

Abstract
Lung cancer accompanied by somatic activating mutations in the epidermal growth factor receptor (EGFR) gene, which is associated with a significant clinical response to the targeted therapy, is frequently found in never‐smoking Asian women with adenocarcinoma. Although this implies genetic factors underlying the carcinogenesis, the etiology remains unclear. To gain insight into the pathogenic mechanisms, we sequenced the exomes in the peripheral‐blood DNA from six siblings, four affected and two unaffected siblings, of a family with familial EGFR‐mutant lung adenocarcinoma. We identified a heterozygous missense mutation in MET proto‐oncogene, p.Asn375Lys, in all four affected siblings. Combined with somatic loss of heterozygosity for MET, the higher allele frequency in a Japanese sequencing database supports a causative role of the MET mutation in EGFR‐mutant lung cancer. Functional assays showed that the mutation reduces the binding affinity of MET for its ligand, hepatocyte growth factor, and damages the subsequent cellular processes, including proliferation, clonogenicity, motility and tumorigenicity. The MET mutation was further observed to abrogate the ERBB3‐mediated AKT signal transduction, which is shared downstream by EGFR. These findings provide an etiological view that the MET mutation is involved in the pathogenesis of EGFR‐mutant lung cancer because it generates oncogenic stress that induces compensatory EGFR activation. The identification of MET in a family with familial EGFR‐mutant lung cancer is insightful to explore the pathogenic mechanism of not only familial, but also sporadic EGFR‐mutant lung cancer by underscoring MET‐related signaling molecules.
Keywords: Epidermal growth factor receptor, exome, familial lung cancer, human MET protein, missense mutation
Lung cancer is the leading cause of cancer death worldwide, with the estimated number of annual deaths around 1.4 million.1, 2, 3 Cigarette smoking is a predominant etiological factor of lung carcinogenesis, especially for small cell lung cancer and squamous cell lung cancer.2, 4 Increasing attention is being paid to a growing number of lung carcinomas occurring in never‐smokers, which are estimated to account for approximately 25% of lung cancers worldwide.2, 5 Such lung cancers in never‐smokers are more likely to have oncogenic mutations, such as epidermal growth factor receptor (EGFR) and anaplastic lymphoma kinase (ALK), which can be precisely targeted for killing the tumor cells by small molecular inhibitors of the internal tyrosine kinase.3, 5 EGFR is of significant interest because the mutations occur more frequently in adenocarcinomas of East Asian women with a history of never smoking, suggesting that the EGFR‐mutant tumor is a biologically distinct disease with a genetic susceptibility.5 Despite these insights from epidemiological evidence, the genetic etiology of EGFR‐mutant lung cancer remains unknown.
Epidermal growth factor receptor is a member of the v‐erb‐b2 erythroblastic leukemia viral oncogene (ERBB) family, commonly referred to as the human epidermal growth factor (HER) family, and is also designated as ERBB1 or HER1.2, 6 EGFR and its family members serve as receptor tyrosine kinases and contribute to carcinogenesis through MAP‐kinase and PI3‐kinase (PI3K) signaling pathways that modulate cell proliferation and cell survival, respectively.7 The PI3K pathway is particularly important in lung cancer as a major signaling pathway downstream of EGFR, which confers multiple PI3K‐binding sites by heterodimerization with ERBB3.8 In this regard, amplification of MET, coding another receptor tyrosine kinase, clinically yields acquired resistance to EGFR‐targeted therapy by forming the MET‐ERBB3 complex to reinstate the PI3K signaling diminished by the targeted therapy for somatic activating mutations in EGFR.8, 9
In the present study, we identified a germline mutation in MET by exome sequencing of whole‐blood DNA from a pedigree consisting of ≥3 family members affected by EGFR‐mutant lung adenocarcinomas. All lung cancer patients, but not the unaffected siblings, carried the missense MET mutation that damages the biological function via impaired binding to the ligand, hepatocyte growth factor (HGF). Notably, the functional consequence of the MET mutation is an attenuation of the ERBB3‐PI3K pathway in lung cancer cells, thereby potentially activating the EGFR‐ERBB3‐PI3K signaling axis as an oncogenic stress to EGFR‐mutant lung cancer.
Materials and Methods
Exome capture and sequencing
Blood samples for DNA extraction were collected according to the protocol approved by the Institutional Review Board at Tohoku University Graduate School of Medicine (Sendai, Japan).
Exome capture was performed with the SureSelect Human All Exon V4 (Agilent Technologies, Santa Clara, CA, USA). Exome capture libraries were sequenced by HiSeq 2000 (Illumina, San Diego, CA, USA) to obtain at least 83 million paired‐end (2 × 101 base pairs) reads per sample. A targeted mean coverage depth of 106–198 × with 74–91% of bases covered at ≥50 × was achieved for 51 Mb exome libraries (Table S1). Additional detail on the method is provided in Supplementary Document S1).
Statistical analysis
Two data sets were compared using Student's unpaired two‐tailed t‐test. For multiple comparisons, post hoc analysis was performed by Tukey's honestly significant difference test. P‐values less than 0.05 were considered statistically significant. Error bars in the graphical data represent the means ± standard error.
