

Abstract
Self-assembly, the autonomous organization of components to form patterns or structures, is a prevalent process in nature at all scales. Particularly, biological systems offer remarkable examples of diverse structures (as well as building blocks) and processes resulting from self-assembly. The exploration of bioinspired assemblies not only allows for mimicking the structures of living systems, but it also leads to functions for applications in different fields that benefit humans. In the last several decades, efforts on understanding and controlling self-assembly of small molecules have produced a large library of candidates for developing the biomedical applications of assemblies of small molecules. Moreover, recent findings in biology have provided new insights on the assemblies of small molecules to modulate essential cellular processes (such as apoptosis). These observations indicate that the self-assembly of small molecules, as multifaceted entities and processes to interact with multiple proteins, can have profound biological impacts on cells. In this review, we illustrate that the generation of assemblies of small molecules in cell milieu with their interactions with multiple cellular proteins for regulating cellular processes can result in primary phenotypes, thus providing a fundamentally new molecular approach for controlling cell behavior. By discussing the correlation between molecular assemblies in nature and the assemblies of small molecules in cell milieu, illustrating the functions of the assemblies of small molecules, and summarizing some guiding principles, we hope this review will stimulate more molecular scientists to explore the bioinspired self-assembly of small molecules in cell milieu.
Graphical abstract
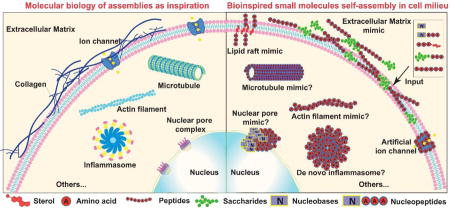
This review provides new insights and approaches for exploring bioinspired self-assembly of small molecules in cellular milieu.
Introduction
Nature, an inexhaustible source, inspires us to explore the world we live in. Chemists, materials scientists, as well as biologists are all interested in dispelling the mysterious veil of nature (e.g., living organisms, especially cells) at molecular levels because nature has evolved elaborate machineries1 that largely consist of supramolecular assemblies of biomacromolecules and carry out sophisticated biological tasks in all organisms. The advances in molecular cell biology have contributed to the development of a new subject (or strategy)—bioinspiration, which involves multiple disciplines to nucleate new ideas for research.2 To mimic the properties of biological systems by non-living systems, many disciplines use the concept of self-assembly, which is a prevalent process in cells and a common phenomenon of nature. In the context of molecules and cells, self-assembly is the autonomous organization of individual components into patterns and functional nanostructures through non-covalent interactions with the balance of both thermodynamic and global (or local) equilibrium. With the increasing understanding of biological systems (especially biomacromolecules assemblies1) and the development of new technologies (e.g., cryo-EM), more and more innovative materials are bio-inspired molecular assemblies. Over the last two decades, inspired by biological organisms, from cells to sub-cell organelles, chemists and material scientists have extensively explored artificial functional macromolecules (especially functional artificial proteins and polymers3) for bottom-up self-assembly to form specific nanostructures.
The understanding of the assemblies of biomacromolecules (e.g., DNA, RNA, and proteins) at a molecular level and the intrinsic forces for the self-assembly of small molecules of cells (e.g., lipids or cholesterols) have provided useful insights on how these simple components self-assemble to form highly ordered and precisely (both spatially and temporally) controlled structures (Fig. 1) in an organism,1 which has stimulated the exploration of assemblies of small molecules to mimic the properties and structures of living systems for applications in different fields to benefit humans, such as liposomes for drug delivery. Many efforts have focused on understanding and controlling self-assemblies of small molecules with non-biological stimuli in vitro (i.e., cell free setting) over the last several decades, and the progress of these studies has resulted in a large library of candidates that promise biomedical applications for the assemblies of small molecules, as documented in several reviews.4 Moreover, recent findings in biology have provided exciting insights for using the assemblies of small molecules to modulate essential cellular processes. For example, lipid rafts modulating apoptosis (i.e., the process of programmed cell death)5 or antibiotics inhibiting bacteria6 are endogenous or naturally occurring processes. They have inspired the development of self-assembly of man-made small molecules in cell milieu to act as multifaceted entities that interact with multiple proteins and control the fates of cells. In fact, such developments have progressed considerably to warrant a review to illustrate the concepts and the design principles for exploring self-assemblies of man-made small molecules in cell milieu.
Fig. 1.
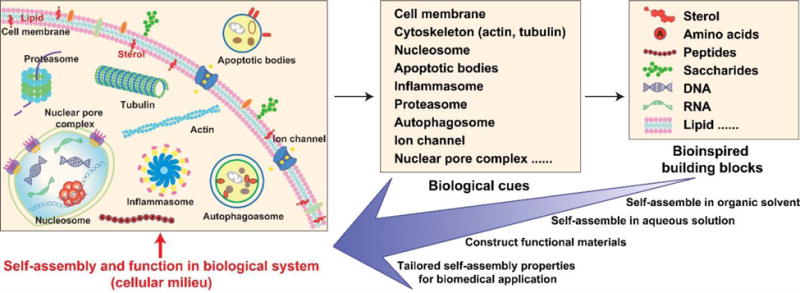
Schematic of endogenous molecular assemblies and their building blocks for inspiring self-assembly of small molecules in cell milieu.
In this review, we first briefly introduce endogenous self-assembly and the corresponding building blocks of self-assembly along with the importance of assemblies of small molecules of cells as bioinspiration. Then, we discuss how the assemblies of small molecules interact with the cells and illustrate new strategies to explore these interactions. After that, we describe the recent advances of in-situ formation (both pericellular and intracellular) of assemblies of small molecules in cell milieu for controlling cell behavior or for imaging specific bio-markers. Following the discussion of two important strategies that enhance the effects of in-situ assemblies of small molecules on cells, we summarize a few guidelines for designing the self-assembly of small molecules in the context of cells. Finally, we discuss the challenges and future promises regarding bioinspired assemblies of small molecules in cell milieu.
Endogenous self-assembly as an inspiration
The existing phenomena in nature, such as the adhesive behavior of mussels, the footpads of a gecko, the hydrophobic property of spider web, and infective ability of virus, etc., have inspired scientists to develop a myriad of materials that largely mimic the structures in nature for achieving similar functions as the solutions for certain problems.2 With the increased understanding of molecular cell biology, the research on bioinspiration has begun to focus on the cell level, in which the system (e.g., cells) offers endless examples of functional self-assembly as an excellent blueprint to construct functional materials. Cells, like factory assembly lines, contain all the components together in a highly specific manner.1 Each individual component acts as a part of the molecular machinery that consists of sophisticated nanostructures1 with spatiotemporal control. Endogenous biological assemblies formed by the interactions of different components in cells, such as protein-protein interaction and protein-nucleic acids interaction, are the most stimulating inspiration. Fig. 1 illustrates the examples of biological architectures resulting from molecular self-assembly. For example, actins and tubulins self-organize7 to generate nanostructures as cytoskeletons, ion channel association proteins self-assemble to form nanostructures to maintain homeostasis of ions in cells, and nucleoporins self-assemble to produce nuclear pore complexes to regulate the flow of macromolecules between the nucleus and cytoplasm. Indeed, materials scientists have been employing self-assembly of receptors or epitopes discovered from those machineries to mimic the structure and function of biomacromolecules, which usually create the molecular structures resembling the organization observed in cells.8
Two well-investigated cellular assemblies are eukaryotic ribosomes and proteasomes (20S and 26S), which are involve in protein formation and protein degradation.1 Ribosome, the place for protein biosynthesis, is a complex molecular machine that consists of ribosomal RNA and proteins. Elucidation of the molecule structures of ribosome provides biological and mechanical cues9 for engineering protein based biomaterials in the applications of drug delivery, controlling cell behaviour, and as molecular recognition and sensor platforms. Proteasome is a highly conserved molecular machine that collaborates with other protein assemblies, such as chaperones and ubiquitin systems.1 Understanding the self-assembly process of proteasome and the mechanism of the ubiquitin-proteasome proteolytic pathway provides biological clues and promising therapeutic targets for treating various diseases, including cancer10 and neurodegenerative diseases.11 The molecular understanding of proteasome, on the other hand, also provides opportunities for researchers to focus on self-assembly in biomedical applications, such as using the chaperones to assist the self-assembly of peptides. This progress may afford guidelines for designing suitable peptidic biomaterials for modulating protein degradation, especially the degradation of aberrant proteins for treating neurodegenerative disease, which remains a big challenge. The building blocks of these systems are largely proteins, which self-assemble through weak interactions (e.g., hydrophobic interactions, hydrogen bonds, van der Waals interactions, and ionic bonds). Until now, most research has centered on using protein building blocks to construct biomaterials with function and/or responsiveness to varieties of stimuli (non-biological and biological) in vitro.4 To advance these efforts in vivo would be exciting research down the road.
Besides leading to biomolecular assemblies for cellular functions, the interactions of proteins and nucleic acids in nucleosomes and spliceosomes also have inspired the generation of materials based on peptide nucleic acid (PNA), which have found applications in cancer therapy, DNA (RNA) biosensors, protein engineering as well as molecular tools for nanotechnology. In addition, individual molecules of DNA or RNA also serve as building blocks to generate nanoscale assemblies (e.g., DNA origami12 and DNA hydrogel), which have high hopes for developing many applications, as suggested in several excellent reviews.13, 14
Largely being the consequence of the self-assembly of small molecules of cells (i.e., endogenous small molecules such as lipids and cholesterols), membranes, including cell and organelle membranes, are the place where the cells build functional signaling complexes and respond to external or endogenous stimuli. The fluid matrix like property of cell membranes can easily regulate the local organization of its constituents and maintain homeostasis. The formation of apoptotic bodies, autophagosomes, extracellular vesicles (exosomes and microvesicles) as well as endosomes (endocytic machinery), just to list a few, are all examples of what the cell membrane involves in the cell response to environments. Inspired by these endogenous assemblies of functional lipid domains, scientists have developed biomaterials based on sterol compounds, especially phosphatidylserine and cholesterol (the basic component of the membrane in molecular machinery15). These sterol derivatives can easy self-assemble in aqueous solvents to form a hydrogel or micelle-like structure. Such biomaterials finding applications in several fields, like drug delivery, analyte detection, membrane theory development, mimicking prototypic living systems, and as immune adjuvant. Besides employing these sterol compounds as building blocks outside cells, another fascinating aspect of the cell membrane is how to generate functional lipid domains and protein clusters on or within the cell membrane. Although much less explored, it is an important field for material scientists because mimicking these properties by rational design of precursors that self-assemble in cell milieu provides new ways to recruit proteins and execute functions at the plasma membrane.
