

Abstract
Mitochondrial dysfunction and neuroinflammation occur in Alzheimer’s disease (AD). The causes of these pathologic lesions remain uncertain, but links between these phenomena are increasingly recognized. In this review, we discuss data that indicate mitochondria or mitochondrial components may contribute to neuroinflammation. While, mitochondrial dysfunction could cause neuroinflammation, neuroinflammation could also cause mitochondrial dysfunction. However, based on the systemic nature of AD mitochondrial dysfunction as well as data from experiments we discuss, the former possibility is perhaps more likely. If correct, then manipulation of mitochondria, either directly or through manipulations of bioenergetic pathways, could prove effective in reducing metabolic dysfunction and neuroinflammation in AD patients. We also review some potential approaches through which such manipulations may be achieved.
Keywords: Alzheimer’s disease, bioenergetics, damage associated molecular pattern, inflammation, mitochondria
Introduction
Mitochondrial dysfunction and neuroinflammation are observed in Alzheimer’s disease (AD) [1–12]. It is increasingly recognized that mitochondrial dysfunction and inflammation are interdependent lesions [11, 13]. Overall, though, the underlying cause(s) of these changes are poorly understood.
Mitochondria in many ways represent remnants of proteobacteria, and they contain immunogenic molecules. Moreover, mitochondrial dysfunction induces inflammation, while the converse, inflammation results in mitochondrial dysfunction, is also true [11, 13–15]. In either case, bioenergetic failure may ultimately result, and combinations of mitochondrial dysfunction, neuroinflammation, and bioenergetic failure may contribute to the development or progression of neurodegeneration and neurodegenerative diseases such as AD [11, 13]. Approaches that target mitochondrial dysfunction, neuroinflammation, or both may, therefore, ultimately prove beneficial for the treatment of AD.
Neuroinflammation
Notable hallmarks of AD include increased numbers of activated microglia and astrocytes [16–20]. Numerous inflammatory pathways appear activated in AD brains, and interestingly elevated cytokines are observed both within AD subject brain tissue and patient sera [11, 16, 18, 21, 22]. Inflammatory signals classically initiate in response to a pathogen, or “foreign” agent. Novel data now suggest mitochondria and/or mitochondrial components could mimic a pathogen and lead to a damage/danger response [23, 24].
Mitochondrial lysates induce inflammation in several cell types, whereas nuclear fractions fail to do so [25, 26]. We recently showed that in a mouse microglial cell line, mitochondrial lysates induced the expression of messenger RNA (mRNA) for the cytokines tumor necrosis factor α (TNFα) and interleukin 8 (IL-8), as well as for matrix metalloproteinase 8 (MMP-8) [23]. Further, triggering receptor expressed on myeloid cells 2 (TREM2), a protein which functions to inhibit cytokine production, was reduced at the mRNA level. These effects appeared to be mediated by the activation of p38 mitogen activated protein kinase (MAPK) signaling through the pro-inflammatory transcription factor nuclear factor κ light chain enhancer of B cells (NFκB) (Figure 1).
Figure 1. Inflammation induced by mitochondrial-derived DAMP molecules.
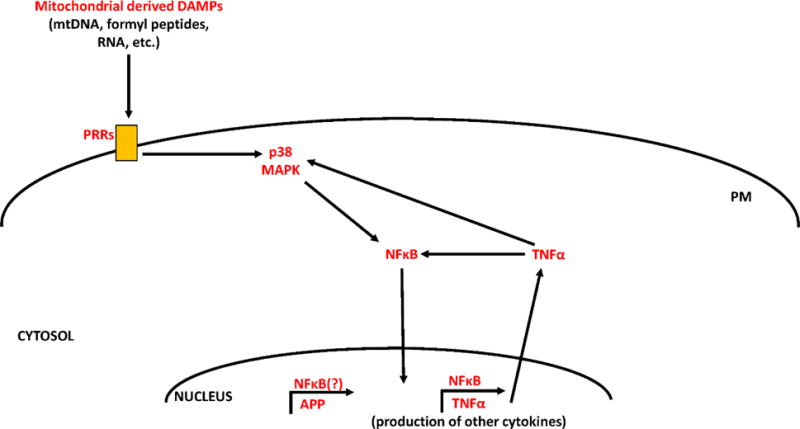
Mitochondrial-derived DAMPs (including mtDNA, formyl peptides, RNA, ATP, certain mitochondrial-localized proteins, and cardiolipin) activate pattern recognition receptors (PRRs) such as toll-like receptors, formyl peptide receptors, or the NLRP3 inflammasome. Activation of these receptors leads to downstream phosphorylation and activation of p38 MAPK. Activated p38 MAPK can then activate NFκB, inducing its translocation to the nucleus. NFκB facilitates the transcription of cytokines such as TNFα. TNFα can further amplify an inflammatory cascade by independently activating p38 MAPK and NFκB signaling. NFκB may also initiate APP transcription.
Of further interest to the AD field, mitochondrial lysates increased amyloid precursor protein (APP) mRNA and protein levels [23]. APP is the parent peptide from which amyloid beta (Aβ) is generated, and Aβ generates fibrils that aggregate to form amyloid plaques in the brains of AD subjects. Therefore, our data implicate mitochondrial lysates as mediators of neuroinflammation, and suggest that through this effect mitochondrial components may influence APP biology.
Mitochondria, in fact, contain numerous pro-inflammatory molecules including, but not limited to, mitochondrial DNA (mtDNA), adenosine triphosphate (ATP), cardiolipin, mitochondrial transcription factor A (TFAM), cytochrome c, formyl peptides, and RNA. The immunogenic properties of these molecules have been previously reviewed [11].
In particular, mtDNA has increasingly been implicated in inflammation. In specific relation to neuroinflammation, oxidatively modified mtDNA induces inflammatory signaling in astrocytes [24]. Oxidant-modified mitochondrial polynucleotides, when transferred to mouse primary astrocytes, stimulate the expression of interleukin 6 (IL-6), monocyte chemotactic protein-1 (MCP-1), interleukin 1β (IL-1β), and TNFα [24]. mtDNA damage can be initiated with hydrogen peroxide [24], and evidence indicates this type of DNA oxidative damage is relatively specific to mitochondria [27].