Results
Germline variants in a family with EGFR‐mutant lung cancer
We previously reported a Japanese family in which three siblings (subjects 2, 5 and 6 in Fig. 1a) were affected by lung adenocarcinoma with EGFR mutations.10 The inconsistent EGFR mutation pattern in the three siblings (subjects 2 and 5, a point mutation L858R on exon 21; subject 4, an in‐frame deletion of exon 19) suggested that their genetic bases contained a vulnerability for EGFR‐mutant lung cancer. After the report was published, another sibling within the family (subject 1), an 80‐year‐old woman who never smoked, came to Tohoku University Hospital (Sendai, Japan) for bulbar paralysis. Given the imaging examination and her family history, she was clinically diagnosed with (probably EGFR‐mutant) lung cancer and brain metastasis, although no pathologic diagnosis was achieved because of her extremely poor general condition (Fig. S1).
Figure 1.
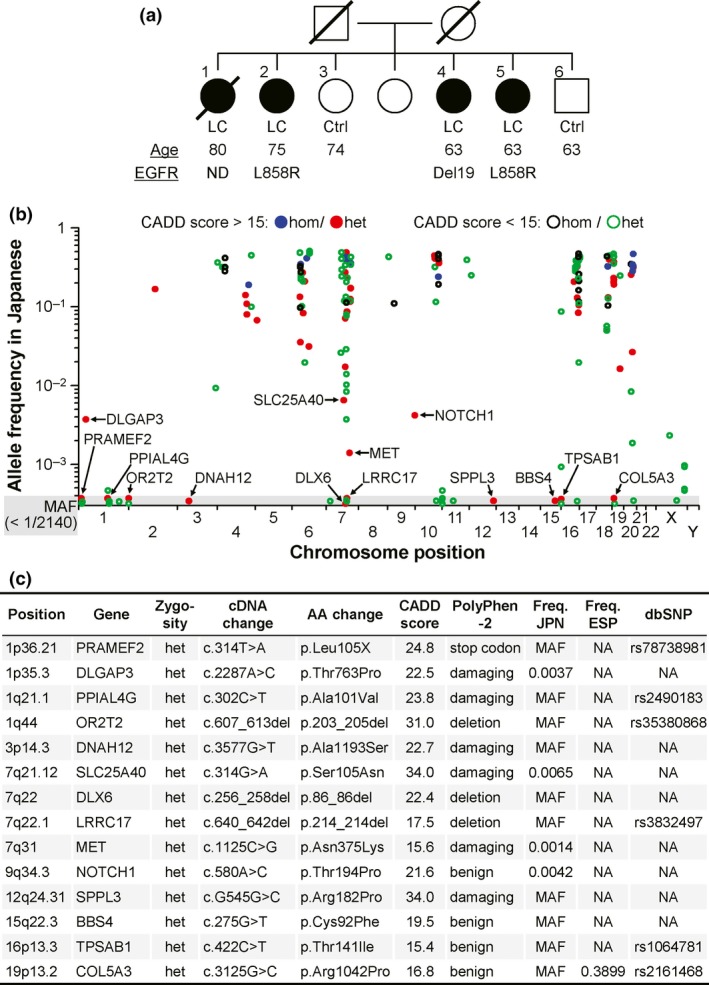
Germline alterations identified in whole‐exome sequencing of family members with EGFR‐mutant lung cancer. (a) Abridged pedigree of familial EGFR‐mutant lung cancer. Filled symbols, individuals affected by lung cancer (LC); open symbols, unaffected control individuals (control). Symbols with slashes are deceased individuals. Exome‐sequenced individuals are numbered at the upper left of the symbols. Age shown in years is the age at diagnosis for lung cancer cases and the age at examination for unaffected siblings. Del19 and L858R of EGFR alterations in lung cancer indicate an in‐frame deletion on exon 19 and a point mutation L858R on exon 21, respectively; ND, not determined. (b) Regional plots of 185 candidate variants in all four patients, but not in the unaffected siblings. The allele frequencies in Japanese individuals (n = 1070) are plotted across the human genome. The homozygous (hom) and heterozygous (het) variants are shown with the pathogenic impact predicted as deleterious or benign by the CADD scores over or below 15, respectively. (c) Details of 14 variants selected as rare (allele frequency <10−2) and deleterious (CADD score >15). AA, amino acid; Freq. JPN, allele frequency in Japanese individuals; Freq. ESP, allele frequency in American individuals of the NHLBI GO Exome Sequencing Project; NA, not available. In (a) and (c), MAF denotes minor allele frequency (<1/2140); the frequency is not specified in the gray area (b), because the variant was not detected in the database.
To clarify the genetic etiology of the familial EGFR‐mutant lung cancer, we performed exome capture followed by deep sequencing on four affected individuals (subjects 1, 2, 4 and 5) and two unaffected control individuals (subjects 3 and 6). An average of 22 944 exonic variants per individual were noted in the genomic DNA from the peripheral blood of the family members, and an average of 1436 variants were novel according to databases of the dbSNP and the NHLBI GO Exome Sequencing Project (Table S2).
