

Abstract
Beginning in the early 2000s, the accumulation of biospecimens linked to electronic health records (EHRs) made possible genome-phenome studies (i.e., comparative analyses of genetic variants and phenotypes) using only data collected as a by-product of typical health care. In addition to disease and trait genetics, EHRs proved a valuable resource for analyzing pharmacogenetic traits and developing reverse genetics approaches such as phenome-wide association studies (PheWASs). PheWASs are designed to survey which of many phenotypes may be associated with a given genetic variant. PheWAS methods have been validated through replication of hundreds of known genotype-phenotype associations, and their use has differentiated between true pleiotropy and clinical comorbidity, added context to genetic discoveries, and helped define disease subtypes, and may also help repurpose medications. PheWAS methods have also proven to be useful with research-collected data. Future efforts that integrate broad, robust collection of phenotype data (e.g., EHR data) with purpose-collected research data in combination with a greater understanding of EHR data will create a rich resource for increasingly more efficient and detailed genome-phenome analysis to usher in new discoveries in precision medicine.
Keywords: phenome-wide association study, genome-wide association study, electronic health record, phenotyping
INTRODUCTION
The ultimate goal of genomic investigation is to determine the molecular drivers underlying human traits and diseases. Discovery of the genomic basis of disease proceeded slowly until the publication of the first human reference genome sequence in 2003 and has dramatically accelerated in the last decade with the advent of increasingly efficient methods of interrogating the human genome. Large-scale, hypothesis-free methods for studying genomic variants—notably genome-wide association studies (GWASs) and technologies such as whole-genome and whole-exome sequencing—have led to numerous discoveries of the genomic bases of both rare and common diseases. Typically, these methods have focused on a single disease or a small set of diseases at a time in order to answer specific research questions, and traditional data sets with genetic data have ascertained a limited number of phenotypes to allow for study. In cohort studies, phenotypes are carefully and accurately ascertained but may have limited longitudinal information and are expensive to accrue.
In the mid-to-late 2000s, a few health care systems began to collect patient biospecimens, typically DNA, linked to electronic health record (EHR) data. The promise of these resources led to the funding of the Electronic Medical Records and Genomics (eMERGE) network in 2007 by the National Human Genome Research Institute (NHGRI) (23, 40). By 2010, the first successful genomic studies using EHR data had demonstrated that these data, collected as a by-product of clinical care, could be used to replicate known genomic associations using a conventional phenotype-to-genotype study design. They also pointed to a new class of study: the phenome-wide association study (PheWAS). Foreshadowed in earlier commentaries (22, 31), PheWAS is a reverse genetics approach that begins with a genotype and then systematically queries a large number of phenotypes (Figure 1). The PheWAS method was first demonstrated with EHR data in a 2010 study that used a systematic approach to replicate known associations (18). Subsequent studies have explored larger populations, richer and different modalities of phenotype definitions, and the use of PheWAS techniques with collections of research data (i.e., non-EHR data).
Figure 1.
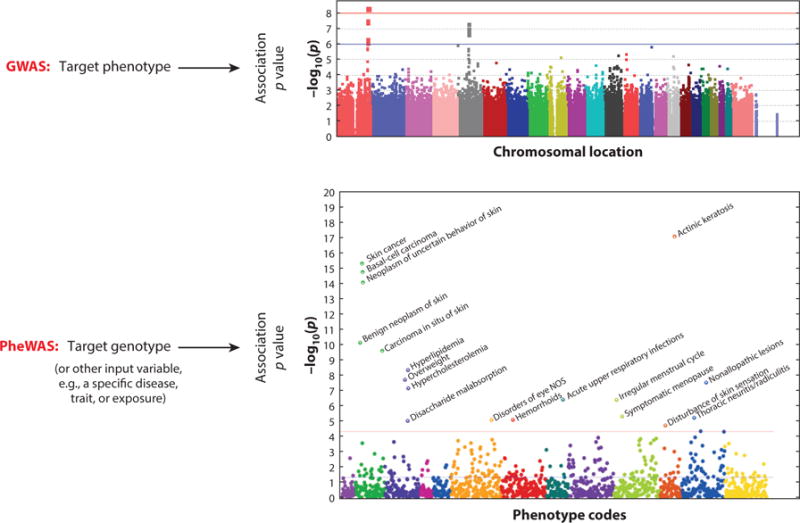
Genome-wide association studies (GWASs) versus phenome-wide association studies (PheWASs). Whereas GWASs usually study a single target phenotype across many genotypes (usually more than 500,000), PheWASs start with a single target genotype (or other independent variable) and analyze many phenotypes (usually more than 1,000). Adapted with permission from Reference 16 with permission from Nature Biotechnology.
The explosion of US and international biobanking efforts portends an exciting future in which massive amounts of EHR data, linked to genomic and other molecular data, will become available for millions of individuals. In addition to their use by the ten eMERGE network institutions, EHR data have been included in the UK Biobank (73) and China Kadoorie Biobank (6) and form the major source of longitudinal phenome information in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort (36), the US Department of Veterans Affairs’ Million Veteran Program (MVP) (http://www.research.va.gov/mvp), and the forthcoming US Precision Medicine Initiative Cohort Program (62). In the past five years, investigators using PheWAS methods have discovered numerous new associations, validated others, and uncovered true pleiotropy in the human genome. Phenome-wide approaches may also lead to new understandings of biology, uncover new therapeutic targets and predictions of side effects, and add to our understanding of diseases and prognosis.
This article reviews the use of EHR data for genomic research, PheWAS methods and applications, and prospects for the future use of phenome-wide approaches to advance our understanding of human disease. Additionally, we review some of the current challenges and future opportunities for PheWAS methods.
ELECTRONIC HEALTH RECORDS AS A TOOL FOR GENOMIC INVESTIGATION
EHR systems were initially designed to facilitate patient registration, transaction processing, and billing rather than to create a persistent record of clinical care. However, the capacity of these systems quickly grew as they became a clinical tool for recording, communicating, and securing care. The availability of clinical data combined with algorithms led to improved care through computerized decision support, diagnostic aids, population surveillance and health management, and improved compliance with recommendations. The earliest computerized decision support systems were being used to improve care before personal computers were commonly available (41, 43). The aggregation of robust, longitudinal clinical records and large populations quickly gave rise to the potential of EHRs as a tool for clinical research as well (42, 82). Much of this research has been done using administrative (billing) data, but other projects highlight the advantages of using detailed laboratory or narrative data (e.g., from physician notes or radiology reports) to tailor antibiotic therapy, identify medication side effects (44), or examine the impact of physical exam findings (15) and laboratory results (39).
EHR-based genomic research, however, is a more recent practice, with the first successful study published in 2010 (79). To be useful for genomic research, EHR data must be linked to a biospecimen resource. The scale of genomic research, often involving thousands or even tens of thousands of individuals, requires automated methods to accrue such samples. Many such efforts have been undertaken by single institutions or networks (e.g., Vanderbilt University, Kaiser Permanente, and Geisinger Health System), and broader national efforts have also been explored (e.g., the UK Biobank, China Kadoorie Biobank, and MVP). Each of these biobanks contains EHR data of varying depths, and several of them (e.g., the UK Biobank) collect robust participant-collected phenotypic data as well.
Among the research cohorts using EHR data, the sites of the eMERGE network have been among the most prolific in conducting genomic research studies based solely on EHR data. An initial study using Vanderbilt’s DNA biobank, BioVU, deployed algorithms to identify individual cases and controls for five diseases [multiple sclerosis, rheumatoid arthritis (RA), Crohn’s disease, type 2 diabetes, and atrial fibrillation] and tested for replication with 21 single-nucleotide polymorphisms (SNPs) already known to be associated with these diseases (66). All adequately powered associations were replicated, and each disease was represented among these replications. GWASs conducted by eMERGE researchers identified associations with red and white blood cell indices, type 2 diabetes, cardiac conduction, dementia, and platelet size and volume, among others (10). EHR data linked to extant genotypes were evaluated for a novel phenotype of primary hypothyroidism, which identified a new genetic locus, forkhead box E1 (FOXE1), associated with this disease (17). This final result demonstrates that, once genotyped for a given condition, the samples could be reused to identify associations with other conditions, which considerably extends the utility of these types of cohorts.
EHR data also have proven utility for studying drug-response traits. Such traits, especially adverse drug reactions, require longitudinal data, can require large population sizes, and may have potentially lethal outcomes, making prospective assessment essential. EHR data have been used to replicate known associations with clopidogrel drug response (13) and warfarin stable dose (64). Novel drug-response associations have been identified using EHR data for vancomycin stable dose (74), bleeding events associated with chronic warfarin use (34), heparin-induced thrombocytopenia (32), and angiotensin-converting enzyme (ACE) inhibitor–induced cough (49). A large meta-analysis combined both clinical trial data and EHR data to identify genetic loci associated with the cholesterol-lowering effect of statins, demonstrating that EHR and trial data can be combined and show similar effects for pharmacologic endpoints (61). Importantly, process analyses suggest that EHRs can be faster, more efficient, and more cost effective than traditional research models for pharmacogenetic analyses (2).
