

Abstract
B‐cell non‐Hodgkin lymphoma (B‐NHL) is the most frequent hematological malignancy. Although refined chemotherapy regimens and several new therapeutics including rituximab, a chimeric anti‐CD20 monoclonal antibody, have improved its prognosis in recent decades, there are still a substantial number of patients with chemorefractory B‐NHL. Anti‐CD19 chimeric antigen receptor (CAR) T‐cell therapy is expected to be an effective adoptive cell treatment and has the potential to overcome the chemorefractoriness of B‐cell leukemia and lymphoma. Recently, several clinical trials have shown remarkable efficacy of anti‐CD19 CAR T‐cell therapy, not only in B‐acute lymphoblastic leukemia but also in B‐NHL. Nonetheless, there are several challenges to overcome before introduction into clinical practice, such as: (i) further refinement of the manufacturing process, (ii) further improvement of efficacy, (iii) finding the optimal infusion cell dose, (iv) optimization of lymphocyte‐depleting chemotherapy, (v) identification of the best CAR structure, and (vi) optimization of toxicity management including cytokine release syndrome, neurologic toxicity, and on‐target off‐tumor toxicity. Several ways to solve these problems are currently under study. In this review, we describe the updated clinical data regarding anti‐CD19 CAR T‐cell therapy, with a focus on B‐NHL, and discuss the clinical implications and perspectives of CAR T‐cell therapy.
Keywords: Adoptive cell therapy, B‐cell non‐Hodgkin lymphoma, CAR T, CD19, chimeric antigen receptor
B‐cell non‐Hodgkin lymphoma (B‐NHL) is the most frequent hematological malignancy. Although refined chemotherapy regimens and several new therapeutic agents including rituximab, a chimeric anti‐CD20 monoclonal antibody, improved its prognosis in the recent decades, there are still a substantial number of patients with chemorefractory B‐NHLs.
Anti‐CD19 chimeric antigen receptor (CAR) T‐cell (CD19‐CAR‐T) therapy is an effective adoptive cell treatment and has the potential to overcome the chemorefractoriness of B‐cell leukemia and lymphoma. Several studies have shown its remarkable efficacy in patients with B‐cell acute lymphoblastic leukemia (B‐ALL),1, 2, 3 and it is designated as a “breakthrough therapy” by the US FDA. Furthermore, recent clinical trials of CD19‐CAR‐T therapy have revealed high efficacy in relapsed/refractory B‐NHL.4
In this review, first, we describe the basic mechanism and overview of CD19‐CAR‐T therapy. Then, we summarize the current clinical developments, clinical implications, and perspectives of CD19‐CAR‐T therapy, focusing on B‐NHL.
Structure of the Anti‐CD19 CAR
CD19 is a B‐cell‐receptor‐associated protein expressed on the B‐cell surface. It is thought to be an optimal therapeutic target because (i) it is uniformly expressed on malignant B cells, and (ii) it is expressed in the B‐cell lineage, not in other lineages or other tissues. There are a multitude of CAR‐T therapies that are tested in clinical trials, with the majority of them utilizing CD19 as a therapeutic target.
Basically, the anti‐CD19 CAR is a recombinant molecule consisting of three parts: (i) a single‐chain variable domain (scFv) derived from an anti‐CD19 monoclonal antibody, (ii) a transmembrane domain, and (iii) the signal transduction domain of T‐cell receptor (TCR) (CD3ζ; Fig. 1a).5, 6, 7 When a CAR‐T recognizes a specific antigen, the cell is activated via the intracellular signal transduction domain and exerts target cell toxicity. Nonetheless, first‐generation CAR‐T showed limited expansion and antitumor efficacy because the CAR‐T expansion was solely dependent on interleukin (IL)‐2 production.8 In contrast, physiological in vivo T‐cell activation is caused by interaction between antigen‐presenting cells via T‐cell receptor and several costimulatory receptors such as CD28 and 4‐1BB (Fig. 2). To improve CAR‐T‐cell expansion capacity and antitumor activity, the second‐generation CAR that contains a costimulatory domain, such as CD289 or 4‐1BB,10, 11 has been studied (Fig. 1b). Because it involves an additional costimulatory domain, second‐generation CAR‐T therapy shows better in vivo expansion. The most recent clinical trials of CAR‐T therapy have used second‐generation CAR‐T. Each academic institution or industry has developed slightly different second‐generation CAR structure, and the details are summarized in Table 1.
Figure 1.
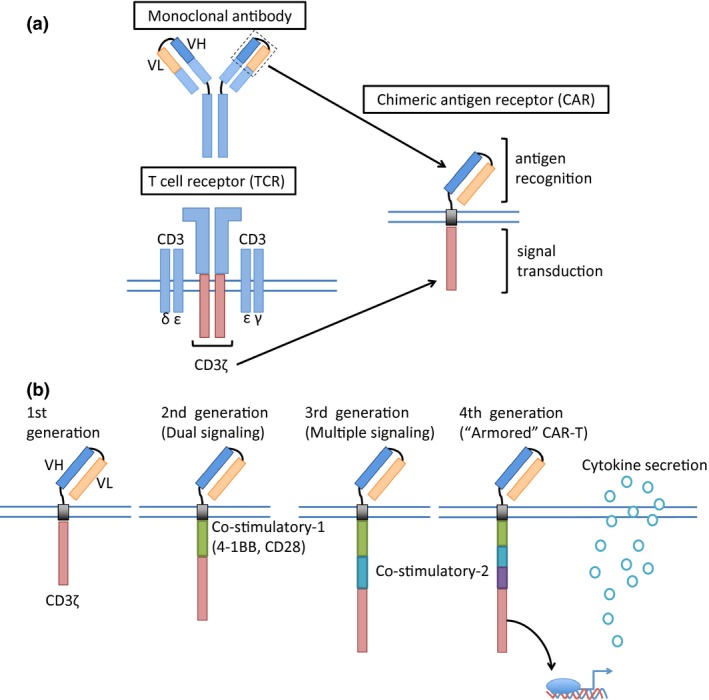
Schematic structure of a chimeric antigen receptor. Chimeric antigen receptor (CAR) consists of a single‐chain variable domain derived from a monoclonal antibody, a transmembrane domain, and a signal transduction domain of T‐cell receptor (CD3ζ) (a). To improve the CAR T‐cell expansion capacity, CAR structure was refined gradually (b). VH, heavy chain variable region; VL, light chain variable region.
Figure 2.