Other studies have tested the effects of mitochondrial derived damage-associated molecular pattern (DAMP) molecules on glial cells. Mixed cultures of glial cells exposed to ATP increase expression of interleukin 6 (IL-6) and chemokine C-X-C motif ligand 1 (CXCL1) [28]. Medium from human primary microglia, when primed with interferon γ (IFNγ) and incubated with TFAM (or mitochondrial proteins), is toxic to SH-SY5Y neuroblastoma cells [29]. These IFNγ and TFAM activated microglia also secrete elevated levels of IL-6.
It is important to consider the question of how mitochondrial components may ultimately be released from neurons. Of potential relevance to this question, a recent study reported that mitochondria are indeed normally released from neurons at axon terminals. These released mitochondria are then degraded by surrounding glial cells. This previously undescribed process is speculated to represent a form of transcellular mitochondrial degradation referred to as “transmitophagy” [30]. Other previously recognized modes of mitochondrial release include cell death events that proceed via necrosis or necroptosis [11].
A second and equally important question has to do with what factors or events might induce (as opposed to facilitate) the release of mitochondrial components from cells. In regards to this question, it is perhaps relevant that a link between mitochondrial dysfunction and inflammation is increasingly recognized. If mitochondrial function is inhibited, inflammation occurs [11, 13, 14]. Conversely, upon the induction of inflammation, mitochondrial function is altered [11, 13, 15]. As discussed in the next section, mitochondrial dysfunction occurs in AD. When considering the relationship between mitochondrial dysfunction and neuroinflammation in AD, it is worth considering which of these two related phenomena might represent the upstream pathology, and which may represent the downstream pathology.
Mitochondrial Dysfunction
Mitochondrial dysfunction occurs in AD and is felt by many to be disease-relevant and a promising therapeutic target [8]. In AD brains various mitochondria-localized enzymes show reduced activity, and in most neurons intact mitochondria are numerically reduced [8]. Glucose utilization, as demonstrated by fluoro-deoxyglucose positron emission tomography (FDG PET), is also reduced in the AD brain [31] and could indirectly reflect a consequence of mitochondrial impairment.
Beyond AD, these changes are also observed in aging. However, when compared to aged individuals, AD subjects show further reductions in the activities of cytochrome oxidase (COX), pyruvate dehydrogenase complex, and α-ketoglutarate dehydrogenase complex [1, 4, 6, 8]. These changes can be observed in the brain and systemically.
The cytoplasmic hybrid (cybrid) model, in which subject platelets are fused with a cell line devoid of mtDNA, replicate mitochondrial changes observed in AD patients [2, 3, 5, 7, 10]. This technique was recently reviewed elsewhere [12]. The only known perpetuating material transferred during the generation of a cybrid is mtDNA, and cybrid cell lines can be generated so that the different lines share equivalent nuclear DNA backgrounds. Cybrid studies indicate mtDNA itself could be responsible for mitochondrial deficits observed in AD subjects.
The majority of (if not all) mtDNA is inherited in a maternal manner. Children of parents that have an AD-affected mother have a greater risk of developing AD than children of parents that have an AD-affected father [32–34]. Further, deficits in brain glucose metabolism that occur before cognitive changes are observed occur to a greater extent in individuals whose mothers have AD than they do in individuals whose fathers have AD [34]. Overall, it is abundantly clear that bioenergetic changes occur in AD, and these changes may be accentuated in those with AD mothers.
One implication of the studies discussed above is that mitochondrial dysfunction could represent a relatively upstream AD pathology [35, 36]. This view is supported by the systemic nature of some AD mitochondrial defects, the apparent ability of mtDNA to account at least in part for some of these defects, and the finding that mtDNA inheritance may influence AD risk. While some inflammation markers may also be upregulated outside the brain in AD subjects [37–42], it is difficult to envision a scenario under which inflammation would account for cybrid data or a maternal inheritance bias. We therefore feel that if in fact a relationship between mitochondrial dysfunction and neuroinflammation exists, and one pathology drives the other, then mitochondrial dysfunction may represent the more upstream event.
Bioenergetic Medicine
Bioenergetic status is determined by rates of, and ultimately the balance of, energy production and consumption. Pathways that influence bioenergetics are characterized by the passage of carbon molecules between pathway intermediates. The movement of carbon through these pathways comprise and define fluxes. Flux pathways are compartmentalized within cells, and may reside within the cytoplasm, the mitochondria, or both.
Mitochondrial medicine utilizes methods which will directly correct or minimize the effects of mitochondrial dysfunction [9, 43]. A related construct, bioenergetic medicine, recognizes that direct manipulation of mitochondrial function may not be required to correct energy metabolism deficits [44]. After all, mitochondria can be manipulated indirectly by targeting bioenergetic pathways that are anatomically external to mitochondria themselves. Mitochondria can, therefore, be manipulated by manipulating metabolic fluxes that reside outside mitochondria. Altering bioenergetic fluxes, either within or outside of mitochondria, can further alter the expression of genes or the post-translational modification of proteins that sense and are sensitive to bioenergetic fluxes.
For the reasons discussed in the preceding section, it is suggested that in the case of AD increasing mitochondrial mass, mitochondrial respiration, and glucose utilization are all well-justified bioenergetic medicine approaches and investigators are currently exploring ways to accomplish these endpoints [44]. In our case, efforts have been informed by caloric restriction, which promotes liver mitochondrial biogenesis in some but not all studies, and exercise, which promotes muscle mitochondrial biogenesis [44, 45]. While we and others find caloric restriction and exercise do affect the brain in specific ways, and may even induce some degree of brain mitochondrial biogenesis, our impression is that these interventions at best affect brain mitochondrial biogenesis less robustly than they affect liver and muscle. We therefore feel approaches that directly and robustly enhance bioenergetic fluxes (glycolysis and respiration-related) and brain mitochondrial biogenesis pathways are needed, and predict such approaches will benefit persons with AD. Some of these approaches are discussed below and summarized in Figure 2.
Figure 2. Potential bioenergetic medicine approaches.
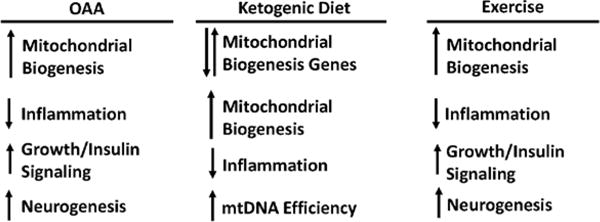
In the brain, OAA activates molecules and pathways that facilitate mitochondrial biogenesis, activates factors that favor cell/tissue growth, enhances insulin signaling, and promotes neurogenesis while countering inflammation. A ketogenic high fat diet favorably affects some parameters that favor mitochondrial biogenesis, counters inflammation, and may increase mtDNA transcription efficiency. Exercise (potentially through lactate for some parameters) activates molecules and pathways that facilitate mitochondrial biogenesis, activates factors that favor cell/tissue growth, and promotes neurogenesis while countering inflammation.