These studies form a carefully validated and diverse story of the use of EHR-linked biobanks for genomic research. They also help elucidate EHR data elements that compose the EHR phenome for study via PheWAS methods, which are discussed in the following sections.
FINDING RESEARCH-GRADE PHENOTYPES IN ELECTRONIC HEALTH RECORDS
Research from the eMERGE network has demonstrated the effectiveness of an iterative approach to creating research-quality phenotypes, a finding that has been corroborated by experiences at other sites and within other networks (54, 79). The development of more than 40 of these phenotypes through eMERGE has shown that high-quality phenotypes tend to leverage multiple classes of EHR data, including four major types of EHR data: billing codes [from the International Classification of Diseases (ICD) and Current Procedural Terminology (CPT)], medication histories, laboratory and test results, and clinical narratives (14, 79). The latter three data types often require text mining and/or natural language processing methods (see below) to extract meaningful and structured information from the narrative text often found in these types of clinical data. Each of these five elements contains a wealth of information, as summarized in Table 1.
Table 1.
Large data classes available in electronic health records (EHRs)
| ICD codes | CPT codes | Laboratory data | Medication records | Clinical documentation | |
|---|---|---|---|---|---|
| Recall | Medium | Poor | Medium | Inpatient: High Outpatient: Variable |
Medium |
| Precision | Medium | High | High | Inpatient: High Outpatient: Variable |
Medium high |
| Query method | Structured | Structured | Mostly structured | Structured, text queries, and natural language processing | Text queries, natural language processing, rarely structured |
| Strengths | Easy to query Serves as a good first pass of disease status |
Easy to query High precision |
Value depends on test High data validity |
Can have high validity and marker of severity | Best record of what providers thought |
| Weaknesses | Disease codes are often used for screening Accuracy is hindered by billing realities and clinic workflow |
Most susceptible to missing data errors Influenced by patient and payer factors |
Normal ranges and units may change over time Aligning labs can be challenging |
Often requires interfacing inpatient and outpatient records Medications prescribed are not necessary taken |
Requires natural language processing May suffer from significant cut and paste May be self-contradictory |
| Has been used in PheWASs to date | Yes (often) | No | Limited | No | In pilot studies |
| Current challenges in PheWASs | Transitioning from ICD9 to ICD10 in the United States | Missing data | Aligning across sites and identifying normal values | Combining brand and generic names Mapping different forms (ophthalmic versus intravenous) |
Extracting robust sets of concepts from notes |
With the advent of the meaningful-use requirements and linked incentive payments from the Centers for Medicare and Medicaid Services, adoption of EHRs capable of logging each of the data types shown in this table has increased significantly; US adoption rates of certified EHRs currently exceed 90% for large hospitals (55). Abbreviations: CPT, Current Procedural Terminology; ICD, International Classification of Diseases; ICD9/10, International Classification of Diseases ninth and tenth editions, respectively; PheWAS, phenome-wide association study.
Natural language processing is the application of computer algorithms to abstract computable “facts” from unstructured narrative documents (e.g., clinical notes, electrocardiogram interpretations, or radiology reports). To be useful, medical natural language processing often involves recognizing medical concepts from narrative text and matching them to controlled vocabularies such as the Systematized Nomenclature of Medicine (SNOMED) or Unified Medical Language System (UMLS) (29), the latter of which is an interlingua of more than 100 distinct controlled vocabularies, including SNOMED. Both SNOMED and UMLS group like terms (e.g., congestive heart failure and its abbreviation, CHF) into single concepts. Natural language processing algorithms have also been devised to accurately determine concept negation (e.g., “no chest pain”) (5), detect about whom a concept refers (e.g., “her father had a myocardial infarction”) (19, 26), and extract detailed medication signature information (e.g., “metformin 500 mg bid”) (83).
Among these phenotype elements, ICD diagnostic billing codes remain the most commonly used element in electronic phenotype algorithms. Combining these codes with medication exposures or laboratory results can improve the positive predictive value of a phenotype algorithm without significantly altering sensitivity (i.e., the true positive rate) (66, 80). Certain phenotypes, however, are not expressed within billing codes and can be found only via natural language processing, in laboratory data, or by combining multiple elements. These include the drug-related traits discussed above, endophenotypes (such as detailed electrographic traits or red blood cell size), or specific diseases that may not be adequately represented by billing codes.
METHODS AND VALIDATION FOR PHENOME-WIDE ASSOCIATION STUDIES IN ELECTRONIC HEALTH RECORDS
The essential process in a PheWAS is to identify a large list of phenotypes, ideally collected systematically (and not restricted to phenotypes of predefined interest). Transformations may need to be performed on the phenotype data to generate cases and controls, after which the sets of cases and controls are tested for SNPs using analyses similar to those in other genetic studies (Figure 2).
Figure 2.
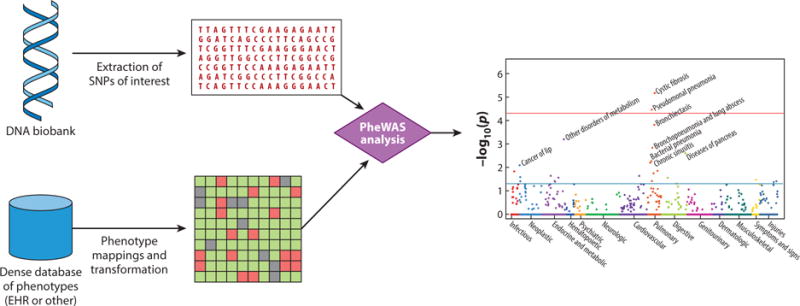
Phenome-wide association studies (PheWASs). A PheWAS begins with identification of a genetic variant of interest, such as a single-nucleotide polymorphism (SNP). For a PheWAS using electronic health records (EHRs), phenotypes are then extracted, and transformations are often made to map raw EHR data to defined cases and controls for analysis. A typical transformation would take ~14,000 diagnostic billing codes and identify ~1,600 distinct case phenotypes, each matched to a control group. A PheWAS analysis is then performed to test for associations between the SNP and each phenotype, using typical statistical genetics methods.
Although the majority of PheWAS investigations have been performed with EHR data, other cohorts with robustly collected observational data can be used (these are discussed in more detail in the next section). Having a broad set of phenotypes collected by a method unbiased toward prespecified outcome classes is essential, as narrow groups of phenotypes or preselected phenotype domains limit the ability of PheWASs to discover truly novel and unexpected associations. A clinical trial may collect large numbers of variables, but they may also all be closely related to the outcomes of interest to the trial.
To date, the most common data modality used for PheWASs has leveraged billing information in the form of ICD codes from either the ninth edition (ICD9, used in the United States until October 2015) or the tenth edition (ICD10, generally used internationally and in the United States after October 2015). This is likely due to the ubiquity of these codes in EHR systems, their generally robust coverage of human disease, and their ease of use, particularly in cross-institutional studies. Despite concerns about their accuracy, given that they are typically collected to facilitate remuneration, ICD codes have proven to be effective for PheWASs.
ICD codes were used in the first demonstration of the PheWAS method in 2010, in which 6,005 individuals in an EHR-linked biobank were assigned case or control status for 744 phenotypes using a custom grouping of ICD9 codes (18). These 744 derived phenotypes have become known as phecodes. The individuals were genotyped for five SNPs with known genetic associations. For example, the ICD9 code system includes multiple type 1 and type 2 diabetes codes (all in the 250.* range). Each phecode has a control definition that specifies similar conditions that should not be present in controls. Thus, for the type 2 diabetes phecode, individuals with any of the diabetes codes or related codes such as secondary diabetes mellitus (249.*) or other abnormal blood glucose (790.29) cannot serve as controls because these individuals may indeed have diabetes. Another example of disparate ICD codes aggregated into a single phecode is tuberculosis, which occurs in ICD9 codes 010 to 018 (primary tuberculosis), 137 (late effects of tuberculosis), and 647.3 (tuberculosis complicating the peripartum period). Figure 3 shows an example of these mappings. After a set of phenotypes (cases and controls) are identified, the PheWAS approach is to test each phenotype serially against a given genotype in much the same manner as GWAS tests many genetic variants against a single phenotype (Figure 1). In this initial PheWAS, four of the seven previously known associations were replicated, thus demonstrating the potential of the method. The results also highlighted several potential novel associations, one of which (erythematous conditions, which included rosacea) was subsequently replicated (28).
Figure 3.
