion channel. We previously showed that the levels of CLIC1 are increased in human gallbladder metastasis and that elevated expression of CLIC1 can promote invasion and migration in gallbladder cancer cells.19, 20 Thus, we sought to explore the role of CLIC1 in GBC cells treated with metformin.
In the present study, we investigated the antineoplastic activity of metformin in two gallbladder cancer cell lines (NOZ and GBC‐SD) and explored the underlying molecular mechanisms involved. We found that metformin can exert growth‐suppressive effects on these cell lines via inhibition of p‐Akt and Bcl‐2 signaling. Notably, either dysfunction or downregulation of CLIC1 can partially decrease the antineoplastic effects of metformin while upregulation of CLIC1 can increase the drug sensitivity. This study could provide experimental evidence for applying metformin as an antitumor drug for treating gallbladder carcinoma.
Materials and Methods
Cell culture and transfection
The two human GBC cell lines used in this study were as follows: NOZ (obtained from the Health Science Research Resources Bank, Osaka, Japan) and GBC‐SD (obtained from the Cell Bank of the Chinese Academy of Sciences, Shanghai, China). NOZ cells were grown in William's medium (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin‐streptomycin (HyClone, Logan, UT, USA). GBC‐SD cells were grown in high‐glucose DMEM (Gibco) supplemented with 10% fetal bovine serum (Gibco) and 1% penicillin‐streptomycin (HyClone). All cell lines were maintained at 37°C in a humidified atmosphere containing 5% CO2. The siRNAs were synthesized by Biomics (Nantong, China). The siRNA sequences were as follows: Hs‐CLIC1 siRNA1 positive‐sense strand: 5′‐CACUCAAUGACAAUCUGGAdTdT‐3′, anti‐sense strand: 5′‐UCCAGAUUGUCAUUGAGUGdTdT‐3′; Hs‐CLIC1 siRNA2 positive‐sense strand: 5′‐CUGUUGCCAAAGUUACACAdTdT‐3′, anti‐sense strand: 5′‐UGUGUAACUUUGGCAACAGdTdT‐3′; and Hs‐CLIC1 siRNA3 positive‐sense strand: 5′‐CAGAAUGAUUAAACGACCAdTdT‐3′, anti‐sense strand: 5′‐UGGUCGUUUAAUCAUUCUGdTdT‐3′. We chose the siRNA1 sequence for all subsequent experiments. The overCLIC1 plasmid was purchased from Long Qian biotechnology (Shanghai, China). Either siRNAs or plasmids were transfected into the cells using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer's instructions. Transfected cells were cultured for at least 36 h after siRNA or plasmid treatment prior to further experimentation.
Reagents and cell treatments
Metformin was purchased from Yuanmu Biotechnology (Shanghai, China). It was dissolved in normal culture medium as a stock solution (200 mmol/L) and stored at 4°C. IAA94 was purchased from Sigma‐Aldrich (St. Louis, MO, USA). It was dissolved in dimethyl sulfoxide as a stock solution (5.6×104 μmol/L) and stored at 4°C. For working solutions, the stock solution was further diluted with normal culture medium to yield the desired concentration. Control cells were treated with an equal volume of vehicle. The DMSO concentration was kept below 0.5% in cell culture, and no detectable effects on either cell growth or cell death were observed. Cell lines (8×104/well for NOZ cells and 1×105/well for GBC‐SD cells) were seeded into six‐well plates (Corning, Corning, NY, USA) and divided into the control, IAA94, control + metformin and IAA94 + metformin groups. After the cells adhered to the plates, the control and control + metformin groups were treated with normal medium while the IAA94 and IAA94 + metformin groups were treated with IAA94 (100 μmol/L)‐containing medium for 24 h. Then, the control and IAA94 groups were treated with normal medium for 24 h. Meanwhile, the control + metformin and IAA94 + metformin groups were treated with 20 mmol/L metformin for 24 h. After treatment, these four groups were assessed using additional tests. The moment that NOZ and GBC‐SD cells were transfected with siRNA and plasmids, they were classified as siNC, siCLIC1, siNC + metformin, siCLIC1 + metformin, overNC, overCLIC1, overNC + metformin and overCLIC1 + metformin depending on the transfection type. The siNC, siCLIC1, overNC and overCLIC1 groups were treated with normal medium for 24 h. The siNC + metformin and siCLIC1 + metformin groups were treated with 20 mmol/L metformin‐containing medium while the overNC + metformin and overCLIC1 + metformin groups were treated with 15 mmol/L metformin‐containing medium. After the treatments, these groups were assayed in further experiments.
Cell viability assay
Cell viability was analyzed using the Cell Counting Kit‐8 (CCK8, Dojindo, Japan) assay according to the manufacturer's instructions. Briefly, NOZ (600/well) and GBC‐SD (800/well) cells were seeded into 96‐well culture plates, incubated overnight, and treated with metformin at final concentrations of 0, 5, 10, 15, 25 and 30 mmol/L for 24, 48, or 72 h. The numbers of seeded cells for the IAA94, siRNA and plasmid groups were the same (600/well of NOZ cells and 800/well of GBC‐SD cells). After the respective treatments, CCK‐8 was then added to each well, and the cells were incubated at 37°C for 3 h. The optical densities (ODs) were measured at a wavelength of 450 nm with a microplate reader (μQuant, Bio‐Tek Instruments, Winooski, VT, USA).
Colony formation assay
NOZ and GBC‐SD cells in the logarithmic growth phase were resuspended into single‐cell suspensions, and 500 cells were seeded into each well of a six‐well plate. After the cells adhered to the plate, they were treated with metformin (0, 1 and 3 mmol/L for both cell lines) for 24 h. The metformin‐containing medium was then removed, and the cells were grown in complete medium for 14 days to encourage colony formation. The cells were then fixed with 4% paraformaldehyde for 15 min and stained with 0.1% crystal violet (Sigma‐Aldrich) for 30 min. After washing, the plates were air‐dried, and the stained colonies were photographed using a microscope (Leica, Wetzlar, Germany). The total number of colonies (50 cells/colony) was manually counted.
