

Abstract
Homeostasis is a fundamental property of living organisms enabling the human body to withstand internal and external insults. In several chronic diseases, and especially in cancer, many homeostatic mechanisms are deranged. Pancreatic cancer, in particular, is notorious for its ability to invoke an intense fibro-inflammatory stromal reaction facilitating its progression and resistance to treatment. In the past decade, a number of seminal discoveries have elucidated previously unrecognized modes of commandeering the host’s defense systems. Here, we review novel discoveries in pancreatic cancer immunobiology and attempt to integrate the notion of deranged homeostasis in the pathogenesis of this disease. We also highlight areas of controversy and obstacles that need to be overcome, hoping to further our mechanistic insight into this malignancy.
Keywords: Pancreatic ductal adenocarcinoma, inflammation, cancer-associated fibroblasts, stroma, immunity
Primer: Cancer Thrives Amongst Chaos
The human body responds to aberrations of normal physiology in numerous ways, attempting to maintain or re-establish balance – this is the essence of homeostasis. The immune system and associated cascades such as the complement system and various tissue regeneration processes, is a vital component of homeostatic responses to tissue stress. If dysregulated, it may perpetuate disease rather than abate it. This holds true for almost all pathologies, including cancer (Key Figure, Figure 1). The importance of understanding homeostatic responses in carcinogenesis stems from two critical determinants in the course of neoplastic diseases: first, that homeostatic responses may be subverted in ways that propagate – rather than counteract – carcinogenesis; and second, that anti-cancer therapies may be rendered ineffective by the bystander effects of homeostatic responses. The timeliness of this topic derives from the recent explosion in high-throughput technologies that can generate an immense amount of information pertaining to the biology of a given tumor. These have brought tremendous excitement and have heralded the era of personalized medicine through the identification of pathways specifically deranged in cancer cells; yet, they often neglect, or fail to take into consideration the non-transformed aspect of a tumor, or “stroma”, which results from aberrant homeostatic responses. Indeed, it is vital, not only to the pathophysiology of cancer, but also to the mechanisms of tumor resistance to many novel targeted therapies.
Key Figure, Figure 1. Homeostatic Responses to Tissue Stress in the Context of Pancreatic Dysfunction and Malignancy.
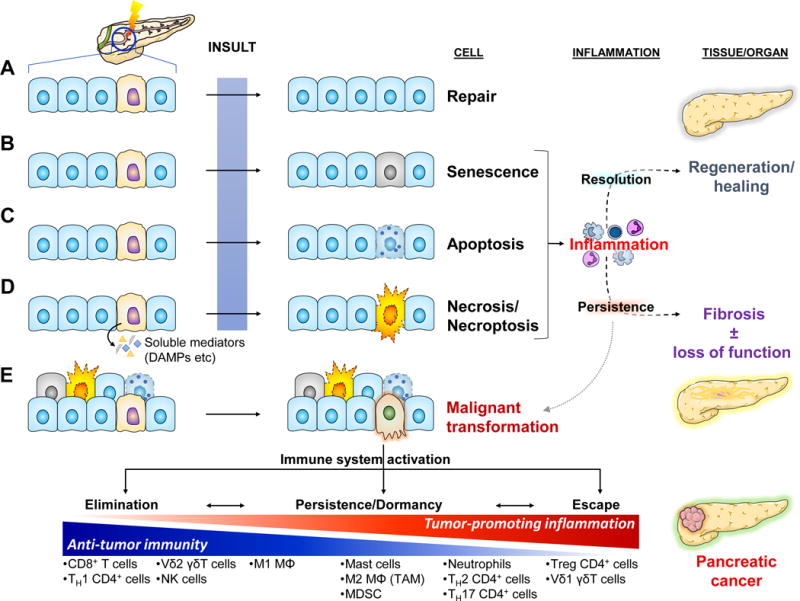
Cellular stress can be invoked by a great variety of internal (e.g. wear and tear/aging, reactive oxygen species, hypoxia, etc) and external causes (e.g. toxins, radiation, infectious agents, trauma, etc). Some can be mutagenic, potentially leading to cancer. Stressed/damaged cells activate defensive systems that attempt to repair the injury. If successful, the cell returns to its normal state and usually minimal or no inflammatory response is induced (a).