When comparing the sequenced family members, we found 185 candidate variants that altered the amino acid sequence and were shared by all four lung cancer patients (subjects 1, 2, 4 and 5), but were not observed in the unaffected siblings (subjects 3 and 6); there were 34 homozygous and 151 heterozygous variants; and there were 176 missense, 3 nonsense and 6 insertion and/or deletion (indel) variants (Tables S3–S8). Among these candidates, the causative mutation is expected to be a relatively rare variant in the Japanese general population with a predicted functional consequence. After annotation by both an allele frequency database of 1070 Japanese individuals and a prediction program CADD of pathological impact, 14 heterozygous variants of 185 variants were defined as rare (allele frequency <10−2) and deleterious (CADD score > 15); there were 10 missense, 1 nonsense and 3 indel variants (Fig. 1b,c).
From the 14 rare and deleterious variants, 4 were excluded as benign variants predicted by another program, polyphen‐2, and 3 (i.e. DLGAP3, SLC25A40 and MET) remained under the following criteria: the variant is observed in the Japanese database, but is de novo in the NHLBI GO Exome Sequencing Project (ESP) and dbSNP database, given the genetic susceptibility of the Asian population including Japanese to EGFR‐mutant lung cancer (Fig. 1c).3 Notably, of the remaining 3 variants, the p.Asn375Lys substitution of MET, known to be implicated in EGFR signaling, has been observed with the lowest frequency in the Japanese database, suggesting that the MET variant could potentially cause EGFR‐mutant lung cancer.2, 8
The altered residue in genetic and protein structures of MET
MET, a receptor tyrosine kinase for HGF, consists of a disulfide‐linked heterodimer of 50‐kDa α‐chain and 140‐kDa transmembrane β‐chain subunits that are cleaved from a 1390 amino acid precursor form (Fig. 2a). The altered residue, asparagine at amino acid 375, locates in the semaphorin‐like (Sema) domain that is generally regarded as responsible for binding to HGF and is highly conserved across species, including chimpanzee, dog, cow, mouse, rat and chicken (Fig. 2b). Close examination of the altered residue in the MET crystal structure revealed that the position is encompassed by a particular section, amino acids 367–426, termed extrusion with a distinctive feature, which is involved in dimerization contacts of MET α‐chains and β‐chains as well as HGF binding of the Sema domain (Fig. 2c).11 Subsequent Sanger sequencing of the encoding region showed that the blood of affected individuals was heterozygous for the relevant single‐nucleotide change (c.1125C>G), while the tumor of subject 5 was homozygous for the variant, indicating somatic loss of heterozygosity (LOH) at this locus (Fig. 2d). Although the similar LOH was not confirmed in the tumors of other affected individuals (subjects 1, 2, and 4) due to quantitative and/or qualitative insufficiency of the clinical samples, these results support the possibility that the missense variant of MET at amino acid 375 represents a pathogenic mutation that disrupts the protein function.
Figure 2.

Location of the MET mutation found in familial EGFR‐mutant lung cancer. (a) Schematic representation of the MET protein. The transmembrane protein consists of the semaphorin‐like (Sema) domain, the plexin‐semaphorin‐integrin (PSI) domain, the immunoglobulin‐plexin‐transcription factor (IPT) 1–4 domains, the juxtamembrane (JM) domain, and the kinase domain. The precursor is cleaved into α‐chains and β‐chains, thus yielding the disulfide‐linked heterodimer. The NP_000236.2 protein was used as a reference for the positions. AA, amino acid. (b) Protein sequence alignment of MET (amino acid residues 371–390) in vertebrate species. Altered residues are colored red. The residues corresponding to the missense mutation found in familial EGFR‐mutant lung cancer are boxed. (c) The 3‐D structure of HGF‐MET complex. The modeling was generated using the Protein Data Bank (PDB) ID 1SHY; grey, HGF β‐chain; green, MET α‐chain; blue, Sema and PSI domains of MET β‐chain; orange, Asn375 in MET β‐chain. (d) Sanger sequencing chromatograms for MET (amino acid residues 371–380) in blood and tumor cells from the individual affected by familial EGFR‐mutant lung cancer. Blood cells from an unaffected control individual were used as a control.
Hepatocyte growth factor‐binding capacity impaired by the conformational change of the mutant MET
We carried out 3‐D modeling of MET protein to examine whether the missense mutation in MET may influence the structure (Fig. 3a). This modeling showed that the MET p.Asn375Lys (c.1125C>G) mutation is likely to affect helical and loop structures of the extrusion section in the Sema domain.11 The Asn375 residue in MET forms hydrogen bonds with Arg280 and Asn371, the former of which is disrupted by the alteration of this asparagine residue to lysine. Thus, the p.Asn375Lys mutation is predicted to increase the conformational flexibility of the Sema domain, potentially impairing the HGF‐binding surface and having an effect on the interactions with HGF.
Figure 3.