The development of self-assembly, like the evolution of cells, has progressed from simple to complex and then to homeostatic systems (Fig. 1) to respond to various stimuli by tuning themselves. Over the past several decades, the focus of self-assembly has shifted from designing building blocks and studying mechanisms of assemblies in organic and aqueous solutions to exploring their potential functions in biological settings. Many different building blocks, including amino acids, peptides, saccharides, DNA, RNA, and sterol compounds, self-assemble to form various nanostructures. These self-assembly based materials are being explored in many research frontiers. Several excellent reviews4, 13, 16, 17 have summarized this progress. With the development of chemistry, molecular biology, and new techniques to characterize nanostructures (e.g., crystal structures of protein) in vitro and in vivo, researchers have developed an increasing number of novel molecular assemblies with organized adaptive nanostructures and tunable functions for controlling cell behaviors. Despite the rapid growth and increased understanding of these fields, the needs for modern biomedical therapies continue to pose challenges. As an approach to meet these challenges, bioinspired self-assembly of small molecules in cell milieu, relying on synthesis, enzymatic reactions, molecular imaging, and molecular cell biology assays, have led to important new findings, such as employing self-assemblies of small molecules to inhibit cancer cells selectively or to promote proliferation of stem cells. These results not only coincide with new insights into the assemblies of small molecules in essential cellular processes, such as lipid rafts modulating apoptosis and generating nanostructures within or on cell surfaces, but they also establish that the self-assembly of small molecules, as multifaceted entities to interact with multiple proteins, can have profound biological impacts on cells. Thus, it is appropriate and useful to review the progress on bio-inspired self-assembly in cell milieu, as briefly discussed in the following sections, for the future development of this exciting research frontier.
Assemblies of small molecules interacting with cells
The serendipitous discovery of the inverse comorbidity between cancer and neurodegenerative diseases implies that nanofibers formed by molecular assemblies can inhibit cancer cells, even in humans.18 This notion (Fig. 2A) apparently is plausible, as revealed by the study of the cytotoxicity of the nanofibers formed by Nap-FF (1 in Fig. 2B), especially that the nanofibers inhibit the growth of glioblastoma cells without detrimental effects to neuronal cells.19 In that study, a TEM image confirmed that Nap-FF was able to self-assemble in water to form nanofibers at concentrations below its critical gelation concentration (i.e., 400 μM) (Fig. 2 C and inset). Above the threshold concentration of the forming nanofibers, 1 significantly decreased the viability of HeLa cells to less than 20% within 48 h (Fig. 2D). At the concentration below the threshold, 1 was unable to form nanofibers and exhibited little cytotoxicity to HeLa cells. A cell migration assay (Fig. 2E) also confirmed that the nanofibers of 1 delayed the migration of HeLa cells. While the untreated T98G cells displayed a long microtubule network stretching through the cell body (Fig. 2F), tubulins in the cells treated by 1 (above the threshold) aggregated to form clusters with scattered microtubule filaments (Fig. 2G), indicating that the nanofibers of 1 disrupted the dynamics of the microtubules. According to these experimental results, a proposed possible mechanism is that 1 self-assembles to form soluble nanofibers that disrupt the dynamics of microtubules and cause apoptosis of glioblastoma cells (Fig. 2A). This study is the first case to establish that assemblies of small molecules exhibit properties drastically different from those of the individual molecules when they are in cell milieu.
Fig. 2.
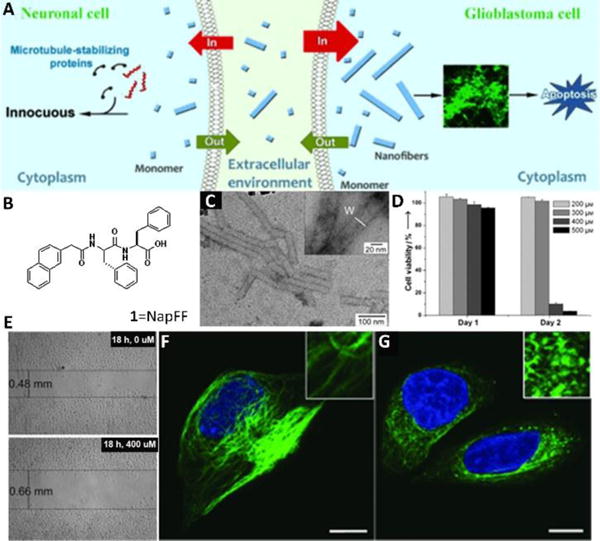
(A) Proposed mechanism for the selective inhibition of the growth of glioblastoma cells by the self-assembled nanofibers of 1. (B) Molecular structure of 1. (C) Negatively stained TEM images of a solution of 1 (400 μM) in PBS buffer. (D) Cell-viability assays of HeLa cells after treatment for 24 and 48 h with 1 at different initial concentrations. (E) Cell-migration assay of HeLa cells treated with 1. Confocal images of tubulin-stained T98G cells treated with a medium containing 1 at a concentration of 0 (F) or 400 μM (G) for 24 h. Insets are images that have been enlarged further by a factor of 3. Scale bar: 10 μm. Adapted with permission from ref. 19. Copyright 2013, Wiley-VCH.
Molecular self-assembly has resulted in many structurally well-defined assemblies such as nanofibers, nanotubes, and nanospheres, and the biological functions of these assemblies likely differ greatly from the individual molecules, as evidenced by Fig. 2. Such a distinction is different from the multivalent presentation of the epitopes by the nanofibers of the peptides for enhancing the bioactivities of the epitopes. Therefore, it is necessary to identify the proteins (besides the targets of the epitopes) that interact with the nanofibers. However, this aspect of molecular nanofibers was unexplored due to the lack of proper assays. A recent assay20 (Fig. 3A) offered a simple way to identify the interaction of the self-assembled nanofibers with cytosol proteins, which is based on the binding affinity between proteins and the molecular assemblies of small molecules in hydrogels. The underlying presumption of the assay is that the hydrogels, which are intrinsically similar to the cellular environments, preserve the non-covalent interactions between proteins and the assemblies of small molecules. This assay, consisting of four major steps (Fig. 3A), is able to analyse and to identify the protein that interacts with the aggregates. For example, a photoleucine-containing gelator (2) self-assembled to form the hydrogel (Fig. 2B) that consisted of the nanofibers of 2. After the retained proteins in the hydrogels were subjected to gel electrophoresis analysis, the major protein bands in the SDS-PAGE gel were then identified by proteomics (Fig. 3B). This analysis, indicating that the molecular nanofibers of 2 interacted with tubulins, actins, and several other proteins, was further confirmed by western blot (Fig. 3C). This result also validated the washing step, effectively removing nonspecific bound proteins from the hydrogel (Fig. 3D). Later study indicated that photoleucine was unnecessary for retaining the proteins.21 This pull-down assay provides a facile and direct way to elucidate the interactions between assemblies of small molecules and protein targets, which contributes to establishing the molecular foundation for exploring assemblies of small molecules in the cell milieu.
Fig. 3.
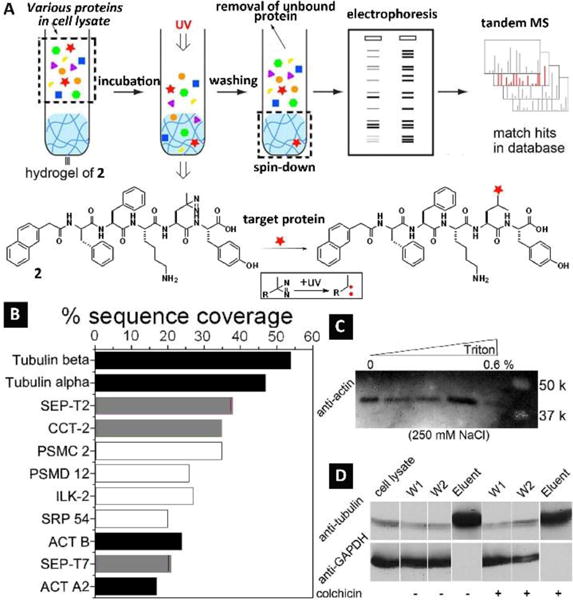
(A) Illustration of the hydrogel protein pull-down assay coupled with electrophoresis and tandem mass spectrometry for the identification of the cytosolic proteins that bind to molecular nanofibers. The expanded box shows the photoreaction between the hydrogelator and the proteins bound to the molecular nanofibers. (B) Tandem MS analysis indicates the most abundant hydrogel bound cytosolic proteins by sequence coverage. Black: proven binding partners; grey: partners that bind to cytoskeleton protein/fibers; white: proteins that were identified with no known association with the verified proteins. Validation of protein–nanofibers interactions: (C) cell lysate was incubated with the hydrogel and thoroughly washed under the conditions shown on the top and subjected to western blotting with antibody to actin; (D) cell lysate was incubated with the hydrogel and thoroughly washed under the conditions of 250 mM of NaCl, 0.6% triton, w/o colchicine, and subjected to western blotting with antibodies to tubulin and GAPDH (another abundant protein expressed inside mammalian cells). Adapted with permission from ref. 20. Copyright 2012, Royal Society of Chemistry.
In-situ formed assemblies of small molecules in cell milieu
The works described above demonstrated the use of assemblies of man-made small molecules to control cell fate or investigate cellular proteins that interact with the assemblies of small molecules. However, in biological systems (i.e., cells), locally formed assemblies are responsible for spatiotemporally regulating diverse cellular functions. So, a more attractive approach is to make individual molecules outside cells and their assemblies inside the cells, like what a virus does to a host cell. Inspired by nature, researchers have started to employ enzymes, which are prevalent in cells, to trigger self-assembly of small molecules.22 In fact, the first example of enzyme-instructed self-assembly (EISA) of small molecules in cell milieu occurred inside cells (Fig. 4A).23 As shown in Fig. 4B, a dipeptidic precursor (3a), containing an enzyme (i.e., esterase) cleavable site (i.e., an ester bond), turned into hydrogelator 4a upon the treatment of esterase, which self-assembled to form nanofibers. Incubating 3a with HeLa cells, known to express esterases, caused cell death. Breaking the collected dead cells (which float) resulted in the formation of a hydrogel consisting of nanofibers. This result indicated the intracellular hydrogelation inside HeLa cells (Fig. 4C). After treating with 3a for one day, the number of HeLa cells decreased, suggesting the intracellular hydrogelation caused cell death (Fig. 4D). Cell viability tests verified that 3a inhibited HeLa cells, and the cytotoxicity increased with higher concentrations of 3a (Fig. 4E). This work established the feasibility for the self-assembly of man-made small molecules to control cell fate.