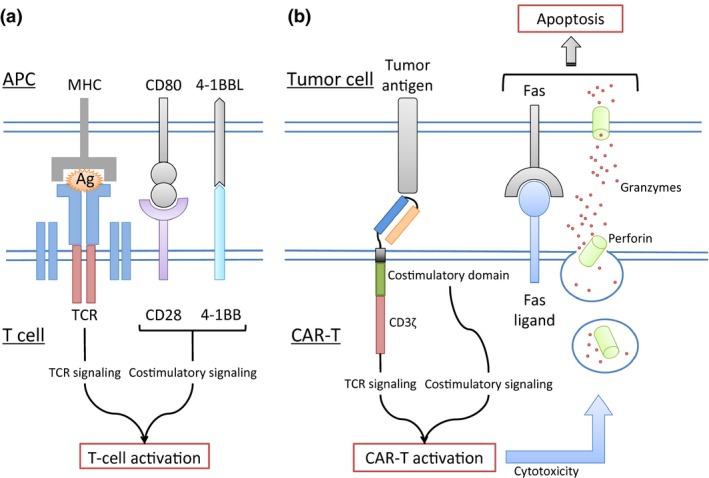
Cytotoxicity of CAR‐T against tumor cells. Normal T cells interact with antigen‐presenting cells (APCs) such as dendritic cells to be activated via the T‐cell receptor (TCR) and other costimulatory domains (a). TCR‐mediated antigen recognition depends on the peptides displayed on the major histocompatibility complex (MHC) molecules. Nevertheless, a CAR‐T can recognize target antigens via the antigen‐recognition domain and is not dependent on MHC (b). When a CAR‐T recognizes a specific antigen, the cell is activated via the intracellular signal transduction domain and exerts target cell toxicity. Ag, antigen; CAR‐T, chimeric antigen receptor T cell.
Table 1.
Structure of selected anti‐CD19 CARs
| Type of CAR‐T cell | CTL019 | KTE‐C19 | JCAR014 | JCAR017 | JCAR015 | Product of BCM | Product of MDACC |
|---|---|---|---|---|---|---|---|
| Academic institute | UPenn | NCI | FHCRC | FHCRC/SCRI | MSKCC | BCM | MDACC |
| Collaborating Company | Novartis | Kite | Juno | Juno | Juno | Celgene/Bluebird | Ziopharma |
| Binding domain | FMC63 (murine) | FMC63 (murine) | FMC63 (murine) | FMC63 (murine) | SJ25C1 (murine) | FMC63 (murine) | FMC63 (murine) |
| Hinge | CD8 | CD28 | IgG4 | IgG4 | CD28 | IgG1 | IgG4 |
| Transmembrane | CD8 | CD28 | CD28 | IgG4 | CD28 | CD4 | CD28 |
| Costimulatory | 4‐1BB | CD28 | 4‐1BB | 4‐1BB | CD28 | CD28 | CD28 |
| Production‐starting cell population | PBMC | PBMC | CD4+/CD8+ CM | CD4+/CD8+ | PBMC | PBMC | PBMC |
| Vector | Lentivirus | Retrovirus | Lentivirus | Lentivirus | Retrovirus | Retrovirus | Transposon |
BCM, Baylor College of Medicine; CAR, chimeric antigen receptor; CM, central memory T cell; FHCRC, Fred Hutchinson Cancer Research Center; MDACC, MD Anderson Cancer Center; MSKCC, Memorial Sloan Kettering Cancer Center; NCI, National Cancer Institute; PBMC, peripheral blood mononuclear cell; UPenn, University of Pennsylvania; SCRI, Seattle Children's Research Institute.
To further augment the antitumor activity, the third‐generation CAR that is equipped with multiple costimulatory domains,12 and the fourth‐generation CAR that contains a transduction domain to promote production of a T‐cell‐activating cytokine such as IL‐12 (so‐called “armored” CAR‐T) are currently being researched.13, 14
An Outline of CAR‐T Therapy
This outline is shown in Figure 3. First, leukocyte apheresis using a blood cell separator is performed, and the patient's autologous mononuclear cells are collected from peripheral blood. The apheresis product is transferred to a cell‐processing center, and selected T cells are activated in a proliferative environment with IL‐2 or anti‐CD3 antibodies. CAR genes are transfected into T cells using retroviral or lentiviral vectors, and then this cell clone is expanded. The newly created CAR‐T product is transferred back to the hospital and is infused into the patient. This manufacturing process takes at least 2–3 weeks in general.15 Prior to the CAR‐T infusion, lymphodepletion‐chemotherapy is administered to the patient. Lymphodepletion‐chemotherapy decreases the numbers of T cells in vivo, including regulatory T cells, and consequently upregulates cytokines such as IL‐7 and IL‐15.16 These cytokines promote T‐cell expansion including CAR‐T and promote the antitumor activity.
Figure 3.
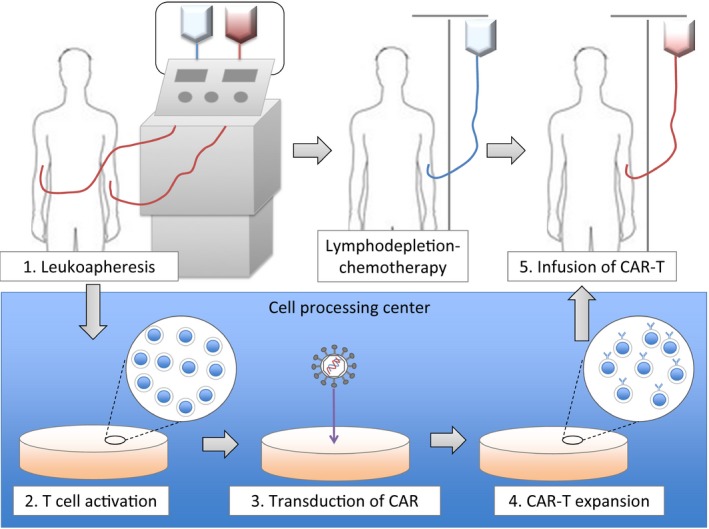
The outline of CAR T‐cell therapy. CAR, chimeric antigen receptor.
Adverse Effects of CD19‐CAR‐T Therapy
(i) Cytokine‐release syndrome
The most prevalent severe adverse effect after CAR‐T infusion is cytokine‐release syndrome (CRS), which occurs several hours to 14 days following the infusion.17 Although there is no clear definition of CRS, it is used as a general term for adverse events related to immune activation. When CAR‐T recognizes a specific antigen on the tumor cell surface, CAR‐T is activated and secretes proinflammatory cytokines such as interferon (IFN)‐γ, IL‐6, and IL‐10. These cytokines promote CAR‐T expansion and subsequent antitumor activity. Nonetheless, too much cytokines leads to severe CRS, meaning that the immune response of CAR‐T therapy is a double‐edge sword. In fact, the trials of the first‐generation CAR‐T therapy showed insufficient antitumor activity without any CRS.5, 6, 7 On the other hand, several subsequent clinical trials showed CRS after infusion of second‐generation CAR‐T. There is no correlation between clinical efficacy and severity of CRS. Nevertheless, the majority of patients who respond to CAR‐T therapy exhibit at least mild CRS.18
Symptoms and signs of CRS include high‐grade fever, fatigue, nausea, anorexia, tachycardia, hypotension, and capillary leak. Clinical grading of CRS caused by CAR‐T therapy has been drawn up based on a grading system of CRS for antibody therapy that is contained in the Common Terminology Criteria for Adverse Events ver.4 (Fig. 4). This grading system is currently widely used in the clinical trials.17 Real‐time quantitative monitoring of serum cytokines may help to assess the severity of CRS precisely. It is, however, not a realistic approach in clinical practice, considering the cost and technical difficulties. Quantification of serum C‐reactive protein (CRP), which is produced by hepatocytes in response to IL‐6, may be useful for estimation of in vivo IL‐6 concentration and severity of CRS.1
Figure 4.
