

Abstract
Pancreatic neuroendocrine tumors (PanNET) are rare cancers that generally have a poor prognosis. Accurate diagnosis and proper treatment of these tumors requires a better understanding of the molecular mechanisms underlying the development of PanNET. It has been shown that the mTOR inhibitor everolimus can improve the progression‐free survival of PanNET patients, suggesting that inhibition of the PI3K‐Akt‐mTOR pathway may suppress the progression of PanNET. PHLDA3 is a novel tumor suppressor protein that inhibits Akt activation by competition for binding to PIP 3. Our analysis of PanNET revealed frequent loss‐of‐heterozygosity and DNA methylation at the PHLDA3 locus, resulting in strong suppression of PHLDA3 transcription. Such alterations in the PHLDA3 gene were also frequently found in lung neuroendocrine tumors (NET), suggesting the possibility that various types of NET have in common the functional loss of the PHLDA3 gene.
Keywords: AKT, neuroendocrine tumors, p53, PanNET, PHLDA3
Neuroendocrine tumors (NET) are malignancies derived from diffuse neuroendocrine systems (DNES): for example, lung, pancreas, pituitary, stomach, duodenum and the small intestine. Among these, PanNET is a rare cancer that affects 0.32 in 1 000 000 people per year, but has a very poor prognosis compared to other NET cancers, with a 5‐year survival rate of 27–43%.1, 2 The drug everolimus, which targets the mammalian target of rapamycin (mTOR), has been shown to improve the progression‐free survival of patients with advanced PanNET.3 Everolimus was first approved for cancer therapy in 2009 in the USA and 2010 in Japan. mTOR is a critical regulator that activates cell proliferation, growth and anti‐apoptosis pathways. The efficacy of everolimus against PanNET strongly indicates that PanNET cells proliferate in a manner that involves the mTOR cascade.
We have previously shown that Pleckstrin homology‐like domain family A, member 3 (PHLDA3) is a novel p53‐regulated repressor of Akt.4 In addition, we found that loss‐of‐heterozygosity (LOH) as well as hyper‐methylation at the PHLDA3 gene are frequently observed in PanNET specimens.5 In this paper, we will review the significance and molecular modes of action of PHLDA3 and Akt in neuroendocrine tumors.
PI3K‐Akt‐mTOR Cascade
Phosphatidylinositols (PI) are pivotal factors that control the PI3K (phosphoinositide 3‐kinase)‐Akt (also known as protein kinase B [PKB]) pathway.6 PI are phospholipids that contain an inositol ring and are a significant lipid component of the cellular membrane. Combinations of phosphorylated 3′‐, 4′‐ or 5′‐hydroxyl groups on the inositol ring define the different PIP subtypes (phosphatidylinositol phosphates). PI(3,4,5)P3 (phosphatidylinositol‐3,4,5‐trisphosphate) has a particularly important role as a biologically active lipid. PI3K are lipid kinases that catalyze the conversion of PI(4,5)P2 (phosphatidylinositol‐4,5‐bisphosphate) to PI(3,4,5)P3.7, 8, 9 Conversely, PTEN (phosphatase and tensin homolog) is a lipid phosphatase that converts PI(3,4,5)P3 back to PI(4,5)P2.10 Proteins with PH, PX or ENTH domains localize to the inner membrane via binding to PIP.11 Akt possesses one PH domain that specifically binds to PIP with high affinity (Fig. 1a).12
Figure 1.
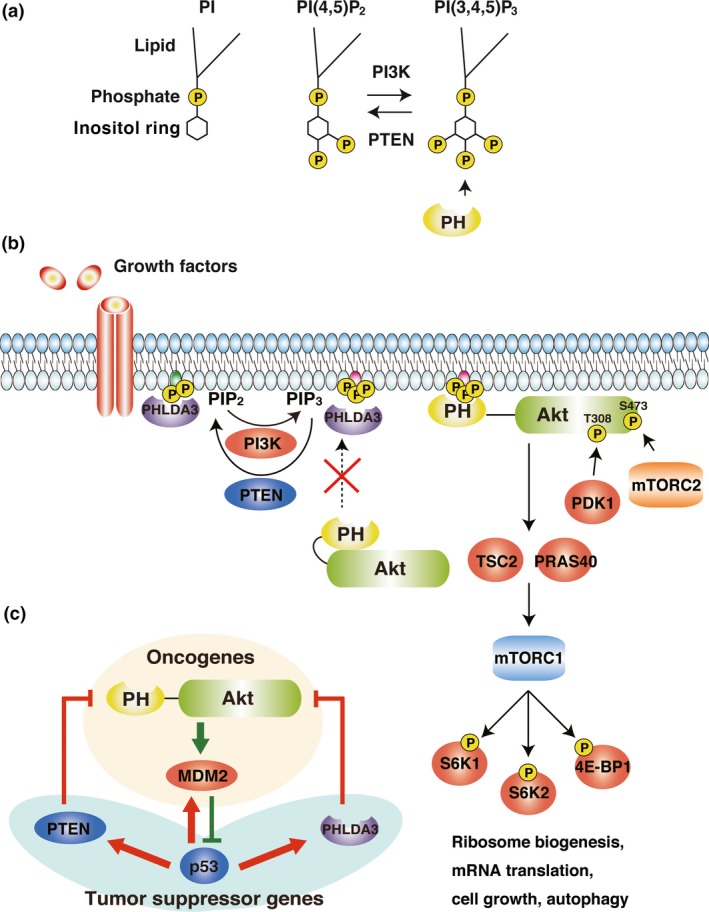
PI3K‐Akt‐mTOR cascades and the p53‐Akt network. (a) Schematic models of PI, PI(4,5)P2 and PI(3,4,5)P3. (b) PI3K‐Akt‐mTOR cascade and the model of competitive Akt suppression by PHLDA3. (c) The Akt‐p53 pathway. Akt and p53 regulate each other. The molecules shown here are either oncoproteins or tumor suppressor proteins.