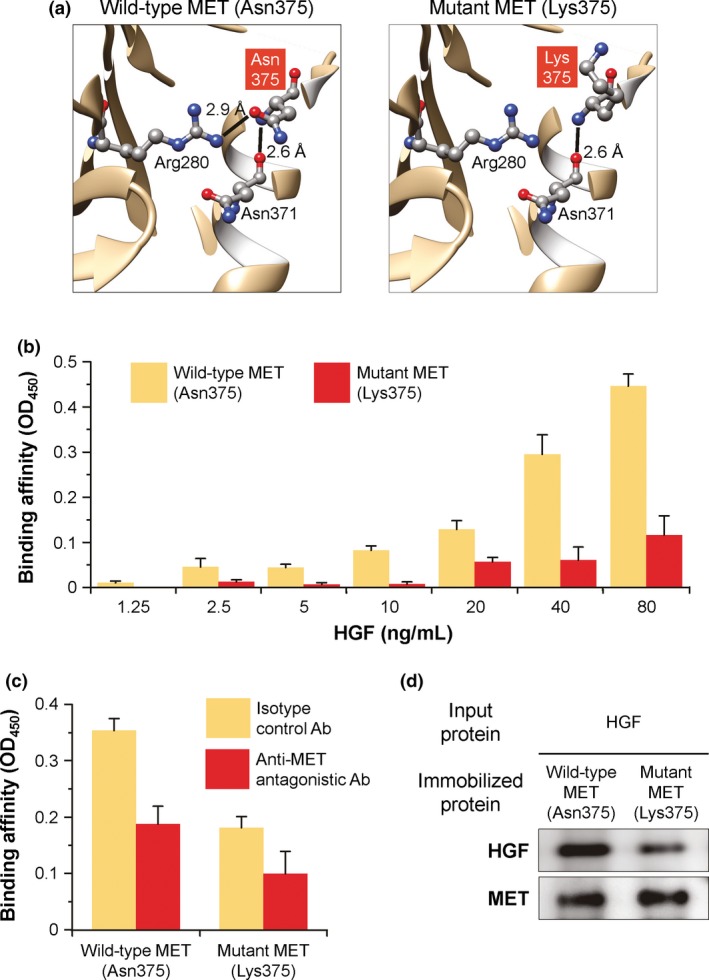
Structure and function of wild‐type (Asn375) and mutant (Lys375) MET ectodomains. (a) Ribbon representation of the MET ectodomains. Asn375 and Lys375 are shown with the surrounding residues by ball‐and‐stick models. Lines represent hydrogen bonds with the distances; gray, carbon; red, oxygen; blue, nitrogen. (b) HGF‐binding capacity of the MET ectodomains. MET‐Fc fusion protein was immobilized to ELISA plates and incubated with increasing concentrations of recombinant human HGF protein. The bound HGF was detected by anti‐HGF Ab and colorimetrically quantified as MET‐HGF binding affinity by the absorbance value at OD 450. (c) Binding specificity of the MET ectodomains to HGF. The study was identical to that described in (b), except that the immobilized MET‐Fc protein was pretreated with anti‐MET antagonistic Ab or isotype control Ab before incubation with 40 ng/mL HGF. (d) Coprecipitation analysis of the MET ectodomains with HGF. Recombinant human HGF protein was coprecipitated with MET‐Fc fusion protein immobilized on protein G‐Sepharose beads. The precipitates were resolved by SDS‐PAGE and then visualized by immunoblotting with anti‐HGF or anti‐MET Ab. For (b) and (c), data are presented as the mean ± standard error of n = 4 (b) or n = 3 (c) per group.
Accordingly, we sought to investigate the impact of the identified MET mutation on the binding affinity to HGF by using recombinant MET‐Fc fusion proteins, in which wild‐type or mutant MET ectodomains were fused to the Fc region of human IgG1 (Fig. 3b–d). Based on the ELISA assay of recombinant HGF protein bound to the MET‐Fc fusion protein, the amount of HGF bound to wild‐type MET, but not to p.Asn375Lys‐mutant MET, sharply increased in a manner that was dependent on the concentration of the applied HGF (wild‐type vs mutant MET: 1.25–20 ng/mL, P > 0.4; 40–80 ng/mL, P < 0001; Fig. 3b). Evidence for the specificity of the observed binding was provided by the finding that pretreatment with anti‐MET antagonistic antibody significantly neutralized the HGF binding of wild‐type MET‐Fc fusion protein, but not p.Asn375Lys‐mutant MET‐Fc fusion protein (anti‐MET vs control antibodies: wild‐type MET, P < 0.05; mutant MET, P > 0.2; Fig. 3c). Consistent with these ELISA data, coprecipitation of the HGF protein followed by western blot analysis displayed a markedly decreased level of HGF bound to p.Asn375Lys‐mutant MET‐Fc fusion protein as compared to wild‐type MET‐Fc fusion protein (Fig. 3d). Taken together, these results demonstrated that the HGF‐binding capacity of the MET ectodomain was abrogated in the p.Asn375Lys alteration of MET.