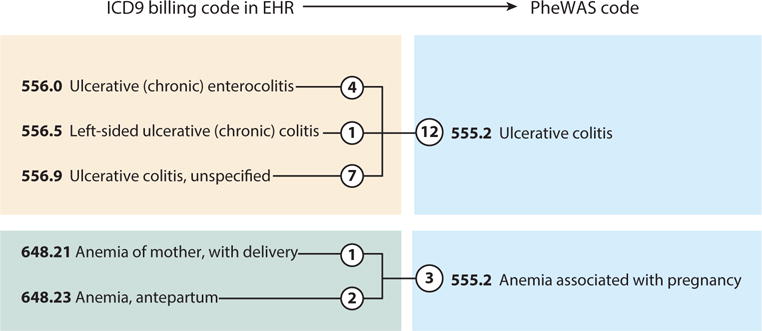
Phecode mappings of codes from the ninth edition of the International Classification of Diseases (ICD9). In this example, the individual has five unique ICD9 codes that map to two phenome-wide association study (PheWAS) phecodes. The circled numbers indicate the number of occurrences of each code in the individual’s electronic health record (EHR). Typically, a code must be billed two or three times in order to be considered a case for the phecode.
Since the 2010 study (18), the PheWAS approach has been revised to include more than 1,700 hierarchical phecodes by (a) including personal history and accident codes (the V and E codes in the ICD9 coding schema); (b) redesigning the code system to include hierarchical relationships, such that one phenotype could be a parent of another child phenotype (e.g., cardiac arrhythmias is a parent of atrial fibrillation, atrial flutter, and other arrhythmias); and (c) including more granular phenotypes in the coding system (e.g., type 1 diabetes with ketoacidosis, which has a parent phenotype of type 1 diabetes) (16). The development of parent hierarchical phenotypes included the creation of phenotypes not present in the ICD9 billing hierarchy, such as inflammatory bowel disease as the parent phenotype for Crohn’s disease and ulcerative colitis. The example in Figure 3 is of the current hierarchy (version 1.2, downloadable from http://phewascatalog.org). Versions of this hierarchical code grouping have been used for many PheWASs, including those involving children (51, 52) and adults (4, 11, 12, 16–18, 48, 53, 67, 70), for subtyping disease (see below) (21), and for other uses (84).
PheWASs can also be performed using raw, ungrouped ICD codes. Based on studies at the Marshfield Clinic, Hebbring et al. (28) published the second PheWAS, using data from the Personalized Medicine Research Project’s EHR-linked biobank to performed a PheWAS on HLA-DRB1*1501, a variant also tested in the first PheWAS. This study used raw ICD9 codes and ICD9 codes grouped into their natural three-digit groupings (e.g., 250.01 → 250). Their results replicated known associations, validated another association suggested in the first PheWAS (erythematous conditions, as mentioned above), and provided evidence that the approach is transportable.
Neuraz et al. (53) at the Hôpital Européen Georges-Pompidou mapped ICD10 codes to phe-codes to correlate thiopurine methyltransferase (TPMT) activity with phenotypes in patients with inflammatory bowel disease, noting that those with increased TPMT activity were more likely to have outcomes associated with inadequate treatment with thiopurine immunosuppressants. They found that mapping ICD10 codes to the original phecode groupings, via an intermediate step of mapping ICD10 to ICD9 codes, yielded more informative results compared with using the “natural” ICD10 groupings.
Pathak et al. (57) demonstrated another method of grouping ICD9 codes for a PheWAS—the Agency for Healthcare Research and Quality’s Clinical Classifications Software (CCS) (9). They leveraged semantic web technologies to identify patients matching known eMERGE case/control phenotype definitions and then mapped ICD9 codes to 285 mutually exclusive diagnoses and 231 procedure categories. CCS supports both single-level groupings and 727 multilevel hierarchical groupings. The CCS code mappings aggregate at higher levels than phecodes, which results in more general child concepts. For example, although there are separate phecodes for type 1 and type 2 diabetes, CCS groups these codes together under 3.2 (diabetes mellitus without complication) and 3.3 (diabetes mellitus with complications). Several common diseases that are represented among phecodes (e.g., gastroesophageal reflux disease) are grouped into nonspecific CCS groupings (e.g., other esophageal disorders).
Studies have demonstrated that requiring multiple instances of a diagnosis code on different days (because a given billing code can occur multiple times during the same visit, for instance, to pay for both a visit and a laboratory test) improved the precision of the phenotypes in PheWASs. In an analysis of ten diseases, using two or more ICD codes (billed on different days) improved the average positive predictive value from 0.71 to 0.84 (80). These results echo findings implicit in several phenotype algorithms that use ICD code counts as a feature [e.g., for Crohn’s disease (66), peripheral artery disease, and RA (38)], and other research studies on administrative data have also implemented such code count thresholds. As a result, most PheWAS analyses have required two or three of the matching ICD codes within a code grouping (i.e., a phecode) to be considered a case for that phenotype. In this approach, individuals with fewer than the target number of codes for a given phenotype are considered neither a case nor a control and thus are excluded from the association test.
A systematic validation of the PheWAS method to replicate known associations tested all known GWAS-discovered associations that were reported in the NHGRI GWAS catalog at the time (16). This PheWAS of 3,141 SNPs in 13,835 individuals of European ancestry replicated 210 of 751 (28%) known SNP-disease associations, including 66% (51 of 77) of the associations for which the analysis was adequately powered (Figure 4). Using a reference standard derived from phenotypes reported in the NHGRI GWAS catalog, the area under the receiver operator characteristic curve for the PheWAS approach was 0.83.
Figure 4.
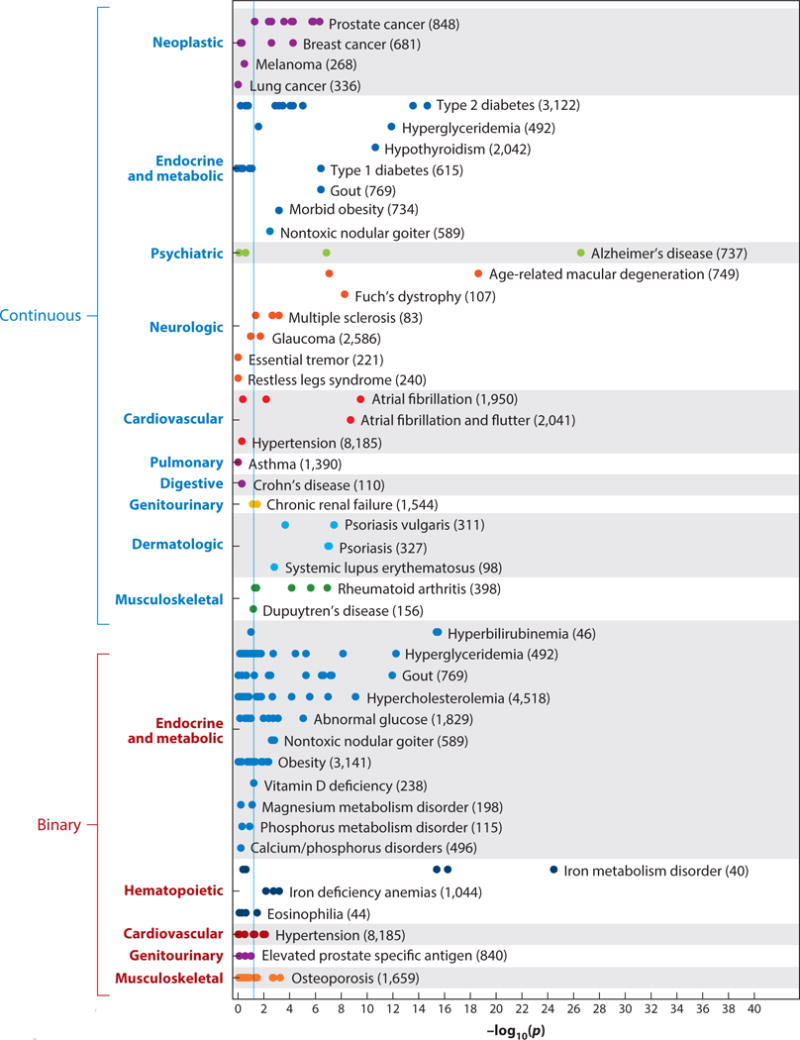
Replication of known single-nucleotide polymorphism (SNP)–phenotype associations by a phenome-wide association study (PheWAS). Each point represents a distinct SNP tested for the phenotype indicated. The numbers in parentheses represent the sample size within the PheWAS data set. The vertical blue line represents p = 0.05. Adapted from Reference 16 with permission from Nature Biotechnology.
PheWASs can be implemented using billing codes in several ways. The original PheWAS was performed using a Perl program, although this is no longer supported in favor of a package for the R statistical language, which can support a variety of different phenotypes, both continuous and categorical independent variables (e.g., for PheWASs of laboratory values and other nongenetic input), more advanced statistical approaches, and graphical outputs (4). The mappings of billing codes to phecodes and the R PheWAS package are available at http://phewascatalog.org. The R PheWAS package can generate cases and controls for PheWAS phenotypes according to prior published code mappings and can also support alternative ones, and users can specify their own code count threshold to instantiate cases (e.g., requiring three matching codes on different days to be a case).