Flow cytometric analysis of proliferation and apoptosis
After the respective treatments, adherent cells were lifted by trypsinization, and floating cells were harvested. After washing the cells twice with cold phosphate‐buffered saline (PBS), they were resuspended at a density of 1×106 cells/mL. Next, 100 μL of binding buffer containing 5 μL of annexin V‐FITC and 5 μL of a working solution of PI (100 μg/mL) was added to the cells followed by a 30‐min incubation in the dark, after which 400 μL of binding buffer was added to the suspension. The samples were then immediately analyzed using flow cytometry (BD Biosciences, San Diego, CA, USA).
Quantitative real‐time PCR
Total RNA was isolated from cell lines using TRIzol Reagent (Invitrogen, Shanghai, China) according to the manufacturer's instructions. cDNA was generated using a Reverse Transcription System Kit. The qRT‐PCR reactions were performed using an ABI7500 System and SYBR Green PCR Master Mix (Takara, Dalian, China). The sequencing service was provided by BioSune (Shanghai, China). The primer sequences for CLIC1 were 5′‐GACTGAAGGAGGAGGACAAAG‐3′ (forward) and 5′‐ATCATGAAGAGCCTCTGTGAAA‐3′ (reverse). Each assay was performed in triplicate, and the average was calculated. The CLIC1 expression levels were normalized to those of GAPDH. The relative expression levels of the target genes were calculated and normalized to the relative expression detected in the corresponding control cells, which were set at 1.0.
Western blot analyses
Following the respective treatments, the cells were harvested and lysed in RIPA buffer (Cell Signaling, Danvers, MA, USA). The protein concentrations were determined using a bicinchoninic acid (BCA) protein assay (Thermo Scientific, Rockford, IL, USA). Equal amounts of protein were separated on 10% SDS‐polyacrylamide gels. The proteins were transferred to a PVDF membrane and probed with antibodies targeting CLIC1 (Abcam, UK; 1:1000 dilution), PI3K 110α (Cell Signaling Technology, USA; 1:1000 dilution), p‐Akt (Cell Signaling Technology, USA; 1:1000 dilution), Akt (Cell Signaling Technology, USA; 1:1000 dilution), Bcl‐2 (Abways, China; 1:1000 dilution), Bax (Abways, China; 1:1000 dilution) and β‐actin (Abways, China; 1:1000 dilution). The blots were then incubated with HRP‐conjugated secondary antibodies followed by enhanced chemiluminescence (ECL) detection.
Statistical analysis
Continuous data were expressed as the mean ± SD. Significant differences between two groups were analyzed using anova. The data were considered statistically significant when P‐values were less than 0.05. Statistical tests were carried out with SPSS 18.0 software (SPSS, Chicago, IL, USA).
Results
Metformin inhibits cell growth and induces apoptosis in gallbladder cancer cells
To explore the effects of metformin on gallbladder carcinoma, we chose two GBC cell lines (NOZ and GBC‐SD) and performed the CCK8 assay to determine changes in cell proliferation. Treatment with metformin led to a marked decrease in the viability of NOZ and GBC‐SD cells in a dose‐ and time‐dependent manner (Fig. 1a). The half maximal inhibitory concentration (IC50) of these two cell lines after a 48‐h treatment was approximately 15 mmol/L. Based on the dose‐response curve, we chose 10, 15, and 20 mmol/L as the concentration range for both cell lines in the subsequent experiments. As the results of the metformin treatment after 48 h were more obvious and stable than at either 24 and 72 h, 48 h was chosen as the incubation time to detect changes in molecular events for all subsequent experiments. The ability of NOZ and GBC‐SD cells to form colonies in the presence of metformin was tested using the flat plate colony formation assay (Fig. 1b). A marked inhibition of the colony forming ability was observed. The colony count indicated that metformin exerted a dose‐dependent decrease on the colony forming ability of these cells (Fig. 1c). These findings demonstrated that metformin apparently inhibits the proliferation and viability of NOZ and GBC‐SD cells. We investigated the apoptotic effects of metformin on NOZ and GBC‐SD cells by measuring annexin V‐FITC/PI double staining. As shown in Figure 2(a), the flow cytometry results indicated that metformin reduced the number of surviving cells and increased the number of early apoptotic cells in a dose‐dependent manner (Fig. 2b).
Figure 1.
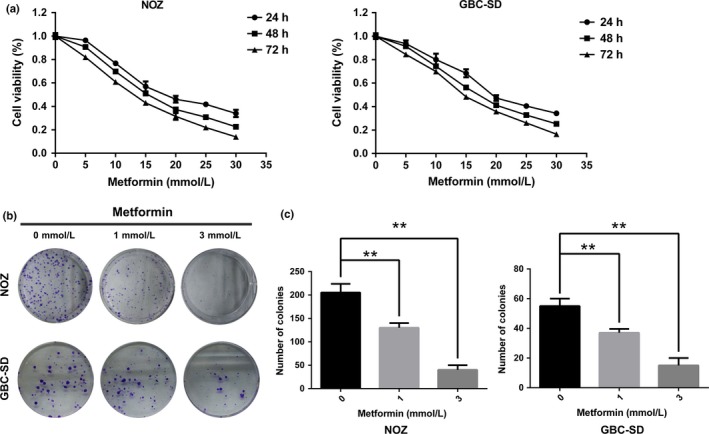
Metformin inhibits the proliferation and reduces the viability of gallbladder cancer cells. (a) NOZ and GBC‐SD cells were treated with the indicated concentrations of metformin (0–30 mmol/L) for the indicated times, and the cell count was assessed by the CCK8 assay. Data are presented as the mean ± SD (n = 3). (b) NOZ and GBC‐SD cells were pretreated with metformin (0, 1 or 3 mmol/L) for 24 h and then allowed to form colonies in fresh medium for 14 days. Representative crystal violet staining pictures are shown. (c) The colony numbers (mean ± SD, n = 3) are shown. *P, 0.05, **P, 0.01 versus the control group (no metformin).
Figure 2.