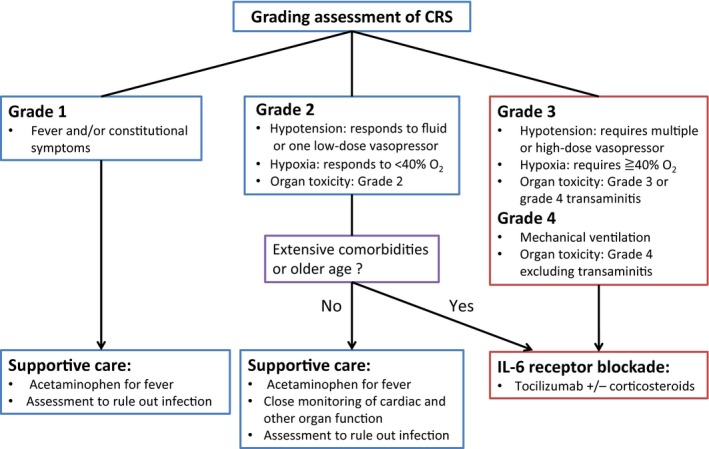
The grading system and treatment algorithm for CRS after CAR T‐cell infusion. CAR, chimeric antigen receptor; CRS, cytokine release syndrome.
Although the management of CRS contains several arguable points, the treatment algorithm based on the modified CRS grading assessment is generally recommended nowadays (Fig. 4). In patients with grade 1–2 CRS, conventional supportive care such as acetaminophen and fluid resuscitation is recommended. It is thought that corticosteroids are possibly effective but should be avoided for low‐grade CRS because they may interfere with in vivo CAR‐T expansion and thereby limit the clinical efficacy. Grade 3–4 CRS is life‐threatening, and prompt optimal management is required. Tocilizumab, a monoclonal antibody for blockade of IL‐6 receptor, causes immediate reversal of severe CRS.19, 20, 21 Considering its remarkable efficacy, tocilizumab is generally accepted as a first‐line treatment of severe CRS.17, 19 Nonetheless, tocilizumab use for low‐grade CRS and prophylactic use are discouraged mainly because of insufficient data.
(ii) Neurologic toxicity
Several clinical trials have shown neurologic toxicity including delirium, aphasia, and transient high‐order brain functional disorders after CAR‐T therapy. Some patients have signs consistent with leukoencephalopathy on imaging although these changes are reversible in most cases. The pathogenesis of neurologic toxicity remains unclear, but similar events are reported among patients who receive high‐dose IL‐2 treatment22 or blinatumomab, a bispecific T‐cell engager,23 suggesting that these adverse effects may be caused by some sort of immunological mechanism.
(iii) B‐cell aplasia
Because CD19 is expressed on normal B cells as well, normal B‐lineage cells are also eliminated after CD19‐CAR‐T infusion. This phenomenon is typically called an “on‐target off‐tumor effect.” Subsequent B‐cell aplasia results in long‐lasting hypogammaglobulinemia, and intermittent immunoglobulin replacement is occasionally required to prevent severe infections.24, 25
Clinical Trials of CD19‐CAR‐T Therapy Against B‐NHLs (Including B‐cell Chronic Lymphocytic Leukemia; B‐CLL)
Several research groups in the US have conducted clinical trials of CAR‐T therapy in concert with pharmaceutical companies. Recently reported clinical trials of CD19‐CAR‐T therapy against B‐NHL are listed in Table 2.
Table 2.
Selected clinical trials of anti‐CD19 CAR T‐cell therapy against B‐NHL
| Studies/Authors | Company/Sponsor | Institute | Type of CAR‐T cell | Disease (No. patients) | Lymphodepletion‐chemo (No. patients) | Infused CAR‐T cell dose | Response (%) | CRS (%) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| ORR | %CR | All Gr | Gr 3/4 | |||||||
|
Kalos et al.27
Porter et al.28 |
Novartis | UPenn | CTL019 | B‐CLL (14) |
Pentostatin/CY (5) FLU/CY (3)/Bend (6) |
0.14–11 × 108 | 58 | 29 | 64 | 43 |
| Schuster et al.29, 30, 31 | Novartis | UPenn | CTL019 |
DLBCL (15) FL (13) MCL (2) |
Various regimens† | 1.79–5.0 × 108 |
47 73 NA |
20 36.5 NA |
53 | 7 |
| Kochenderfer et al.24 | – | NCI | KTE‐C19 | FL (3)/SMZL (1)/B‐CLL (4) | FLU/CY | 3.0–30 × 106/kg | 85 | 14 | NA | NA |
| Kochenderfer et al.32 | – | NCI | KTE‐C19 |
PMBL (4) + DLBCL (5) LG‐NHL (2) + B‐CLL (4) |
FLU/CY | 1.0–5.0 × 106/kg |
85 100 |
57 67 |
NA | 27 |
| ZUMA‐133 | Kite | Multi‐center | KTE‐C19 | DLBCL (51) | FLU/CY | 1.0–2.0 × 106/kg | 76 | 47 | NA | 20 |
| Turtle et al.37 | Juno | FHCRC | JCAR014 | B‐NHL (32) [DLBCL (21‡)/BL (1)/LG‐NHL (6)/MCL (4)] |
CY +/− etoposide (12) FLU/CY (20) |
2 × 105–7/kg |
50 72 |
8 50 |
63 | 13 |
| Turtle et al.38 | Juno | Multi‐center | JCAR017 | B‐NHL (26) | FLU/CY | 2 × 106/kg | 73 | 46 | NA | 12 |
†Details of LD‐chemo (number of patients in parentheses): CY (16), bendamustine (9), EPOCH (3), GEM (1), FLU/CY (1). ‡Details of the histological subtype of DLBCL: de novo DLBCL (7), T‐cell rich DLBCL (1), PMBL (2), histological transformation from LG‐NHL (11). B‐CLL, B‐cell chronic lymphocytic leukemia; B‐NHL, B‐cell non‐Hodgkin lymphoma; CAR, chimeric antigen receptor; CR, complete remission; CRS, cytokine‐release syndrome; CY, cyclophosphamide; DLBCL, diffuse large B‐cell lymphoma; EPOCH, etoposide, prednisone, vincristine, cyclophosphamide and doxorubicin; FHCRC, Fred Hutchinson Cancer Research Center; FL, follicular lymphoma; FLU, fludarabine; GEM, gemcitabine; LG‐NHL, low‐grade non‐Hodgkin lymphoma; NA, not applicable; NCI, National Cancer Institute; ORR, overall response rate; PMBL, primary mediastinal large B‐cell lymphoma; PR, partial response; SMZL, splenic marginal zone lymphoma; UPenn, the University of Pennsylvania.