Akt acts as an oncogene that stimulates cell proliferation.6 In quiescent cells, in the absence of mitogen stimulation, Akt is catalytically inactive, and its activation involves multiple steps. The first step involves the activation of PI3K by one or more signaling events, such as ligand binding to RTK (receptor tyrosine kinases), activation of G‐protein‐coupled receptors or activation of Ras.13 In the second step, activated PI3K selectively converts PI(4,5)P2 to PI(3,4,5)P3.14, 15, 16 Next, Akt binds to PI(3,4,5)P3 via its PH domain and re‐localizes to the plasma membrane. There, two residues of Akt, Thr308 and Ser473, are phosphorylated by PDK1 (3‐phosphoinositide‐dependent kinase 1) and mTORC2 (mTOR complex 2), respectively.17, 18 Once phosphorylated, Akt is active and, in turn, activates multiple proteins related to cell proliferation, cell growth and suppression of apoptosis (Fig. 1b).19
Among the targets activated by Akt are TSC2 and PRAS40, which are responsible for the activation of mTORC1.20, 21 mTORC1 is a rapamycin‐sensitive protein complex that activates S6K1, S6K2 and 4E‐BP1, which regulate ribosome biogenesis, mRNA translation, cell growth and autophagy (Fig. 1b).20, 22, 23, 24 Akt also activates MDM2, a ubiquitin E3 ligase, that leads to degradation of p53 protein via the proteasome system.25, 26 p53 regulates the transcription of PTEN and PHLDA3, both of which are responsible for the suppression of Akt activity.4, 27 Thus, the Akt oncogenic pathway and the p53 tumor suppressive pathway regulate each other to fine‐tune cell proliferation (Fig. 1c).
PHLDA3 Suppresses the Akt Signal Pathway
The transcription factor p53 regulates several genes related to the suppression of cancer progression.28 Various stresses such as DNA damage, hypoxia and oncogene activation can trigger p53 activation. We became interested in PHLDA3 as it was identified in a screen for p53 target genes.29 Murine PHLDA3 was first identified in 1999 (then designated Tih1) as the closest paralog of the imprinted gene Ipl.30 PHLDA3 is comprised of 127 amino acids, and contains one PH domain. We found that p53 localizes to the transcription start site of PHLDA3 and that p53 transcriptionally activates PHLDA3.4 We also found that overexpression of PHLDA3 results in an increase in the apoptotic cell fraction. We further demonstrated the important relationship between the PH domain of Akt and the PHLDA3 protein as follows.
- An in vitro phosphatidylinositol phosphate (PIP) binding assay revealed that PHLDA3 binds to all combinations of PIP (PI(3)P, PI(4)P, PI(5)P, PI(3,4)P2, PI(4,5)P2, PI(3,5)P2 and PI(3,4,5)P3), whereas the PH domain of Akt selectively binds to PI(3,4)P2 and PI(3,4,5)P3 (Fig. 2a).
Figure 2.
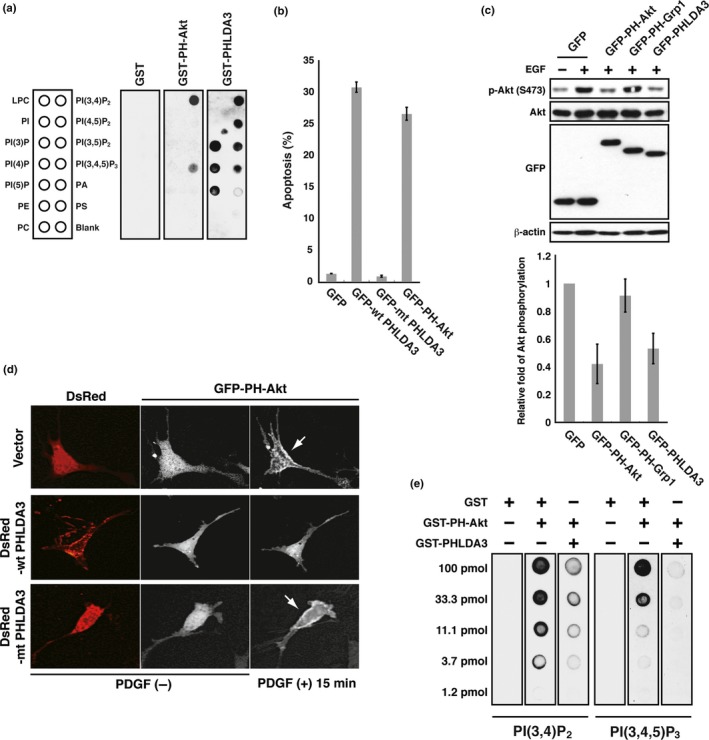 PHLDA3 competes with the PH domain of Akt. (a) Binding of GST‐PHLDA3, GST‐PH‐Akt or GST to immobilized PIP was assessed by protein‐lipid overlay assay. Nitrocellulose membranes spotted with 100 pmol of different phospholipids were used. Bound proteins were detected with anti‐GST antibody. Note that GST alone produced no signal under the conditions employed. LPC, lysophosphatidylcholine; PA, phosphadic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine. (b) 293T cells were transfected with GFP, GFP‐WT PHLDA3, GFP‐mtPHLDA3 (a PHLDA3 mutant with a small deletion within the PH domain), or GFP‐PH‐Akt and analyzed for GFP‐positive cells 48 h post‐transfection. The apoptotic rate, measured by PI‐positive cells (cells stained with PI without fixation), is shown. Mean apoptotic rates ± SD from three experiments are shown. (c) PHLDA3 inhibits Akt activation. COS7 cells were transfected with the indicated fusion proteins for 24 h and subsequently stimulated with EGF for 5 min. Induction of Akt phosphorylation upon EGF treatment was detected in control cells expressing GFP. Akt activity after EGF treatment was analyzed by western blotting, and Akt activity relative to the GFP‐transfected control was calculated. The mean ± SD from three experiments is shown. GFP fusion protein levels were also analyzed by western blotting. (d) Akt translocation to the plasma membrane upon PDGF treatment was analyzed by live‐cell imaging. NIH 3T3 cells were transfected with GFP‐PH‐Akt together with DsRed, DsRed‐WT PHLDA3 or DsRed‐mtPHLDA3. GFP‐PH‐Akt subcellular localization was monitored before and after PDGF treatment (15 min). Note that Akt is localized at the plasma membrane in cells expressing DsRed or DsRed‐mtPHLDA3 (shown by arrows). (e) PHLDA3 inhibits PH‐Akt binding to PIP 2 and PIP 3. Binding of GST‐PH‐Akt to immobilized PIP was assessed by protein‐lipid overlay assay. Nitrocellulose membranes spotted with serially diluted PIP 2 and PIP 3 were incubated with the indicated proteins. While GST did not interfere with Akt binding to PIP, PHLDA3 significantly interfered. Bound Akt was detected with anti‐Akt PH domain antibody.