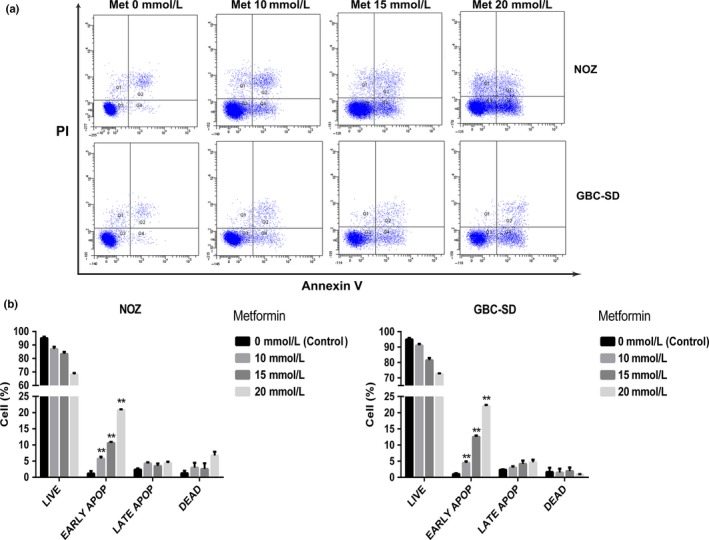
Metformin induces apoptosis in gallbladder cancer cells. (a) NOZ and GBC‐SD cells were treated with metformin (0, 10, 15, or 20 mmol/L) for 48 h. Then, the appearance of apoptotic cells was assessed by flow cytometry using annexin V/propidium iodide (PI) double staining. The cells positive for annexin V alone reflects early apoptosis, and the fraction positive for annexin V and PI double staining indicates late apoptosis. (b) The percentages of apoptotic cells are shown (mean ± SD, n = 3). *P, 0.05, **P, 0.01 versus the control group (no metformin).
Metformin exerts antitumor effects via regulation of PI3K‐Akt signaling and the Bcl‐2 family
It is well known that proteins in the PI3K‐Akt pathway and Bcl‐2 family play pivotal roles in cell proliferation and the apoptotic process.21 However, it has been shown that some membrane proteins can influence the antitumor effects of metformin.17, 18 Chloride intracellular channel 1 (CLIC1) is a membrane protein that has chloride ion channel activity. High levels of CLIC1 expression were associated with metastasis in gallbladder carcinoma.19, 20 Therefore, we investigated how the PI3K‐Akt axis, the Bcl‐2 family of proteins and CLIC1 affect the underlying molecular mechanism of metformin‐induced inhibition of cell proliferation and promotion of apoptosis in NOZ and GBC‐SD cells. The expression levels of these proteins were measured by Western blot analysis after treating the cell lines with various concentrations of metformin for 48 h. As shown in Figure 3(a), upregulation of Bax and downregulation of p‐Akt and Bcl‐2 were induced by metformin in a dose‐dependent manner. However, the expression levels of CLIC1, PI3K, and Akt were not affected by metformin. The level of Akt phosphorylation apparently decreased (Fig. 3b). The ratio of Bax to Bcl‐2 reflects the occurrence and severity of apoptosis in the presence of apoptotic stimuli. An increased Bax to Bcl‐2 ratio promotes cell apoptosis, whereas a decreased Bax to Bcl‐2 ratio promotes cell survival.22 Based on our Western blot assay, we found that the ratio of Bax to Bcl‐2 in gallbladder cancer cells treated with metformin was obviously increased in a dose‐dependent manner (Fig. 3c). Taken together, these data suggest that metformin is involved in modulating the PI3K‐Akt pathway and Bcl‐2 family in gallbladder cancer cells.
Figure 3.
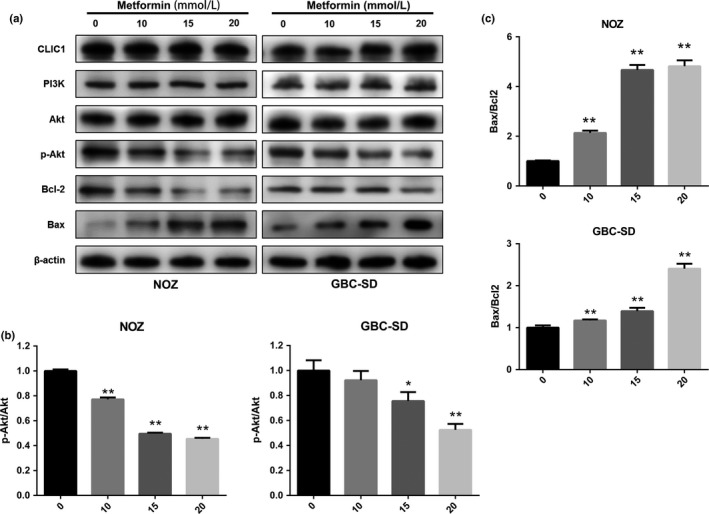
Metformin inhibits proliferation and induces apoptosis via regulation of PI3K‐AKT signaling and Bcl‐2 family activity in gallbladder cancer cells. (a) Cellular proteins were collected from NOZ and GBC‐SD cells cultured with metformin (0, 10, 15, or 20 mmol/L) for 48 h. Western blotting was performed to examine the expression levels of CLIC1, PI3K, Akt, p‐Akt, Bcl‐2, and Bax with β‐actin as a loading control. (b) The relative ratio of p‐Akt to Akt. (c) The relative ratio of Bax to Bcl‐2. All the data are presented as the mean ± standard deviation from three independent experiments. *P, 0.05, **P, 0.01 versus the control group (no metformin).
Dysfunction or silencing of CLIC1 increases GBC survival in the presence of metformin
Although metformin does not influence the protein expression of CLIC1, we found that using the CLIC1 selective inhibitor IAA94 can reduce metformin cytotoxicity to a certain degree. As shown in Figure 4(a), a 24‐h treatment with IAA94 significantly reduced the effects of metformin on NOZ and GBC‐SD cell viability as evaluated by the CCK8 assay, while IAA94 slightly affected basal cell proliferation. CLIC1 protein dysfunction also increased the colony forming abilities (Fig. 4b) and decreased the number of early apoptotic cells (Fig. 4c) in the presence of metformin. Furthermore, we performed Western blotting to measure the expression of related proteins. Figure 4(d) shows that compared to the cells that did not receive IAA94, the treated cells presented increased levels of p‐Akt and a decreased ratio of Bax to Bcl‐2. There were no marked effects on the expression of PI3K, Akt and CLIC1. According to these data, it is suggested that CLIC1 dysfunction can increase the resistance of NOZ and GBC‐SD cells to metformin. To further elucidate this conclusion, we performed RNA interference to knock down CLIC1. The mRNA and protein levels of CLIC1 were successfully silenced in NOZ and GBC‐SD cells treated with CLIC1‐specific siRNA (Fig. 5a). Silencing CLIC1 had no effect on cell proliferation or viability but impaired the cytotoxicity of metformin (Fig. 5b,c,d). Similar to the results observed upon CLIC1 dysfunction, CLIC1 silencing increased the levels of p‐Akt and lowered the ratio of Bax to Bcl‐2 in the presence of metformin (Fig. 5e). Finally, inhibition of CLIC1 can increase the resistance of GBC to metformin.