(i) Reports on CTL019
Investigators at the University of Pennsylvania (UPenn) collaborate with Novartis and have developed second‐generation CD19‐CAR‐T named CTL019 involving a CAR consisting of a murine anti‐CD19 scFv, a CD8 hinge and a transmembrane domain, 4‐1BB (costimulatory molecule), and CD3ζ. They reported the first patient with refractory B‐CLL who achieved complete remission (CR) after infusion of CTL019.26 Subsequently, a pilot study to assess the feasibility of CTL019 in patients with relapsed/refractory B‐CLL was conducted.27, 28 Among 14 patients who received the infusion, four patients (29%) achieved CR, four patients (29%) showed a partial response (PR), and the overall response rate (ORR) was 58% (8/14). One patient who achieved PR relapsed, and lymphadenopathy developed rapidly 9 months after the infusion. A lymph node biopsy revealed Richter's transformation, and the tumor cells were negative for CD19. Furthermore, CTL019 cells were eliminated both from peripheral blood and from bone marrow at the time of the relapse. In vivo expansion magnitude of CTL019 was assessed using a quantitative polymerase chain reaction assay. In four patients who achieved CR, there was a higher peak‐expansion level (median 73 237 copies/μg, range 25 070–409 645) with persistent CTL019 proliferation (range 14–19 months). On the other hand, patients without objective therapeutic responses showed significantly lower expansion magnitude (median 420 copies/μg, range 6.5–13 876; P = 0.013). These data are suggestive of a correlation between the therapeutic response and peak expansion level of CAR‐T. CRS was observed in nine patients (64%) and presented 1–14 days after the infusion (median 7 days). Six patients experienced grade 3–4 CRS, and four of them required supportive care in an intensive care unit.
Recently, interim results of a phase IIa trial for relapsed/refractory B‐NHL (excluding B‐CLL) were presented at the 57th annual meeting of the American Society of Hematology (ASH‐2015).29 Forty‐three patients with relapsed/refractory B‐NHL, including diffuse large B‐cell lymphoma (DLBCL; n = 26), follicular lymphoma (FL; n = 14), and mantle cell lymphoma (MCL; n = 3), were enrolled. Only 30 of 43 patients received a CAR T‐cell infusion. Thirteen patients did not receive the infusion because of disease progression (n = 4), production failure (n = 6), or withdrawal of consent (n = 3). In the 28 evaluable patients (DLBCL, n = 15; FL, n = 12; MCL, n = 1), the ORR of each histological subtype at 3 months after infusion was 47% in DLBCL (CR, n = 3; PR, n = 4), and 73% in FL (CR, n = 4; PR, n = 4). Of note, six patients who showed PR during the response assessment at 3 months achieved CR 6 months after the infusion. These findings suggest that the best response to CTL019 therapy is observed later than with conventional chemotherapy. Longer follow‐up data were presented at the ASH‐2016, and the patients who achieved CR showed durable responses.30, 31 CRS was observed in 16 patients (53%), and most of them were of grade 2 (grade 2, n = 14; grade 3, n = 1; grade 4, n = 1). The prevalence of severe CRS is relatively lower as compared to patients with B‐ALL receiving CD19‐CAR‐T therapy.1, 2, 3
Although there remain several unsolved problems, and most clinical data are preliminary, these results point to high efficacy of CTL019 against relapsed/refractory B‐NHL with manageable adverse effects. At present, a global phase II trial (including Japan) of CTL019 therapy for relapsed/refractory DLBCL is in progress (JULIET; NCT02445248).
(ii) Reports on KTE‐C19
Investigators of the NCI in the US developed CD19‐CAR‐T where CAR contains an anti‐CD19 scFv, CD28 (hinge, transmembrane, and costimulatory domains), and CD3ζ. This construct is now developed by Kite Pharma and is named KTE‐C19.
The NCI group reported a patient with refractory FL who achieved durable PR after CD19‐CAR‐T therapy.24 Subsequently, the investigators conducted a phase I trial of CD19‐CAR‐T in patients with relapsed/refractory B‐NHL.32 Among the seven evaluable patients with aggressive B‐NHL, four patients (57%) achieved CR, and two (28%) showed PR.
Currently, Kite Pharma is conducting several multicenter clinical trials of this construct, KTE‐C19, against B‐cell leukemia and lymphoma. The ZUMA‐1 trial (NCT02348216) is a multicenter phase I/II study of KTE‐C19 in patients with relapsed/refractory DLBCL conducted by Kite Pharma. In the phase I portion, seven patients with refractory DLBCL received KTE‐C19 at a target dose of 2 × 106 cells/kg subsequent to the lymphodepletion‐chemotherapy.33 Five of seven patients (71%) achieved an objective response within a month after the infusion, with four of seven (57%) achieving CRs. Six of seven patients experienced CRS; 71% (5/7) experienced grade 1–2 CRS, and 14% (1/7) experienced grade 4 CRS, which is a dose‐limiting toxicity (DLT). All evaluable patients experienced at least one neurologic toxicity, with 43% (3/7) having maximum grade 3, and 14% (1/7) having a maximum grade 4 (occurring in the same patient with a DLT). Except for the one patient with a DLT, CRS and neurologic toxicity were self‐limiting and reversible (median duration was 7–8 days). Based on these results, the subsequent pivotal phase II portion was conducted.
The early results of a phase II portion of ZUMA‐1 were presented in the late‐breaking abstract session of ASH‐2016.34 In total, 101 patients received KTE‐C19, and 51 were eligible for analysis at the time. KTE‐C19 was successfully manufactured for 99% of the patients enrolled. Average turnaround time from apheresis to receipt of KTE‐C19 at the clinical site was 17.4 days. This is a relatively short period as compared to other trials because the NCI group has developed a new rapid cell expansion procedure for KTE‐C19, making it possible to implement a 6‐ to 8‐day process of manufacturing KTE‐C19.35
The ORR was 76% (47% CRs and 29% PRs) and was significantly higher as compared to the historical control36: a primary endpoint of this study. The estimated progression‐free survival at 1 and 3 months was 92% and 56%, respectively. Grade 3 or higher CRS and neurologic toxicity developed in 20% and 29% of patients, respectively. KTE‐C19 expanded within 14 days after infusion, and the peak expansion level was associated with responses at 3 months after infusion (P = 0.008). Severe neurologic toxicity was associated with increased serum concentrations of IL‐15, IL‐6, IL‐10, and IFN‐γ‐inducible protein 10 (IP‐10).