PheWAS-View (60) and PhenoGram (81) are stand-alone programs that can graphically represent PheWAS results for given genetic variants and along chromosomal ideograms, respectively. The R PheWAS package also enables some simple graphical displays of results. Additionally, standard genetic analysis tools such as PLINK and PLATO also support the testing of multiple phenotypes against genotypes (24, 63).
DISCOVERIES USING PHENOME-WIDE ASSOCIATION STUDIES
Early PheWAS analyses were primarily designed not to reveal new findings but to develop the methodology and validate the approach. In the process of developing methods, several studies noted new associations at nonsignificant thresholds; statistical challenges in analysis of PheWAS data are discussed below. More recently, larger PheWASs have yielded new discoveries. The PheWAS examining known NHGRI GWAS catalog variants identified 63 new associations surpassing a false discovery rate of <0.1; 6 of 7 were replicated with a separate EHR-linked data set using physician-validated natural language processing–based phenotype algorithms (16). Novel associations included interferon regulatory factor 4 (IRF4) variants with actinic keratosis (p = 4.1 × 10−26), telomerase reverse transcriptase (TERT) variants with seborrheic keratosis (p = 1.6 × 10−7), and IRF4 and tyrosinase (TYR) variants with nonmelanoma skin cancers (p < 3 × 10−10). This PheWAS also made it clear that IRF4 is associated with multiple phenotypes associated with skin pigmentation (Figure 5). A recent analysis replicated these associations with actinic keratosis and extended the study with a full body skin examination (30). Adjusting the analysis for pigmentation suggested a pleiotropic effect of IRF4, TYR, and melanocortin 1 receptor (MC1R) on both pigmentation and processes that lead to actinic keratosis, a precancerous skin lesion.
Figure 5.
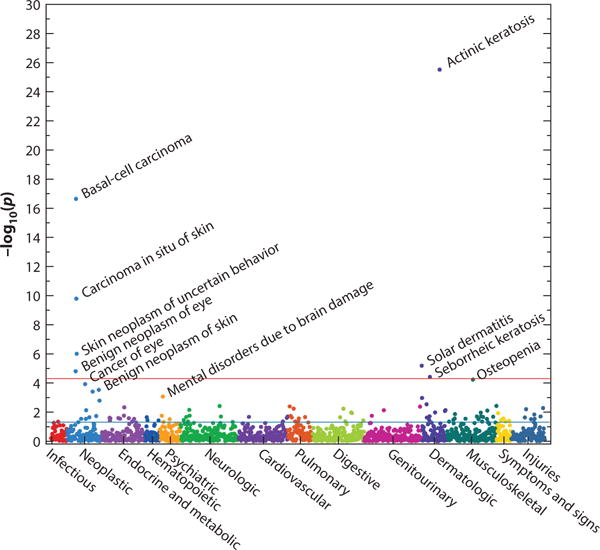
A phenotype-wide association study (PheWAS) plot of rs12203592 in IRF4. The horizontal red line indicates a Bonferroni correction for the number of phenotypes tested in this PheWAS (p = 0.05/1,358 = 3.7 × 10−5); the horizontal blue line indicates p = 0.05. The analysis shows that this single-nucleotide polymorphism is associated with several phenotypes related to sun exposure, such as actinic keratosis, basal cell carcinomas, osteopenia, and solar dermatitis (sunburns); these were new discoveries in this PheWAS. Figure drawn using data derived from Reference 16 with permission from Nature Biotechnology.
Most PheWASs have been performed using common genetic variants. Ye et al. (85) performed a PheWAS on stop-gain and stop-loss variants, identifying a nonsense variant in age-related maculopathy susceptibility 2 (ARMS2; rs2736911) associated with age-related macular degeneration. A large study of 2,476 variants in pediatric populations replicated several known associations and identified new associations between variants near NEDD4 family–interacting protein 1 (NDFIP1) and mental retardation, between phospholipase C–like 1 (PLCL1) and developmental delays, and between a cluster of SNPs in the interleukin 5 (IL5)–IL13 region and eosinophilic esophagitis (51). Other new discoveries have been made in conjunction with GWASs and through observational cohorts (both discussed in more detail below).
Simonti et al. (71) used the PheWAS method to explore the phenotypic influence of Neandertal admixture in modern humans. The authors tested alleles derived from a Neandertal lineage in 28,416 adults and found associations with neurologic, psychiatric, immunologic, and dermatologic phenotypes. Individual Neandertal alleles were associated with hypercoagulable states and tobacco use. In addition to performing individual SNP association tests, this study explored the aggregate influence of Neandertal alleles on diverse phenotypes using mixed linear models, demonstrating associations with depression and actinic keratosis. Overall, Neandertal alleles were associated with more neurologic and psychiatric phenotypes and fewer digestive phenotypes than randomly chosen SNPs. This study highlights unexpected use of clinically annotated data sets to investigate basic science questions—in this case, evolutionary biology.
USING PHENOME-WIDE ASSOCIATON STUDIES IN CONJUNCTION WITH GENOME-WIDE ASSOCIATION STUDIES
One use of the PheWAS method is to better characterize genetic variants discovered via a GWAS for a given trait or disease. Along these lines, investigators performed a GWAS of cardiac conduction in heart-healthy individuals and then analyzed 23 variants associated with cardiac conduction duration using the PheWAS method (67). This PheWAS demonstrated that SNPs in the sodium channel, voltage gated, type X alpha subunit (SCN10A) gene also predict future development of atrial fibrillation. A time-to-event analysis with a Cox proportional hazards model verified this finding in the original, heart-healthy population. Shameer et al. (70) performed a GWAS of platelet count and size, noting that these genetic variants had pleiotropic associations with myocardial infarction, autoimmune diseases, and hematologic disorders. Similarly, PheWASs have been used to confirm GWAS results for hypothyroidism (17) and herpes zoster (13). Table 2 summarizes these associations and their findings.
Table 2.
Phenome-wide association studies (PheWASs) used as an adjunct to genome-wide association studies (GWASs)
| Study | GWAS phenotype | GWAS-identified loci | PheWAS finding from GWAS loci |
|---|---|---|---|
| Denny et al. 2011 (17) | Hypothyroidism | FOXE1 | Hypothyroidism, Hashimoto’s thyroiditis, goiter |
| Denny et al. 2013 (16) | NHGRI GWAS catalog associations (numerous) | 3,141 SNPs | 63 novel results, including actinic keratosis |
| Ritchie et al. 2013 (67) | QRS duration in normal hearts | SCN10A (and other loci) | SCN10A also associated with atrial fibrillation; other loci not |
| Shameer et al. 2014 (70) | Platelet count and volume | 5 regions influencing count and 8 influencing volume | Myocardial infarction, autoimmune disorders, hematologic disorders |
| Karol et al. 2015 (33) | Steroid-associated osteonecrosis | GRIN3A | Several ischemic vascular phenotypes |
| Namjou et al. 2015 (52) | Liver function tests | 5 loci, including UGT1A and TA7 | Suggestive association between TA7 and cerebral ischemia |
| Crosslin et al. 2015 (12) | Herpes zoster | HCP5 | Herpes zoster and suggestive associations with other inflammatory diseases |
Abbreviations: NHGRI, National Human Genome Research Institute; SNP, single-nucleotide polymorphism.
Before 2014, many studies had identified associations between intronic variants in the fat mass and obesity associated (FTO) gene and obesity, which is also associated with type 2 diabetes. Cronin et al. (11) used a PheWAS to investigate exonic and intronic FTO variants. Only SNPs in linkage disequilibrium with the known intronic obesity-associated SNP were associated with obesity; other variants had weak associations with non-obesity-related traits. Conclusive studies published in 2014 and 2015 demonstrated that the presence of these intronic variants was associated with obesity via regulation of Iroquois homeobox 3 (IRX3) and IRX5, not via a change in function in FTO itself (7, 72).
USING PHENOME-WIDE ASSOCIATION STUDIES TO DEFINE COMORBIDITIES AND PRECISION SUBSETS OF DISEASE
Phenome-wide analyses of EHR data are not limited to using genetic data as the input function. The availability of a curated human phenome enables PheWASs to identify comorbidities associated with a given trait or disease (1, 21), to identify associations with laboratory results (39, 53, 75, 77), to identify subtypes of diseases using the PheWAS as a vector of defined comorbidities (21), and to enable population-based health-service-type research.
The ability to rapidly and comprehensively characterize individuals’ disease profiles over time enables rapid characterization of a population for potential subtypes of disease. Doshi-Velez et al. (21) at Harvard Medical School found differential comorbidity landscapes among autism spectrum disorder patients. Repeating this process at Vanderbilt identified the same subgroups of autism spectrum patients, as represented in Figure 6.
Figure 6.