Fig. 4.
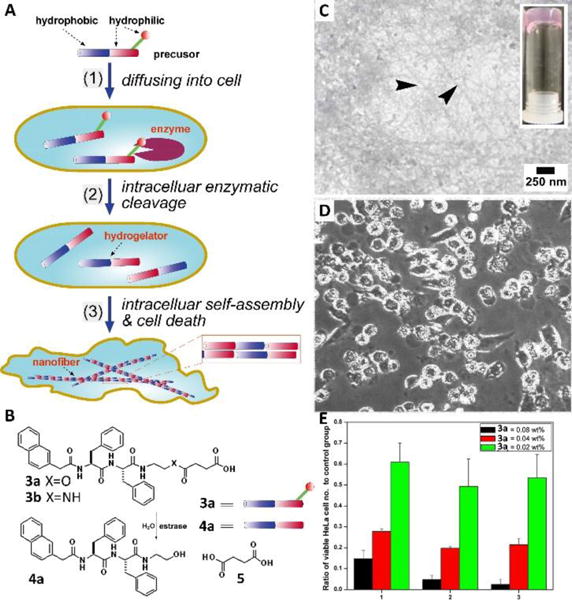
(A) Schematic intracellular formation of nanofibers that leads to hydrogelation and cell death. (B) The chemical structures and graphic representations of the precursor (3a), the control molecule (3b), and the hydrogelator (4a). (C) TEM of the hydrogels formed by the dead HeLa cells after culturing with 3a for three days (arrows indicated the nanofibers formed by 4a). (D) Optical images (x100) of the growth of HeLa cells after being cultured in the media containing 0.08 wt% of 3a in 1 day. (E) MTT assays of HeLa cells treated with 3a at concentrations of 0.08 wt%, 0.04 wt%, and 0.02 wt%. Adapted with permission from ref. 23. Copyright 2007, Wiley-VCH.
Besides using intracellular enzymes to instruct (i.e., triggering) self-assembly, a rather unexpected result is that ectoenzymes instruct the self-assembly of small molecules on the cell surface to control cell behavior (Fig. 5A).24 Precursor 6, a phosphotyrosine containing D-tripeptide derivative, turned into self-assembling molecule 7 upon catalytic dephosphorylation by alkaline phosphatase (ALP)(Fig. 5B). As an ectoenzyme overexpressed on certain cancer cells (e.g., HeLa25), ALP dephosphorylated 6 to produce 7 on the cell surface to increase the local concentration of 7, thus resulting in the formation of fibrous nanonets on the cell surface, which led to the build up a layer of visible hydrogel around the cancer cells (Fig. 5C). This hydrogel blocked the mass exchange and disrupted cell communication, leading to cell apoptosis. Congo red, a dye for β-sheet like nanofibers, revealed that the hydrogel/nanofibers only formed around the HeLa cells treated with 6 (Fig. 5D), establishing the pericellular nanonet formation (Fig. 5A). DAPI, a dye for the nucleus, co-localized with Congo red instead of entering the nucleus of the HeLa cells treated with 6, further proving that the pericellular nanonets prevented the mass exchange. In contrast to HeLa cells, no nanonets formed on the surface of the Ect1/E6E7 cells, an immortalized normal human cervical epithelial cell line, indicating that 6 only formed pericellular nanonets on the cancer cells with overexpressed ALP. One key feature to ensure the formation of pericellular nanofibers is to use a D-tripeptide, which is proteolytic resistant in cell milieu.
Fig. 5.
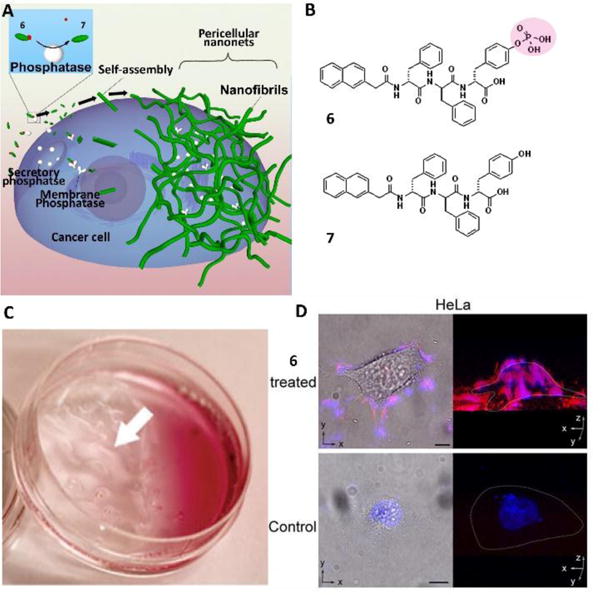
(A) Enzyme-catalyzed formation of pericellular hydrogel/nanonets to induce cell death. (B) Molecular structures of the precursor (6) and the hydrogelator (7). (C) Optical images of the HeLa cells incubated with 6 at 560 μM. White arrows point to the hydrogel/nanonets. (D) Overlaid images and 3D stacked z-scan images of Congo red and DAPI stained HeLa cell treated by 6 or the culture medium as a control for 12 h. HeLa cells treated by 6 at 280 μM. Scale bar=10 mm. White dots outline the cells. Adapted with permission from ref. 24. Copyright 2014, Wiley-VCH.
While the dogmatic ligand-receptor interaction (e.g., an inhibitor binding to an enzyme) is thermodynamic in nature, the self-assembly of small molecules is inherently kinetic, which is particularly suitable for regulating cells via kinetic control. Such kinetic control has been achieved in cell milieu by simply tailoring the number of tyrosine phosphates on the substrates of EISA.26 For example, precursors 8-2p, 9-2p and 10-2p were tetrapeptide derivatives with two phosphotyrosine residues (2p), while 8-1p, 9-1p and 10-1p only contained one phosphotyrosine (1p) (Fig. 6A). Upon ALP catalyzing, the rates of forming the corresponding hydrogelators from mono-phosphorylated precursors were faster than from the di-phosphorylated precursors because the di-phosphorylated precursors required dephosphorylation twice. Generating self-assembling molecules faster than the di-phosphorylated precursors, the mono-phosphorylated precursors exhibited higher inhibition efficacy on cancer cells, suggesting the feasibility of tailoring the number of enzyme responsive sites on EISA precursors for controlling the extent of inhibition (Fig. 6B), which may be advantageous for enhancing selectivity to target the diseased cells.
Fig. 6.
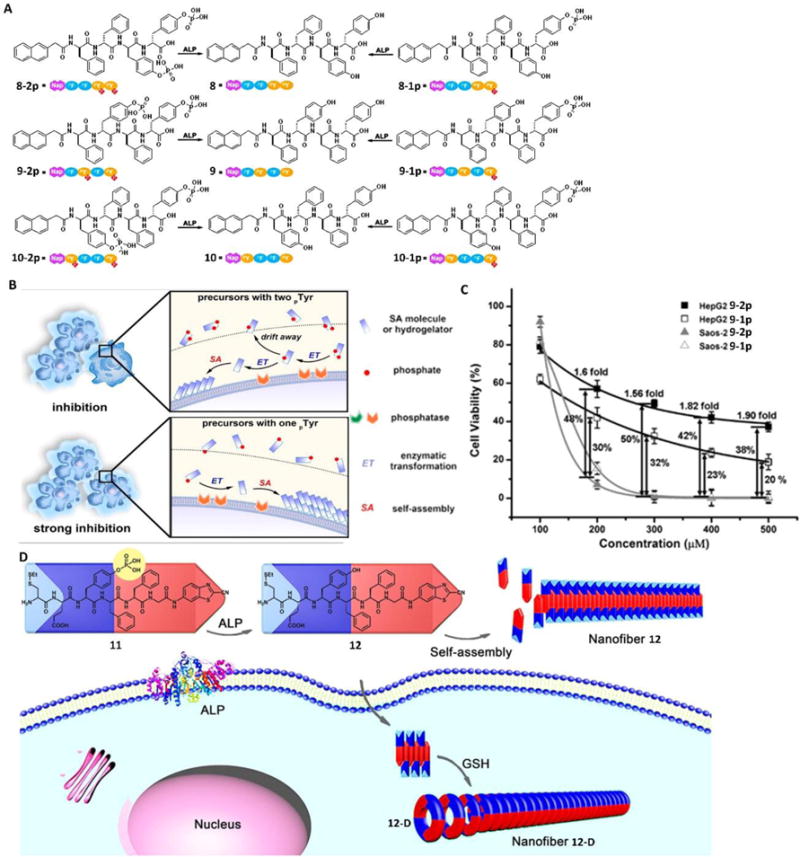
(A) Molecular structures of the precursors (8-2p, 9-2p, 10-2p, 8-1p, 9-1p, and 10-1p) that have one or two phosphotyrosine residues and the corresponding self-assembling D-peptides (i.e., hydrogelators 8, 9, and 10). (B) EISA to inhibit cancer cells. (C) 48-hour cytotoxicity of 9-2p and 9-1p on HepG2 and Saos-2 cells at different concentrations. The viability differences between the two cell lines are labelled as well. (D) Schematic illustration of ALP-directed self-assembly of 11 into nanofiber 12 in extracellular environment, and GSH-controlled condensation of 12 to yield the cyclic amphiphilic dimer 12-D, which self-assembles into nanofiber 12-D in an intracellular environment. Blue parts indicate the hydrophilic structures, and red parts indicate the hydrophobic structures. Adapted with permission from ref. 45, 46 and 47. Copyright 2016, American Chemical Society (ref. 26 and 28). Copyright 2016, Wiley-VCH (ref. 27).
In fact, regulating the rate of molecular self-assembly affords selective inhibition of cancer cells by amplifying the differences of the expression levels of enzymes on the targeted cells and non-targeted cells, even when those cells express the same enzymes. A recent study27 demonstrated this concept in the case of Saos-2 and HepG2 cells, both expressing tissue non-specific alkaline phosphatase (TNAP), but the former had a significantly higher level of TNAP on the cell membrane than the later. Cell viability tests revealed that the precursor 9-2p was much less cytotoxic against HepG2 cells (known to express TNAP) compared with 9-1p since 9-1p had a faster self-assembly rate (Fig. 6C). However, the significantly higher level of TNAP on the cell membrane of the Saos-2 cells enabled rapid dephosphorylation of 9-2p to generate hydrogelator 9 before the intermediates diffused away, resulting in similar inhibitory efficacy of these two precursors (9-2p and 9-1p) on Saos-2 cells. So, at a certain concentration (e.g., 300 mM), 9-2p killed (almost) all the Saos-2 cells, but spared about 60% of the HepG2 cell. These studies established that EISA is a multiple-step and kinetic process for selectively controlling the fates of cells.