(iii) Reports on JCAR014 and JCAR017
Investigators at Fred Hutchinson Cancer Research Center (FHCRC), the Memorial Sloan Kettering Cancer Center (MSKCC), and Seattle Children's Research Institute founded a venture, Juno Therapeutics, and conducted several clinical trials of CD19‐CAR‐T products: JCAR014, JCAR015, JCAR017, JCAR021, and others. Among them, the results on JCAR014 and JCAR017 in B‐NHL have been published.
JCAR014 involves a CAR consisting of a murine anti‐CD19 scFv, an IgG4 hinge, the CD28 transmembrane domain, 4‐1BB costimulatory domain, and CD3ζ. JCAR014 is also transfected with truncated epidermal growth factor receptor, which is used for detection, selection, or eradication of CAR‐T. It is produced from separate subsets of T cells (CD4+ and CD8+ central memory [CM] T cells) to ensure a defined ratio of CD4+/CD8+ CM‐CAR‐T at 1:1. In a preclinical study, FHCRC investigators reported that the CAR‐T generated from a different subset show a different function in vivo.37 For example, CD8+ CM‐CAR‐T exert a potent direct antitumor activity, and CD4+‐CAR‐T have a mild activity compared to that of CD8+ CM‐CAR‐T. Instead, CD4+‐CAR‐T produce several inflammatory cytokines, and after infusion of CD8+ CM‐CAR‐T, synergistic enhancement of proliferation is observed. Based on these findings, JCAR014 is produced from separate subsets of CD4+ and CD8+ CM‐T cells.
The FHCRC group conducted a phase I trial of JCAR014 in relapsed/refractory B‐NHL.38 In contrast to other studies, that study revealed a significant relation between the regimen of lymphodepletion‐chemotherapy or cell dose and responses or adverse effects. For example, among 30 evaluable patients, 18 patients who received cyclophosphamide and fludarabine (CY/FLU) as lymphodepletion‐chemotherapy showed a significantly higher CR rate as compared to the 12 patients who received CY ± etoposide (CR 50% vs. 8%, P = 0.02). In that study, three cell doses (2 × 105/kg, 2 × 106/kg, and 2 × 107/kg) were evaluated, and the ORR at each dose in patients who received CY/FLU was compared. The 2 × 106/kg cohort achieved the highest ORR among them (ORR: 1 of 3 [33%] for 2 × 105/kg; 9 of 11 [82%] for 2 × 106/kg; and 3 of 4 [75%] for 2 × 107/kg), and the 2 × 107/kg cohort showed a tendency to develop severe CRS more frequently (0 of 3 [0%] in group 2 × 105/kg; 1 of 11 [9%] in group 2 × 106/kg; 3 of 6 [50%] in group 2 × 107/kg).
Subsequently, CD19‐CAR‐T made from CD4+ and CD8+ subsets with a defined ratio of 1:1 were constructed (JCAR017). Juno Therapeutics conducted a multicenter phase I trial of JCAR017 for relapsed/refractory B‐NHL.39 Twenty‐six B‐NHL patients (including 22 patients with aggressive histology) were treated with CY/FLU‐lymphodepletion‐chemotherapy followed by 2 × 106/kg CAR‐T. The ORR was 73%, and the CR rate was 46%. Twelve percent of the patients experienced either grade 3–4 CRS and/or grade 3 neurologic toxicity; no patients with CRS required vasopressors. Detailed characterization of early biomarkers of CRS and neurologic toxicity was also carried out in that study. Compared to patients with grade 0–2 CRS, those with grade 3–5 CRS had significantly higher peak levels of IL‐15, IL‐6, IL‐2, IFN‐γ, CRP, and ferritin. These cytokine parameters may help to identify patients at a higher risk of CRS or neurologic toxicity. Further research with a larger sample size is needed.
Unsolved Problems in CAR‐T Therapy
Despite its promising efficacy, CD19‐CAR‐T therapy has several problems to be solved before introduction into clinical practice. The problems awaiting solutions are summarized in Table 3.
Table 3.
Problems to be solved in anti‐CD19 CAR T‐cell therapy
| Problems | Possible way to overcome |
|---|---|
| 1) Disease control during the CAR‐T cell production | |
| 2) Production failure | |
| 3) Healthy B‐cell depletion; on‐target off‐tumor effect |
|
| 4) Poor expansion and early elimination of CAR‐T cells |
|
| 5) Insufficient activity of CAR‐T cells | |
| 6) CD19 negative conversion | |
| 7) Optimal management of CRS | |
| 8) Optimal management of neurologic toxicity |
|
CAR, chimeric antigen receptor; CLL, chronic lymphocytic leukemia; CRS, cytokine release syndrome.
First, disease control during the CAR‐T manufacture is difficult, especially in patients with aggressive lymphomas, because most patients who need CAR T‐cell therapy are refractory to conventional cytotoxic chemotherapy. In the US or Europe, several small molecules such as ibrutinib and lenalidomide are used as bridging therapies prior to CAR‐T infusion in clinical trials. Refinement of the process of manufacturing CAR‐T is also needed because a simplified rapid production method can ensure shorter turnaround time from apheresis to infusion and may decrease the risk of production failure. Actually, the KTE‐C19 phase II trial involved a rapid manufacturing method (designed by the NCI group) and achieved short turnaround time with a low production failure rate.34, 35 Generally, open‐tissue culture vessels are utilized for CAR‐T production, and human serum is required for the cell processing. However, such a complex “open” system takes long time and is difficult to further scale up. The NCI investigators used a cell processing device that enables automated and “closed” cell processing in bags (Sepax II manufactured by Biosafe America). They also developed a serum‐free culture system using alternative solutions (OpTimizer CTS with 2.5% TSCR). These simplified manufacturing processes enabled a rapid production and a low production failure rate.35
Patients and physicians must wait for the CAR‐T production because it is custom‐made for each patient. Furthermore, there is the risk of production failure especially in heavily pretreated patients who do not have a sufficient number of healthy T cells. Qasim and colleagues recently reported the possibility of “off‐the‐shelf” CAR‐T, which can overcome these issues.40 They used transcription activator‐like effector nuclease (TALEN) to disrupt the expression of TCRαβ and simultaneously transduced the CAR gene into cells using a lentiviral vector. Thus, these CAR‐T cells can evade the host immunity of human leukocyte antigen (HLA)‐unmatched recipients. They have made a bank of non‐HLA‐matched universal CD19‐CAR‐T from a healthy female donor. Two infants with relapsed/refractory B‐ALL received infusions of these “off‐the‐shelf” CAR‐T cells and achieved molecular remission. These results may suggest the usefulness of a universal CAR‐T bank and further investigation is necessary.