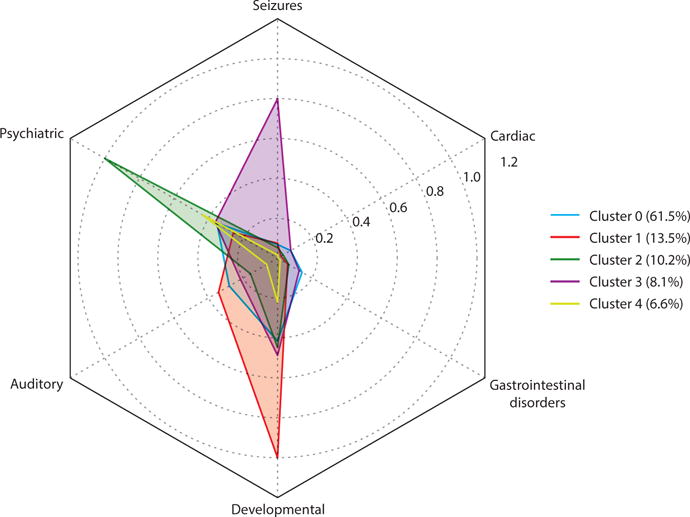
Clustering of autism spectrum disorders (ASDs) based on phenome-wide association study (PheWAS) comorbidities in a Vanderbilt study. This study used the approach applied in Reference 21 to identify different subpopulations of ASD patients by their comorbidities. Unsupervised hierarchical clustering was performed on all individuals identified as having an ASD. The five identified clusters are represented by the types of codes found in each cluster. For example, cluster 2 identifies 10.2% of patients with an ASD, and nearly all of these individuals had a psychiatric phecode. In cluster 3, 80% of the individuals had a seizure phecode.
PheWASs may also be used to explore the relationship between traits and clinical outcomes. Boland et al. (1) at Columbia University used a PheWAS method to find comorbid conditions associated with periodontal disease, noting associations with diabetes, hypertension, and hypercholesterolemia. This group later performed a PheWAS with birth month as the independent variable and analyzed for subsequent development of diseases, finding that the lifetime disease risk of 55 diseases was affected by birth month. Liao et al. (39) used a PheWAS to investigate associations with autoantibodies. Warner & Alterovitz (75) applied the PheWAS method to discover specific ranges of white blood cell counts that were correlated with specific diseases. Other studies by Warner and colleagues noted diseases predicting longer lengths of stay (77) and treatment-related complications of multiple myeloma (76).
In one experiment showing genetic differences between disease subclasses, Li et al. (37) used topological analyses of diverse clinical comorbidities (using CCS codes) of type 2 diabetes patients to identify three distinct groups of diabetic patients. One subtype clustered the commonly associated type 2 diabetes complications of nephropathy and retinopathy, whereas the other two contained malignancies and cardiovascular disease, among other diseases. Importantly, they identified distinct SNPs and genes associated with each disease subtype. Such results would need to be replicated but provide an important demonstration that genetic risk could suggest future comorbid disease risk or define new disease subtypes that could lead to personalized treatment and monitoring.
PHENOME-WIDE ASSOCIATION STUDIES MAY PREDICT APPROPRIATE MEDICATION AND ADVERSE DRUG REACTIONS
The cost of generating new therapeutics has risen dramatically over the past 60 years, with each new drug costing approximately 80 times as much in 2010 as it did in 1960, in inflation-adjusted terms (69). Thus, the promise of high-throughput computational approaches to drug discovery and repurposing is attractive.
A growing body of evidence suggests that genetic association data for a given disease may predict drug targets for that disease. Sanseau et al. (68) found that 15.6% of genes identified in GWASs are existing drug targets (compared with 5.7% of the genome as a whole). In support of this finding, a multiethnic GWAS of 103,638 cases and controls for RA noted 101 risk loci; these loci identified 18 of 27 current RA drug target genes directly or via protein-protein interactions and identified three approved cancer medications that may be active against RA (56).
Existing evidence for currently marketed drugs further suggests the validity of using genetic variants (especially loss-of-function variants) to predict effective drug targets. Perhaps the best known example of this is proprotein convertase subtilisin/kexin type 9 (PCSK9). Loss-of-function variants in PCSK9 dramatically reduce low-density lipoprotein cholesterol and coronary disease (8), and medications inhibiting PCSK9 that were developed after these genetic studies have proven very effective at reducing low-density lipoprotein cholesterol and recently won regulatory approval. Similar studies have shown that ezetimibe, via loss-of-function variants in its target Niemann-Pick C1–like 1 (NPC1L1), should also reduce the risk of coronary disease (50). A randomized clinical trial recently validated this association (3).
Thus, evidence suggests that PheWASs may be able to identify disease indications and possible adverse drug effects for a target gene (e.g., one for which an agonist or inhibitor has been developed). As a proof of concept, Diogo et al. (20) sequenced candidate immunogenes to discover variants predisposing to RA, identifying loss-of-function variants in tyrosine kinase 2 (TYK2) associated with RA. The authors then studied these loss-of-function variants in 29,377 individuals via a PheWAS, replicating protective associations with RA. Potential side effects of TYK2 inhibition could be identified by phenotypes whose risk was increased by these variants. The strongest risk phenotype was pneumonia (odds ratio = 1.54, p = 0.004), which was not significant but nonetheless could represent a side effect of TYK2 inhibition.
In a broad-scale test of the potential for PheWASs to uncover drug indications, Rastegar-Mojarad et al. (65) evaluated the online PheWAS catalog (16) for medications that could potentially be repurposed. They used DrugBank to link genes from drug targets to the genes studied in the PheWAS catalog, then looked for co-occurrence between the drug and disease in MEDLINE abstracts and clinical trial data. By permutation analysis, they found that the PheWAS results significantly enhanced the probability of finding MEDLINE evidence for the indication. Overall, they identified 127 known drug-indication pairs and 2,583 strongly supported drug-indication pairs, significantly more than were found in the NHGRI GWAS catalog using similar methods.
PHENOME-WIDE ASSOCIATION STUDIES USING NON–ELECTRONIC HEALTH RECORD PHENOTYPES
The PheWAS methodology of simultaneously testing many phenotypes for individual SNPs has also been applied to non-EHR-based, population-based studies with predefined phenotypes. The first such framework and pipeline to leverage research data for a PheWAS was in the Population Architecture Using Genomics and Epidemiology (PAGE) network, which comprises diverse populations collected from research cohorts with research-collected phenotypes (59). Pendergrass et al. (58) later studied more than 70,061 individuals across the PAGE network using 4,706 phenotypes. They noted 33 novel phenotypes at p < 0.01 in two or more PAGE sites. Other studies have looked at mitochondrial SNPs (46), human immunodeficiency virus (HIV) clinical trial data (47), and multiethnic populations within the National Health and Nutrition Examination Survey (NHANES) (25). Collectively, these studies have shown the value of the systematic analysis of research-collected data to discover novel associations and better characterize existing ones.
Mendelian randomization is an approach by which genetic variants are used as an instrument variable to test whether an association between an exposure or trait and an outcome is causal or not. For example, this approach has demonstrated that low-density lipoprotein levels are causally associated with cardiovascular disease, whereas high-density lipoprotein levels are likely not causal. Millard et al. (45) used a PheWAS approach as a modality for performing Mendelian randomization among phenotypes potentially associated with body mass index (BMI). They evaluated the association of 172 phenotypic outcomes from an observational cohort with a BMI genetic risk score and found that 21 of the 172 (12%) were associated with the genetic BMI risk at p < 0.05. Among the strongest associations were lipid levels and blood pressure, validating that high BMI is indeed causal for these outcomes.
PheWASs using EHR and non-EHR research data highlight both advantages and disadvantages of each approach. Research data are often systematically collected, typically at prescribed intervals, from the population. The protocols for obtaining laboratory measurements, phenotype information, and disease definitions are standardized across sites, which is not true of EHR data. Despite these quality differences, research studies using EHR data consistently align with those from traditional research studies, as detailed above, including through phenome-wide approaches. A disadvantage of research phenotypes is that they are often limited to those considered of value to the research questions being asked. Thus, phenomes explored in research studies are often not as broad or as balanced as those collected via EHRs. Additionally, although EHR data are often not generated at regularly scheduled intervals, the frequency of contact in health care settings is often greater, and the number of observations may be much greater. For example, it would not be unusual for a single patient’s EHR to include hundreds or thousands of blood pressure measurements—far more than would be expected for a participant in a typical prospective research study. Finally, research studies may explore measurements (e.g., skin fold thickness), phenotypes (e.g., hair color), and analytes (e.g., trans fatty acid levels) not routinely acquired for EHRs. However, EHRs do routinely include robust and expensive testing that is not feasible except in the costliest of research studies.
Given the complementary nature of EHR and research-collected data—where a weakness in one approach is compensated by a strength in the other—one ought not consider these approaches to data collection mutually exclusive. Indeed, future studies, such as those envisioned with the US Precision Medicine Initiative Cohort Program, will likely incorporate both routine collection of EHR data and systematic collection of participant-provided research data.