Oxaloacetate
The ideal agent would ideally be systemically safe, cross the blood brain barrier, access neurons and astrocytes, activate mitochondrial biogenesis, increase respiratory capacity, and increase glycolysis capacity. Oxaloacetate (OAA), a Krebs cycle and gluconeogenesis intermediate, putatively meets these criteria [46]. The structure of OAA is shown in Figure 3. OAA forms a redox pair with malate and is reduced to malate in a reaction that consumes NADH and generates NAD+. This would predictably shift cell redox balances towards more oxidized states, an effect reported to initiate mitochondrial biogenesis and mediate at least some health benefits of caloric restriction and exercise [47]. OAA, following its reduction to malate, can also access mitochondria and thereby supply carbon to the Krebs cycle and respiratory chain. Finally, OAA is converted to phosphoenolpyruvate (PEP) by PEP carboxykinase (PEPCK), a reaction that comprises part of the gluconeogenesis and glyceroneogenesis pathways. Muscle PEPCK overexpression, possibly due to enhanced glyceroneogenesis, induces a profound muscle mitochondrial biogenesis, enhances respiratory fitness, and robustly extends lifespan [48].
Figure 3.
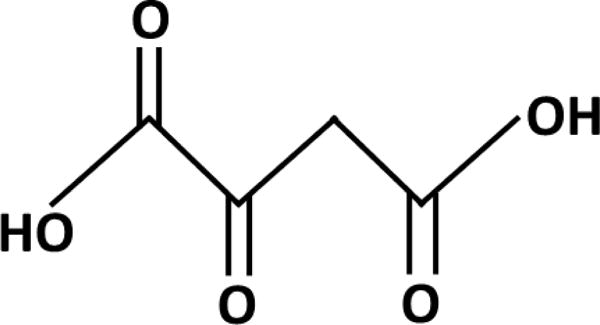
Structure of OAA.
In one study using C. elegans, OAA extended median and maximum lifespan through AMP kinase (AMPK) and forkhead O-box (FOXO) transcription factor-dependent pathways [49]. In mammals, it can be inferred that systemically administered OAA accesses the brain. This inference is based on rodent studies that report OAA prevents kainate-induced seizures and neuron damage, decreases ischemia-induced stroke volume, decreases cortical impact-induced traumatic brain injury, and slows glioma growth rates [50–53].
The literature currently reports only one human study of OAA, a 1968 clinical trial that evaluated its potential anti-hyperglycemic effects. This study, performed in human diabetics, reported OAA was well-tolerated and also provided limited pharmacokinetic data [54]. Levels were not increased 30 minutes but were increased 60 minutes after 200 mg were orally administered to three subjects. On average, OAA levels rose from 0 μg/100 ml to 2.3 μg/100 ml.
We recently tested the effects of high-dose OAA administration on brain bioenergetic infrastructures [46]. In these studies, male C57Bl/6 mice received intraperitoneal (IP) OAA, 1–2 g/kg once per day for 1–2 weeks. Both doses induced brain changes; we did not determine a minimum effective dose. Observed changes were consistent with an activation of mitochondrial biogenesis pathways. Brain peroxisome proliferator activated receptor γ coactivator 1α (PGC1α) and peroxisome related coactivator (PRC) mRNA levels increased, and brain PGC1α and PRC protein within cells redistributed such that their nucleus:cytoplasm ratios increased. mRNA levels of nuclear respiratory factor 1 (NRF1) and TFAM increased. OAA treatment also increased cytochrome oxidase subunit 4 isoform 1 (COX4I1) mRNA and protein. AMPK Thr172 phosphorylation, p38 MAPK phosphorylation, and cAMP-response element binding (CREB) Ser133 phosphorylation each increased.
OAA treatment enhanced the brain insulin signaling pathway activation state. Akt Ser473, mTOR Ser2448, and P70S6K Thr389 phosphorylation increased. OAA treatment also reduced markers of neuroinflammation. Hippocampal C-C motif chemokine 11 (CCL11) mRNA, hemisphere nuclear NFκB protein levels, and NFκB nucleus-to-cytoplasm ratios were lower than in control mice.
It was previously reported in mice that CCL11 retards hippocampal dentate gyrus subgranular layer neurogenesis [55]. Because CCL11 gene expression was reduced in the hippocampus of OAA-treated mice, and because Akt and mTOR activation promotes cell growth, we evaluated hippocampal neurogenesis and found enhanced hippocampal neurogenesis. Hippocampal vascular endothelial growth factor A (VEGF) mRNA levels increased. Hippocampal mRNA and protein levels of doublecortin (DCX), which is commonly used as a surrogate measure of new neuron formation, increased. The number of intensely DCX-positive neurons increased. Relative to the control group, DCX-positive neurites were longer in the OAA-treated mice.
We further probed for changes in metabolic fluxes by using proton magnetic resonance spectroscopy (1H-MRS) to measure in vivo levels of detectable neurochemicals. 1H-MRS was performed on mice before receiving any treatment, and on the same mice after they received 2 g/kg/day OAA IP for one week (the 1 g/kg/day dose was not studied). OAA induced changes in several neurochemicals. After one week of OAA, brain lactate, GABA, and glutathione (GSH) levels were respectively 21%, 15%, and 27% higher than they were on the pre-treatment scans.
We are currently conducting in vitro experiments to determine the precise mechanisms through which OAA affects neuronal bioenergetics. We have also commenced human studies to provide more insight into OAA pharmacokinetics, and to determine whether clinically feasible doses can demonstrably manipulate brain bioenergetics.
Dietary Interventions and Ketone Bodies
It is also possible to manipulate brain bioenergetics through dietary maneuvers. When considering such maneuvers, it is important to note that the brain is a nutritionally privileged organ, and other tissues will work to keep the brain in a state of energy homeostasis. However, even in the absence of changing levels of brain energy substrates, diet can clearly affect brain bioenergetic infrastructures. For example, we placed young adult mice on an alternate day fasting diet. Hypoglycemia did not occur on the fasting day and we hypothesize this was due to liver gluconeogenesis. However, in the brains of the alternate day fasted mice we nevertheless observed evidence of reduced insulin signaling pathway activity, which presumably reflected reduced peripheral insulin production [56].