Designing precursors for forming assemblies with sophisticate spatiotemporal control is important and challenging. Liang and co-workers28 recently reported an ingenious design towards meeting such a challenge. They designed precursor 11, which could efficiently transform to amphiphilic molecule, 12, upon dephosphorylation by ALP. Further reacting with glutathione (GSH), 12 turns to 12-D. A key feature is that 12 and 12-D self-assemble to form two different nanofibers in different cellular locations. As shown in Fig. 6D, extracellular ALP dephosphorylated 11 from 12, which then self-assembled into nanofiber 12 in the pericellular space. After being up-taken by cells via endocytosis, the disulphide bond in hydrogelator 12 was reduced by intracellular GSH to expose the reactive 1,2-aminothiol group, which then reacted with a cyanobenzothiazole (CBT) motif on another molecule 12 to form the cyclic dimer 12-D. 12-D further self-assembled to form nanofibers of 12-D, which was longer and denser than the nanofibers of 12. The demonstration of a precursor (e.g., 11) to differentiate cell locations and to result in different nanostructures likely will provide a way to use two biological cues to modulate the assemblies of small molecules in cell milieu. It would be interesting to further explore the biological applications of this promising result.
The concept of EISA is general and applicable to molecules other than peptides for selectively inhibiting cancer cells without harming normal cells, as elegantly demonstrated by Ulijn and Pries et al.29 They reported that a simple carbohydrate amphiphile, after being dephosphorylated by ALP, was able to self-assemble on cancer cells (Saos-2) to induce cell death. Upon dephosphorylation, precursor 13a turned into hydrogelator 13b (Fig. 7A), which then self-assembled to form a typical hydrogel. The authors incubated the precursor 13a with cell suspension or cell monolayers, in which condition they found macroscopic gels formed in the pericellular space of the Saos-2 cells, but not the ATDC5 cells that expressed low levels of ALP (Fig. 7B). The formed gel in the pericellular space blocked metabolite exchange between the cells, resulting in cell apoptosis. This study implies that a wide range of small, amphiphilic molecules may serve as the substrates for EISA.
Fig. 7.
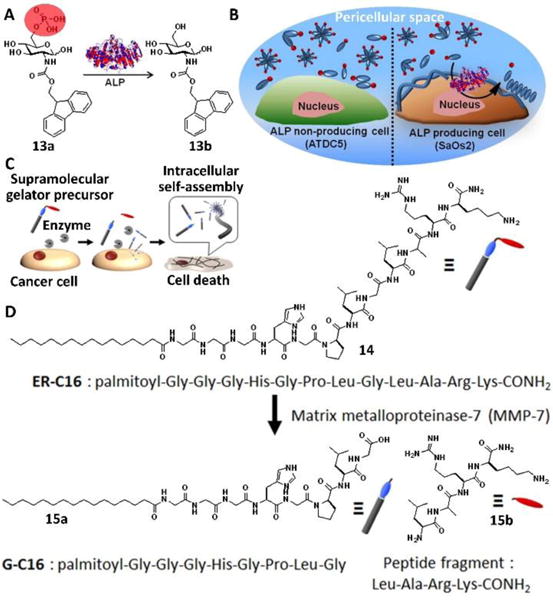
Illustration of enzymatic transformation of 13a to 13b upon enzymatic activity of phosphatases (e.g., ALP): (A) Molecular structures and (B) in situ biocatalytic self-assembly. Precursor 13a is converted into the self-assembling hydrogelator 13b in situ by the Saos-2 membrane-bound ALP. Upon conversion, 13b generates a cytotoxic nanonet/hydrogel “cage” surrounding the cells, while innocuous to ATDC5, which express lower membrane bound ALP. (C) Cancer cell death induced by EISA of hydrogelators: Cancer cells secrete excessive amounts of MMP-7, which converted the precursor 14 into a supramolecular gelator 15a prior to its uptake by the cells. Once inside the cells, the supramolecular gelator formed a gel via molecular self-assembly, exerting vital stress on the cancer cells. (D) Molecular structures of ER-C16, G-C16, and Leu-Ala-Arg-Lys-CONH2 (peptide fragment). Adapted with permission from ref. 29 and 30. Copyright 2015, American Chemical Society.
The concept of EISA also is applicable to other enzymes, as demonstrated by Maruyama et al.30 They reported the first case of using matrix metalloproteinase-7 (MMP-7), an excessively produced and secreted protease by various cancer cells, to trigger intracellular molecular self-assembly of a peptide lipid conjugate (14). Precursor 14 exhibited cytotoxicity against five different cancer cell lines, while being innocuous to normal cells (Fig, 7D). After being incubated with 14, cancer cells up-took 14 and converted it to a hydrogelator (15a), which self-assembled to form nanofibers that critically impaired cellular function and killed the cancer cells (Fig. 7C). These excellent studies have underscored the promises of using assemblies of small molecules in cell milieu for controlling the fate of cells for developing anticancer therapeutics.
Imaging assemblies of small molecules in cell milieu
The development of molecular imaging has provided a convenient strategy for investigating the self-assembly of small molecules in cell milieu. Although it is reasonable to introduce a fluorophore into a small molecule for imaging the location of the assemblies in a biological system, it is difficult to distinguish individual fluorescent molecules and corresponding assemblies due to the minimal difference of these two cases, and aggregation always induces fluorescence quenching of fluorophores. Recently, this difficulty was resolved by using a molecule with an environmentally sensitive fluorophore, which caused the precursors and corresponding self-assembling molecules (e.g., hydrogelators) to exhibit no or low fluorescence in the monomer state but to be brightly fluorescent in the colloidal state31 (Fig. 8A). Under this condition, the high concentration of fluorophores in the colloids compensates the loss due to the intermolecular interaction induced quenching. Fig. 8B showed the molecule structure of precursor 16, which consisted of a fluorophore, 4-nitro-2,1,3-benzoxadiazole (NBD), and a small peptide phosphorylated at the tyrosine residue. Dephosphorylation of 16 in the solution of 16 by ALP generated hydrogelator 17 and resulted in a hydrogel. This gelation process was accompanied by changes in the fluorescence from weak in solution to bright in the gel state.31 The incubation of HeLa cells with 16 also caused the changes of fluorescence inside the cells over time (Fig. 8C), which indicated the self-assembly process of 17 and the distribution of assemblies of 17 in cell milieu. As is the case for studying the assemblies (or self-assembly process) of small molecules inside live cells that involve cellular processes to construct the designed assemblies in situ, this work may lead to a model system in a phenotype assay to screen the inhibitors of fibrillar aggregates inside cells.
Fig. 8.
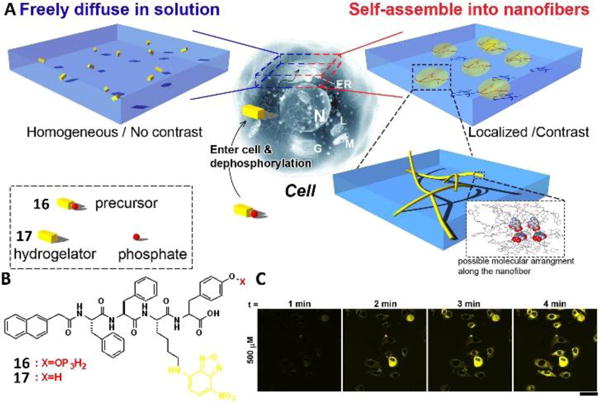
(A) Principle of imaging enzyme-triggered supramolecular self-assembly inside cells. (B) Molecular structure of the precursor 16 and the corresponding hydrogelator 17. (C) Fluorescent confocal microscope images show the time course of fluorescence emission inside the HeLa cells incubated with 500 μM of 16 in PBS buffer, which shows the different distribution of fluorophores inside the living cells. Scale bar is 50 μm. Adapted with permission from ref. 31. Copyright 2012, Nature Publishing Group.
The above work successfully demonstrates the incorporation of an environment sensitive fluorophore into a small molecule to enable real-time imaging of molecular self-assembly in live cells. However, the introduction of a fluorescent label to small molecules self-assembling in cell milieu caused undesired problems in most reported cases, such as changing the self-assembling ability of the original molecules, alternating the interaction of assemblies with cellular components, and requiring additional characterization of the fluorophore. Heterotypic self-assembly of small molecules provided a feasible approach to probing self-assembly of non-fluorescent small molecules inside live cells, as illustrated by the study32 shown in Fig. 9A. A rationally designed molecule 18a and its fluorescent analogue 19a with the modification of the dansyl (DNS) group upon the addition of ALP turned to the corresponding hydrogelator 18b (or 19b), which formed a nanofibrous network and entrapped water to generate a typical hydrogel (Fig. 9B). Sharing the same self-assembling motif, hydrogelators 18b and 19b mixed at their optimal concentrations to result in heterotypic molecular assemblies, which showed similar properties with individual molecules of 18a, thus allowing imaging of the cellular distribution of the self-assembly of parent molecule 18a by fluorescence imaging. As shown in Fig. 9C and 9D, co-incubating HeLa cells with the mixture of 18a and 18b revealed the distribution of assemblies of 18b, since 18b itself hardly showed fluorescence at optimal concentrations. Further experiments also indicated that assemblies of 18b localized to the endoplasmic reticulum (ER) and were likely processed via the cellular secretory pathway (i.e., ER–Golgi–lysosomes/secretion). As a feasible approach to designing adaptive heterotypic assemblies of small molecules in cell milieu with spatiotemporal control, this study offered a useful way to evaluate the fate of assemblies of small molecules in cell milieu.
Fig.9.