CHALLENGES AND LIMITATIONS FOR PHENOME-WIDE ASSOCIATION STUDIES
PheWAS approaches are limited by several challenges. One of the biggest is achieving statistical significance given the large number of multiple hypotheses tested, especially when many genetic variants are tested. Although the number of phenotypes is relatively small compared with the number of possible genotypes tested in a typical GWAS, the Bonferroni correction grows with respect to the number of phenotypes (on the order of 103) × genotypes (on the order of 106) tested. Using less aggregated code systems (such as raw ICD codes) produces even more phenotypes, although the increase is typically less than a factor of 10. However, just as many genotypes are in linkage disequilibrium and thus not truly independent, many phenotypes are highly correlated. Taking this lack of independence into consideration may ease overly stringent correction thresholds. To this end, we must attain a deeper understanding of the true number of independent phenotypes. Methods such as principal component analysis have been used to discover the size of the phenotype space, but more work is required (11).
Another challenge (and opportunity) is to develop methods that can distinguish pseudopleiotropy from true pleiotropy. Pseudopleiotropy can be defined as differences along a causal pathway, such as intronic FTO variants associating with type 2 diabetes via their effect on obesity (11) or a SNP influencing lung cancer and chronic obstructive pulmonary disease via its effect on smoking behavior. In these situations, PheWASs have a distinct advantage over traditional GWASs in that multiple phenotypes can be tested simultaneously in the same population and then mutually adjusted for in order to identify true independent associations. This process is analogous to that used to interpret GWAS data in which SNPs may be tested for independence within a linkage disequilibrium block. Tests for independence have been conducted in prior PheWAS investigations to separate signals that were truly pleiotropic from those sharing clinical comorbidity (16, 17).
Another challenge is the accuracy of the phenotypes derived via PheWASs, especially for EHR data. Much prior work using EHR data has used purpose-defined and validated phenotyping algorithms; the PheWAS approach uses approximate methods that will necessarily vary in their precision. Combining multiple data sources and further refinements in the applied methods should improve performance (80), although this hypothesis has not been tested on a broad scale.
FUTURE DIRECTIONS
To date, PheWAS methods have been deployed primarily using billing code data. However, other rich clinical data are contained within EHRs, including laboratory tests, reports and imaging studies, and narrative documents such as clinical notes. A brief survey of the nearly 215,000 individuals in BioVU shows that the average person has 94 ICD9 codes but 132 clinical notes, 601 drug references, 596 laboratory tests, and 10 radiographic tests over a mean follow-up period of 5.7 years. A total of 5,948 distinct lab tests are included in BioVU. Thus, it is evident that a richer EHR phenome is available for investigation than has been explored. Combining these elements from EHR systems could prove highly informative by providing insights into the pathology of phenotypes and allowing the dissection of pleiotropy and pseudopleiotropy. Indeed, detailed results from expensive testing, such as imaging tests and cardiovascular procedures, although collected nonrandomly, provide an exciting modality to expand the types of information available for research.
Efforts to utilize greater portions of EHRs have been undertaken. In the first demonstration of the value of EHR text data, Hebbring et al. (27) identified 23,384 one- to four-word phrases occurring in clinical narratives that matched to medical concepts in the UMLS. In this approach, which they called a TextWAS, they replicated known associations for five SNPs with similar performance to using ICD9 codes for these diseases. Their approach highlighted certain other possible phenotypes seen via narratives that have not been part of prior PheWAS analyses, such as medications (e.g., Visudyne, a drug used to treat age-related macular degeneration, is associated with rs1061170, an age-related macular degeneration risk SNP). However, their work also highlights some of the remaining challenges of using text data, including difficulties related to identifying controls, a lack of term specificity, and textual ambiguity. For example, certain top-ranked text strings lacked clinical specificity, such as “of atrial” for rs220733 (associated with atrial fibrillation) and “spondylitis” for rs9501572 (associated with ankylosing spondylitis). Use of natural language processing tools to map text to concepts and incorporate negation detection and section tagging will ameliorate some of these concerns, but much work remains to be done.
The best results will likely come from combining multiple modalities of information into a single phenomic system. For example, a highly accurate algorithm for type 2 diabetes includes billing codes, laboratory results, and medication data (35), and resources have been created that link medications to their diseases (78). Similar resources do not yet exist for laboratory data and the logic necessary to link them. However, the systematic combination of data classes and their logic likely will not prove trivial. For example, although a fasting glucose exceeding 126 mg/dL may be part of the common diabetes definition, most fasting blood glucose values included in EHRs may not truly be fasting.
CONCLUSIONS
Numerous studies have demonstrated the value of using EHR data for genomic research. An outgrowth of research on this dense clinical data set was the observation that such data allow exploration of diverse phenotypes through phenomic scans, which have been fruitful in discovering new associations and differentiating between genetic pleiotropy and clinical comorbidity. The key use of PheWASs is investigating genetic variants of interest, derived either from another genomic study or through primary genomic investigation (e.g., cataloging the impact of nonsense variants). Such investigations hold promise for accelerating drug discovery. In addition, investigations such as PheWASs have opened up the possibility of subdividing the phenome, leading to a more nuanced and genetically informed classification of disease.
Goals such as disease redefinition and drug discovery are among those envisioned for the US Precision Medicine Initiative Cohort Program. This program will merge longitudinal EHR data with participant-generated research data collection, opening up new possibilities for joint phenomic approaches and enabling PheWASs on much larger populations than has heretofore been possible.
Currently, the disease and trait phenome has been the most explored. However, EHRs contain many more rich traits, including signs and symptoms from narrative documents, medication treatments, laboratory results, and image-defined traits. These phenotypes may form more elemental components of diseases than our current syndrome-oriented disease classifications, which are rooted in millennia of clinical observation.
Acknowledgments
This work was supported by the National Library of Medicine (R01 LM 010685), the National Institutes of Health’s Pharmacogenomics Research Network (U19 HL65962 and P50 GM115305), and the eMERGE network (U01 HG04603 and U01 HG008672).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
The Annual Review of Genomics and Human Genetics is online at genom.annualreviews.org
LITERATURE CITED
- 1.Boland MR, Hripcsak G, Albers DJ, Wei Y, Wilcox AB, et al. Discovering medical conditions associated with periodontitis using linked electronic health records. J Clin Periodontol. 2013;40:474–82. doi: 10.1111/jcpe.12086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bowton E, Field JR, Wang S, Schildcrout JS, Van Driest SL, et al. Biobanks and electronic medical records: enabling cost-effective research. Sci Transl Med. 2014;6:234cm3. doi: 10.1126/scitranslmed.3008604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–97. doi: 10.1056/NEJMoa1410489. [DOI] [PubMed] [Google Scholar]
- 4.Carroll RJ, Bastarache L, Denny JC. R PheWAS: data analysis and plotting tools for phenome-wide association studies in the R environment. Bioinformatics. 2014;30:2375–76. doi: 10.1093/bioinformatics/btu197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chapman WW, Bridewell W, Hanbury P, Cooper GF, Buchanan BG. A simple algorithm for identifying negated findings and diseases in discharge summaries. J Biomed Inform. 2001;34:301–10. doi: 10.1006/jbin.2001.1029. [DOI] [PubMed] [Google Scholar]
- 6.Chen Z, Chen J, Collins R, Guo Y, Peto R, et al. China kadoorie biobank of 0.5 million people: survey methods, baseline characteristics and long-term follow-up. Int J Epidemiol. 2011;40:1652–66. doi: 10.1093/ije/dyr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Claussnitzer M, Dankel SN, Kim K-H, Quon G, Meuleman W, et al. FTO obesity variant circuitry and adipocyte browning in humans. N Engl J Med. 2015;373:895–907. doi: 10.1056/NEJMoa1502214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72. doi: 10.1056/NEJMoa054013. [DOI] [PubMed] [Google Scholar]
- 9.Cowen ME, Dusseau DJ, Toth BG, Guisinger C, Zodet MW, Shyr Y. Casemix adjustment of managed care claims data using the clinical classification for health policy research method. Med Care. 1998;36:1108–13. doi: 10.1097/00005650-199807000-00016. [DOI] [PubMed] [Google Scholar]