Figure 4.
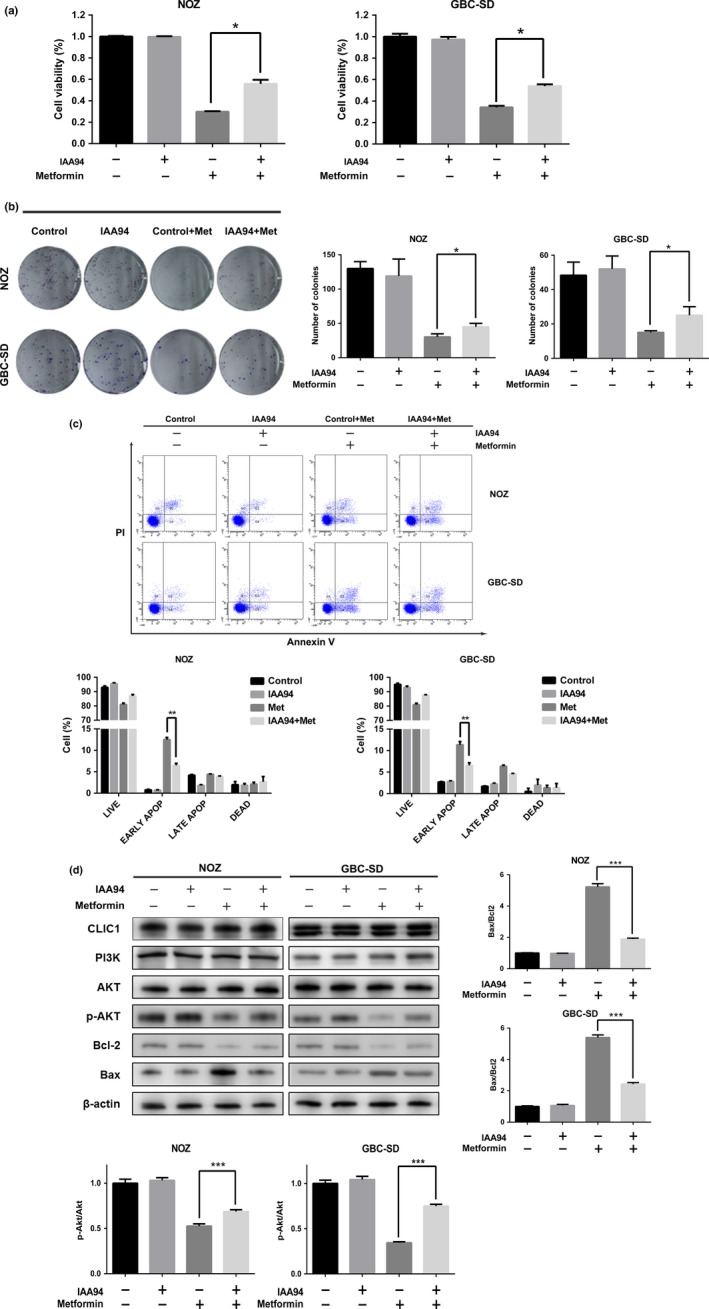
The CLIC1 inhibitor IAA94 increases GBC survival in the presence of metformin. NOZ and GBC‐SD cells were divided into four groups: control, IAA94, control + metformin and IAA94 + metformin. Cells were pretreated with IAA94‐containing medium for 24 h and then treated with metformin for 24 h. After these treatments, these four groups were assessed in further experiments. (a) Cell viability was measured by CCK8 assay. Data are presented as the mean ± SD (n = 3). (b) All the groups were allowed to form colonies in fresh medium in the absence of metformin for 14 days. The colony numbers (mean ± SD, n = 3) are shown. (c) The appearance of apoptotic cells was assessed by flow cytometry. The percentages of apoptotic cells are shown (mean ± SD, n = 3). (d) Western blotting was performed to examine the expression levels of CLIC1, PI3K, Akt, p‐Akt, Bcl‐2, and Bax with β‐actin as a loading control. The relative ratios of p‐Akt to Akt and Bax to Bcl‐2 are presented as the mean ± SD (n = 3). All the data are presented as the mean ± standard deviation from three independent experiments. *P, 0.05, **P, 0.01, ***P, 0.001 versus the control group.
Figure 5.