PHLDA3 competes with the PH domain of Akt. (a) Binding of GST‐PHLDA3, GST‐PH‐Akt or GST to immobilized PIP was assessed by protein‐lipid overlay assay. Nitrocellulose membranes spotted with 100 pmol of different phospholipids were used. Bound proteins were detected with anti‐GST antibody. Note that GST alone produced no signal under the conditions employed. LPC, lysophosphatidylcholine; PA, phosphadic acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine. (b) 293T cells were transfected with GFP, GFP‐WT PHLDA3, GFP‐mtPHLDA3 (a PHLDA3 mutant with a small deletion within the PH domain), or GFP‐PH‐Akt and analyzed for GFP‐positive cells 48 h post‐transfection. The apoptotic rate, measured by PI‐positive cells (cells stained with PI without fixation), is shown. Mean apoptotic rates ± SD from three experiments are shown. (c) PHLDA3 inhibits Akt activation. COS7 cells were transfected with the indicated fusion proteins for 24 h and subsequently stimulated with EGF for 5 min. Induction of Akt phosphorylation upon EGF treatment was detected in control cells expressing GFP. Akt activity after EGF treatment was analyzed by western blotting, and Akt activity relative to the GFP‐transfected control was calculated. The mean ± SD from three experiments is shown. GFP fusion protein levels were also analyzed by western blotting. (d) Akt translocation to the plasma membrane upon PDGF treatment was analyzed by live‐cell imaging. NIH 3T3 cells were transfected with GFP‐PH‐Akt together with DsRed, DsRed‐WT PHLDA3 or DsRed‐mtPHLDA3. GFP‐PH‐Akt subcellular localization was monitored before and after PDGF treatment (15 min). Note that Akt is localized at the plasma membrane in cells expressing DsRed or DsRed‐mtPHLDA3 (shown by arrows). (e) PHLDA3 inhibits PH‐Akt binding to PIP 2 and PIP 3. Binding of GST‐PH‐Akt to immobilized PIP was assessed by protein‐lipid overlay assay. Nitrocellulose membranes spotted with serially diluted PIP 2 and PIP 3 were incubated with the indicated proteins. While GST did not interfere with Akt binding to PIP, PHLDA3 significantly interfered. Bound Akt was detected with anti‐Akt PH domain antibody. We found high rates of apoptosis in cells expressing either GFP‐PHLDA3 or GFP‐PH‐Akt (PH domain of Akt) compared to controls (Fig. 2b). The amount of phosphorylated Akt (S473) was consistently lower in both cells (Fig. 2c).
Overexpression of PHLDA3 results in inhibition of Akt translocation to the plasma membrane (Fig. 2d).
Protein‐lipid overlay assays revealed that PHLDA3 binding to PI(3,4)P2 and PI(3,4,5)P3 inhibits the binding of Akt to these PIP (Fig. 2e).
PHLDA3 appears to function as if it is an isolated PH domain of Akt, and thereby acts as a dominant‐negative form of Akt. Thus, we concluded that PHLDA3 inhibits Akt activity via competitive binding to PIP (Fig. 1b).
Neuroendocrine Tumors, Endocrine Cells and Akt
In many cancer cells, factors related to the PI3K‐Akt‐mTOR cascade are highly activated and contribute to rampant cell proliferation.31, 32 Accordingly, suppression of this pathway may be an effective approach for cancer therapy and molecules targeting this cascade may be candidate anticancer drugs. Indeed, several inhibitors against PI3K‐Akt‐mTOR pathway have been examined for PanNET therapy.33 Everolimus and temsirolimus, which are derivatives of rapamycin, specifically inhibit the kinase activity of mTORC1. Everolimus (RAD001) has been tested on patients with PanNET, metastatic renal cell carcinoma and subependymal giant cell astrocytoma accompanied with tuberous sclerosis complex.3, 34, 35 In RADIANT‐3, a double‐blind phase 3 study, PanNET patients were randomly assigned to treatment with oral everolimus or a matching placebo.3 Progression‐free survival was significantly prolonged in patients receiving everolimus (11.0 months) compared to patients receiving placebo (4.6 months), representing a 65% reduction in the estimated risk of progression or death.3 The efficacy of everolimus against PanNET strongly indicates that PanNET cell proliferation depends on the PI3K‐Akt‐mTOR cascade.