Biological function damaged by the missense change of MET
To reveal the biological impact of the identified mutation in MET, we retrovirally transduced H1299 lung cancer cells to stably express wild‐type or p.Asn375Lys‐mutant MET (Fig. 4). Both H1299 transductants with the wild‐type and mutant MET cDNA expressed comparable protein levels of MET, whereas the control transductant with no transgene (i.e. null vector) did not express MET at a detectable protein level (Fig. 4a). Despite these MET expression patterns, the H1299 transductant expressing wild‐type MET, but not the p.Asn375Lys‐mutant MET, showed increased cell proliferation as early as day 2 of the HGF‐supplemented culture and until at least day 4, as compared with the control null transductant (wild‐type versus mutant MET: P < 0.05 on day 2, P < 0.0001 on day 4; wild‐type MET versus null: P < 0.05 on day 2, P < 0.0005 on day 4; mutant MET versus null: P > 0.9 on day 2, P > 0.8 on day 4; Fig. 4b). The levels of in vitro colony formation, an indicator of the clonogenic potential, were also significantly higher in the H1299 transductant with wild‐type MET than in that with the p.Asn375Lys‐mutant MET, which exhibited similar levels to those of the control null transductant (wild‐type versus mutant MET, P < 0.005; wild‐type MET versus null, P < 0.005; mutant MET versus null, P > 0.4; Fig. 4c). As determined by the gap closure of the scratched cell monolayer in the HGF‐supplemented culture, the cell motility was significantly augmented upon transduction with wild‐type MET, but remained unchanged or was only mildly reduced upon transduction with p.Asn375Lys‐mutant MET compared to transduction with no transgene (wild‐type versus mutant MET, P < 0.0005; wild‐type MET vs null, P < 0.005; mutant MET vs null, P > 0.4; Fig. 4d). These observations supported the premise that the p.Asn375Lys alteration mediates the functional damage to MET and could be a potential mechanism in the development of EGFR‐mutant lung tumors.
Figure 4.
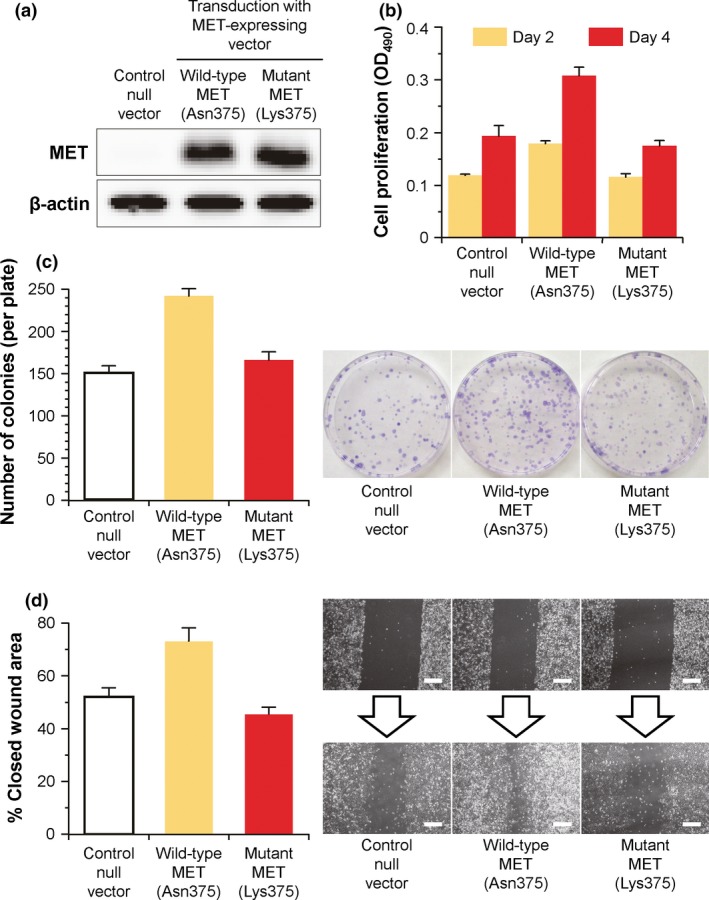
In vitro properties of H1299 lung cancer cells retrovirally transduced to stably express wild‐type (Asn375) and mutant (Lys375) MET. (a) MET expression analysis by western blot. The cell lysates were prepared from transduced H1299 cells and immunoblotted for MET and β‐actin (loading control). (b) Proliferative capacity. Transduced H1299 cells were starved overnight for serum, and then cultured in low‐serum media supplemented with 5 ng/mL recombinant human HGF protein. After 2 and 4 days of the culture, the number of viable cells was colorimetrically assessed by the absorbance value at OD 490. (c) Clonogenic capacity. The study was similar to that in (b), but, 2 weeks after the culture, the colonies were stained with crystal violet for counting the number in each plate. (d) Motile capacity. The study was similar to that in (b), but the serum‐starved cells were scraped (day 0) and cultured for 2 days. The closed wound area on day 2 was quantified as the percentage of the open wound area on day 0. Scale bar, 200 μm. For all figure parts, controls included H1299 cells transduced with control null vector that is identical to the MET‐expressing retrovirus vectors but contains no transgene. Data are presented as the mean ± standard error of n = 3 (b,c) or n = 6 (d) per group, except for (a).