Dietary maneuvers can still be used, though, to alter brain energy substrate levels. The ketogenic diet is one such maneuver. Experience with ketogenic diets goes back multiple decades, as it has long been recognized such diets reduce seizure frequency in children with epilepsy [57, 58]. A ketogenic diet was also recently tested in human subjects meeting criteria for a mild cognitive impairment (MCI) syndrome, and found to benefit cognition [59]. Notably, while ketogenic diets typically restrict carbohydrate and even protein intake while dramatically increasing fat intake, ketosis can also be induced through the administration of medium chain triglycerides in the absence of any adjustments to dietary carbohydrate:protein:fat ratios. In two relevant studies in which AD subjects were administered a medium chain triglyceride supplement as the sole dietary intervention, evidence consistent with improved memory or cognitive performance were reported [60, 61]. A larger study intended to more definitively confirm or refute these reported results is currently underway.
Studies have compared caloric restriction (CR) to a ketogenic diet in juvenile rats [62]. Rats were fed a diet of normal chow (ad libitum), subjected to CR, or to CR with a ketogenic diet for 7 days. Both CR and CR with ketogenic diet-treated rats had reduced glucose levels and decreased brain insulin like growth factor (IGF) mRNA. Insulin like growth factor receptor 1 (IGFR1) mRNA was reduced with CR but increased with the combination of CR and a ketogenic diet. Insulin like growth factor binding protein 3 (IGFBP-3) mRNA was increased in the group receiving CR with the ketogenic diet. Glut3 mRNA was decreased with CR but increased with the CR plus ketogenic diet.
In another study, rats were maintained for 22 days on a CR plus ketogenic diet and compared to rats maintained on an ad libitum standard chow diet [63]. Microarray analysis of hippocampal transcripts depicted increased expression of metabolic genes, including those involved in glycolysis, the citric acid cycle, and oxidative phosphorylation in the CR plus ketogenic diet-treated rats. However, no changes in enzyme activity levels were observed. Hippocampal mitochondria content was elevated, suggesting increased mitochondrial biogenesis [63].
We recently used mice to compare the effects of a ketogenic high fat diet to those of a non-ketogenic, Western high fat diet. In this study, young adult C57Bl/6J mice were placed on a ketosis-inducing high-fat ketogenic diet, a non-ketosis inducing high fat Western diet, or a standard chow diet for one month [64]. Mice on the ketogenic diet experienced an approximate five-fold increase in their serum β-hydroxybutyrate level, while mice on the Western diet did not increase their serum β-hydroxybutyrate level. Relative to mice on the ketogenic diet, fasting blood glucose levels increased in mice on the Western diet. Peripheral homeostatic model assessment of insulin resistance (HOMA-IR) calculations revealed peripheral insulin resistance in the Western diet-fed mice was greater than it was in ketogenic diet-fed mice. However, over this one month period no changes in AKT, GSK3β, or mTOR phosphorylation were observed with either diet. This suggests that during the limited course of this experiment, the brain insulin signaling pathway was able to maintain homeostasis.
Despite this, bioenergetic pathways appeared to be affected. In both groups mRNA levels of CREB, PGC1α, and NRF2 increased. mRNA levels of NRF1, TFAM, and COX4I1 decreased with both diets. The ketogenic diet, but not the Western diet, increased PGC1β mRNA. With both diets brain mtDNA was reduced, with no change in the levels of the nuclear DNA-encoded mitochondrial-localized translocase of the outer mitochondrial membrane (TOMM20) and COX4I1 proteins, or of the mtDNA-encoded COX2 protein. COX2 mRNA levels were also maintained. Brain mRNA levels of TNFα, a marker of neuroinflammation, decreased in mice on the ketogenic diet.
Overall, changes observed in mice kept for a limited period on both ketogenic high fat and non-ketogenic high fat diets suggest these diets can modify some aspects of brain bioenergetics. Further, these diets may enhance mtDNA transcription efficiency, as shown by reductions in mtDNA levels in the setting of preserved mtDNA-encoded COX2 transcript and COX2 protein levels. The surprising finding that many of the observed molecular changes in brain bioenergetic infrastructure occurred in the presence of high fat ketogenic and non-ketogenic (Western) diets suggests increased fat as opposed to ketone bodies may account for some changes. On the other hand, changes specific to the ketogenic diet, such as decreased TNFα and increased PGC1β mRNA levels, may represent direct ketone body-related changes. Future studies are warranted to determine the exact mechanisms that underlie and mediate these findings.
Exercise
Aerobic training increases a muscle’s mitochondrial content, which enhances endurance [65, 66]. It appears that mitochondrial-produced oxidative stress to some extent mediates this effect as the concomitant administration of antioxidants blunts the effect [67, 68]. By increasing energy consumption and perhaps other mechanisms, exercise also shifts muscle cell redox values through the oxidation of NADH to NAD+, and especially due to an increase in NAD+ [69].
Exercise’s effect on cell infrastructures, though, is not limited to muscle. For example, hepatocyte Cori cycle capacity increases in aerobically trained mice [70]. A likely goal of this adaptation is to prevent exercise-induced hypoglycemia, as it enhances the ability of the liver to convert muscle-generated lactate back to glucose via gluconeogenesis. To accomplish this, hepatocytes increase their monocarboxylate transporter (MCT) content, at least for the lactate-importing MCT2 [70]. Hepatocyte PGC1α mRNA levels also increase [70, 71].
Exercise also affects the brain. In rodents, exercise has been associated with increased levels of brain derived neurotrophic factor (BDNF), which is believed to mediate many of its brain effects including increased hippocampal neurogenesis [72, 73]. Exercise appears to mitigate age-related declines in brain respiratory chain function [74, 75], and increase co-transcription factors that drive mitochondrial biogenesis [71, 76]. We have also found that following a course of aerobic exercise training, mice have reduced brain TNFα mRNA levels [70, 71]. Further, an inverse correlation between plasma CCL11 (an inflammatory cytokine) levels and hippocampal DCX (a neurogenesis marker) mRNA levels that was observed in aged, sedentary mice was not observed in age-matched, aerobically trained mice [77]. As CCL11 was previously shown to inhibit hippocampal neurogenesis [55], this finding suggests aerobic exercise may benefit the brain by reducing the presence or mitigating the adverse consequences of chronic inflammation.