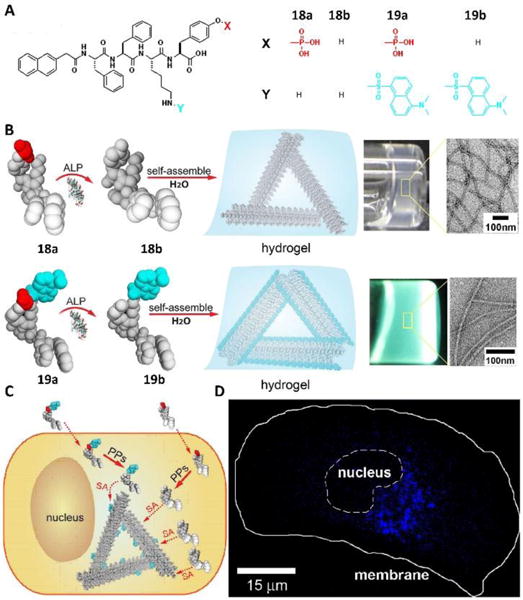
(A) Molecular structures of precursors 18a and 19a and their corresponding hydrogelators 18b and 19b, respectively. (B) Illustrations showing the basic steps of EISA and formation of molecular assemblies and hydrogels: the precursor molecules 18a and 19a are converted to hydrogelators 18b and 19b by dephosphorylation catalyzed by alkaline phosphatase (ALP); these hydrogelators self-assemble in water to form molecular assemblies that generate hydrogels. Toward the right, optical images of hydrogels of the small molecules 18b and 19b (ALP-catalyzed conversion of 18a (0.2 wt %, 2.35 mM) and ALP-catalyzed conversion of 19a (0.6 wt %, 5.5 mM). The zoom-in transmission electron microscope (TEM) images on the very right show the detailed structure of negatively stained molecular assemblies of 18b and 19b. Illustrations (C) and fluorescence images (D) of two experiments that use a precursor mixture of 18a and 19a. Adapted with permission from ref. 32. Copyright 2013, American Chemical Society.
In-situ self-assembly for imaging enzyme activities of living cells
The use of molecular imaging to detect the activities of enzymes is more successful for intracellular enzymes than for ectoenzymes because the molecule imaging probes are easier to confine intracellularly than extracellularly. The self-assembly of small molecules does not favor the diffusion of the probes, which makes the molecular imaging of ectoenzymes possible, as recently demonstrated by imaging ALPs on live cells.33 This method is particularly useful for detecting the activity of ALPs on cancer cells because some of them overexpress ALPs. As shown in Fig. 10, the designed precursor 20a, consisting of D-peptide, turned to 20b to form nanofibers in the presence of ALP. CLSM images revealed that 20a mainly self-assembled on the pericellular space of cancer cells (e.g., HeLa cells in Fig. 10C as an example), but not on normal cells (e.g., HS-5). The fluorescence around HeLa cells increased when the cells were incubated with 20a for a longer time. The correlative light and electron microscopy (CLEM) provided direct visualization of the local formation of assemblies of 20b on the cell surface (Fig. 10D, E, F). This study not only validated a spatiotemporal approach to visualizing enzyme activity on live cells at the single-cell level, but it also offered new paradigms for understanding the functions of different forms of ALPs (isoenzymes, isoforms, membrane bound and soluble forms) in live cells.
Fig. 10.

(A) Molecular structures of pNDP1 and NDP1 and the conversion catalyzed by ALP under physiological conditions (orange part is the fluorophore, NBD; red part is the enzymatic trigger, phosphate). (B) Illustration of the EISA of NDP1 to form fluorescent pericellular nanofibrils in co-culture. (C) Merged image (differential interference contrast and fluorescence microscopy images) of treated HeLa cells (grown on marked Aclar discs and incubated for 6 h with 500 μM pNDP1) recorded only a few minutes before the sample was high-pressure frozen for TEM; note the highest intensity of the fluorescence signal on the cell surface, indicating a high abundance of nanofibrils in that region of the cell. (D) High-magnification electron microscopy image shows a slice through the same cell depicted in (C). Note that this is a montage image that consists of more than 200 individual high magnification image tiles. (E) Higher-magnification electron micrograph of the cell region highlighted by the red box in (C) and (D). This region of the cell also corresponds to the area of highest fluorescence signal in (C). (F) High-magnification electron micrographs of the red boxed area in (E). Note the presence of nanofibrils (white arrowheads) containing NDP1s. Adapted with permission from ref. 33. Copyright 2016, Elsevier.
Certainly, it is feasible to use assemblies of small molecules for imaging the activity of intracellular enzymes, as reported by Rao and co-workers.34 They demonstrated an approach for controlling the covalent synthetic process (i.e., condensation reaction) within live cells. Based on the fact that fireflies create luciferin = through a condensation between CBT and the 1,2-aminothiol in D-cysteine, Rao et al., designed a short library of compounds in which CBT was attached to a protected 1,2-aminothiol group by a small spacer (either a single amino acid or a short peptide). To fulfil the purpose of in vivo reaction and self-assembly, the amine and thiol groups of precursor 22 were protected with protecting groups that were sensitive to pH change, reduction conditions, or enzyme catalysis. Under the above conditions, precursor 22 was de-protected to yield 23, which formed a dimer (24) and oligomers (25) via a condensation reaction to result in nanostructures. To visualize the condensation products in live cells, they used biotin-tagged monomers to allow fluorescence imaging. Fig. 11B and 11C show two representative examples of in-situ condensation reactions, which were triggered by the reductive environment in the HeLa cells (Fig. 11B) and the activity of furin and reductive environment in breast cancer cells (MDA-MB-468 cells) (Fig. 11C). Further study also indicated that attaching an anti-cancer drug to monomer 2235 can inhibit drug resistant cancer cells.
Fig. 11.
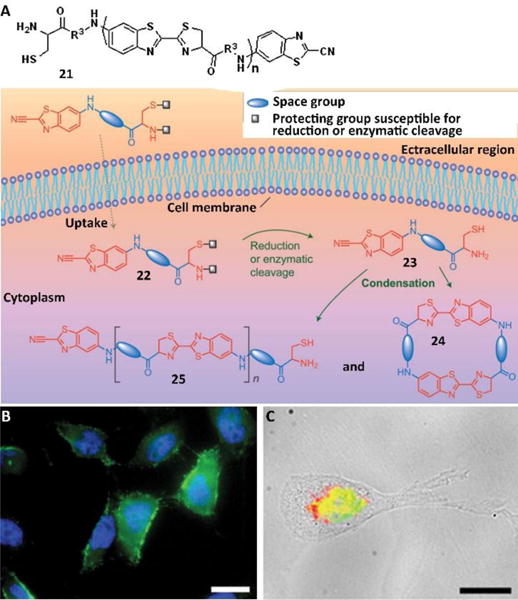
(A) Condensation reaction carried out in living cells. The precursor (22), inside a cell, is converted into the monomer (23), which self-condensates to form the dimer (24) and oligomers (25), which can in turn assemble into nanostructures. (B) Fluorescence images of HeLa cells incubated with 200 μM of monomer based on 21 (R1=SCH2CH3, R2=H, R3=Biotin-Lys). Scale bars: 20 μm. (C) Merged fluorescence and transmitted light images of MDA-MB-468 cells incubated with monomer based on 21 (R1= SC(CH3)3, R2= Ac-Arg-Val-Arg-Arg, R3=Biotin-Lys, 200 μM) for 8 hours. Scale bars: 20 μm. Adapted with permission from ref. 34. Copyright 2010, Nature Publishing Group.
Cell membrane related in-situ assemblies of small molecules
Cholesterol, composing 30% of animal plasma membranes, plays critical roles in maintaining the integrity and fluidity of cell membranes as well as interacting with proteins to assist cellular functions. Taking advantage of the in-situ enzymatic reactions to generate assemblies on or inside cancer cells, a recent demonstration used a simple conjugate of cholesterol and tyrosine phosphate 26a (Fig. 12A) to generate assemblies of the dephosphorylate product 26b on cells and inside cells, which potently and selectively inhibited platinum-resistant ovarian cancer cells (A2780cis) by activating extrinsic and intrinsic cell death signalling.36 Dephosphorylation of 26a yielded 26b to resulting in nanoparticles (Fig. 12C). The IC50 value of 26a against A2780cis cells (13 μM) was five times lower than the IC50 value of the clinic drug cisplatin (71 μM). The treatment of 26a with A2780cis cells significantly enhanced the clustering of cell death receptor DR5 (Fig. 12D), which was consistent with the increase in the microheterogeneity of the cell membrane induced by 26a (Fig. 12E). In addition, after entering the cells, the assemblies of 26b disrupted the dynamics of cytoskeletons (i.e., F-actin, microtubule (Fig. 12E)) to cause cell death. This work provided a useful guidance for modifying bioactive small molecules to interact with multiple cellular targets.
Fig. 12.
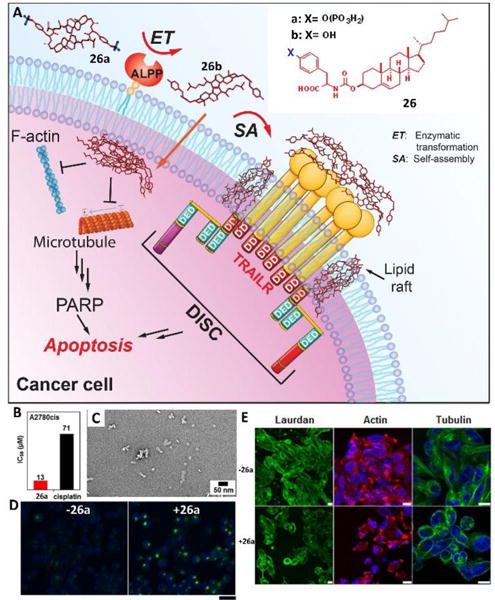
(A) EISA of the tyrosine-cholesterol conjugate activates extrinsic and intrinsic cell death signaling. (B) IC50 of 26a and cisplatin against A2780cis cell lines for 48 h. (C) TEM images of 24a (100 μM) treated with ALP (1 U/mL) in PBS (pH7.4) after 24 h; bar is 50 nm. (D) Confocal images of A2780cis cells treated with anti-DR5 without or with the addition of 26a (12.5 μM) for 24 h. Scale bar = 30 μm. (E) Confocal images of A2780cis cells stained with laurdan (10 μM), Alexa Fluor 633 phalloidin (F-actin, red), and Hoechst (nuclei, blue) or tubulin tracker (green) without or with the addition of 26a (12.5 μM) for 12 h (scale bar is 10 μm). Adapted with permission from ref. 36. Copyright 2016, American Chemical Society.