Healthy B‐cell depletion is an “on‐target off‐tumor effect” that cannot be avoided in CD19‐CAR‐T therapy. Although B‐cell aplasia and subsequent hypogammaglobulinemia might be less serious toxicities compared to other immune toxicities such as CRS, a substantial number of patients require immune substitutions for years after treatment. To protect healthy B cells, a novel therapeutic target more specific to tumor cells was investigated. Faitschuk and colleagues reported preclinical data on immunoglobulin M Fc receptor (FcμR)‐specific CAR‐T and demonstrated FcμR‐CAR‐T derived from CLL patients purged autologous CLL cells in vitro without reducing healthy B cells.41 FcμR is highly and consistently expressed on CLL cells, while healthy B cells express only low levels. Therefore, FcμR can be an alternative target for CAR‐T therapy in patients with CLL.
Increasing the CAR‐T expansion magnitude and achieving durable in vivo proliferation are necessary to obtain clinically meaningful antitumor activity. Further improvement of lymphodepletion‐chemotherapy to increase CAR‐T expansion is currently in progress. In addition, further improvement of CAR structure itself is also important as described in the section “Structure of the anti‐CD19 CAR” above. Moreover, because the antigen recognition domain of CAR is usually derived from murine antibodies, it is believed that immune responses against CAR partly cause CAR‐T elimination in the human body. NCI investigators are currently designing CD19‐CAR‐T by means of a fully human CAR, and they presented the first report of efficacy in eight patients with B‐NHL at the ASH‐2016.42 Juno Therapeutics is also studying fully human CD19‐CAR‐T, JCAR021. Because these data are still preliminary, the clinical implications of fully human CAR remain unclear. Further research is expected.
In comparison with the remarkable efficacy of CAR‐T therapy against B‐ALL, efficacy against B‐NHL is slightly lower. This situation may be explained by a difference in the tumor microenvironment and the expression of immune checkpoint molecules.43, 44 CAR‐T, just as normal T cells, expresses programmed death 1 (PD‐1) on the cell surface. Therefore, CAR‐T can be downregulated by immune checkpoint proteins, such as programmed death ligand 1 (PD‐L1), that are frequently expressed on tumor cells as an escape mechanism. Based on these observations, combined or sequential use of immune checkpoint inhibitors is actively being studied.45 Investigators at UPenn recently started a phase I/II trial to evaluate the feasibility and efficacy of an anti‐PD‐1 antibody, pembrolizumab, in patients failing to respond to (or relapsing after) CTL019 therapy for B‐NHL (NCT02650999). FHCRC and Juno Therapeutics have also started a phase I trial of JCAR014 in combination with an anti‐PD‐L1 antibody, durvalumab, in patients with relapsed/refractory B‐NHL (NCT02706405). Such an approach may pose a risk of increased prevalence and severity of CRS. Careful research should be conducted on this approach.
Even if CD19‐CAR‐T treatment led to objective responses, some patients experience relapse with a loss of CD19 expression on the tumor cells.27, 28 CD22, CD20, and CD123 are being actively studied as alternative CAR‐T targets.46 CD123 is expressed in several hematologic malignancies, including B‐ALL and acute myeloid leukemia.47 Ruella and colleagues tested anti‐CD123 CAR‐T in vitro and in vivo, and observed its efficacy against B‐ALL cells obtained from patients with B‐ALL that had relapsed with loss of CD19 after the CD19‐CAR‐T therapy.48 Subsequently, Ruella et al. confirmed the efficacy of CD19‐CAR‐T in combination with anti‐CD123 CAR‐T in a murine model of B‐ALL without CD19‐negative relapse. Another group reported the efficacy of bispecific CAR‐T targeting both CD19 and CD20 in a murine model of B‐cell malignancy.49 Dual targeting CAR‐T therapy might be a promising strategy for preventing antigen escape and further investigation is required.
The optimal management of CRS is still debatable because it is the most frequent and serious adverse effect of CAR‐T therapy. Although tocilizumab may be an effective treatment, its optimal timing or influence on the CAR‐T expansion remain unclear. As described above, some cytokine parameters may help to identify patients at a high risk of CRS; early intervention strategies based on these parameters are a promising approach.34, 39 To reduce the risk of CRS, combined use of Bruton's tyrosine kinase inhibitor, ibrutinib, is a novel and promising strategy. The investigators at UPenn have developed a xenograft model of CRS and compared the cytokine levels and survival after infusion of CAR‐T alone or CAR‐T in combination with ibrutinib. As a result, the mice receiving CAR T cells and ibrutinib showed better survival with mild upregulation of inflammatory cytokines.50 Although ibrutinib's influence on clinical efficacy remains unclear, it may be worth further research because ibrutinib itself is an effective and less toxic agent for several subtypes of B‐NHL.
As another method for management of severe immune‐system‐related adverse effects, integration of a “suicide gene” or “elimination genes” into the CAR structure is under development. Inducible caspase 9 (iCasp9) is another suicide system that has been studied in the clinic. iCasp9 is a monomer of caspase 9 combined with a binding domain for a specific small molecule that plays a role of a “dimerizer.” Administration of the dimerizer promotes iCasp9 dimerization, and consequently, caspase 9 is activated. Subsequently, caspases 3, 6, and 7 are activated and induce apoptosis.51, 52, 53 Several clinical trials of CAR‐T with iCasp9 are currently in progress (NCT01822652, NCT02247609, and NCT02274584).
The “elimination gene” is a gene for expression of a selective antigen that can serve as a target of clinically available antibody therapy, such as CD20 or EGFR.54, 55 After administration of a monoclonal antibody, the engineered cells expressing its target molecule can be eliminated rapidly. This is an attractive method for clinicians, but adverse effects associated with the antibody itself may pose another problem.
Conclusions
Several recent studies have shown encouraging efficacy of CD19‐CAR‐T therapy in patients with relapsed/refractory B‐NHL. Nonetheless, most trials contained only a small number of patients. Larger‐scale clinical trials for evaluation of efficacy are necessary to incorporate CAR‐T therapy into clinical practice. Furthermore, there are multiple factors that contribute to its clinical efficacy, such as the type of vector, culture conditions, CAR design, cell type, lymphodepletion‐chemotherapy, derivation of autologous or allogeneic T cells, and the infused cell dose. To determine the optimal protocol of CAR‐T therapy, further research and accumulation of data are needed. Because CAR‐T therapy involves a more complex methodology as compared to conventional chemotherapy, CAR‐T therapy implies sufficient multi‐disciplinary support, for example, from intensive‐care unit doctors, well‐educated nurses, and technicians qualified to manipulate cells. Therefore, preparing such resources for CAR‐T therapy is also necessary. Although there are several problems awaiting solutions before introduction of CAR‐T therapy into clinical practice as mentioned in this manuscript, there are definite unmet medical needs among patients with chemorefractory B‐NHL. CAR T‐cell therapy holds promise to defeat such chemorefractory diseases, and further efforts are warranted.