Consistent with the view that exercise may influence the brain through bioenergetic medicine principles, we have shown that injecting mice with IP lactate recapitulates several bioenergetic infrastructure changes that occur in the brains of aerobically trained mice [71]. This raises the possibility that with exercise vigorous enough to exceed the lactate threshold (the point at which muscle-generated lactate begins to collect in the blood more rapidly than it can be removed), lactate is delivered to the brain. In neurons, lactate is postulated by some to constitute an important, perhaps even critical, bioenergetic fuel [78–81]. Overall, these experiments suggest exercise may alter brain bioenergetics, mitochondrial infrastructure, and neuroinflammation at least partly by increasing brain lactate levels.
Conclusion
“Energy (or active exercise) of the mind is the essence of life”. This quote, attributed to the ancient Greek philosopher Aristotle, in some ways applies to the search for AD therapeutics. All cellular processes are driven by the availability of energy. Bioenergetic pathways are required for the formation of cellular energy stores. Neuroinflammation and mitochondrial function, two commonly observed lesions in AD, are intricately linked to bioenergetic pathways. Therefore, bioenergetic medicine may reveal strategies that will ultimately modify the course of AD and perhaps other neurodegenerative disorders that feature mitochondrial dysfunction.
Acknowledgments
Conflict of Interest
This work was supported by the University of Kansas Alzheimer’s Disease Center (P30AG035982), the Frank and Evangeline Thompson Alzheimer’s Treatment Program Fund, the Hugh and Betty Libby Foundation, the Greater Kansas City Automobile Dealers Association, the Gene and Marge Sweeney Chair, University of Kansas Alzheimer’s Disease Center Pilot program, Landon Center for Aging, Frontiers the Heartland Institute for Clinical and Translational Research, the Kansas IDeA Network of Biomedical Research Excellence, and the KUMC Biomedical Research Training Program.
Abbreviations
- Aβ
amyloid beta
- AD
Alzheimer’s disease
- AMPK
AMP kinase
- APP
Amyloid precursor protein
- ATP
adenosine triphosphate
- CR
caloric restriction
- CCL11
C-C motif chemokine 11
- CXCL1
chemokine C-X-C motif ligand 1
- COX
cytochrome oxidase
- COX4I1
cytochrome oxidase subunit 4 isoform 1
- CREB
cAMP response element binding protein
- DCX
doublecortin
- FDG PET
fluoro-deoxyglucose positron emission tomography
- FOXO
forkhead O box
- GSH
glutathione
- HOMA-IR
homeostatic model assessment of insulin resistance
- IGF
insulin like growth factor
- IGFR1
insulin like growth factor receptor 1
- IGFBP3
insulin like growth factor binding protein 3
- IL-1β
interleukin 1β
- IL-6
interleukin 6
- IL-8
interleukin 8
- IFN-γ
interferon γ
- IP
intraperitoneal
- MAPK
mitogen activated protein kinase
- MCP-1
monocyte chemotactic protein 1
- MCT
monocarboxylate transporter
- mtDNA
mitochondrial DNA
- MMP-8
matrix metalloproteinase 8
- MRS
magnetic resonance spectroscopy
- NFκB
nuclear factor κ light chain enhancer of B cells
- NRF1
nuclear respiratory factor 1
- OAA
oxaloacetate
- PEP
phosphoenolpyruvate
- PEPCK
PEP carboxykinase
- PGC1-α
proliferator activated receptor γ coactivator α
- PRC
peroxisome related coactivator
- TFAM
mitochondrial transcription factor A
- TOMM20
translocase of the outer mitochondrial membrane 20
- TREM2
triggering receptor expressed on myeloid cells 2
- TNFα
tumor necrosis factor α
- VEGF
vascular endothelial growth factor
References
- 1.Bosetti F, Brizzi F, Barogi S, Mancuso M, Siciliano G, Tendi EA, Murri L, Rapoport SI, Solaini G. Cytochrome c oxidase and mitochondrial F1F0-ATPase (ATP synthase) activities in platelets and brain from patients with Alzheimer’s disease. Neurobiology of aging. 2002;23(3):371–376. doi: 10.1016/s0197-4580(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 2.Cardoso SM, Santana I, Swerdlow RH, Oliveira CR. Mitochondria dysfunction of Alzheimer’s disease cybrids enhances Abeta toxicity. Journal of neurochemistry. 2004;89(6):1417–1426. doi: 10.1111/j.1471-4159.2004.02438.x. [DOI] [PubMed] [Google Scholar]
- 3.Khan SM, Cassarino DS, Abramova NN, Keeney PM, Borland MK, Trimmer PA, Krebs CT, Bennett JC, Parks JK, Swerdlow RH, Parker WD, Jr, Bennett JP., Jr Alzheimer’s disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Annals of neurology. 2000;48(2):148–155. [PubMed] [Google Scholar]
- 4.Maurer I, Zierz S, Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiology of aging. 2000;21(3):455–462. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 5.Onyango IG, Bennett JP, Jr, Tuttle JB. Endogenous oxidative stress in sporadic Alzheimer’s disease neuronal cybrids reduces viability by increasing apoptosis through pro-death signaling pathways and is mimicked by oxidant exposure of control cybrids. Neurobiology of disease. 2005(19):1–2. 312–322. doi: 10.1016/j.nbd.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 6.Parker WD, Jr, Filley CM, Parks JK. Cytochrome oxidase deficiency in Alzheimer’s disease. Neurology. 1990;40(8):1302–1303. doi: 10.1212/wnl.40.8.1302. [DOI] [PubMed] [Google Scholar]
- 7.Silva DF, Selfridge JE, Lu JEL, Roy N, Hutfles L, Burns JM, Michaelis EK, Yan S, Cardoso SM, Swerdlow RH. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Human molecular genetics. 2013;22(19):3931–3946. doi: 10.1093/hmg/ddt247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swerdlow RH. Mitochondria and cell bioenergetics: increasingly recognized components and a possible etiologic cause of Alzheimer’s disease. Antioxidants & redox signaling. 2012;16(12):1434–1455. doi: 10.1089/ars.2011.4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Swerdlow RH. Mitochondrial Medicine and the Neurodegenerative Mitochondriopathies. Pharmaceuticals. 2009;2(3):150–167. doi: 10.3390/ph2030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trimmer PA, Keeney PM, Borland MK, Simon FA, Almeida J, Swerdlow RH, Parks JP, Parker WD, Jr, Bennett JP., Jr Mitochondrial abnormalities in cybrid cell models of sporadic Alzheimer’s disease worsen with passage in culture. Neurobiology of disease. 