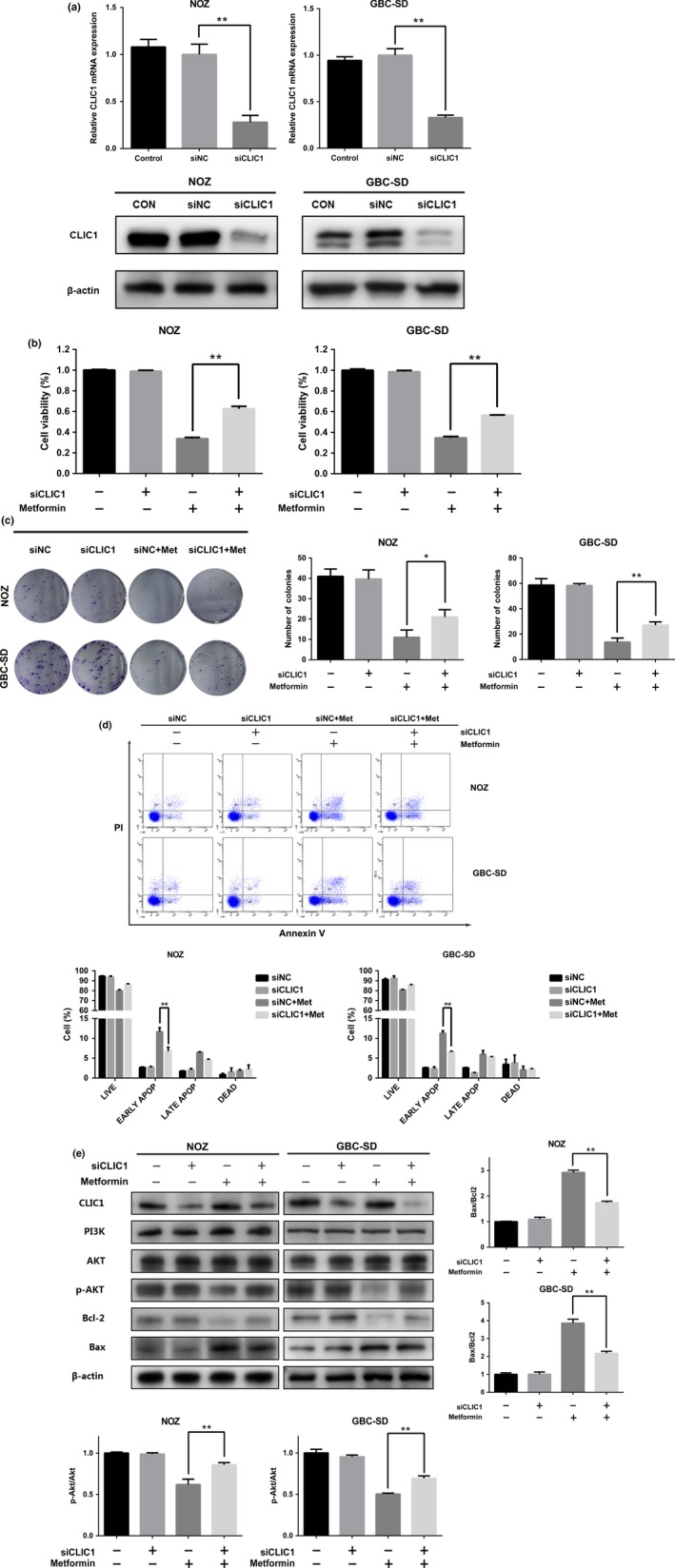
Downregulation of CLIC1 increases GBC survival in the presence of metformin. NOZ and GBC‐SD cells were divided into four groups: siNC, siCLIC1, siNC + metformin and siCLIC1 + metformin. Cells were transfected with siRNA for 36 h and then treated with metformin for 24 h. (a) The mRNA and protein expression levels of CLIC1 after siRNA knockdown. (b) Cell viability was measured using the CCK8 assay. Data are presented as the mean ± SD (n = 3) (c) All groups were allowed to form colonies in fresh medium in the absence of metformin for 14 days. The colony numbers (mean ± SD, n = 3) are shown. (d) The number of apoptotic cells was assessed by flow cytometry. The percentages of apoptotic cells are shown (mean ± SD, n = 3). (e) Western blotting was performed to examine the expression levels of CLIC1, PI3K, Akt, p‐Akt, Bcl‐2, and Bax with β‐actin as a loading control. The relative ratios of p‐Akt to Akt and Bax to Bcl‐2 are presented as the mean ± SD (n = 3). All the data are presented as the mean ± standard deviation from three independent experiments. * P, 0.05, ** P, 0.01 versus the control group.
Overexpression of CLIC1 increases the antitumor effects of metformin in GBC
According to our previous results, we concluded that the function of CLIC1 might be important for the antitumor effects of metformin and that CLIC1 dysfunction might partially impair the effects of metformin. Conversely, overexpression of CLIC1 might increase the cytotoxicity of metformin. To test this hypothesis, we transfected plasmids to overexpress CLIC1. The mRNA and protein levels of CLIC1 are shown in Figure 6(a). Upregulation of CLIC1 expression did not affect basal cell proliferation or viability but significantly increased the cytotoxic effects of metformin (Fig. 6b,c). Overexpression of CLIC1 increased the number of early apoptotic cells in the presence of a lower concentration of metformin (Fig. 6d). As opposed to CLIC1 silencing, upregulation of CLIC1 decreased the levels of p‐Akt while increasing the ratio of Bax to Bcl‐2 (Fig. 6e). Our results showed that upregulation of CLIC1 can increase the effects of metformin as an anti‐proliferative agent.
Figure 6.
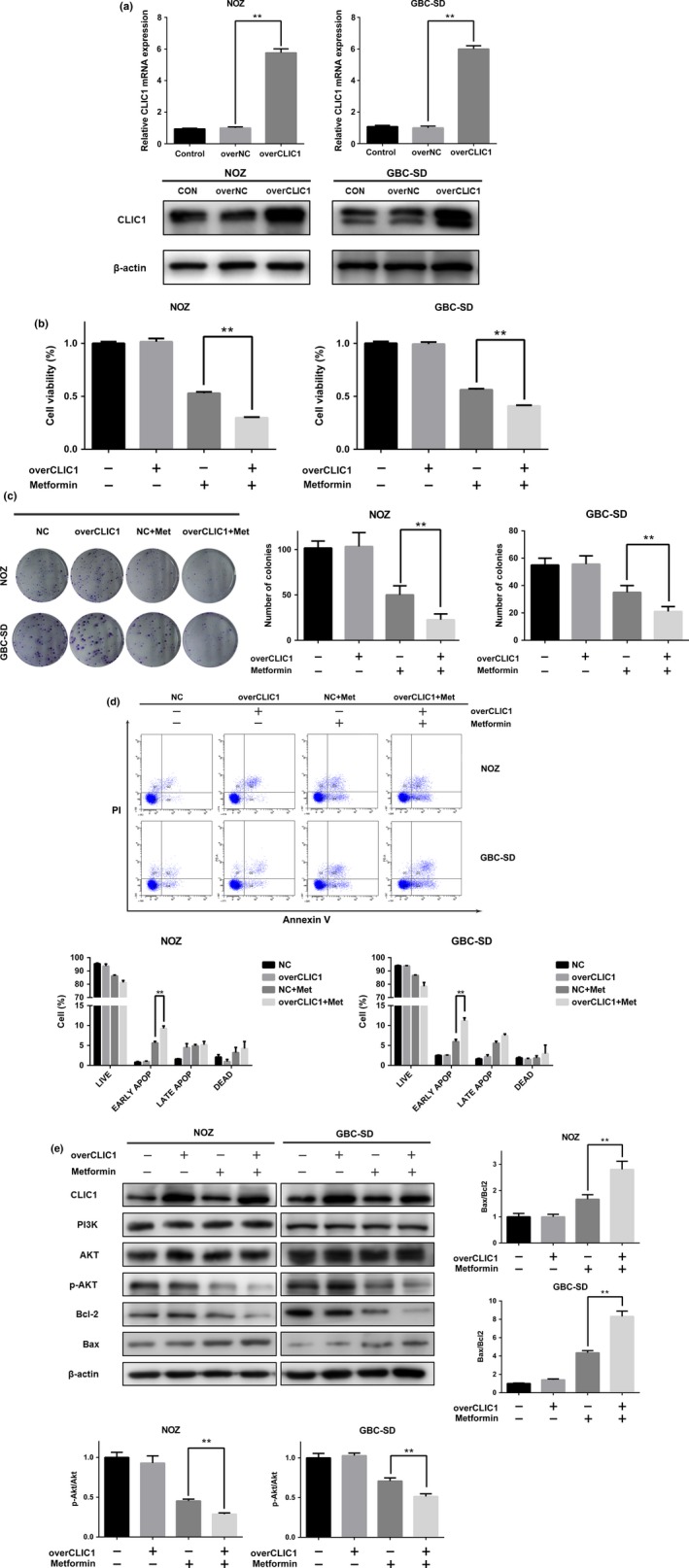
Overexpression of CLIC1 increases GBC survival in the presence of metformin. NOZ and GBC‐SD cells were divided into four groups: NC, overCLIC1, NC + metformin and overCLIC1 + metformin. Cells were transfected with plasmids for 36 h and then treated with metformin for 24 h. (a) The mRNA and protein expression levels of CLIC1 after plasmid transfection. (b) Cell viability was measured using the CCK8 assay. Data are presented as the mean ± SD (n = 3) (c) All the groups were allowed to form colonies in fresh medium in the absence of metformin for 14 days. The colony numbers (mean ± SD, n = 3) are shown. (d) The appearance of apoptotic cells was assessed by flow cytometry. The percentages of apoptotic cells are shown (mean ± SD, n = 3). (e) Western blotting was performed to examine the expression levels of CLIC1, PI3K, Akt, p‐Akt, Bcl‐2, and Bax with β‐actin as a loading control. The relative ratios of p‐Akt to Akt and Bax to Bcl‐2 are presented as the mean ± SD (n = 3). All the data are presented as the mean ± standard deviation from three independent experiments. *P, 0.05, **P, 0.01 versus the control group.
Discussion