Defective tumorigenicity and the vital signaling downstream of the mutant MET
Finally, we investigated how the stable expression of wild‐type or mutant MET in lung cancer cells affects the tumorigenic potential and the relevant molecular pathway of MET (Fig. 5). Consistent with our in vitro results, stable transduction of H1299 lung cancer cells with wild‐type MET significantly increased the tumor growth on the flanks of implanted mice at time points from day 44 to day 54, as compared to that with the p.Asn375Lys‐mutant MET (as measured by tumor volume, P < 0.005, Fig. 5a). At the end of the experiment on day 54, mice implanted with H1299 cells expressing wild‐type MET had a greater tumor burden compared to mice implanted with H1299 cells expressing p.Asn375Lys‐mutant MET (photographic inset, Fig. 5a; 2.7‐fold increase, as measured by tumor weight, P < 0.005, Fig. 5b). We next verified that stimulation with recombinant HGF protein enhanced the amount of phosphorylated MET (P‐MET), which was greater in H1299 cells expressing wild‐type MET than in those expressing p.Asn375Lys‐mutant MET in either condition with or without the HGF stimulation (Fig. 5c). Recently, among complex signaling events that follow the MET ligation by HGF, the functional crosstalk of MET with ERBB3‐PI3K‐AKT signaling has been shown to be a crucial mechanism in the progression of lung cancer.9, 12 To determine whether the ERBB3‐PI3K‐AKT pathway is influenced by the identified mutation of MET, we analyzed the phosphorylation of ERBB3 and p70 S6 kinase (a downstream mediator of AKT). H1299 cells expressing wild‐type MET augmented the phosphorylation of both ERBB3 and p70 S6 kinase upon HGF stimulation, whereas H1299 cells expressing p.Asn375Lys‐mutant MET were crippled not only for the phosphorylation but also for the protein expression of both pathway members with compromised reactivity in response to the HGF stimulation (Fig. 5c). Moreover, in the published data, EGFR‐mutant lung adenocarcinomas were less likely to express MET mRNA at a markedly increased level than those negative for sensitizing EGFR mutations (4 vs 16%, Table S9).13, 14 These results reinforced our notion that the missense mutation abolishes the ability of MET to activate the ERBB3‐PI3K‐AKT pathway associated with tumorigenicity, thereby prompting lung cells to acquire constitutively activating mutations of EGFR (also known as ERBB1) for replenishment of the disrupted AKT activation.
Figure 5.

Tumor growth and MET signaling of H1299 lung cancer cells retrovirally transduced to stably express wild‐type (Asn375) and mutant (Lys375) MET. (a) Tumor volume. Transduced H1299 cells (2.5 × 106) were injected subcutaneously in the right flank of female BALB/c nu/nu mice on day 0. On and after day 26, the volume of each tumor was assessed two or three times weekly. The tumors on day 54 are shown in the photographic inset; scale bar, 10 mm. (b) Tumor weight. The weight of the tumors shown in the inset of (a) was assessed. (c) Altered downstream signaling. Transduced H1299 cells were treated with 50 ng/mL recombinant human HGF protein (rHGF) or vehicle control for 15 min and lysed. The cell lysates were resolved by SDS‐polyacrylamide gel electrophoresis and then visualized by immunoblotting for phospho‐MET (P‐MET), phospho‐ERBB3 (P‐ERBB3), and phospho‐p70 S6 kinase (P‐p70S6). Total MET, ERBB3 and p70S6 in the lysates were used as loading controls, respectively. Data are presented as the mean ± standard error of n = 3 per group, except for (c).
Discussion
To determine the genetic factors underlying EGFR‐mutant lung cancer, we focused on familial cases and performed exome sequencing on their germline DNA. We then identified the heterozygous c.1125C>G (p.Asn375Lys) variant in MET, which encodes a receptor tyrosine kinase that could mutually complement EGFR to activate the downstream PI3K‐AKT signaling in lung cancer. The missense mutation affects a highly conserved amino acid residue in the extracellular HGF‐binding Sema domain, thus damaging the ligation by HGF and the subsequent MET activation that leads to cellular responses such as proliferation, clonogenicity, motility and tumorigenicity. Moreover, the somatic loss of heterozygosity for the MET mutation bolsters our view that the heterozygous inactivating mutation mediates an oncogenic stress to EGFR‐mutant (i.e. EGFR‐addicted) lung cancers through a haploinsufficiency that reduces the amount of wild‐type MET protein.