2004;15(1):29–39. doi: 10.1016/j.nbd.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 11.Wilkins HM, Carl SM, Greenlief AC, Festoff BW, Swerdlow RH. Bioenergetic dysfunction and inflammation in Alzheimer’s disease: a possible connection. Frontiers in aging neuroscience. 2014;6:311. doi: 10.3389/fnagi.2014.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilkins HM, Carl SM, Swerdlow RH. Cytoplasmic hybrid (cybrid) cell lines as a practical model for mitochondriopathies. Redox biology. 2014;2C:619–631. doi: 10.1016/j.redox.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Filippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P. Mitochondria and the link between neuroinflammation and neurodegeneration. Journal of Alzheimer’s disease : JAD. 2010;20(Suppl 2):S369–379. doi: 10.3233/JAD-2010-100543. [DOI] [PubMed] [Google Scholar]
- 14.Brown GC. Nitric oxide inhibition of cytochrome oxidase and mitochondrial respiration: implications for inflammatory, neurodegenerative and ischaemic pathologies. Molecular and cellular biochemistry. 1997(174):1–2. 189–192. [PubMed] [Google Scholar]
- 15.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in L RP3 inflammasome activation. Nature. 2011;469(7329):221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 16.Akiyama H. Inflammatory response in Alzheimer’s disease. The Tohoku journal of experimental medicine. 1994;174(3):295–303. doi: 10.1620/tjem.174.295. [DOI] [PubMed] [Google Scholar]
- 17.Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer disease and associated disorders. 2000;14(Suppl 1):S47–53. doi: 10.1097/00002093-200000001-00008. [DOI] [PubMed] [Google Scholar]
- 18.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer’s disease. Neurobiology of aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358(9280):461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 20.Versijpt JJ, Dumont F, Van Laere KJ, Decoo D, Santens P, Audenaert K, Achten E, Slegers G, Dierckx RA, Korf J. Assessment of neuroinflammation and microglial activation in Alzheimer’s disease with radiolabelled PK11195 and single photon emission computed tomography. A pilot study. European neurology. 2003;50(1):39–47. doi: 10.1159/000070857. [DOI] [PubMed] [Google Scholar]
- 21.Cartier L, Hartley O, Dubois-Dauphin M, Krause KH. Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain research Brain research reviews. 2005;48(1):16–42. doi: 10.1016/j.brainresrev.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 22.Swardfager W, Lanctot K, Rothenburg L, Wong A, Cappell J, Herrmann N. A meta-analysis of cytokines in Alzheimer’s disease. Biological psychiatry. 2010;68(10):930–941. doi: 10.1016/j.biopsych.2010.06.012. [DOI] [PubMed] [Google Scholar]
- 23.Wilkins HM, Carl SM, Weber SG, Ramanujan SA, Festoff BW, Linseman DA, Swerdlow RH. Mitochondrial Lysates Induce Inflammation and Alzheimer’s Disease-Relevant Changes in Microglial and Neuronal Cells. Journal of Alzheimer’s disease : JAD. 2014 doi: 10.3233/JAD-142334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathew A, Lindsley TA, Sheridan A, Bhoiwala DL, Hushmendy SF, Yager EJ, Ruggiero EA, Crawford DR. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. Journal of Alzheimer’s disease : JAD. 2012;30(3):617–627. doi: 10.3233/JAD-2012-120145. [DOI] [PubMed] [Google Scholar]
- 25.Crouser ED, Shao G, Julian MW, Macre JE, Shadel GS, Tridandapani S, Huang Q, Wewers MD. Monocyte activation by necrotic cells is promoted by mitochondrial proteins and formyl peptide receptors. Critical care medicine. 2009;37(6):2000–2009. doi: 10.1097/CCM.0b013e3181a001ae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Julian MW, Shao G, Bao S, Knoell DL, Papenfuss TL, VanGundy ZC, Crouser ED. Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. Journal of immunology. 2012;189(1):433–443. doi: 10.4049/jimmunol.1101375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abramova NE, Davies KJ, Crawford DR. Polynucleotide degradation during early stage response to oxidative stress is specific to mitochondria. Free radical biology & medicine. 2000;28(2):281–288. doi: 10.1016/s0891-5849(99)00239-7. [DOI] [PubMed] [Google Scholar]
- 28.Savage CD, Lopez-Castejon G, Denes A, Brough D. NLRP3-Inflammasome Activating DAMPs Stimulate an Inflammatory Response in Glia in the Absence of Priming Which Contributes to Brain Inflammation after Injury. Frontiers in immunology. 2012;3:288. doi: 10.3389/fimmu.2012.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Little JP, Simtchouk S, Schindler SM, Villanueva EB, Gill NE, Walker DG, Wolthers KR, Klegeris A. Mitochondrial transcription factor A (Tfam) is a pro-inflammatory extracellular signaling molecule recognized by brain microglia. Molecular and cellular neurosciences. 2014;60:88–96. doi: 10.1016/j.mcn.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, Marsh-Armstrong N. Transcellular degradation of axonal mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(26):9633–9638. doi: 10.1073/pnas.1404651111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, Pirraglia E, De Santi S, Reisberg B, Wisniewski T, de Leon MJ. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. European journal of nuclear medicine and molecular imaging. 2009;36(5):811–822. doi: 10.1007/s00259-008-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC. Increased risk of dementia in mothers of Alzheimer’s disease cases: evidence for maternal inheritance. Neurology. 1996;47(1):254–256. doi: 10.1212/wnl.47.1.254. [DOI] [PubMed] [Google Scholar]
- 33.Honea RA, Vidoni ED, Swerdlow RH, Burns JM. Alzheimer’s Disease Neuroimaging, I. Maternal family history is associated with Alzheimer’s disease biomarkers. Journal of Alzheimer’s disease : JAD. 2012;31(3):659–668. doi: 10.3233/JAD-2012-120676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mosconi L, Brys M, Switalski R, Mistur R, Glodzik L, Pirraglia E, Tsui W, De Santi S, de Leon MJ. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(48):19067–19072. doi: 10.1073/pnas.0705036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochimica et biophysica acta. 2014;1842(8):1219–1231. doi: 10.1016/j.bbadis.2013.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Swerdlow RH, Khan SM. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Medical hypotheses. 2004;63(1):8–20. doi: 10.1016/j.mehy.2003.12.045. [DOI] [PubMed] [Google Scholar]