- 10.Crawford DC, Crosslin DR, Tromp G, Kullo IJ, Kuivaniemi H, et al. Emerging progress in genomics—the first seven years. Front Genet. 2014;5:184. doi: 10.3389/fgene.2014.00184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cronin RM, Field JR, Bradford Y, Shaffer CM, Carroll RJ, et al. Phenome-wide association studies demonstrating pleiotropy of genetic variants within FTO with and without adjustment for body mass index. Appl Genet Epidemiol. 2014;5:250. doi: 10.3389/fgene.2014.00250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crosslin DR, Carrell DS, Burt A, Kim DS, Underwood JG, et al. Genetic variation in the HLA region is associated with susceptibility to herpes zoster. Genes Immun. 2015;16:1–7. doi: 10.1038/gene.2014.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delaney JT, Ramirez AH, Bowton E, Pulley JM, Basford MA, et al. Predicting clopidogrel response using DNA samples linked to an electronic health record. Clin Pharmacol Ther. 2012;91:257–63. doi: 10.1038/clpt.2011.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denny JC. Chapter 13: Mining electronic health records in the genomics era. PLOS Comput Biol. 2012;8:e1002823. doi: 10.1371/journal.pcbi.1002823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Denny JC, Arndt FV, Dupont WD, Neilson EG. Increased hospital mortality in patients with bedside hippus. Am J Med. 2008;121:239–45. doi: 10.1016/j.amjmed.2007.09.014. [DOI] [PubMed] [Google Scholar]
- 16.Denny JC, Bastarache L, Ritchie MD, Carroll RJ, Zink R, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31:1102–11. doi: 10.1038/nbt.2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Denny JC, Crawford DC, Ritchie MD, Bielinski SJ, Basford MA, et al. Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome- and phenome-wide studies. Am J Hum Genet. 2011;89:529–42. doi: 10.1016/j.ajhg.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Denny JC, Ritchie MD, Basford MA, Pulley JM, Bastarache L, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26:1205–10. doi: 10.1093/bioinformatics/btq126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denny JC, Spickard A, Johnson KB, Peterson NB, Peterson JF, Miller RA. Evaluation of a method to identify and categorize section headers in clinical documents. J Am Med Inform Assoc. 2009;16:806–15. doi: 10.1197/jamia.M3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diogo D, Bastarache L, Liao KP, Graham RR, Fulton RS, et al. TYK2 protein-coding variants protect against rheumatoid arthritis and autoimmunity, with no evidence of major pleiotropic effects on non-autoimmune complex traits. PLOS ONE. 2015;10:e0122271. doi: 10.1371/journal.pone.0122271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doshi-Velez F, Ge Y, Kohane I. Comorbidity clusters in autism spectrum disorders: an electronic health record time-series analysis. Pediatrics. 2014;133:e54–63. doi: 10.1542/peds.2013-0819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghebranious N, McCarty C, Wilke R. Clinical phenome scanning. Pers Med. 2007;4:175–82. doi: 10.2217/17410541.4.2.175. [DOI] [PubMed] [Google Scholar]
- 23.Gottesman O, Kuivaniemi H, Tromp G, Faucett WA, Li R, et al. The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet Med. 2013;15:761–71. doi: 10.1038/gim.2013.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grady BJ, Torstenson E, Dudek SM, Giles J, Sexton D, Ritchie MD. Finding unique filter sets in PLATO: a precursor to efficient interaction analysis in GWAS data. Pac Symp Biocomput. 2010;2010:315–26. [PMC free article] [PubMed] [Google Scholar]
- 25.Hall MA, Verma A, Brown-Gentry KD, Goodloe R, Boston J, et al. Detection of pleiotropy through a phenome-wide association study (PheWAS) of epidemiologic data as part of the Environmental Architecture for Genes Linked to Environment (EAGLE) study. PLOS Genet. 2014;10:e1004678. doi: 10.1371/journal.pgen.1004678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harkema H, Dowling JN, Thornblade T, Chapman WW. ConText: an algorithm for determining negation, experiencer, and temporal status from clinical reports. J Biomed Inform. 2009;42:839–51. doi: 10.1016/j.jbi.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hebbring SJ, Rastegar-Mojarad M, Ye Z, Mayer J, Jacobson C, Lin S. Application of clinical text data for phenome-wide association studies (PheWASs) Bioinformatics. 2015;31:1981–87. doi: 10.1093/bioinformatics/btv076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hebbring SJ, Schrodi SJ, Ye Z, Zhou Z, Page D, Brilliant MH. A PheWAS approach in studying HLA-DRB1*1501. Genes Immun. 2013;14:187–91. doi: 10.1038/gene.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Humphreys BL, Lindberg DA, Schoolman HM, Barnett GO. The unified medical language system: an informatics research collaboration. J Am Med Inf Assoc. 1998;5:1–11. doi: 10.1136/jamia.1998.0050001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacobs LC, Liu F, Pardo LM, Hofman A, Uitterlinden AG, et al. IRF4, MC1R and TYR genes are risk factors for actinic keratosis independent of skin color. Hum Mol Genet. 2015;24:3296–303. doi: 10.1093/hmg/ddv076. [DOI] [PubMed] [Google Scholar]
- 31.Jones R, Pembrey M, Golding J, Herrick D. The search for genenotype/phenotype associations and the phenome scan. Paediatr Perinat Epidemiol. 2005;19:264–75. doi: 10.1111/j.1365-3016.2005.00664.x. [DOI] [PubMed] [Google Scholar]
- 32.Karnes JH, Cronin RM, Rollin J, Teumer A, Pouplard C, et al. A genome-wide association study of heparin-induced thrombocytopenia using an electronic medical record. Thromb Haemost. 2014;113:772–81. doi: 10.1160/TH14-08-0670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karol SE, Yang W, Van Driest SL, Chang TY, Kaste S, et al. Genetics of glucocorticoid-associated osteonecrosis in children with acute lymphoblastic leukemia. Blood. 2015;126:1770–76. doi: 10.1182/blood-2015-05-643601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawai VK, Cunningham A, Vear SI, Van Driest SL, Oginni A, et al. Genotype and risk of major bleeding during warfarin treatment. Pharmacogenomics. 2014;15:1973–83. doi: 10.2217/pgs.14.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kho AN, Hayes MG, Rasmussen-Torvik L, Pacheco JA, Thompson WK, et al. Use of diverse electronic medical record systems to identify genetic risk for type 2 diabetes within a genome-wide association study. J Am Med Inform Assoc. 2012;19:212–18. doi: 10.1136/amiajnl-2011-000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kvale MN, Hesselson S, Hoffmann TJ, Cao Y, Chan D, et al. Genotyping informatics and quality control for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics. 2015;200:1051–60. doi: 10.1534/genetics.115.178905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li L, Cheng W-Y, Glicksberg BS, Gottesman O, Tamler R, et al. Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci Transl Med. 2015;7:311ra174. doi: 10.1126/scitranslmed.aaa9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liao KP, Cai T, Gainer V, Goryachev S, Zeng-Treitler Q, et al. Electronic medical records for discovery research in rheumatoid arthritis. Arthritis Care Res. 2010;62:1120–27. doi: 10.1002/acr.20184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liao KP, Kurreeman F, Li G, Duclos G, Murphy S, et al. Associations of autoantibodies, autoimmune risk alleles, and clinical diagnoses from the electronic medical records in rheumatoid arthritis cases and non-rheumatoid arthritis cases. Arthritis Rheum. 2013;65:571–81. doi: 10.1002/art.37801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCarty CA, Chisholm RL, Chute CG, Kullo IJ, Jarvik GP, et al. The eMERGE network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genom. 2011;4:13. doi: 10.1186/1755-8794-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonald CJ. Protocol-based computer reminders, the quality of care and the non-perfectability of man. N Engl J Med. 1976;295:1351–55. doi: 10.1056/NEJM197612092952405. [DOI] [PubMed] [Google Scholar]
- 42.McDonald CJ, Tierney WM. Computer-stored medical records: their future role in medical practice. JAMA. 1988;259:3433–40. [PubMed] [Google Scholar]
- 43.McDonald CJ, Wilson GA, McCabe GP. Physician response to computer reminders. JAMA. 1980;244:1579–81. [PubMed] [Google Scholar]