In addition, Jiao et al. performed whole exomic sequencing to analyze genetic alterations in 68 non‐familial PanNET samples. They revealed that 44% of the samples had mutations in MEN1 (encoding menin, a component of a histone methyltransferase), and 43% had mutations in either of the two subunits of the transcription/chromatin remodeling complex DAXX (death‐domain‐associated protein) and ATRX (α thalassemia/mental retardation syndrome X‐linked). They also revealed that 15% of the specimens harbored mutations in genes related to the mTOR pathway, such as PTEN or TSC2.36 This report consistently indicates that aberrant activations in mTOR pathway are related to PanNET tumorigenicity.
Several studies in mouse models have further indicated the importance of the PI3K‐Akt‐mTOR pathway in the regulation of pancreatic islet cell proliferation. Overexpression of active Akt1 in mouse islet β cells resulted in increased cell mass and hyperplasia,37, 38 and a conditional PTEN deletion in islet β cells resulted in increased β cell proliferation, cell size and mass.39, 40 These reports indicate the possibility that the proliferation of pancreatic endocrine cells is dependent on Akt signaling, suggesting that aberrant activation of PI3K‐Akt‐mTOR leads to PanNET oncogenicity.
Neuroendocrine Tumors and Akt‐PHLDA3
PHLDA3 suppresses activity of the Akt oncoprotein, suggesting the possibility that PHLDA3 is a tumor suppressor gene. It is known that activation of the PI3K‐Akt pathway is related to oncogenicity in lung cancer.41 Using several kinds of lung cancer specimens, we investigated copy number alterations in the PTPN7 gene locus, which is adjacent to PHLDA3 (Fig. 3a,b).4 We found frequent chromosome loss at the PTPN7 locus in 12 out of 29 large‐cell neuroendocrine carcinoma (LCNEC), 8 out of 13 carcinoid samples, and 1 out of 8 small cell lung carcinomas (21 out of 50 lung NET in all). However, copy number alteration at this locus was very rare in other kinds of lung cancers (Fig. 3b).4 Consistently, PHLDA3 mRNA abundance was significantly reduced in 10 out of 11 LCNEC (Fig. 3c). Elevated phospho‐Akt staining was detected in 27 out of 32 LCNEC (Fig. 3d). These observations are consistent with our hypothesis that aberrant cell proliferation of lung neuroendocrine tumor/carcinoma depends on Akt hyper‐activation resulting from PHLDA3 inactivation. This indicates that PHLDA3 is a tumor suppressor gene in LCNEC. We also analyzed the epistatic relationship between p53 and PHLDA3. We found that 63% (5/8) of the samples with WT p53 showed LOH at PHLDA3, whereas only 13% (3/24) of the samples with nonfunctional p53 showed LOH at PHLDA3. These results further support the notion that PHLDA3 is an important downstream mediator of p53 in tumor suppression.
Figure 3.
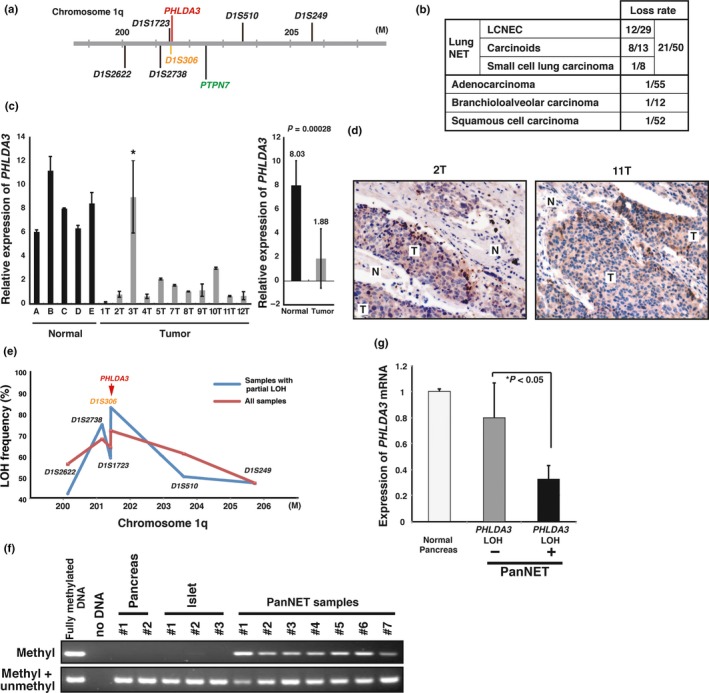
PHLDA3 locus is lost and PHLDA3 expression is downregulated in neuroendocrine tumors (NET). (a) Chromosomal locations of the PHLDA3 gene and microsatellite markers used in the study. D1S306 is located just next to the PHLDA3 gene (32 kb upstream). (b) Chromosome copy number alterations analyzed by MCG cancer array‐800 CGH. (c) Expression of PHLDA3 was analyzed by quantitative RT‐PCR. Total RNA were prepared from normal lung tissues (derived from patients A–E) and LCNEC (derived from patients 1T–12T). In the right column, the mean expression ± SD of PHLDA3 expression in normal lungs and tumor samples is shown. (d) LCNEC tumor sections were subjected to immunohistochemistry to detect activated Akt. Stronger positive brown signals were detected in the tumor regions (T) compared to normal tissue regions (N). (e) Loss‐of‐heterozygosity (LOH) frequency for each microsatellite marker. Frequencies from all samples (shown by red line) and frequencies from samples showing LOH partially within the analyzed region (shown by blue line) are described. (f) Methylation status of the PHLDA3 promoter in normal pancreas, normal isolated islets and PanNET (samples showing LOH at the PHLDA3 locus were analyzed). Genomic DNA from the indicated samples were analyzed by methylation‐specific PCR. (g) PHLDA3 gene expression in PanNET. Total RNA were prepared from normal pancreas and PanNET. RNA was pooled from five normal pancreases for the normal controls. RNA was isolated from PanNET samples with (10 samples) or without LOH (7 samples). Gene expression was quantitated by RT‐PCR and normalized to GAPDH.