Cell membranes carry out many functions, and they even promote formation of supramolecular hydrogels, as elegantly illustrated be Eelkema et al.37 They developed two precursors, 27 and 28, which formed the hydrogelator 29 in aqueous media at pH 7.0 (Fig. 13). The formation rate of 29 could be tuned by applying acid or nucleophilic aniline catalysis, which affected the gelation rate and the resulting morphology. Impressively, negatively charged lipid membranes, which created a proton gradient in their peripheral space through electrostatic interactions, changed the pH to result in hydrogelation. Addition of DPPG and DOPG, two negatively charged lipids, to the mixture of 27 and 28, resulted in quick gelation and decreased MGC of 29. Further studies using two fluorescence dyes, 30 and 31, which specifically label the hydrogelator and liposome, respectively, revealed the mechanism of such system. Instead of using a negatively charged liposome, the authors reported that co-incubation of 27, 28 and 30 with HeLa cells led to the formation of gel networks on the surface of live cells. The study provided an alternative strategy for in-situ self-assembly of small molecules in cell milieu by the mere presence of cells.
Fig. 13.
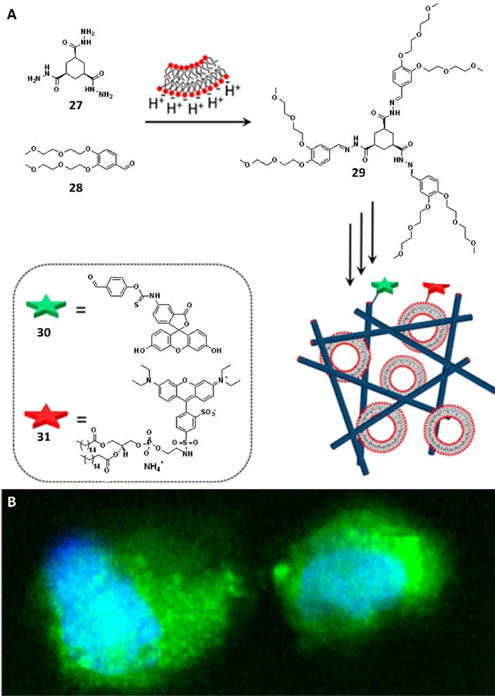
(A) Catalysis by negatively charged membranes; the surface of a negatively charged lipid membrane attracts protons, leading to a local decrease in the pH that can catalyze the formation of hydrogelator 29 from nonassembling precursors 27 and 28. To visualize the gel fibers and lipid membranes, probes 30 and 31, respectively, are used. (B) Confocal images showing the formation of gel fibers in HeLa cells; 3D reconstruction of a z-stack, revealing the formation of gel fibers throughout the cell. The gel fibers were stained with 30 (green), whereas the nuclei were visualized with Hoechst nuclear dye (blue). Adapted with permission from ref. 37. Copyright 2016, American Chemical Society.
Assemblies of small molecules as functional mimics of proteins
Glycosylation, one of the most ubiquitous forms of posttranslational modification, plays a crucial role in biology. For example, glycoproteins, as an essential component to a variety of biological events in living cells, exhibit a pivotal role in immune response, cell communication, neuronal development, protein folding, and fertilization. Although glycoconjugates (especially glycoproteins), as mediators of complex cellular events, have intrigued researchers from different fields for two decades, the difficulties in chemical and biological synthesis of the desired glycoproteins or glycopeptides have limited their developments and further applications. An alternative strategy to avoid the laborious synthesis of glycoproteins is to use self-assembly of small glycoconjugates for mimicking the functions of glycoproteins.38 Fig. 14 showed a designed glycoconjugate 32, which consisted of a nucleobase, peptide RGD, and a saccharide (glycosamine), 32, was able to form assemblies in the PBS buffer, in the cell culture medium, or on the cell medium, which allowed its application for mES cell culture. The results showed that the assemblies of 32 promoted the proliferation of mES cells and the development of zygotes into blastocysts of mice. This work indicated that the assemblies of small molecular conjugates, consisting of multiple fundamental biological building blocks, could modulate the cell behaviors, thus providing a simple and feasible strategy for mimicking the functional glycoconjugate in cell milieu. Such a strategy also led to supramolecular glycosylation for dissociating peptide nanofibrils,39 which provides insight for accelerating degradation of the aggregates of aberrant proteins.
Fig. 14.
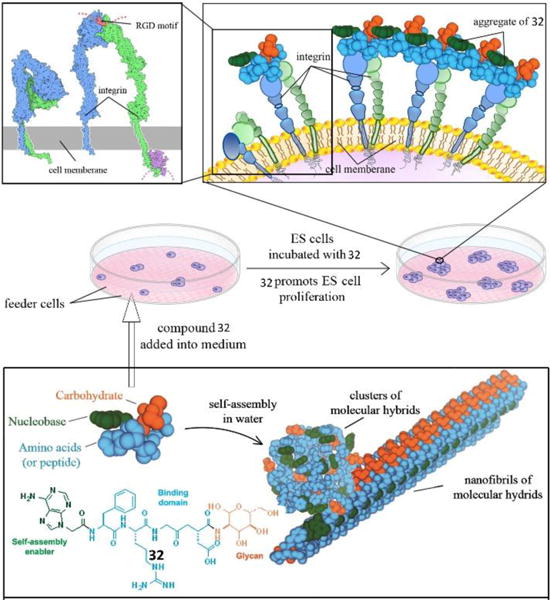
Molecule of 32 promoting the proliferation of mES Cells via plausible interactions of supramolecular assemblies with integrins. Adapted with permission from ref. 38. Copyright 2014, American Chemical Society.
Enhancing the activities of the assemblies of small molecules
Although bioactive molecules, including hydrophobic drugs, protein drugs, and short peptides (both L and D enantiomer) exhibit promising therapeutic efficacy, the poor cellular uptake of these agents has been a challenge that limits their clinical applications. Current strategies, such as employing cationic oligopeptides to improve cellular uptake still remain problematic and inefficient due to metabolic degradation, poor cellular compatibility, lack of cellular selectivity, and uncontrolled diffusion. The use of self-assembly of small molecules in cell milieu promises a solution for meeting the challenge, as demonstrated recently by conjugating taurine to a D-peptide.40 As shown in Fig. 15A, an esterase substrate, 33, consisted of the fluorophore of NBD (for visualizing cellular uptake and distribution), D-diphenylalanine peptide (for self-assembly and protease resistance), an ester bond (for enzymatic cleavage), and taurine (for boosting cellular uptake). Once the precursor entered into the cell, intracellular esterases removed the taurine group from 33 to form the hydrogelator 34, which then self-assembled to form assemblies. With increasing incubation time, the assemblies accumulated and remained inside the cell. Further mechanistic study indicated that the enhanced uptake of 33, being independent of the taurine transporter, likely involved both dynamin-dependent endocytosis and macropinocytosis. This design, in fact, provided a strategy for promoting the uptake of D-peptides to form nanofibers inside cells for boosting the activity of cisplatin against drug-resistant ovarian cancer cells.41
Fig. 15.
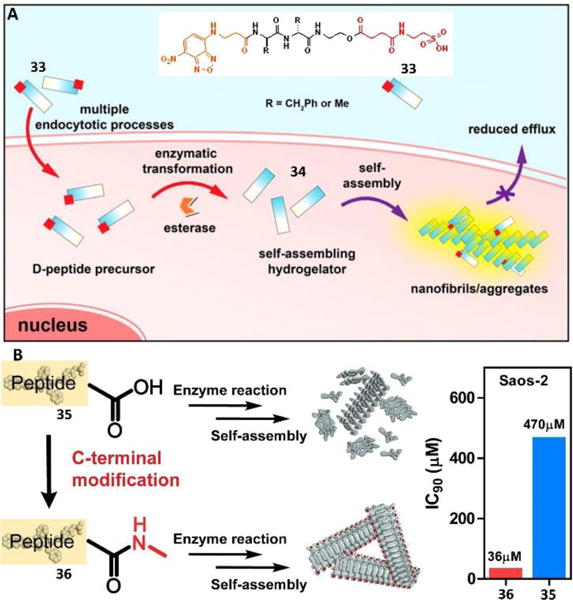
(A) Molecular structures of precursor 34 and the illustration of taurine conjugation boosts cellular uptake of d-peptide precursors and subsequent enzyme-instructed self-assembly to form nanofibers, accumulating inside cells. (B) Illustration of the C-terminal modification enhancing self-assembly and their IC50 against cancer cell line of Saos-2. The sequence of peptide present represents Napffpy. Adapted with permission from ref. 40 and 42. Copyright 2015, American Chemical Society (ref. 40). Copyright 2016, Royal Society of Chemistry (ref. 42).
While it is applicable to enhance the efficacy of assemblies of the small molecules in cell milieu by increasing the uptake of small molecules, this strategy is less useful for the assemblies of small molecules that are formed in the pericellular space. Increasing the self-assembling ability of small molecules would be a more attractive approach. This logical approach, in fact, has been demonstrated recently.42 For example, a simple method to enhance the self-assembling ability and boost anticancer efficacy was through minimal modification of the C-terminus of a small D-peptide. Such modification, only changing the molecular weight by 2%, increased the inhibitory activity by over an order of magnitude (IC50 from 431 to 23 μM) towards an osteosarcoma cell line (Fig. 15B). This work also indicated that a simple modification of molecular structures could have a profound impact on multiple length scales, from nanometers to microns, which would help explore the emergent properties of assemblies of small molecules in cell milieu.
Besides increasing cellular uptake, organelle targeting is another efficient strategy to boost the efficacy of EISA by enhancing the local concentration of assemblies. A study demonstrated that combining enzymatic self-assembly and mitochondria targeting enabled selective inhibition of cancer cells (i.e., Saos-2 cells) with a high expression level of phosphatase.43 Precursor 37a (Fig. 16A), consisting of both the ALP substrate and mitochondria targeting motif (i.e., triphenyl phosphinium (TPP)), turned into fluorescent assemblies upon treatment of ALP due to the environment-sensitive fluorophore (i.e., NBD). Generated on the cell membrane of Saos-2 cells via EISA, these assemblies were internalized via multiple endocytosis modes—caveolae/lipid raft-mediated endocytosis and clathrin-mediated endocytosis. CLSM images (Fig. 16B) indicated the escape of assemblies from lysosomes and mainly accumulated in mitochondria (red) (Fig. 16C), confirming the targeting of mitochondria. In addition, some assemblies remained on the cell surface (Fig. 16C), proving their generation on the cell membrane. Enriched in mitochondria, these assemblies disrupted the mitochondria and resulted in the leakage of cytochrome c, thus, activating death signaling pathways. Notably, unlike traditional protein inhibitors, the strategy of employing assemblies to kill cancer cells via multiple targeting hardly caused an acquired drug resistance, a promising observation that deserves further development.
Fig. 16.