Disclosure Statement
Shinichi Makita has no conflict of interest; Kiyoshi Yoshimura received research funding from Noile‐Immune Biotech; Kensei Tobinai received research funding from AbbVie, Celgene, Chugai, Eisai, GlaxoSmithKline, Janssen, Kyowa Hakko Kirin, Mundipharma, Novartis, Ono Pharmaceutical, Servier, and Takeda; Kensei Tobinai received honoraria from Janssen, HUYA Bioscience, Takeda, and Zenyaku Kogyo.
Abbreviations
- ASH‐2015
57th annual meeting of the American Society of Hematology
- B‐ALL
B‐cell acute lymphoblastic leukemia
- B‐CLL
B‐cell chronic lymphocytic leukemia
- B‐NHL
B‐cell non‐Hodgkin lymphoma
- CAR
chimeric antigen receptor
- CM
central memory
- CR
complete remission
- CRS
cytokine release syndrome
- CY
cyclophosphamide
- DLBCL
diffuse large B‐cell lymphoma
- FHCRC
Fred Hutchinson Cancer Research Center
- FL
follicular lymphoma
- FLU
fludarabine
- HLA
human leukocyte antigen
- FcμR
immunoglobulin M Fc receptor
- iCasp9
inducible caspase 9
- IFN
interferon
- IL
interleukin
- MSKCC
Memorial Sloan Kettering Cancer Center
- ORR
overall response rate
- PD‐1
programmed death 1
- PD‐L1
programmed death ligand 1
- PFS
progression‐free survival
- PR
partial response
- scFv
single‐chain variable domain
- TCR
T‐cell receptor
- UPenn
University of Pennsylvania
Acknowledgment
This work was supported in part by the National Cancer Center Research and Development Fund (26‐A‐4 and 27‐A‐2).
Cancer Sci 108 (2017) 1109–1118
Funding Information
National Cancer Center Research and Development Fund (26‐A‐4 and 27‐A‐2).
References
- 1. Maude SL, Frey N, Shaw PA et al Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371: 1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lee DW, Kochenderfer JN, Stetler‐Stevenson M et al T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose‐escalation trial. Lancet 2015; 385: 517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Davila ML, Riviere I, Wang X et al Efficacy and toxicity management of 19‐28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6: 224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Batlevi CL, Matsuki E, Brentjens RJ, Younes A. Novel immunotherapies in lymphoid malignancies. Nat Rev Clin Oncol 2016; 13: 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody‐binding domains and the gamma or zeta subunits of the immunoglobulin and T‐cell receptors. Proc Natl Acad Sci USA 1993; 90: 720–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Annenkov AE, Moyes SP, Eshhar Z et al Loss of original antigenic specificity in T cell hybridomas transduced with a chimeric receptor containing single‐chain Fv of an anti‐collagen antibody and Fc epsilonRI‐signaling gamma subunit. J Immunol 1998; 161: 6604–13. [PubMed] [Google Scholar]
- 7. Haynes NM, Snook MB, Trapani JA et al Redirecting mouse CTL against colon carcinoma: superior signaling efficacy of single‐chain variable domain chimeras containing TCR‐zeta vs Fc epsilon RI‐gamma. J Immunol 2001; 166: 182–7. [DOI] [PubMed] [Google Scholar]
- 8. Brocker T. Chimeric Fv‐zeta or Fv‐epsilon receptors are not sufficient to induce activation or cytokine production in peripheral T cells. Blood 2000; 96: 1999–2001. [PubMed] [Google Scholar]
- 9. Savoldo B, Ramos CA, Liu E et al CD28 costimulation improves expansion and persistence of chimeric antigen receptor‐modified T cells in lymphoma patients. J Clin Invest 2011; 121: 1822–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Milone MC, Fish JD, Carpenito C et al Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased antileukemic efficacy in vivo. Mol Ther 2009; 17: 1453–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Long AH, Haso WM, Shern JF et al 4‐1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med 2015; 21: 581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haso W, Lee DW, Shah NN et al Anti‐CD22‐chimeric antigen receptors targeting B‐cell precursor acute lymphoblastic leukemia. Blood 2013; 121: 1165–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL‐12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen‐independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res 2011; 71: 5697–706. [DOI] [PubMed] [Google Scholar]
- 14. Yeku OO, Brentjens RJ. Armored CAR T‐cells: utilizing cytokines and pro‐inflammatory ligands to enhance CAR T‐cell anti‐tumour efficacy. Biochem Soc Trans 2016; 44: 412–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kochenderfer JN, Rosenberg SA. Treating B‐cell cancer with T cells expressing anti‐CD19 chimeric antigen receptors. Nat Rev Clin Oncol 2013; 10: 267–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Klebanoff CA, Khong HT, Antony PA et al Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell‐mediated tumor immunotherapy. Trends Immunol 2005; 26: 111–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee DW, Gardner R, Porter DL et al Current concepts in the diagnosis and management of cytokine release syndrome. Blood 2014; 124: 188–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T‐cell therapy. Mol Ther Oncolytics 2016; 3: 16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davila ML, Sauter C, Brentjens R. CD19‐targeted T cells for hematologic malignancies: clinical experience to date. Cancer J 2015; 21: 470–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grupp SA, Kalos M, Barrett D et al Chimeric antigen receptor‐modified T cells for acute lymphoid leukemia. N Engl J Med 2013; 368: 1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maude SL, Barrett D, Teachey DT, Grupp SA. Managing cytokine release syndrome associated with novel T cell‐engaging therapies. Cancer J 2014; 20: 119–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rosenberg SA, Lotze MT, Yang JC et al Experience with the use of high‐dose interleukin‐2 in the treatment of 652 cancer patients. Ann Surg 1989; 210: 474–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Topp MS, Gökbuget N, Stein AS et al Safety and activity of blinatumomab for adult patients with relapsed or refractory B‐precursor acute lymphoblastic leukaemia: a multicentre, single‐arm, phase 2 study. Lancet Oncol 2015; 16: 57–66. [DOI] [PubMed] [Google Scholar]
- 24. Kochenderfer JN, Wilson WH, Janik JE et al Eradication of B‐lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010; 116: 4099–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kochenderfer JN, Dudley ME, Feldman SA et al B‐cell depletion and remissions of malignancy along with cytokine‐associated toxicity in a clinical trial of anti‐CD19 chimeric‐antigen‐receptor‐transduced T cells. Blood 2012; 119: 2709–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Porter DL, Levine BL, Kalos M et al Chimeric antigen receptor‐modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365: 725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalos M, Levine BL, Porter DL et al T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med 2011; 3: 95ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Porter DL, Hwang WT, Frey NV et al Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med 2015; 7: 303ra139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schuster SJ, Svoboda J, Nasta SD et al Sustained remissions following chimeric antigen receptor modified T cells directed against CD19 (CTL019) in patients with relapsed or refractory CD19+ lymphomas [abstract]. Blood 2015; 126: 183. [Google Scholar]