Adenocarcinoma is the most common lung cancer subtype, representing approximately 40% of all lung cancers, with squamous cell lung cancer constituting 20% of these.1 Relative to adenocarcinoma, squamous cell lung carcinoma has been decreasing, probably due to the stronger epidemiological association with cigarette smoking.2 Meanwhile, the clinical relevance of lung adenocarcinoma has been strengthened not only by the increasing number of cases but also by the genomic diversity.3, 5, 7, 8 Recent molecular profiling studies have revealed that, according to cigarette‐smoking history, distinct genetic abnormalities are found within lung adenocarcinomas as driver mutations in a mutually exclusive pattern; the KRAS mutations are more frequently detected in patients with a history of smoking, whereas the EGFR mutations and the EML4‐ALK rearrangements are more common in light or never‐smokers.5 Notably, EGFR and ALK alterations are recognized to be of great therapeutic significance as predictive biomarkers for the response to tyrosine kinase inhibitors against EGFR and ALK, respectively.1, 3, 7, 8
Besides a robust association with nonsmoking status, Asian ethnicity was also shown to be associated with an increased likelihood of the activating alterations in the tyrosine kinase of EGFR, suggesting that lung adenocarcinomas positive for EGFR mutations have a genetic component to their etiology.3, 5 In this context, genome‐wide association studies proposed several chromosomal regions that might be susceptible to lung cancer in non‐smokers, including chromosomes 3q28, 5p15.33 (telomerase reverse transcriptase, TERT), 6p21.32, 6q22.2, 10q25.2, 13q31.3 (glypican 5, GPC5), 15q25, 17q24.3 and 18p11.22.5, 15, 16 Other epidemiologic studies demonstrated that variant alleles of several genes are associated with increased susceptibility to lung cancer, such as an EGFR germline mutation, and polymorphisms in the cytochrome p450 enzyme genes, DNA mismatch repair genes, and glutathione‐S‐transferase genes.5, 15 However, because the results of these association studies have been inconsistent, the oncogenic mechanisms that enable EGFR‐mutant adenocarcinomas to develop have not been elucidated.
We identified a missense mutation at 375asparagine to lysine (i.e. p.Asn375Lys) within the extracellular Sema domain of the MET proto‐oncogene in lung adenocarcinoma patients. In the screening process of the candidate variants for the causative mutation of the familial EGFR‐mutant lung cancer, we excluded 7 variants owing to registration in the dbSNP database or non‐registration in the Japanese database. Although we could not exclude a possible etiologic significance of these variants without further functional studies, the MET engagement is known to have broad implications for cancer biology, including cellular proliferation, cell survival, invasion, cell motility, metastasis and angiogenesis.12 This knowledge has fostered a systematic analysis of the complete exons of the MET gene for a number of solid tumors, and uncovered the frequent occurrence of MET mutations in the Sema domain, especially for lung cancer.17 The Sema domain is crucial for receptor activation and dimerization, providing a specific binding site for HGF.17, 18 Within the Sema domain, 375asparagine has been recognized as a hot spot of cancer‐associated MET mutations, and the missense germline change to serine (i.e. p.Asn375Ser) is common in smokers with squamous cell lung carcinoma, but not in never‐smokers with lung adenocarcinoma.17 Unlike p.Asn375Ser, we identified the p.Asn375Lys germline mutation of MET in never‐smokers with familial EGFR‐mutant lung adenocarcinoma. Our finding and the findings of others suggest that lung adenocarcinoma and squamous cell lung carcinoma emerge in never‐smokers and ever‐smokers, respectively, based on distinct genetic abnormalities.2, 5, 7, 8
Of note, somatic mutations in the EGFR gene have been identified as driver mutations underlying the oncogenic addiction of lung adenocarcinomas in never‐smokers.5 Analyses of the signal transduction downstream of the activating EGFR mutations revealed the particular efficacy of the EGFR‐ERBB3 heterodimer to stimulate the PI3K‐AKT pathway.8 Further work on the tumor resistance to EGFR‐targeted therapy has shown that MET amplification undergoes a kinase switch under EGFR blockade by driving ERBB3‐dependent activation of the PI3K‐AKT signaling.19 Conversely, activation of EGFR‐ERBB3 signal transduction has been shown to mediate resistance to MET inhibition.20 These studies indicate that EGFR and MET mutually compensate for activating the PI3K‐AKT pathway via interaction with ERBB3. Our study highlights the importance of the compensatory signals in oncogenesis by identifying an inactivating MET germline mutation that theoretically enhances the signaling dependence on the EGFR‐ERBB3‐PI3K axis as the oncogenic potential of familial EGFR‐mutant lung adenocarcinoma.
Disclosure Statement
The authors declare no conflict of interest related to this work.
Supporting information
Doc. S1. Additional detail on the methods.
Fig. S1. Chest CT and brain MRI of subject 1 (an 80‐year‐old woman).
Table S1. Sequencing read information and exome coverage statistics for each sample.
Table S2. Exonic variants called in genomic DNA from peripheral blood.
Table S3. Chromosome 1–5: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S4. Chromosome 6: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S5. Chromosome 7: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S6. Chromosomes 9–15: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S7. Chromosomes 16 and 17: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S8. Chromosomes 18‐X: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S9. Sensitizing EGFR mutations and the levels of MET expression of lung adenocarcinomas among 230 cases.
Acknowledgments
We thank M. Takahashi, A. Santoso, M. Kanehira, R. Funayama, M. Tsuda, M. Kikuchi, M. Nakagawa and K. Kuroda for their technical assistance, and B. Bell for reading the manuscript. We also acknowledge the technical support of the Biomedical Research Core of Tohoku University Graduate School of Medicine. This work was supported, in part, by Grants‐in‐Aid for Scientific Research and the Project for Development of Innovative Research on Cancer Therapeutics from the Ministry of Education, Culture, Sports, Science and Technology (Tokyo, Japan), the Core Research for Evolutional Science and Technology Program, the Adaptable and Seamless Technology Transfer Program from the Japan Science and Technology Agency (Tokyo, Japan), and the Urgent Research Grant Program from the Astellas Foundation for Research on Metabolic Disorders (Tokyo, Japan).