- 44.Melton GB, Hripcsak G. Automated detection of adverse events using natural language processing of discharge summaries. J Am Med Inf Assoc. 2005;12:448–57. doi: 10.1197/jamia.M1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Millard LAC, Davies NM, Timpson NJ, Tilling K, Flach PA, Smith GD. MR-PheWAS: hypothesis prioritization among potential causal effects of body mass index on many outcomes, using Mendelian randomization. Sci Rep. 2015;5:16645. doi: 10.1038/srep16645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mitchell SL, Hall JB, Goodloe RJ, Boston J, Farber-Eger E, et al. Investigating the relationship between mitochondrial genetic variation and cardiovascular-related traits to develop a framework for mitochondrial phenome-wide association studies. BioData Min. 2014;7:6. doi: 10.1186/1756-0381-7-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moore CB, Verma A, Pendergrass S, Verma SS, Johnson DH, et al. Phenome-wide association study relating pretreatment laboratory parameters with human genetic variants in aids clinical trials group protocols. Open Forum Infect Dis. 2015;2:ofu113. doi: 10.1093/ofid/ofu113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mosley JD, Brittain EL, Loyd JE, Denny JC, Austin ED, Larkin EK. Letter by Mosley regarding article, “Iron homeostasis and pulmonary hypertension: Iron deficiency leads to pulmonary vascular remodeling in the rat”. Circ Res. 2015;117:e56–57. doi: 10.1161/CIRCRESAHA.115.306867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mosley JD, Shaffer CM, Van Driest SL, Weeke PE, Wells QS, et al. A genome-wide association study identifies variants in KCNIP4 associated with ACE inhibitor-induced cough. Pharmacogenom J. 2016 doi: 10.1038/tpj.2015.51. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Myocard. Infarct. Genet. Consort. Investig. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371:2072–82. doi: 10.1056/NEJMoa1405386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Namjou B, Marsolo K, Caroll RJ, Denny JC, Ritchie MD, et al. Phenome-wide association study (PheWAS) in EMR-linked pediatric cohorts, genetically links PLCL1 to speech language development and IL5–IL13 to eosinophilic esophagitis. Front Genet. 2014;5:401. doi: 10.3389/fgene.2014.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Namjou B, Marsolo K, Lingren T, Ritchie MD, Verma SS, et al. A GWAS study on liver function test using emerge network participants. PLOS ONE. 2015;10:e0138677. doi: 10.1371/journal.pone.0138677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neuraz A, Chouchana L, Malamut G, Le Beller C, Roche D, et al. Phenome-wide association studies on a quantitative trait: application to TPMT enzyme activity and thiopurine therapy in pharmacogenomics. PLOS Comput Biol. 2013;9:e1003405. doi: 10.1371/journal.pcbi.1003405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Newton KM, Peissig PL, Kho AN, Bielinski SJ, Berg RL, et al. Validation of electronic medical record-based phenotyping algorithms: results and lessons learned from the emerge network. J Am Med Inform Assoc. 2013;20:e147–54. doi: 10.1136/amiajnl-2012-000896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Off. Natl. Coord. Health Inf. Technol. CMS Medicare and Medicaid EHR Incentive Program, electronic health record products used for attestation. Data Set, US Dep. Health Hum. Serv; Washington, DC: 2015. http://www.healthdata.gov/dataset/cms-medicare-and-medicaid-ehr-incentive-program-electronic-health-record-products-used. [Google Scholar]
- 56.Okada Y, Wu D, Trynka G, Raj T, Terao C, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2013;506:736–81. doi: 10.1038/nature12873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pathak J, Kiefer RC, Bielinski SJ, Chute CG. Applying semantic web technologies for phenome-wide scan using an electronic health record linked biobank. J Biomed Semant. 2012;3:10. doi: 10.1186/2041-1480-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pendergrass SA, Brown-Gentry K, Dudek S, Frase A, Torstenson ES, et al. Phenome-wide association study (PheWAS) for detection of pleiotropy within the Population Architecture Using Genomics and Epidemiology (PAGE) Network. PLOS Genet. 2013;9:e1003087. doi: 10.1371/journal.pgen.1003087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pendergrass SA, Brown-Gentry K, Dudek SM, Torstenson ES, Ambite JL, et al. The use of phenome-wide association studies (PheWAS) for exploration of novel genotype-phenotype relationships and pleiotropy discovery. Genet Epidemiol. 2011;35:410–22. doi: 10.1002/gepi.20589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pendergrass SA, Dudek SM, Crawford DC, Ritchie MD. Visually integrating and exploring high throughput phenome-wide association study (PheWAS) results using PheWAS-View. BioData Min. 2012;5:5. doi: 10.1186/1756-0381-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Postmus I, Trompet S, Deshmukh HA, Barnes MR, Li X, et al. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat Commun. 2014;5:5068. doi: 10.1038/ncomms6068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Precis. Med. Initiat. Work. Group. The Precision Medicine Initiative Cohort Program—building a research foundation for 21st century medicine. Rep., Precis. Med. Initiat. Work. Group, Natl. Inst. Health; Bethesda, MD: 2015. http://acd.od.nih.gov/reports/DRAFT-PMI-WG-Report-9-11-2015-508.pdf. [Google Scholar]
- 63.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ramirez AH, Shi Y, Schildcrout JS, Delaney JT, Xu H, et al. Predicting warfarin dosage in European-Americans and African-Americans using DNA samples linked to an electronic health record. Pharmacogenomics. 2012;13:407–18. doi: 10.2217/pgs.11.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rastegar-Mojarad M, Ye Z, Kolesar JM, Hebbring SJ, Lin SM. Opportunities for drug repositioning from phenome-wide association studies. Nat Biotechnol. 2015;33:342–45. doi: 10.1038/nbt.3183. [DOI] [PubMed] [Google Scholar]
- 66.Ritchie MD, Denny JC, Crawford DC, Ramirez AH, Weiner JB, et al. Robust replication of genotype-phenotype associations across multiple diseases in an electronic medical record. Am J Hum Genet. 2010;86:560–72. doi: 10.1016/j.ajhg.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ritchie MD, Denny JC, Zuvich RL, Crawford DC, Schildcrout JS, et al. Genome- and phenome-wide analyses of cardiac conduction identifies markers of arrhythmia risk. Circulation. 2013;127:1377–85. doi: 10.1161/CIRCULATIONAHA.112.000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sanseau P, Agarwal P, Barnes MR, Pastinen T, Richards JB, et al. Use of genome-wide association studies for drug repositioning. Nat Biotechnol. 2012;30:317–20. doi: 10.1038/nbt.2151. [DOI] [PubMed] [Google Scholar]
- 69.Scannell JW, Blanckley A, Boldon H, Warrington B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov. 2012;11:191–200. doi: 10.1038/nrd3681. [DOI] [PubMed] [Google Scholar]
- 70.Shameer K, Denny JC, Ding K, Jouni H, Crosslin DR, et al. A genome- and phenome-wide association study to identify genetic variants influencing platelet count and volume and their pleiotropic effects. Hum Genet. 2014;133:95–109. doi: 10.1007/s00439-013-1355-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Simonti CN, Vernot B, Bastarache L, Bottinger E, Carrell DS, et al. The phenotypic legacy of admixture between modern humans and Neandertals. Science. 2016;351:737–41. doi: 10.1126/science.aad2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smemo S, Tena JJ, Kim K-H, Gamazon ER, Sakabe NJ, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507:371–75. doi: 10.1038/nature13138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sudlow C, Gallacher J, Allen N, Beral V, Burton P, et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLOS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Van Driest SL, McGregor TL, Velez Edwards DR, Saville BR, Kitchner TE, et al. Genome-wide association study of serum creatinine levels during vancomycin therapy. PLOS ONE. 2015;10:e0127791. doi: 10.1371/journal.pone.0127791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Warner JL, Alterovitz G. Phenome based analysis as a means for discovering context dependent clinical reference ranges. AMIA Annu Symp Proc. 2012;2012:1441–49. [PMC free article] [PubMed] [Google Scholar]
- 76.Warner JL, Alterovitz G, Bodio K, Joyce RM. External phenome analysis enables a rational federated query strategy to detect changing rates of treatment-related complications associated with multiple myeloma. J Am Med Inform Assoc. 2013;20:696–99. doi: 10.1136/amiajnl-2012-001355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Warner JL, Zollanvari A, Ding Q, Zhang P, Snyder GM, Alterovitz G. Temporal phenome analysis of a large electronic health record cohort enables identification of hospital-acquired complications. J Am Med Inform Assoc. 2013;20:e281–87. doi: 10.1136/amiajnl-2013-001861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei W-Q, Cronin RM, Xu H, Lasko TA, Bastarache L, Denny JC. Development and evaluation of an ensemble resource linking medications to their indications. J Am Med Inform Assoc. 2013;20:954–61. doi: 10.1136/amiajnl-2012-001431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wei W-Q, Denny JC. Extracting research-quality phenotypes from electronic health records to support precision medicine. Genome Med. 2015;7:1–14. doi: 10.1186/s13073-015-0166-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wei W-Q, Teixeira PL, Mo H, Cronin RM, Warner JL, Denny JC. Combining billing codes, clinical notes, and medications from electronic health records provides superior phenotyping performance. J Am Med Inform Assoc. 2016 doi: 10.1093/jamia/ocv130. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wolfe D, Dudek S, Ritchie MD, Pendergrass SA. Visualizing genomic information across chromosomes with phenogram. BioData Min. 2013;6:18. doi: 10.1186/1756-0381-6-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wyatt J. Clinical data systems, part 1: data and medical records. Lancet. 1994;344:1543–47. doi: 10.1016/s0140-6736(94)90353-0. [DOI] [PubMed] [Google Scholar]
- 83.Xu H, Stenner SP, Doan S, Johnson KB, Waitman LR, Denny JC. MedEx: a medication information extraction system for clinical narratives. J Am Med Inform Assoc. 2010;17:19–24. doi: 10.1197/jamia.M3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yao L, Li Y, Ghosh S, Evans JA, Rzhetsky A. Health ROI as a measure of misalignment of biomedical needs and resources. Nat Biotechnol. 2015;33:807–11. doi: 10.1038/nbt.3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ye Z, Mayer J, Ivacic L, Zhou Z, He M, et al. Phenome-wide association studies (PheWASs) for functional variants. Eur J Hum Genet. 2014;23:523–29. doi: 10.1038/ejhg.2014.123. [DOI] [PMC free article] [PubMed] [Google Scholar]