Even though metformin has been used mainly as a first‐line drug for the treatment of type 2 diabetes, the results from a number of large‐scale epidemiologic and laboratory studies suggest that metformin may also exert anticancer effects through a different mechanism.23 Due to the rarity of gallbladder carcinoma, no data are currently available regarding cancer prevalence and metformin treatment in patients with GBC, nor is there evidence on the concrete effects of metformin on tumor growth. In this study, we not only demonstrated that metformin is capable of inhibiting gallbladder cancer cell viability and inducing apoptosis by inhibiting the PI3K‐Akt axis and the Bcl2 family but also revealed the regulatory roles of chloride intracellular channel 1 (CLIC1) in the resistance of GBC to metformin.
Recent studies have shown that metformin exerts multiple antitumor effects in vivo and in vitro.24, 25, 26 However, these antitumor effects as well as the related underlying mechanisms vary among different cancer cells. In colorectal cancer, metformin suppresses colonic epithelial proliferation by inhibiting the mTOR pathway via AMPK activation.27 In prostate cancer, metformin can exert inhibitory effects on castration‐induced EMT by repressing the COX2/PGE2/STAT3 axis.28 Co‐treatment with metformin and Y27632 can inhibit EMT in breast cancer cell lines.29 In cholangiocarcinoma, metformin exerts anti‐proliferative and anti‐metastatic effects by targeting STAT3 and NF‐ĸB.30 In our study, we demonstrated that metformin could inhibit cell proliferation in the NOZ and GBC‐SD cell lines. Metformin can decrease cell viability of these cells in a dose‐ and time‐dependent manner. Then, we investigated the apoptotic effects of metformin on NOZ and GBC‐SD cells. Metformin reduced the number of surviving cells and mainly increased the number of early apoptotic cells in a dose‐dependent manner. However, the half maximal inhibitory concentration (IC50) of metformin does not exert marked effects on cell cycle arrest. We assumed that the main effects of metformin are not identical in different types of cell lines. For example, metformin mainly affects cell cycle progression in renal cancer cells31 and inhibits castration‐induced EMT in prostate cancer.28 In addition, we speculated that higher concentrations of metformin might exert a more obvious effect on the cell cycle.
To determine the associated signaling pathways, we performed Western blotting and discovered that metformin affected the expression of the Bcl2 family and the levels of phosphorylated Akt. An increased Bax to Bcl‐2 ratio of in gallbladder cancer cells was observed after treatment with metformin. Apoptosis can suppress cell proliferation and inhibit tumorigenesis.32 The relative ratio of pro‐apoptotic proteins, such as Bax, to anti‐apoptotic proteins, such as Bcl‐2, determines cell survival or death.33 Thus, a high Bax/Bcl‐2 ratio is associated with increased vulnerability to apoptotic activation. Based on our Western blot results, we found that the ratio of Bax to Bcl‐2 in NOZ and GBC cells treated with metformin was obviously increased in a dose‐dependent manner. PI3K/Akt signaling contributes to a variety of processes that are crucial in mediating multiple aspects of cellular function, including cell growth and survival.21 The core components of this pathway, namely, phosphatidylinositide 3‐kinases (PI3Ks) and Akt, have been demonstrated to be frequently hyperactivated in the majority of cancers and have been the focus of many studies in this field.34 We found that metformin did not influence either PI3K or Akt expression. However, metformin apparently decreased the levels of phosphorylated Akt. Taken together, these data led us to propose that metformin decreased cell survival by upregulating the Bax/Bcl‐2 ratio and downregulating the levels of p‐Akt in human gallbladder cancer cells.