Cancer Sci 108 (2017) 1263–1270
Funding Information
This work was supported, in part, by Grants‐in‐Aid for Scientific Research and the Project for Development of Innovative Research on Cancer Therapeutics from the Ministry of Education, Culture, Sports, Science and Technology (Tokyo, Japan), the Core Research for Evolutional Science and Technology Program, the Adaptable and Seamless Technology Transfer Program from the Japan Science and Technology Agency (Tokyo, Japan), and the Urgent Research Grant Program from the Astellas Foundation for Research on Metabolic Disorders (Tokyo, Japan).
References
- 1. Cagle PT, Allen TC. Lung cancer genotype‐based therapy and predictive biomarkers: present and future. Arch Pathol Lab Med 2012; 136: 1482–91. [DOI] [PubMed] [Google Scholar]
- 2. de Groot P, Munden RF. Lung cancer epidemiology, risk factors, and prevention. Radiol Clin North Am 2012; 50: 863–76. [DOI] [PubMed] [Google Scholar]
- 3. Nana‐Sinkam SP, Powell CA. Molecular biology of lung cancer: diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest 2013; 143: e30S–9S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yokota J, Shiraishi K, Kohno T. Genetic basis for susceptibility to lung cancer: recent progress and future directions. Adv Cancer Res 2010; 109: 51–72. [DOI] [PubMed] [Google Scholar]
- 5. Couraud S, Zalcman G, Milleron B, Morin F, Souquet PJ. Lung cancer in never smokers–A review. Eur J Cancer 2012; 48: 1299–311. [DOI] [PubMed] [Google Scholar]
- 6. Roskoski R Jr. ErbB/HER protein‐tyrosine kinases: structures and small molecule inhibitors. Pharmacol Res 2014; 87: 42–59. [DOI] [PubMed] [Google Scholar]
- 7. Swanton C, Govindan R. Clinical implications of genomic discoveries in lung cancer. N Engl J Med 2016; 374: 1864–73. [DOI] [PubMed] [Google Scholar]
- 8. Buettner R, Wolf J, Thomas RK. Lessons learned from lung cancer genomics: the emerging concept of individualized diagnostics and treatment. J Clin Oncol 2013; 31: 1858–65. [DOI] [PubMed] [Google Scholar]
- 9. Engelman JA, Zejnullahu K, Mitsudomi T et al MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007; 316: 1039–43. [DOI] [PubMed] [Google Scholar]
- 10. Sakakibara T, Saijo Y, Fukuhara T et al Adenocarcinoma with epidermal growth factor receptor gene mutations in three siblings. J Thorac Oncol 2008; 3: 311–3. [DOI] [PubMed] [Google Scholar]
- 11. Niemann HH. Structural basis of MET receptor dimerization by the bacterial invasion protein InlB and the HGF/SF splice variant NK1. Biochim Biophys Acta 2013; 1834: 2195–204. [DOI] [PubMed] [Google Scholar]
- 12. Gherardi E, Birchmeier W, Birchmeier C, Vande Woude G. Targeting MET in cancer: rationale and progress. Nat Rev Cancer 2012; 12: 89–103. [DOI] [PubMed] [Google Scholar]
- 13. Cancer Genome Atlas Research Network . Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014; 511: 543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cerami E, Gao J, Dogrusoz U et al The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2: 401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marshall AL, Christiani DC. Genetic susceptibility to lung cancer–Light at the end of the tunnel? Carcinogenesis 2013; 34: 487–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shiraishi K, Kunitoh H, Daigo Y et al A genome‐wide association study identifies two new susceptibility loci for lung adenocarcinoma in the Japanese population. Nat Genet 2012; 44: 900–3. [DOI] [PubMed] [Google Scholar]
- 17. Sattler M, Reddy MM, Hasina R, Gangadhar T, Salgia R. The role of the c‐Met pathway in lung cancer and the potential for targeted therapy. Ther Adv Med Oncol 2011; 3: 171–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kong‐Beltran M, Stamos J, Wickramasinghe D. The Sema domain of Met is necessary for receptor dimerization and activation. Cancer Cell 2004; 6: 75–84. [DOI] [PubMed] [Google Scholar]
- 19. Sadiq AA, Salgia R. MET as a possible target for non‐small‐cell lung cancer. J Clin Oncol 2013; 31: 1089–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Corso S, Ghiso E, Cepero V et al Activation of HER family members in gastric carcinoma cells mediates resistance to MET inhibition. Mol Cancer 2010; 9: 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Doc. S1. Additional detail on the methods.
Fig. S1. Chest CT and brain MRI of subject 1 (an 80‐year‐old woman).
Table S1. Sequencing read information and exome coverage statistics for each sample.
Table S2. Exonic variants called in genomic DNA from peripheral blood.
Table S3. Chromosome 1–5: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S4. Chromosome 6: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S5. Chromosome 7: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S6. Chromosomes 9–15: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S7. Chromosomes 16 and 17: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S8. Chromosomes 18‐X: candidate variants identified in all four patients, but not in the unaffected siblings.
Table S9. Sensitizing EGFR mutations and the levels of MET expression of lung adenocarcinomas among 230 cases.