- 37.Soares HD, Potter WZ, Pickering E, Kuhn M, Immermann FW, Shera DM, Ferm M, Dean RA, Simon AJ, Swenson F, Siuciak JA, Kaplow J, Thambisetty M, Zagouras P, Koroshetz WJ, Wan HI, Trojanowski JQ, Shaw LM. Plasma biomarkers associated with the apolipoprotein E genotype and Alzheimer disease. Archives of neurology. 2012;69(10):1310–1317. doi: 10.1001/archneurol.2012.1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Faria MC, Goncalves GS, Rocha NP, Moraes EN, Bicalho MA, Gualberto Cintra MT, Jardim de Paula J, Jose Ravic de Miranda LF, Clayton de Souza Ferreira A, Teixeira AL, Gomes KB, Carvalho M, Sousa LP. Increased plasma levels of BDNF and inflammatory markers in Alzheimer’s disease. Journal of psychiatric research. 2014;53:166–172. doi: 10.1016/j.jpsychires.2014.01.019. [DOI] [PubMed] [Google Scholar]
- 39.Leung R, Proitsi P, Simmons A, Lunnon K, Guntert A, Kronenberg D, Pritchard M, Tsolaki M, Mecocci P, Kloszewska I, Vellas B, Soininen H, Wahlund LO, Lovestone S. Inflammatory proteins in plasma are associated with severity of Alzheimer’s disease. PLoS One. 2013;8(6):e64971. doi: 10.1371/journal.pone.0064971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang R, Miller RG, Madison C, Jin X, Honrada R, Harris W, Katz J, Forshew DA, McGrath MS. Systemic immune system alterations in early stages of Alzheimer’s disease. Journal of neuroimmunology. 2013(256):1–2. 38–42. doi: 10.1016/j.jneuroim.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alsadany MA, Shehata HH, Mohamad MI, Mahfouz RG. Histone deacetylases enzyme, copper, and IL-8 levels in patients with Alzheimer’s disease. American journal of Alzheimer’s disease and other dementias. 2013;28(1):54–61. doi: 10.1177/1533317512467680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee KS, Chung JH, Choi TK, Suh SY, Oh BH, Hong CH. Peripheral cytokines and chemokines in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2009;28(4):281–287. doi: 10.1159/000245156. [DOI] [PubMed] [Google Scholar]
- 43.Luft R. The development of mitochondrial medicine. Proceedings of the National Academy of Sciences of the United States of America. 1994;91(19):8731–8738. doi: 10.1073/pnas.91.19.8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Swerdlow RH. Bioenergetic medicine. British journal of pharmacology. 2014;171(8):1854–1869. doi: 10.1111/bph.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Swerdlow RH. Role and treatment of mitochondrial DNA-related mitochondrial dysfunction in sporadic neurodegenerative diseases. Current pharmaceutical design. 2011;17(31):3356–3373. doi: 10.2174/138161211798072535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilkins HM, Harris JL, Carl SM, E L, Lu J, Eva Selfridge J, Roy N, Hutfles L, Koppel S, Morris J, Burns JM, Michaelis ML, Michaelis EK, Brooks WM, Swerdlow RH. Oxaloacetate activates brain mitochondrial biogenesis, enhances the insulin pathway, reduces inflammation and stimulates neurogenesis. Human molecular genetics. 2014;23(24):6528–6541. doi: 10.1093/hmg/ddu371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin SJ, Guarente L. Nicotinamide adenine dinucleotide, a metabolic regulator of transcription, longevity and disease. Curr Opin Cell Biol. 2003;15(2):241–246. doi: 10.1016/s0955-0674(03)00006-1. [DOI] [PubMed] [Google Scholar]
- 48.Hakimi P, Yang J, Casadesus G, Massillon D, Tolentino-Silva F, Nye CK, Cabrera ME, Hagen DR, Utter CB, Baghdy Y, Johnson DH, Wilson DL, Kirwan JP, Kalhan SC, Hanson RW. Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J Biol Chem. 2007;282(45):32844–32855. doi: 10.1074/jbc.M706127200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Williams DS, Cash A, Hamadani L, Diemer T. Oxaloacetate supplementation increases lifespan in Caenorhabditis elegans through an AMPK/FOXO-dependent pathway. Aging Cell. 2009;8(6):765–768. doi: 10.1111/j.1474-9726.2009.00527.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yamamoto HA, Mohanan PV. Effect of alpha-ketoglutarate and oxaloacetate on brain mitochondrial DNA damage and seizures induced by kainic acid in mice. Toxicol Lett. 2003;143(2):115–122. doi: 10.1016/s0378-4274(03)00114-0. [DOI] [PubMed] [Google Scholar]
- 51.Campos F, Sobrino T, Ramos-Cabrer P, Castillo J. Oxaloacetate: a novel neuroprotective for acute ischemic stroke. Int J Biochem Cell Biol. 2012;44(2):262–265. doi: 10.1016/j.biocel.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 52.Ruban A, Berkutzki T, Cooper I, Mohar B, Teichberg VI. Blood glutamate scavengers prolong the survival of rats and mice with brain-implanted gliomas. Investigational new drugs. 2012 doi: 10.1007/s10637-012-9794-x. [DOI] [PubMed] [Google Scholar]
- 53.Zlotnik A, Sinelnikov I, Gruenbaum BF, Gruenbaum SE, Dubilet M, Dubilet E, Leibowitz A, Ohayon S, Regev A, Boyko M, Shapira Y, Teichberg VI. Effect of glutamate and blood glutamate scavengers oxaloacetate and pyruvate on neurological outcome and pathohistology of the hippocampus after traumatic brain injury in rats. Anesthesiology. 2012;116(1):73–83. doi: 10.1097/ALN.0b013e31823d7731. [DOI] [PubMed] [Google Scholar]
- 54.Yoshikawa K. Studies on the anti-diabetic effect of sodium oxaloacetate. Tohoku J Exp Med. 1968;96(2):127–141. doi: 10.1620/tjem.96.127. [DOI] [PubMed] [Google Scholar]
- 55.Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard-Despres S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, Xie XS, Rando TA, Wyss-Coray T. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477(7362):90–94. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lu J, E L, Wang W, Frontera J, Zhu H, Wang WT, Lee P, Choi IY, Brooks WM, Burns JM, Aires D, Swerdlow RH. Alternate day fasting impacts the brain insulin-signaling pathway of young adult male C57BL/6 mice. Journal of neurochemistry. 2011;117(1):154–163. doi: 10.1111/j.1471-4159.2011.07184.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Huttenlocher PR. Ketonemia and seizures: metabolic and anticonvulsant effects of two ketogenic diets in childhood epilepsy. Pediatric research. 1976;10(5):536–540. doi: 10.1203/00006450-197605000-00006. [DOI] [PubMed] [Google Scholar]