Loss‐of‐heterozygosity is frequently found at the 1q31 locus in PanNET specimens derived from insulinomas and gastrinomas, both of which are pancreatic NET.42, 43 Because we found frequent LOH at the PHLDA3 locus in lung NET, we speculated that the PHLDA3 gene, located at 1q31, may also undergo LOH in PanNET. We analyzed 54 PanNET samples to determine the frequency of LOH.5 We found LOH in 36 out of 50 samples (72%) at the PHLDA3 locus (Fig. 3e). Furthermore, we investigated both genetic and epigenetic alterations at the PHLDA3 locus. Although no mutations were observed in the coding region of PHLDA3, DNA methylation was observed at the exon 1 locus (7 out of 7 samples, which had undergone LOH at the PHLDA3 loci; Fig. 3f). We consistently observed strikingly lower abundance of PHLDA3 mRNA in the PHLDA3 LOH‐positive cells (Fig. 3g). These results indicate that concomitant alterations in LOH and DNA methylation (two‐hit inactivation) result in the suppression of PHLDA3 expression, which, in turn, promotes the development of PanNET. Taken together, these data indicate that PHLDA3 is a tumor suppressor gene in neuroendocrine tumors.
In addition to our study showing importance of PHLDA3 in the suppression of NET, Brady et al. have also reported a tumor suppressive function for PHLDA3.44 The authors constructed knock‐in mice expressing p53 with mutations in the first, second or both of the TAD (transactivation domains). They found that MEF cells with p53 mutated in the first TAD lost the ability to arrest cell cycle at G1 and that apoptosis was significantly decreased in thymus and small intestine in response to acute DNA damage. However, HrasV12‐induced cellular senescence was robustly observed in the MEF cells with p53 mutated in the first TAD. Furthermore, Kras‐driven lung tumorigenesis was significantly suppressed in mice with p53 mutated in the first TAD. These results suggest that p53 mutated in the first TAD can still induce important factors that suppress tumorigenesis. The authors identified several genes related to the inhibition of tumorigenesis, including PHLDA3, which was shown to efficiently suppress tumorigenicity. Overexpression of PHLDA3 in HrasV12;p53 null MEF or human non‐small cell lung carcinoma cells inhibited cell cycle progression. Conversely, knockdown of PHLDA3 in E1A‐HRasV12‐transformed MEF resulted in enhanced tumor growth.44 This report reinforces the idea that p53 regulation of PHLDA3 transcription functions to inhibit tumorigenesis.
PHLDA3 Function in Islet Cells
In islet β cells, cell growth and inhibition of apoptosis depend on Akt signaling.40 We examined mouse MIN6 cells (an insulinoma cell line) to analyze the function of PHLDA3 in islet β cells. We confirmed that PHLDA3 protein levels were quite low, suggesting that PHLDA3 function may be lost in the MIN6 cell line (Fig. 4a). We introduced PHLDA3 into this line and analyzed its effect on the Akt pathway. We found that the phosphorylated forms of Akt, p70 S6K and S6 were significantly reduced in the PHLDA3‐overexpressing cells (Fig. 4a). These lines of evidence indicate that the Akt pathway is suppressed by PHLDA3 in islet cells.
Figure 4.
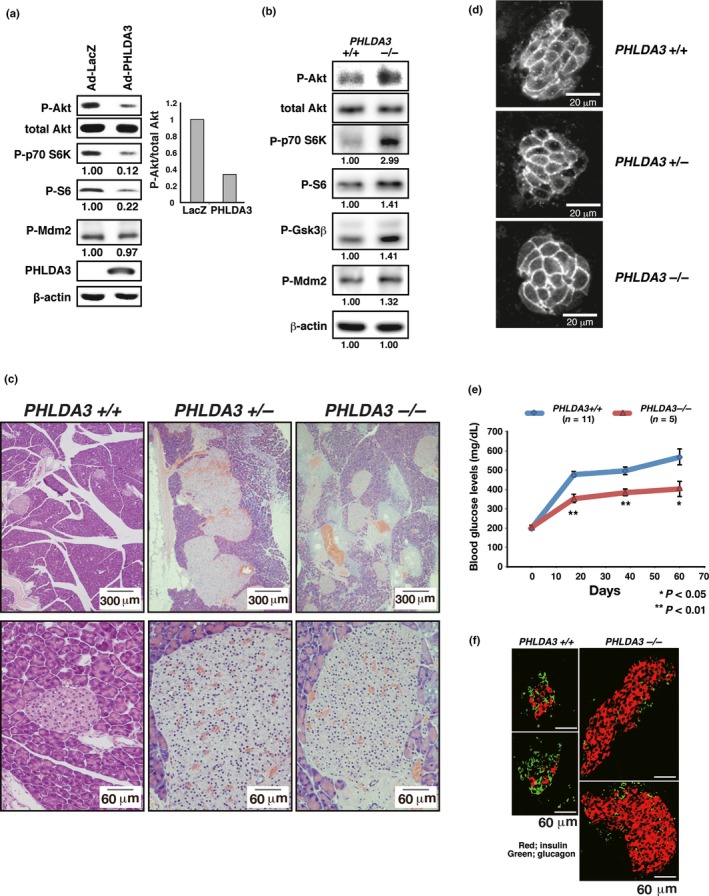
PHLDA3 function in islet cells. (a) Effect of PHLDA3 expression on Akt activity in MIN6 cells. MIN6 cells were transduced with Ad‐LacZ or Ad‐PHLDA3 at a moi of 35, and harvested 30 h post‐infection. Akt activation and phosphorylation of Akt downstream signaling molecules were analyzed by western blotting and quantified by normalization to total Akt levels (P‐Akt) or by β‐actin levels (P‐p70 S6K, P‐S6, P‐Mdm2). (b) Akt activation and phosphorylation of Akt downstream signaling molecules were analyzed by western blotting and quantified by normalization to total Akt levels (P‐Akt, Right) or by β‐actin levels (P‐p70 S6K, P‐S6, P‐GSK3β and P‐Mdm2). (c) HE staining of islets from wild type, heterozygote and PHLDA3‐deficient 10‐month‐old mice. (d) Islet cell size in wild type, heterozygote and PHLDA3‐deficient mice. (e) Blood glucose levels in streptozotocin‐induced diabetic mice. Indicated numbers (n) of PHLDA3 +/+ or PHLDA3 −/− mice were injected i.p. with streptozotocin (STZ) for 5 consecutive days. Blood glucose levels were determined at different time points as indicated after administration of STZ. (f) Distribution of β and α cells in STZ‐treated PHLDA3 +/+ and PHLDA3 −/− mice. Sections were stained with antibody against insulin (β cell marker; red) and glucagon (α cell marker; green) and representative images are shown.