Chloride intracellular channel 1 (CLIC1) is a member of an identified class of Cl channel proteins and is primarily expressed in the nuclear and plasma membrane.35 CLIC1 protein levels are reportedly increased in multiple human cancers and have been proposed as a tumor marker.36 CLIC1 mRNA and protein levels were highly expressed in gallbladder cancer tissues compared with adjacent non‐tumor tissues and associated with poor prognosis.19 It has been reported that membrane proteins such as SPRY2 and OCT‐3 can affect the antineoplastic effects of metformin.17, 18 Previous studies demonstrated that metformin can exert anti‐proliferative effects by inhibiting chloride intracellular channel 1 (CLIC1)‐mediated ion current.16 The Western blot results showed that metformin did not affect the protein expression of CLIC1 in gallbladder cancer cells. However, either dysfunction or downregulation of CLIC1 can partially increase GBC resistance to metformin while attenuating the inhibition of Akt phosphorylation and the reduction of the Bax/Bcl2 ratio. Furthermore, upregulation of CLIC1 can enhance the anti‐proliferative efficacy of metformin. Of note, either upregulating or downregulating CLIC1 did not present obvious effects on cell viability, apoptosis or the signal pathways. The effect of CLIC1 on cell proliferation is controversial. Downregulation of CLIC1 enhanced proliferative activity and decreased the percentage of apoptotic in hepatocellular cancer,35 while downregulation of CLIC1 significantly reduced cell proliferation in pancreatic cancer.37 We assumed that even though CLIC1 expression affects the migration and EMT in gallbladder carcinoma,20 it may not be the key factor in GBC tumorigenesis. However, CLIC1 might be important regarding the antineoplastic role of metformin. Studies have shown that some proteins have facilitatory effects on metformin. The effects of metformin on mitochondria and growth are transduced via mTORC1 the NPC, respectively. RNAi knockdown of NPC components suppresses the effects of metformin.38 Metformin can exert inhibitory activities against growth via both membrane and intracellular proteins17, 27 and CLIC1 exists both as a soluble cytoplasmic protein and membrane‐bound protein. Taken together, these data led us to hypothesize that in GBC, metformin exerts antitumor effects via CLIC1‐mediated ion current or subsequent interacting proteins. Both the phenotype experiments and signaling pathway results indicated that GBC cells with high levels of CLIC1 expression were much more vulnerable to metformin.
Metformin can play a role in inhibiting GBC, especially in conjunction with high levels of CLIC1 expression. We speculate that metformin may be a good treatment option for patients suffering from advanced and metastatic cancer. Above all, this study provides further support for the development of metformin as an effective therapeutic agent for gallbladder carcinoma and shows that CLIC1 is a critical factor in the sensitivity of GBC to metformin.
Disclosure statement
The authors declare no conflicts of interest.
Acknowledgments
This study was supported by the Leading Talent Program of Shanghai affiliated with the Sailing Program of Shanghai Science and Technology Commission (No. 14YF1403000), the National Natural Science Foundation of China (Nos. 31600075, 31620103910, and 91440203), and the Shanghai Science and Technology Commission Medical Guidance Project (Nos. 16JC1400200 and 16411952501).
Cancer Sci 108 (2017) 1240–1252
Funding Information
This study was supported by the Leading Talent Program of Shanghai affiliated with the Sailing Program of Shanghai Science and Technology Commission (No. 14YF1403000), the National Natural Science Foundation of China (Nos. 31600075, 31620103910, and 91440203), and the Shanghai Science and Technology Commission Medical Guidance Project (Nos. 16JC1400200 and 16411952501).
Contributor Information
Wenguang Wu, Email: wuwenguang08@126.com.
Yingbin Liu, Email: liuybphd@126.com.
References
- 1. Shu YJ, Bao RF, Jiang L et al MicroRNA‐29c‐5p suppresses gallbladder carcinoma progression by directly targeting CPEB4 and inhibiting the MAPK pathway. Cell Death Differ 2017; 24: 445–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhao S, Cao Y, Liu SB et al The E545K mutation of PIK3CA promotes gallbladder carcinoma progression through enhanced binding to EGFR. J Exp Clin Cancer Res 2016; 35(1): 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bao RF, Shu YJ, Hu YP et al miR‐101 targeting ZFX suppresses tumor proliferation and metastasis by regulating the MAPK/Erk and Smad pathways in gallbladder carcinoma. Oncotarget 2016; 7: 22339–54. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 4. Shu YJ, Weng H, Ye YY et al SPOCK1 as a potential cancer prognostic marker promotes the proliferation and metastasis of gallbladder cancer cells by activating the PI3K/AKT pathway. Mol Cancer 2015; 14: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ryu S, Chang Y, Yun KE, Jung HS, Shin JH, Shin H. Gallstones and the risk of gallbladder cancer mortality: a cohort study. Am J Gastroenterol 2016; 111: 1476–87. [DOI] [PubMed] [Google Scholar]
- 6. Cao Y, Liang H, Zhang F et al Prohibitin overexpression predicts poor prognosis and promotes cell proliferation and invasion through ERK pathway activation in gallbladder cancer. J Exp Clin Cancer Res 2016; 35: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li HF, Wang XA, Xiang SS et al Oleanolic acid induces mitochondrial‐dependent apoptosis and G0/G1 phase arrest in gallbladder cancer cells. Drug Des Devel Ther 2015; 9: 3017–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kordes S, Pollak MN, Zwinderman AH et al Metformin in patients with advanced pancreatic cancer: a double‐blind, randomised, placebo‐controlled phase 2 trial. Lancet Oncol 2015; 16: 839–47. [DOI] [PubMed] [Google Scholar]
- 9. Choi Y, Kim TY, Oh DY et al The Impact of diabetes mellitus and metformin treatment on survival of patients with advanced pancreatic cancer undergoing chemotherapy. Cancer Res Treat 2016; 48: 171–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pryor R, Cabreiro F. Repurposing metformin: an old drug with new tricks in its binding pockets. Biochem J 2015; 471: 307–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castillo‐Quan JI, Blackwell TK. Metformin: restraining nucleocytoplasmic shuttling to fight cancer and aging. Cell 2016; 167: 1670–1. [DOI] [PubMed] [Google Scholar]