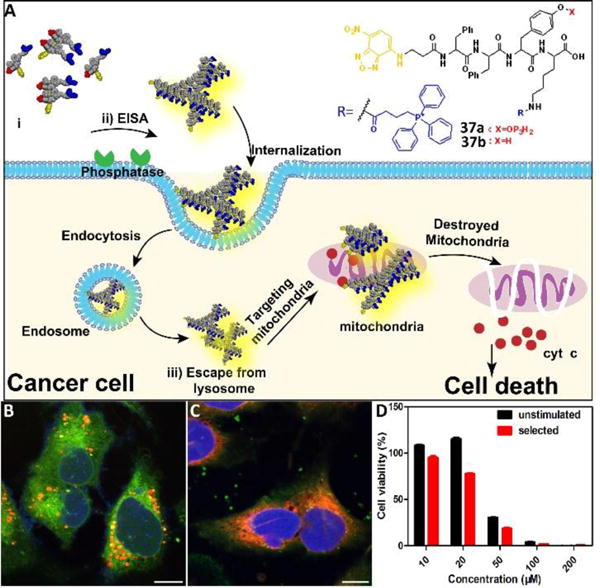
(A) Illustration of enzyme-instructed self-assembly for targeting mitochondria and inducing the death of cancer cells. (B) CLSM images of Saos-2 cells treated with D-37a (50 μM) for 4 h and then stained with Lyso-Tracker. Scale bar is 10 μm. (C) CLSM images of Saos-2 cells treated with D-37a (50 μM) for 4 h and then stained with Mito-tracker. Scale bar is 10 μm. (D) Cell viability of unstimulated Saos-2 cell line or selected Saos-2 cell line (after five weeks treatment of the precursors with gradually increase concentrations) incubated with D-37a at different concentrations for 48 h. Adapted with permission from ref.43. Copyright 2016, American Chemical Society.
A few guidelines for generating assemblies in cell milieu
Balanced hydrophobic and hydrophilic segments in amphiphilic molecules
In recent years, there has been a growing interest in assemblies of small molecules, motivated by not only their potential applications, but also the ease of manipulating these assemblies in the cell environment to mimic cellular machinery and functions. Although most assemblies of small molecules result from serendipitous discoveries, these studies not only generated a large library of small molecules with self-assembling ability, but also provided useful information on prerequisites for designing assemblies of small molecules. One key requirement for generating self-assembling molecules is to balance hydrophobic and hydrophilic segments in amphiphilic molecules, which is essential for enhancing intermolecular interactions in the aqueous phase to achieve self-assembly in the cell milieu.
Suitable biological trigger(s)
Another key requirement is to have suitable biological trigger(s) or que(s) to form in-situ assemblies, which is important to endow selectivity in forming assemblies of small molecules in different cellular environments. When one designs a precursor of a self-assembling molecule, the preferred precursor should be hydrophilic rather than hydrophobic, so that the difficulty of dissolution in water and the non-specific binding in the cell milieu can be minimized. Although most of the studies use an enzymatic reaction as a triggering mechanism. It is conceivable to use ligand-receptor interactions to instruct the self-assembly of small molecules in cell milieu, as demonstrated recently.44 With the increased structural information on protein-protein interactions, it is likely that such knowledge will help design self-assembly of small molecules triggered by the presence of a protein, instead of an enzyme.
Stability of the molecules in vivo
Unlike self-assembly in vitro (i.e., cell free setting), the stability of assemblies of small molecules in cell milieu is essential for their applications. This requirement is particularly important when the molecules are small peptides because the peptides made of L-amino acids are susceptible to proteolytic degradation catalyzed by endogenous enzymes, such as peptidases or amidases. To enhance the stability of assemblies of small peptides in cell milieu, several strategies are useful: i) D-amino acids, the enantiomers of L-amino acids, exhibit unique stereochemistry properties that lead to resistance towards most of the endogenous enzymes. As shown in the works of several labs, replacing L-amino acids with D-amino acids greatly enhances the stability of the peptide towards proteolysis. In fact, this is a strategy used by nature, as shown in peptidic antibiotics that consist of D-amino acid residues.45 ii) The use of β-peptides and staple peptides is an alternative option for in vivo stability, as demonstrated by the reported unusually long half-life in the blood serum.46 iii) Glycosylation is a promising strategy for stabilizing peptides in vivo since nature already uses glycosylation to enhance proteolysis stability, as demonstrated by the case of the ribonuclease that glycosylated RNase B, which is much more resistant toward proteolytic degradation than RNase A, a un-glycosylated pancreatic ribonuclease.47 Taking advantage of this, several reports already show unexpected stability against protease K and functions by incorporating saccharides into small molecules.48
Focusing on kinetics and processes
Based on thermodynamics, the classic pharmacodynamics of a drug (in a concentration dependent manner) is to show the dosage curve of a standard sigmoidal curve in vitro. Different from the thermodynamic equilibrium of self-assembly in vitro, the formation of assemblies in cell milieu is a kinetically controlled process. Considering the cell, itself, is a dynamic system operating far-from equilibrium, it is challenging and exciting to explore self-assembly of small molecules in cell milieu. For example, molecules passing through the cell membrane and entering into the cells are not only controlled by the properties of the molecules and their reactions, but are influenced by the cellular response/states to these molecules. This subtle feature has a profound implication, that is, as a reaction diffusion controlled phenomenon, EISA, acts a multi-step process for controlling cell behaviors.49 Changing substrates or the expression level of enzymes or isoenzymes can affect the resultant assemblies, thus controlling cell behaviors. Besides enzymes, other biological cues such as pH gradients, redox difference, and intracellular ionic strength can trigger in-situ self-assembly in cell milieu.
Spatiotemporal control
Spatiotemporal control is ubiquitous in cellular processes and living organisms. This fundamental fact is a bioinspiration for exploring the self-assembly of small molecules in cell milieu. Since the driving force for self-assembly of small molecules is non-covalent interaction, it is rational to design adaptive assemblies of small molecules, whose structure and function can be reversibly controlled in situ. To achieve spatiotemporal control for functions, three conditions should be met: i) suitable trigger(s) that are capable of reversibly switching the formation of assemblies; ii) generating the assemblies in specific locations; iii) endowing the molecular assemblies with the desired activity.
Molecular validation
The interactions of self-assembly of small molecules with cells, undoubtedly, are complicated. For example, the colloidal aggregates formed by the precursors of the self-assembling molecules affect the in-situ self-assembly of small molecules in cell milieu because the aggregation of the precursors alters the cellular uptake of the precursors or their promiscuous interactions with the cell surface molecules, agnostically or antagonistically, depending on the molecules. Therefore, the best and most reliable way to validate the molecular design for assemblies of small molecules in cell milieu is to generate a series of molecular analogs against the same set of cell lines. This approach minimizes the uncertainty and artifacts introduced by changing the cells genetically or biochemically.
Future and outlook
In the last several decades, the self-assembly of small molecules has been intensively studied and developed in a cell free setting. In recent years, the study in self-assembly of small molecules has advanced to in-situ formation of assemblies in biological systems (i.e., cells). Although modifying the serendipitously obtained precursors can generate new precursors for assemblies, such a strategy only explores a limited molecular space around the original precursors. It is a challenge, as well as an opportunity, to develop novel precursors or methodology for self-assembly in cell milieu. For example, the use of interactions between ligands and receptors and the common phenomenon in nature for catalyzing the formation of assemblies of small molecules may be a promising approach for controlling the fate of cancer cells44 and tuning the behavior of stem cells. The development of biocompatible chemical reactions in aqueous solutions (e.g., bio-orthogonal reactions) also provides a feasible strategy to generate assemblies in cell milieu.
Currently, the concentration of the precursors required for the formation of functional assemblies on cell milieu is relatively high and above the clinical standard. Molecular engineering of the precursors without compromising their activity has successfully lowered the IC50 against cancer cells from submillimolar to a few micromolar. The goal is to lower the IC90 to micromolar or nanomolar, which certainly requires more research and likely is achievable. For example, accumulation of assemblies in subcellular organelles is a promising strategy to improve the efficacy of assemblies of small molecules. Consequently, how to control the location of assemblies of small molecules more selectively and more precisely is another challenge and should be the first consideration for designing the molecules. Currently, by selecting suitable enzymes (e.g., ALP or esterase) that are overexpressed in cancer cells, it is feasible to control assemblies of small molecules either on the pericellular or intracellular space, resulting in changes in cell environments through different mechanisms. But what would be the most effective strategy to generate the assemblies exclusively inside the cell nucleus? Another subtle factor that could influence the resultant assemblies is the reactivity of the trigger, which could influence cell behaviors through reaction diffusion. For example, if intracellular self-assembly is fast, the formation of the corresponding assemblies is enough to prevent diffusing through efflux pumps and to result in higher activity on the cell or vice versa. This kind of detail deserves further investigation.
Besides chemistry, the development of assemblies of small molecules also depends on other disciplines. Although one can characterize the morphologies of given assemblies, it is still difficult to interrogate the atomistic structures of the assemblies of small molecules, even in vitro. There is little information on the morphologies and the structures of the assemblies in cell milieu, which requires the development of new technologies. The rapid growth of cryo-EM and correlative light and electron microscopy in the application of protein structures could be a solution. Obviously, the advance of knowledge of biology and pharmacology will be crucial to explore assemblies of small molecules to control cell behaviors. For example, most protein inhibitors of small molecules show good affinity in vitro and low efficacy or no efficacy in cell milieu, which is partly because of the aggregation of the inhibitors and the complex environment of the cell that influences the targets. Such knowledge would be useful for designing assemblies of small molecules to target multiple processes of cells.
As a research area that is attracting researchers from different disciplines to work together to explore the mystery of cells, the exploration of bioinspired assembly of small molecules in cell milieu is just beginning. Being multifunctional, the assemblies of small molecules are finding applications in various fields, such as cancer therapy, analyte detection, modulating immune response, controlling stem cell behaviors and so on. Currently, one of the most explored applications is cancer therapy since cancer cells will be unlikely to acquire resistance to the molecular assemblies, as shown in a recent study.43 Another promising aspect that deserves exploration is reversible formation of the assemblies of small molecules in cell milieu, which is less explored. The only reported example is employing an enzyme switch (i.e., kinase and phosphatase) to control the self-assembly of a short peptide in vitro.50 To carry out a similar process in vivo and couple it with cellular processes is exciting and challenging. Like any other frontiers of research, some questions still remain and need to be resolved in the coming years. For example, how do these precursors self-assemble in cells and interact with cells? How will one generate assemblies with precise spatiotemporal control? How to identify the morphologies of assemblies on the cell surface or inside the cells? What are the detailed cellular mechanisms for responding to the assemblies? Answering these questions requires more research. Moreover, besides answering the above questions, the research on assemblies of small molecules may help understanding of the origin of life, a holy grail in chemistry.