We further analyzed the molecular effects of PHLDA3 on the Akt pathway using PHLDA3 −/− mice. Consistent with our observations in MIN6 cells, the phosphorylated forms of Akt, p70 S6K and S6 were significantly increased in PHLDA3 −/− mice (Fig. 4b). Enhanced Akt activity is known to lead to cell proliferation, enlarged cell size and resistance to apoptosis.37, 40 Compared to wild‐type mice, there was a significant increase in Ki67‐positive cells in PHLDA3 −/− mice, and both PHLDA3 +/− and PHLDA3 −/− mice developed islet hyperplasia (Fig. 4c). We also found that these hyperplastic islets mainly consisted of hypertrophic β cells. Islet cell size was significantly increased in both PHLDA3 −/− and PHLDA3 +/− mice (Fig. 4d). We next analyzed the effect of PHLDA3 knockout on β cell apoptosis induced by STZ (streptozotocin), a chemical that is specifically toxic to β cells. PHLDA3 −/− β cells were found to be more resistant to STZ as judged by the elevation of blood glucose levels compared to wild‐type mice (Fig. 4e). When we compared the areas of β and α cells in STZ‐treated mice, we observed a significant increase in the areas occupied by β cells in the PHLDA3 −/− mice (Fig. 4f), suggesting that PHLDA3‐deficient β cells are relatively resistant to STZ‐induced apoptosis. Collectively, these data indicate that PHLDA3 represses Akt activity, suppresses cell proliferation and facilitates apoptosis in vivo.
Conclusion
In this paper we have reviewed the importance of PHLDA3 and Akt in neuroendocrine tumors. Akt is an oncogene that facilitates cell proliferation and cell growth, and suppresses apoptosis. Whole exomic sequencing has revealed that 15% of PanNET specimens harbor mutations in the PI3K‐Akt‐mTOR pathway, demonstrating the importance of this pathway in PanNET malignancy. We found very frequent LOH of PHLDA3 in 36 out of 50 PanNET samples (72%) and DNA methylation in 7 out of 7 samples (100%, which underwent LOH), features that are not detectable by exomic sequencing. Because PHLDA3 is frequently inactivated in both lung and pancreatic NET, we deduce that PHLDA3 commonly acts as a tumor suppressor gene for various types of NET (Fig. 5). MEN1 has long been known as a tumor suppressor gene responsible for NET,36, 45, 46 and, indeed, we found that the frequency of LOH in MEN1 and PHLDA3 were both quite high (67% and 72%, respectively), suggesting that these two genes are equally important in PanNET development.5 Interestingly, the frequent occurrence of simultaneous LOH in MEN1 and PHLDA3 indicates that these pathways suppress PanNET tumorigenesis independently.
Figure 5.
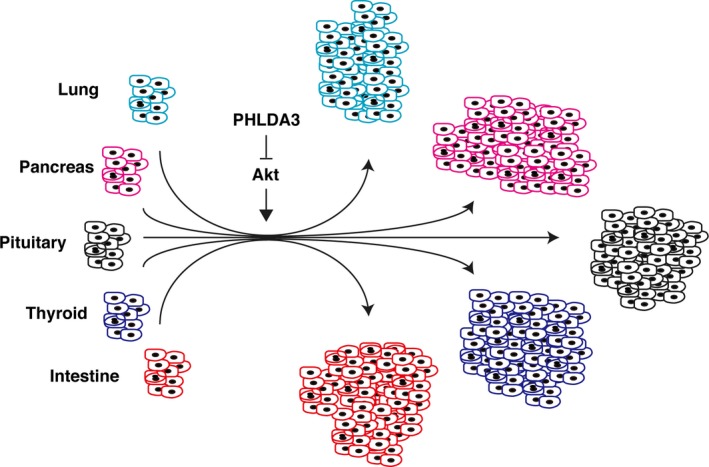
PHLDA3 is a tumor suppressor gene of neuroendocrine tumors (NET). PHLDA3 suppresses proliferation of various types of neuroendocrine cells, such as lung, pancreas, pituitary, thyroid or intestine. Loss of PHLDA3 in these cells results in hyperactivation of Akt and promotes NET development.