- 12. Mayer MJ, Klotz LH, Venkateswaran V. The effect of metformin use during docetaxel chemotherapy on prostate cancer‐specific and overall survival of diabetic castration‐resistant prostate cancer patients. J Urol 2017; 197: 1068–75. [DOI] [PubMed] [Google Scholar]
- 13. Gao ZY, Liu Z, Bi MH et al Metformin induces apoptosis via a mitochondria‐mediated pathway in human breast cancer cells in vitro . Exp Ther Med 2016; 11: 1700–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chowdhury S, Yung E, Pintilie M et al MATE2 expression is associated with cancer cell response to metformin. PLoS ONE 2016; 11: e0165214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ling S, Song L, Fan N et al Combination of metformin and sorafenib suppresses proliferation and induces autophagy of hepatocellular carcinoma via targeting the mTOR pathway. Int J Oncol 2017; 50(1): 297–309. [DOI] [PubMed] [Google Scholar]
- 16. Gritti M, Wurth R, Angelini M et al Metformin repositioning as antitumoral agent: selective antiproliferative effects in human glioblastoma stem cells, via inhibition of CLIC1‐mediated ion current. Oncotarget 2014; 5: 11252–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feng YH, Wu CL, Shiau AL et al MicroRNA‐21‐mediated regulation of Sprouty2 protein expression enhances the cytotoxic effect of 5‐fluorouracil and metformin in colon cancer cells. Int J Mol Med 2012; 29: 920–6. [DOI] [PubMed] [Google Scholar]
- 18. Patel H, Younis RH, Ord RA, Basile JR, Schneider A. Differential expression of organic cation transporter OCT‐3 in oral premalignant and malignant lesions: potential implications in the antineoplastic effects of metformin. J Oral Pathol Med 2013; 42: 250–6. [DOI] [PubMed] [Google Scholar]
- 19. Ding Q, Li M, Wu X et al CLIC1 overexpression is associated with poor prognosis in gallbladder cancer. Tumour Biol 2015; 36(1): 193–8. [DOI] [PubMed] [Google Scholar]
- 20. Wang JW, Peng SY, Li JT et al Identification of metastasis‐associated proteins involved in gallbladder carcinoma metastasis by proteomic analysis and functional exploration of chloride intracellular channel 1. Cancer Lett 2009; 281(1): 71–81. [DOI] [PubMed] [Google Scholar]
- 21. Yu JS, Cui W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 2016; 143: 3050–60. [DOI] [PubMed] [Google Scholar]
- 22. Viant C, Guia S, Hennessy RJ et al Cell cycle progression dictates the requirement for BCL2 in natural killer cell survival. J Exp Med 2017; 214: 491–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Poli G, Cantini G, Armignacco R et al Metformin as a new anti‐cancer drug in adrenocortical carcinoma. Oncotarget 2016; 7: 49636–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xue C, Wang C, Sun Y et al Targeting P‐glycoprotein function, p53 and energy metabolism: combination of metformin and 2‐deoxyglucose reverses the multidrug resistance of MCF‐7/Dox cells to doxorubicin. Oncotarget 2017; 8: 8622–8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seabloom DE, Galbraith AR, Haynes AM et al Fixed‐dose combinations of pioglitazone and metformin for lung cancer prevention. Cancer Prev Res (Phila) 2017; 10: 116–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gonzalez N, Prieto I, Del Puerto‐Nevado L et al 2017 update on the relationship between diabetes and colorectal cancer: epidemiology, potential molecular mechanisms and therapeutic implications. Oncotarget 2017; 8: 18456–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hosono K, Endo H, Takahashi H et al Metformin suppresses azoxymethane‐induced colorectal aberrant crypt foci by activating AMP‐activated protein kinase. Mol Carcinog 2010; 49: 662–71. [DOI] [PubMed] [Google Scholar]
- 28. Tong D, Liu Q, Liu G et al Metformin inhibits castration‐induced EMT in prostate cancer by repressing COX2/PGE2/STAT3 axis. Cancer Lett 2016; 389: 23–32. [DOI] [PubMed] [Google Scholar]
- 29. Leonel C, Ferreira LC, Borin TF et al Inhibition of epithelial‐mesenchymal transition in response to treatment with metformin and Y27632 in breast cancer cell lines. Anticancer Agents Med Chem 2017; 17: DOI:10.2174/1871520617666170102153954. [DOI] [PubMed] [Google Scholar]
- 30. Saengboonmee C, Seubwai W, Cha'on U, Sawanyawisuth K, Wongkham S, Wongkham C. Metformin exerts antiproliferative and anti‐metastatic effects against cholangiocarcinoma cells by targeting STAT3 and NF‐kB. Anticancer Res. 2017; 37(1): 115–23. [DOI] [PubMed] [Google Scholar]
- 31. Xie W, Wang L, Sheng H et al Metformin induces growth inhibition and cell cycle arrest by upregulating MicroRNA34a in renal cancer cells. Med Sci Monit 2017; 23: 29–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jiang Y, Sheng H, Meng L et al RBM5 inhibits tumorigenesis of gliomas through inhibition of Wnt/beta‐catenin signaling and induction of apoptosis. World J Surg Oncol 2017; 15(1): 9 https://doi.org/10.1186/s12957-016-1084-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xi H, Zhang Y, Xu Y et al Caspase‐1 inflammasome activation mediates homocysteine‐induced pyrop‐apoptosis in endothelial cells. Circ Res 2016; 118: 1525–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer 2015; 15(1): 7–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Peretti M, Angelini M, Savalli N, Florio T, Yuspa SH, Mazzanti M. Chloride channels in cancer: focus on chloride intracellular channel 1 and 4 (CLIC1 AND CLIC4) proteins in tumor development and as novel therapeutic targets. Biochim Biophys Acta 2015; 1848(10 Pt B): 2523–31. [DOI] [PubMed] [Google Scholar]
- 36. Setti M, Osti D, Richichi C et al Extracellular vesicle‐mediated transfer of CLIC1 protein is a novel mechanism for the regulation of glioblastoma growth. Oncotarget 2015; 6: 31413–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu J, Dong Q, Zhang B et al Chloride intracellular channel 1 (CLIC1) is activated and functions as an oncogene in pancreatic cancer. Med Oncol 2015; 32: 616. [DOI] [PubMed] [Google Scholar]
- 38. Wu L, Zhou B, Oshiro‐Rapley N et al An ancient, unified mechanism for metformin growth inhibition in C. elegans and cancer. Cell 2016; 167: 1705–18 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]