- 58.Neal EG, Chaffe H, Schwartz RH, Lawson MS, Edwards N, Fitzsimmons G, Whitney A, Cross JH. The ketogenic diet for the treatment of childhood epilepsy: a randomised controlled trial. The Lancet Neurology. 2008;7(6):500–506. doi: 10.1016/S1474-4422(08)70092-9. [DOI] [PubMed] [Google Scholar]
- 59.Krikorian R, Shidler MD, Dangelo K, Couch SC, Benoit SC, Clegg DJ. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiology of aging. 2012;33(2):425, e419–427. doi: 10.1016/j.neurobiolaging.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Reger MA, Henderson ST, Hale C, Cholerton B, Baker LD, Watson GS, Hyde K, Chapman D, Craft S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiology of aging. 2004;25(3):311–314. doi: 10.1016/S0197-4580(03)00087-3. [DOI] [PubMed] [Google Scholar]
- 61.Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond) 2009;6:31. doi: 10.1186/1743-7075-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Cheng CM, Kelley B, Wang J, Strauss D, Eagles DA, Bondy CA. A ketogenic diet increases brain insulin-like growth factor receptor and glucose transporter gene expression. Endocrinology. 2003;144(6):2676–2682. doi: 10.1210/en.2002-0057. [DOI] [PubMed] [Google Scholar]
- 63.Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, Shaw R, Smith Y, Geiger JD, Dingledine RJ. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Annals of neurology. 2006;60(2):223–235. doi: 10.1002/ana.20899. [DOI] [PubMed] [Google Scholar]
- 64.Selfridge JE, Wilkins HM, E L, Carl SM, Koppel S, Funk E, Fields T, Lu J, Tang EP, Slawson C, Wang W, Zhu H, Swerdlow RH. Effect of one month duration ketogenic and non-ketogenic high fat diets on mouse brain bioenergetic infrastructure. Journal of bioenergetics and biomembranes. 2014 doi: 10.1007/s10863-014-9570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hood DA. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle. Appl Physiol Nutr Metab. 2009;34(3):465–472. doi: 10.1139/H09-045. [DOI] [PubMed] [Google Scholar]
- 66.Holloszy JO. Adaptation of skeletal muscle to endurance exercise. Med Sci Sports. 1975;7(3):155–164. [PubMed] [Google Scholar]
- 67.Ristow M, Zarse K, Oberbach A, Kloting N, Birringer M, Kiehntopf M, Stumvoll M, Kahn CR, Bluher M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(21):8665–8670. doi: 10.1073/pnas.0903485106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gomez-Cabrera MC, Domenech E, Romagnoli M, Arduini A, Borras C, Pallardo FV, Sastre J, Vina J. Oral administration of vitamin C decreases muscle mitochondrial biogenesis and hampers training-induced adaptations in endurance performance. Am J Clin Nutr. 2008;87(1):142–149. doi: 10.1093/ajcn/87.1.142. [DOI] [PubMed] [Google Scholar]
- 69.Koltai E, Szabo Z, Atalay M, Boldogh I, Naito H, Goto S, Nyakas C, Radak Z. Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech Ageing Dev. 2010;131(1):21–28. doi: 10.1016/j.mad.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.E L, Lu J, Burns JM, Swerdlow RH. Effect of exercise on mouse liver and brain bioenergetic infrastructures. Experimental physiology. 2013;98(1):207–219. doi: 10.1113/expphysiol.2012.066688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.E L, Lu J, Selfridge JE, Burns JM, Swerdlow RH. Lactate administration reproduces specific brain and liver exercise-related changes. Journal of neurochemistry. 2013;127(1):91–100. doi: 10.1111/jnc.12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cotman CW, Berchtold NC. Exercise: a behavioral intervention to enhance brain health and plasticity. Trends Neurosci. 2002;25(6):295–301. doi: 10.1016/s0166-2236(02)02143-4. [DOI] [PubMed] [Google Scholar]
- 73.van Praag H, Shubert T, Zhao C, Gage FH. Exercise enhances learning and hippocampal neurogenesis in aged mice. J Neurosci. 2005;25(38):8680–8685. doi: 10.1523/JNEUROSCI.1731-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boveris A, Navarro A. Systemic and mitochondrial adaptive responses to moderate exercise in rodents. Free radical biology & medicine. 2008;44(2):224–229. doi: 10.1016/j.freeradbiomed.2007.08.015. [DOI] [PubMed] [Google Scholar]
- 75.Navarro A, Gomez C, Lopez-Cepero JM, Boveris A. Beneficial effects of moderate exercise on mice aging: survival, behavior, oxidative stress, and mitochondrial electron transfer. Am J Physiol Regul Integr Comp Physiol. 2004;286(3):R505–511. doi: 10.1152/ajpregu.00208.2003. [DOI] [PubMed] [Google Scholar]
- 76.Steiner JL, Murphy EA, McClellan JL, Carmichael MD, Davis JM. Exercise training increases mitochondrial biogenesis in the brain. J Appl Physiol. 2011;111(4):1066–1071. doi: 10.1152/japplphysiol.00343.2011. [DOI] [PubMed] [Google Scholar]
- 77.E L, Burns JM, Swerdlow RH. Effect of high-intensity exercise on aged mouse brain mitochondria, neurogenesis, and inflammation. Neurobiology of aging. 2014;35(11):2574–2583. doi: 10.1016/j.neurobiolaging.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Magistretti PJ. Neuron-glia metabolic coupling and plasticity. J Exp Biol. 2006;209(Pt 12):2304–2311. doi: 10.1242/jeb.02208. [DOI] [PubMed] [Google Scholar]
- 79.Pellerin L, Magistretti PJ. Neuroenergetics: calling upon astrocytes to satisfy hungry neurons. Neuroscientist. 2004;10(1):53–62. doi: 10.1177/1073858403260159. [DOI] [PubMed] [Google Scholar]
- 80.Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ, Alberini CM. Astrocyte-neuron lactate transport is required for long-term memory formation. Cell. 2011;144(5):810–823. doi: 10.1016/j.cell.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B. In vivo evidence for lactate as a neuronal energy source. J Neurosci. 2011;31(20):7477–7485. doi: 10.1523/JNEUROSCI.0415-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]