We have emphasized the molecular importance of PHLDA3 in tumor suppressive roles for various types of NET. We suggest that examining possible genetic and epigenetic alterations in the PHLDA3 gene contributes to a more accurate diagnosis of PanNET patients. Finally, accelerating the function of PHLDA3 or inhibiting Akt activity could be a promising strategy for the treatment of various types of NET.
Disclosure Statement
The authors have no conflict of interest to declare.
Acknowledgments
We thank Marc Lamphier for critical reading of the manuscript. This study was partly supported by a Grant‐in‐Aid for Scientific Research (C) (#26430133) (R.O.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Japan Agency for Medical Research and Development (AMED) (R.O.), Applied Research for Innovative Treatment of Cancer from the Ministry of Health, Labour and Welfare (R.O.), Project for Development of Innovative Research on Cancer Therapeutics (P‐DIRECT) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (R.O.), Foundation for Promotion of Cancer Research in Japan (R.O.), Takeda Science Foundation (R.O.), Astellas Foundation for Research on Metabolic Disorders (R.O.), Daiichi‐Sankyo Foundation of Life Science (to R.O.), an Extramural Collaborative Research Grant of the Cancer Research Institute, Kanazawa University, Japan (to R.O.), and the Cooperative Research Program of Institute for Frontier Medical Sciences, Kyoto University, Japan (to R.O.).
Cancer Sci 108 (2017) 1101–1108
Funding Information
This study was partly supported by a Grant‐in‐Aid for Scientific Research (C) (#26430133) (R.O.) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, Japan Agency for Medical Research and Development (AMED) (R.O.), Applied Research for Innovative Treatment of Cancer from the Ministry of Health, Labour and Welfare (R.O.), Project for Development of Innovative Research on Cancer Therapeutics (P‐DIRECT) from the Ministry of Education, Culture, Sports, Science and Technology of Japan (R.O.), Foundation for Promotion of Cancer Research in Japan (R.O.), Takeda Science Foundation (R.O.), Astellas Foundation for Research on Metabolic Disorders (R.O.), Daiichi‐Sankyo Foundation of Life Science (to R.O.), an Extramural Collaborative Research Grant of the Cancer Research Institute, Kanazawa University, Japan (to R.O.), and the Cooperative Research Program of Institute for Frontier Medical Sciences, Kyoto University, Japan (to R.O.).
References
- 1. Hauso O, Gustafsson BI, Kidd M et al Neuroendocrine tumor epidemiology: Contrasting Norway and North America. Cancer 2008; 113: 2655–64. [DOI] [PubMed] [Google Scholar]
- 2. Yao JC, Hassan M, Phan A et al One hundred years after “carcinoid”: Epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 2008; 26: 3063–72. [DOI] [PubMed] [Google Scholar]
- 3. Yao JC, Shah MH, Ito T et al Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011; 364: 514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kawase T, Ohki R, Shibata T et al PH domain‐only protein PHLDA3 is a p53‐regulated repressor of Akt. Cell 2009; 136: 535–50. [DOI] [PubMed] [Google Scholar]
- 5. Ohki R, Saito K, Chen Y et al PHLDA3 is a novel tumor suppressor of pancreatic neuroendocrine tumors. Proc Natl Acad Sci USA 2014; 111: E2404–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Coffer PJ, Jin J, Woodgett JR. Protein kinase B (c‐Akt): A multifunctional mediator of phosphatidylinositol 3‐kinase activation. Biochem J 1998; 335(Pt 1): 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burgering BM, Coffer PJ. Protein kinase B (c‐Akt) in phosphatidylinositol‐3‐OH kinase signal transduction. Nature 1995; 376: 599–602. [DOI] [PubMed] [Google Scholar]
- 8. Franke TF, Yang SI, Chan TO et al The protein kinase encoded by the Akt proto‐oncogene is a target of the PDGF‐activated phosphatidylinositol 3‐kinase. Cell 1995; 81: 727–36. [DOI] [PubMed] [Google Scholar]
- 9. Kohn AD, Kovacina KS, Roth RA. Insulin stimulates the kinase activity of RAC‐PK, a pleckstrin homology domain containing ser/thr kinase. EMBO J 1995; 14: 4288–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5‐trisphosphate. J Biol Chem 1998; 273: 13375–8. [DOI] [PubMed] [Google Scholar]
- 11. Stahelin RV. Lipid binding domains: More than simple lipid effectors. J Lipid Res 2009; 50(Suppl): S299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Takeuchi H, Kanematsu T, Misumi Y et al Distinct specificity in the binding of inositol phosphates by pleckstrin homology domains of pleckstrin, RAC‐protein kinase, diacylglycerol kinase and a new 130 kDa protein. Biochim Biophys Acta 1997; 1359: 275–85. [DOI] [PubMed] [Google Scholar]
- 13. Capdevila J, Tabernero J. A shining light in the darkness for the treatment of pancreatic neuroendocrine tumors. Cancer Discov 2011; 1: 213–21. [DOI] [PubMed] [Google Scholar]
- 14. Cantley LC. The phosphoinositide 3‐kinase pathway. Science 2002; 296: 1655–7. [DOI] [PubMed] [Google Scholar]
- 15. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: Variations on a theme. Oncogene 2008; 27: 5497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3‐kinases as regulators of growth and metabolism. Nat Rev Genet 2006; 7: 606–19. [DOI] [PubMed] [Google Scholar]
- 17. Mora A, Komander D, van Aalten DM, Alessi DR. PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 2004; 15: 161–70. [DOI] [PubMed] [Google Scholar]