- 30. Schuster SJ, Svoboda J, Nasta SD et al Treatment with chimeric antigen receptor modified T cells directed against CD19 (CTL019) results in durable remissions in patients with relapsed or refractory diffuse large B cell lymphomas of germinal center and non‐germinal center origin, “double hit” diffuse large B cell lymphomas, and transformed follicular to diffuse large B cell lymphomas [abstract] Blood 2016; 128: 3026.28034869 [Google Scholar]
- 31. Chong EA, Svoboda J, Nasta SD et al Chimeric antigen receptor modified T cells directed against CD19 (CTL019) in patients with poor prognosis, relapsed or refractory CD19 + follicular lymphoma: prolonged remissions relative to antecedent therapy [abstract] Blood 2016; 128: 1100. [Google Scholar]
- 32. Kochenderfer JN, Dudley ME, Kassim SH et al Chemotherapy‐refractory diffuse large B‐cell lymphoma and indolent B‐cell malignancies can be effectively treated with autologous T cells expressing an anti‐CD19 chimeric antigen receptor. J Clin Oncol 2015; 33: 540–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Locke FL, Neelapu SS, Bartlett NL et al Phase 1 Results of ZUMA‐1: A Multicenter Study of KTE‐C19 Anti‐CD19 CAR T Cell Therapy in Refractory Aggressive Lymphoma. Mol Ther 2017; 25: 285–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Neelapu SS, Locke FL, Bartlett NL et al Kte‐C19 (anti‐CD19 CAR T Cells) induces complete remissions in patients with refractory diffuse large B‐cell lymphoma (DLBCL): results from the pivotal phase 2 ZUMA‐1 [abstract] Blood 2016; 128: LBA‐6. [Google Scholar]
- 35. Lu TL, Pugach O, Somerville RP et al A rapid cell expansion process for production of engineered autologous CAR‐T cell therapies. Hum Gene Ther Methods 2016; 27: 209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Crump M, Neelapu SS, Farooq U et al Outcomes in refractory aggressive diffuse large B‐cell lymphoma (DLBCL): results from the international SCHOLAR‐1 study [abstract] J Clin Oncol 2016; 34 (suppl): 7516. [Google Scholar]
- 37. Sommermeyer D, Hudecek M, Kosasih PL et al Chimeric antigen receptor‐modified T cells derived from defined CD8 + and CD4 + subsets confer superior antitumor reactivity in vivo. Leukemia 2016; 30: 492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Turtle CJ, Hanafi LA, Berger C et al Immunotherapy of non‐Hodgkin's lymphoma with a defined ratio of CD8 + and CD4 + CD19‐specific chimeric antigen receptor‐modified T cells. Sci Transl Med 2016; 8: 355ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Turtle CJ, Hay KA, Juliane G et al Biomarkers of cytokine release syndrome and neurotoxicity after CD19 CAR‐T cells and mitigation of toxicity by cell dose [abstract] Blood 2016; 128: 1852. [Google Scholar]
- 40. Qasim W, Zhan H, Samarasinghe S et al Molecular remission of infant B‐ALL after infusion of universal TALEN gene‐edited CAR T cells. Sci Transl Med 2017; 9: eaaj2013. [DOI] [PubMed] [Google Scholar]
- 41. Faitschuk E, Hombach AA, Frenzel LP, Wendtner CM, Abken H. Chimeric antigen receptor T cells targeting Fc μ receptor selectively eliminate CLL cells while sparing healthy B cells. Blood 2016; 128: 1711–22. [DOI] [PubMed] [Google Scholar]
- 42. Brudno JN, Shi V, Stroncek D et al T cells expressing a novel fully‐human anti‐CD19 chimeric antigen receptor induce remissions of advanced lymphoma in a first‐in‐humans clinical trial [abstract] Blood 2016; 128: 999. [Google Scholar]
- 43. Reiners KS, Topolar D, Henke A et al Soluble ligands for NK cell receptors promote evasion of chronic lymphocytic leukemia cells from NK cell anti‐tumor activity. Blood 2013; 121: 3658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McClanahan F, Hanna B, Miller S et al PD‐L1 checkpoint blockade prevents immune dysfunction and leukemia development in a mouse model of chronic lymphocytic leukemia. Blood 2015; 126: 203–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. John LB, Kershaw MH, Darcy PK. Blockade of PD‐1 immunosuppression boosts CAR T‐cell therapy. Oncoimmunology 2013; 2: e26286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shah NN, Stetler‐Stevenson M, Yuan CM et al Minimal residual disease negative complete remissions following anti‐CD22 chimeric antigen receptor (CAR) in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL) [abstract] Blood 2016; 128: 650.27281794 [Google Scholar]
- 47. Hassanein NM, Alcancia F, Perkinson KR, Buckley PJ, Lagoo AS. Distinct expression patterns of CD123 and CD34 on normal bone marrow B‐cell precursors (“hematogones”) and B lymphoblastic leukemia blasts. Am J Clin Pathol 2009; 132: 573–80. [DOI] [PubMed] [Google Scholar]
- 48. Ruella M, Barrett DM, Kenderian SS et al Dual CD19 and CD123 targeting prevents antigen‐loss relapses after CD19‐directed immunotherapies. J Clin Invest 2016; 126: 3814–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zah E, Lin MY, Silva‐Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res 2016; 4: 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ruella M, Kenderian SS, Shestova O et al Kinase inhibitor ibrutinib to prevent cytokine‐release syndrome after anti‐CD19 chimeric antigen receptor T cells for B‐cell neoplasms. Leukemia 2017; 31: 246–8. [DOI] [PubMed] [Google Scholar]
- 51. Straathof KC, Pulè MA, Yotnda P et al An inducible caspase 9 safety switch for T‐cell therapy. Blood 2005; 105: 4247–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Di Stasi A, Tey SK, Dotti G et al Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med 2011; 365: 1673–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Budde LE, Berger C, Lin Y et al Combining a CD20 chimeric antigen receptor and an inducible caspase 9 suicide switch to improve the efficacy and safety of T cell adoptive immunotherapy for lymphoma. PLoS ONE 2013; 8: e82742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Serafini M, Manganini M, Borleri G et al Characterization of CD20‐transduced T lymphocytes as an alternative suicide gene therapy approach for the treatment of graft‐versus‐host disease. Hum Gene Ther 2004; 15: 63–76. [DOI] [PubMed] [Google Scholar]
- 55. Philip B, Kokalaki E, Mekkaoui L et al A highly compact epitope‐based marker/suicide gene for easier and safer T‐cell therapy. Blood 2014; 124: 1277–87. [DOI] [PubMed] [Google Scholar]