Fig. 17.
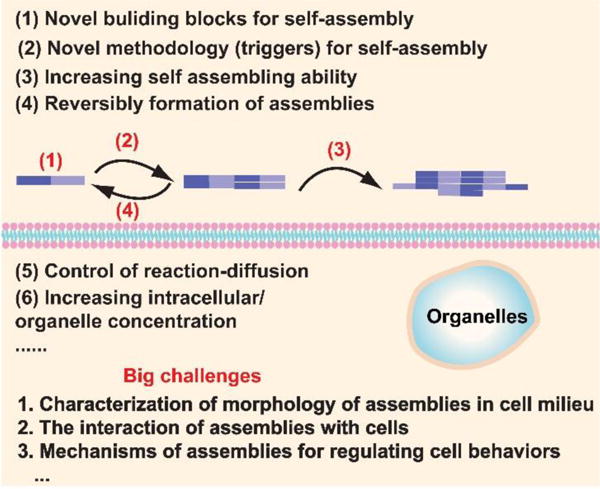
Potential directions of bioinspired self-assembly (SA) of small molecules in cell milieu.
Key learning points.
Self-assembly of small molecules in water is a general phenomenon.
How to generate the self-assembly of small molecules in cell milieu.
How the assemblies of small molecules interact with multiple proteins, regulate cellular processes, and result in primary phenotypes
Guiding principles to design the self-assembly of small molecules to control the fate of cells
Acknowledgments
This work was partially supported by NIH (R01CA142746), NSF (MRSEC-1420382), and International S&T Cooperation Program of China (ISTCP, 2015DFA50310).
Notes and references
- 1.Steven AC, Baumeister W, Johnson LN, Perham RN. Molecular Biology of Assemblies and Machines. Garland Science; 2016. [Google Scholar]
- 2.Whitesides GM. Interface focus. 2015;5:20150031. doi: 10.1098/rsfs.2015.0031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lutolf M, Hubbell J. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 4.Du X, Zhou J, Shi J, Xu B. Chem Rev. 2015;115:13165–13307. doi: 10.1021/acs.chemrev.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lingwood D, Simons K. science. 2010;327:46–50. doi: 10.1126/science.1174621. [DOI] [PubMed] [Google Scholar]
- 6.Breukink E, de Kruijff B. Nat Rev Drug Discovery. 2006;5:321–323. doi: 10.1038/nrd2004. [DOI] [PubMed] [Google Scholar]
- 7.Kueh HY, Mitchison TJ. Science. 2009;325:960–963. doi: 10.1126/science.1168823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ulijn RV, Smith AM. Chem Soc Rev. 2008;37:664–675. doi: 10.1039/b609047h. [DOI] [PubMed] [Google Scholar]
- 9.Wool IG. Annu Rev Biochem. 1979;48:719–754. doi: 10.1146/annurev.bi.48.070179.003443. [DOI] [PubMed] [Google Scholar]
- 10.Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. Clin Cancer Res. 1999;5:2638–2645. [PubMed] [Google Scholar]
- 11.Ciechanover A, Brundin P. Neuron. 2003;40:427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 12.Yonamine Y, Cervantes-Salguero K, Minami K, Kawamata I, Nakanishi W, Hill JP, Murata S, Ariga K. Phys Chem Chem Phys. 2016;18:12576–12581. doi: 10.1039/c6cp01586g. [DOI] [PubMed] [Google Scholar]
- 13.Roh YH, Ruiz RC, Peng S, Lee JB, Luo D. Chem Soc Rev. 2011;40:5730–5744. doi: 10.1039/c1cs15162b. [DOI] [PubMed] [Google Scholar]
- 14.Pandian GN, Sugiyama H. Bull Chem Soc Jpn. 2016;89:843–868. [Google Scholar]
- 15.Simons K, Ikonen E. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 16.Zhang S. Nat Biotechnol. 2003;21:1171–1178. doi: 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- 17.Webber MJ, Appel EA, Meijer E, Langer R. Nat Mater. 2016;15:13–26. doi: 10.1038/nmat4474. [DOI] [PubMed] [Google Scholar]
- 18.Svensson M, Håkansson A, Mossberg A-K, Linse S, Svanborg C. Proc Natl Acad Sci. 2000;97:4221–4226. doi: 10.1073/pnas.97.8.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuang Y, Xu B. Angew Chem, Int Ed. 2013;52:6944–6948. doi: 10.1002/anie.201302658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao Y, Long M, Shi J, Hedstrom L, Xu B. Chem Commun (Cambridge, U K) 2012;48:8404. doi: 10.1039/c2cc33631f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuang Y, Long MJ, Zhou J, Shi J, Gao Y, Xu C, Hedstrom L, Xu B. J Biol Chem. 2014;289:29208–29218. doi: 10.1074/jbc.M114.600288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang Z, Gu H, Fu D, Gao P, Lam JK, Xu B. Adv Mater. 2004;16:1440–1444. [Google Scholar]
- 23.Yang Z, Xu K, Guo Z, Guo Z, Xu B. Adv Mater. 2007;19:3152–3156. [Google Scholar]
- 24.Kuang Y, Shi J, Li J, Yuan D, Alberti KA, Xu Q, Xu B. Angew Chem, Int Ed. 2014;53:8104–8107. doi: 10.1002/anie.201402216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fishman WH. Nature. 1968;219:697–699. doi: 10.1038/219697a0. [DOI] [PubMed] [Google Scholar]
- 26.Zhou J, Du X, Yamagata N, Xu B. J Am Chem Soc. 2016;138:3813–3823. doi: 10.1021/jacs.5b13541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou J, Du X, Xu B. Angew Chem. 2016;128:5864–5869. [Google Scholar]
- 28.Zheng Z, Chen P, Xie M, Wu C, Luo Y, Wang W, Jiang J, Liang G. J Am Chem Soc. 2016;138:11128–11131. doi: 10.1021/jacs.6b06903. [DOI] [PubMed] [Google Scholar]
- 29.Pires RA, Abul-Haija YM, Costa DS, Novoa-Carballal R, Reis RL, Ulijn RV, Pashkuleva I. J Am Chem Soc. 2015;137:576–579. doi: 10.1021/ja5111893. [DOI] [PubMed] [Google Scholar]
- 30.Tanaka A, Fukuoka Y, Morimoto Y, Honjo T, Koda D, Goto M, Maruyama T. J Am Chem Soc. 2015;137:770–775. doi: 10.1021/ja510156v. [DOI] [PubMed] [Google Scholar]
- 31.Gao Y, Shi J, Yuan D, Xu B. Nat Commun. 2012;3:1033. doi: 10.1038/ncomms2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao Y, Berciu C, Kuang Y, Shi J, Nicastro D, Xu B. ACS nano. 2013;7:9055–9063. doi: 10.1021/nn403664n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou J, Du X, Berciu C, He H, Shi J, Nicastro D, Xu B. Chem. 2016;1:246–263. doi: 10.1016/j.chempr.2016.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liang G, Ren H, Rao J. Nat Chem. 2010;2:54–60. doi: 10.1038/nchem.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yuan Y, Wang L, Du W, Ding Z, Zhang J, Han T, An L, Zhang H, Liang G. Angew Chem. 2015;127:9836–9840. doi: 10.1002/anie.201504329. [DOI] [PubMed] [Google Scholar]
- 36.Wang H, Feng Z, Wu D, Fritzsching KJ, Rigney M, Zhou J, Jiang Y, Schmidt-Rohr K, Xu B. J Am Chem Soc. 2016;138:10758–10761. doi: 10.1021/jacs.6b06075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Versluis F, van Elsland DM, Mytnyk S, Perrier DL, Trausel F, Poolman JM, Maity C, le Sage VA, van Kasteren SI, van Esch JH. J Am Chem Soc. 2016;138:8670–8673. doi: 10.1021/jacs.6b03853. [DOI] [PubMed] [Google Scholar]
- 38.Du X, Zhou J, Guvench O, Sangiorgi FO, Li X, Zhou N, Xu B. Bioconjugate Chem. 2014;25:1031–1035. doi: 10.1021/bc500187m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan D, Shi J, Du X, Zhou N, Xu B. J Am Chem Soc. 2015;137:10092–10095. doi: 10.1021/jacs.5b05888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J, Du X, Li J, Yamagata N, Xu B. J Am Chem Soc. 2015;137:10040–10043. doi: 10.1021/jacs.5b06181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J, Kuang Y, Shi J, Zhou J, Medina JE, Zhou R, Yuan D, Yang C, Wang H, Yang Z. Angew Chem. 2015;127:13505–13509. doi: 10.1002/anie.201507157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feng Z, Wang H, Du X, Shi J, Li J, Xu B. Chem Commun. 2016;52:6332–6335. doi: 10.1039/c6cc02282k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang H, Feng Z, Wang Y, Zhou R, Yang Z, Xu B. J Am Chem Soc. 2016 [Google Scholar]
- 44.Shi J, Du X, Huang Y, Zhou J, Yuan D, Wu D, Zhang Y, Haburcak R, Epstein IR, Xu B. J Am Chem Soc. 2014;137:26–29. doi: 10.1021/ja5100417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brückner H. D-amino acids in chemistry, life sciences, and biotechnology. Verlag Helvetica Chimica Acta; 2011. [Google Scholar]
- 46.Frackenpohl J, Arvidsson PI, Schreiber JV, Seebach D. ChemBioChem. 2001;2:445–455. doi: 10.1002/1439-7633(20010601)2:6<445::aid-cbic445>3.3.co;2-i. [DOI] [PubMed] [Google Scholar]
- 47.Rudd PM, Joao HC, Coghill E, Fiten P, Saunders MR, Opdenakker G, Dwek RA. Biochemistry. 1994;33:17–22. doi: 10.1021/bi00167a003. [DOI] [PubMed] [Google Scholar]
- 48.Li X, Du X, Li J, Gao Y, Pan Y, Shi J, Zhou N, Xu B. Langmuir. 2012;28:13512–13517. doi: 10.1021/la302583a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou J, Xu B. Bioconjugate Chem. 2015;26:987–999. doi: 10.1021/acs.bioconjchem.5b00196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang Z, Liang G, Wang L, Xu B. J Am Chem Soc. 2006;128:3038–3043. doi: 10.1021/ja057412y. [DOI] [PubMed] [Google Scholar]