- 18. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor‐mTOR complex. Science 2005; 307: 1098–101. [DOI] [PubMed] [Google Scholar]
- 19. Manning BD, Cantley LC. AKT/PKB signaling: Navigating downstream. Cell 2007; 129: 1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans 2009; 37: 217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Memmott RM, Dennis PA. Akt‐dependent and ‐independent mechanisms of mTOR regulation in cancer. Cell Signal 2009; 21: 656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goberdhan DC, Boyd CA. mTOR: Dissecting regulation and mechanism of action to understand human disease. Biochem Soc Trans 2009; 37: 213–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Porstmann T, Santos CR, Lewis C, Griffiths B, Schulze A. A new player in the orchestra of cell growth: SREBP activity is regulated by mTORC1 and contributes to the regulation of cell and organ size. Biochem Soc Trans 2009; 37: 278–83. [DOI] [PubMed] [Google Scholar]
- 24. Dunlop EA, Tee AR. Mammalian target of rapamycin complex 1: Signalling inputs, substrates and feedback mechanisms. Cell Signal 2009; 21: 827–35. [DOI] [PubMed] [Google Scholar]
- 25. Mayo LD, Donner DB. A phosphatidylinositol 3‐kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA 2001; 98: 11598–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M. Cross‐talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene 2002; 21: 1299–303. [DOI] [PubMed] [Google Scholar]
- 27. Stambolic V, MacPherson D, Sas D et al Regulation of PTEN transcription by p53. Mol Cell 2001; 8: 317–25. [DOI] [PubMed] [Google Scholar]
- 28. Vousden KH, Lu X. Live or let die: The cell's response to p53. Nat Rev Cancer 2002; 2: 594–604. [DOI] [PubMed] [Google Scholar]
- 29. Ohki R, Kawase T, Ohta T, Ichikawa H, Taya Y. Dissecting functional roles of p53 N‐terminal transactivation domains by microarray expression analysis. Cancer Sci 2007; 98: 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Frank D, Mendelsohn CL, Ciccone E, Svensson K, Ohlsson R, Tycko B. A novel pleckstrin homology‐related gene family defined by Ipl/Tssc3, TDAG51, and Tih1: Tissue‐specific expression, chromosomal location, and parental imprinting. Mamm Genome 1999; 10: 1150–9. [DOI] [PubMed] [Google Scholar]
- 31. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene 2005; 24: 7455–64. [DOI] [PubMed] [Google Scholar]
- 32. Luo J, Manning BD, Cantley LC. Targeting the PI3K‐Akt pathway in human cancer: Rationale and promise. Cancer Cell 2003; 4: 257–62. [DOI] [PubMed] [Google Scholar]
- 33. Wolin EM. PI3K/Akt/mTOR pathway inhibitors in the therapy of pancreatic neuroendocrine tumors. Cancer Lett 2013; 335: 1–8. [DOI] [PubMed] [Google Scholar]
- 34. Franz DN, Belousova E, Sparagana S et al Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST‐1): A multicentre, randomised, placebo‐controlled phase 3 trial. Lancet 2013; 381: 125–32. [DOI] [PubMed] [Google Scholar]
- 35. Motzer RJ, Escudier B, Oudard S et al Efficacy of everolimus in advanced renal cell carcinoma: A double‐blind, randomised, placebo‐controlled phase III trial. Lancet 2008; 372: 449–56. [DOI] [PubMed] [Google Scholar]
- 36. Jiao Y, Shi C, Edil BH et al DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011; 331: 1199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tuttle RL, Gill NS, Pugh W et al Regulation of pancreatic beta‐cell growth and survival by the serine/threonine protein kinase Akt1/PKBalpha. Nat Med 2001; 7: 1133–7. [DOI] [PubMed] [Google Scholar]
- 38. Bernal‐Mizrachi E, Wen W, Stahlhut S, Welling CM, Permutt MA. Islet beta cell expression of constitutively active Akt1/PKB alpha induces striking hypertrophy, hyperplasia, and hyperinsulinemia. J Clin Invest 2001; 108: 1631–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stiles BL, Kuralwalla‐Martinez C, Guo W et al Selective deletion of Pten in pancreatic beta cells leads to increased islet mass and resistance to STZ‐induced diabetes. Mol Cell Biol 2006; 26: 2772–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Elghazi L, Bernal‐Mizrachi E. Akt and PTEN: Beta‐cell mass and pancreas plasticity. Trends Endocrinol Metab 2009; 20: 243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Engelman JA, Cantley LC. The role of the ErbB family members in non‐small cell lung cancers sensitive to epidermal growth factor receptor kinase inhibitors. Clin Cancer Res 2006; 12: 4372–6s. [DOI] [PubMed] [Google Scholar]
- 42. Yang YM, Liu TH, Chen YJ et al Chromosome 1q loss of heterozygosity frequently occurs in sporadic insulinomas and is associated with tumor malignancy. Int J Cancer 2005; 117: 234–40. [DOI] [PubMed] [Google Scholar]
- 43. Chen YJ, Vortmeyer A, Zhuang Z, Huang S, Jensen RT. Loss of heterozygosity of chromosome 1q in gastrinomas: Occurrence and prognostic significance. Cancer Res 2003; 63: 817–23. [PubMed] [Google Scholar]
- 44. Brady CA, Jiang D, Mello SS et al Distinct p53 transcriptional programs dictate acute DNA‐damage responses and tumor suppression. Cell 2011; 145: 571–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Norton JA, Krampitz G, Jensen RT. Multiple endocrine neoplasia: Genetics and clinical management. Surg Oncol Clin N Am 2015; 24: 795–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Corbo V, Dalai I, Scardoni M et al MEN1 in pancreatic endocrine tumors: Analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr Relat Cancer 2010; 17: 771–83. [DOI] [PubMed] [Google Scholar]