(b–d) If the injury is extensive and irreparable, the cell may either go into a senescence – a “safe mode” characterized by cell cycle arrest and slowing of cellular functions [101] – or may activate programmed cell death by apoptosis or necroptosis. If the injury is sudden and overwhelming, it may induce cell rupture and resultant necrosis. Both senescence and the various modes of cell death are characterized by altered expression of cell surface molecules as well as release of soluble mediators that activate the immune system [101, 102]. The resultant inflammatory response attempts to clear off the debris and support tissue regeneration. If the normal homeostatic mechanisms malfunction, the inflammation may persist leading to a futile cycle of further tissue injury, fibrosis, and potentially carcinogenesis.
(e) Malignant transformation usually arises de novo secondary to serial mutagenic insults to normal cells. The immune system usually recognizes single transformed cells and clears them off (elimination). Occasionally, some transformed cells may go unrecognized by the immune system and persist. As they accumulate additional mutations, they may initiate an inflammatory response that can clear them off. At the same time though, the inflammatory response places a selection pressure on the transformed cells which may enable the emergence of “resistant” clones and eventual escape from immunosurveillance mechanisms [103]. One of the ways the immune system escapes is by maintaining an immune infiltrate permissive to tumor growth through release of pro-tumorigenic mediators and simultaneous exclusion of cytotoxic cells (see Fig. 4). Finally, the sustained intratumoral inflammatory response may induce collateral damage to surrounding non-transformed epithelial cells. Activation of the responses to cell injury described above leads to release of additional pro-inflammatory mediators and further perpetuation of tumor-associated inflammation.
Pancreatic ductal adenocarcinoma (PDAC; the most common form of pancreatic cancer) is probably the best example of an inflammation-driven neoplastic process [1]. Notably, its incidence is climbing even though our ability to treat it effectively has improved only meagerly in the past few decades. As such, it is projected that PDAC will soon become the second most lethal cancer in the developed world [2]. PDAC is remarkable for its overwhelming stromal infiltrate and the resultant fibrotic reaction (also known as “desmoplasia”) – which exemplifies how homeostatic responses can be detrimental when curtailed by cancer. In the past decade, a number of seminal studies have elucidated previously under-appreciated aspects of the pathogenesis of PDAC-associated stromal reaction and its fundamental role in pancreatic carcinogenesis. However, they have also fueled considerable debate since some of the most recent evidence contradicts earlier doctrines (such as the anti-tumorigenic role of Toll-like receptors). In this review, we attempt to bring an alternative perspective to pancreatic carcinogenesis (and by extension, to all solid tumors), by focusing on the non-transformed aspect of the tumor that is fueled by aberrant homeostatic responses to tissue stress, characterized by dysregulated inflammatory responses and failed regeneration, thus affecting the body as a whole.
Tissue Stress, Inflammation and Carcinogenesis
Inflammation involves the coordinated activation of several components including the immune system, the complement system, the coagulation cascade, and the wound healing/tissue regeneration program which crosstalk to each other in a coordinated manner. The ultimate goal is to eliminate the noxious stimuli that lead to the initiation of an inflammatory response and restore balance, ideally with reversal of the compromised function of affected tissues. Therefore, activation of tissue resident immune cells (e.g. macrophages, intraepithelial lymphocytes) and other supportive cells, as well as the sequential recruitment of innate (e.g. neutrophils, mast cells, monocytes, NK cells, γδ T cells) and adaptive immune cells (B and T lymphocytes) are required to fight off exogenous (microbes) or endogenous (e.g. stressed/transformed cells) offenders. At the same time, immune and other supportive cells release soluble mediators, including cytokines such as interleukin-6 (IL-6) and tumor necrosis factor (TNF), that activate complementary loco-regional responses as well as systemic programs to conserve energy and create an organism-wide state that is less conducive to infection. For example, phenomena such as febrile reactions, decreased iron availability, and altered hepatic metabolism, among others, aim to protect the host from side effects of tissue stress, and prevent or curtail various deleterious effects. The complement system is activated early during the inflammatory response and contributes to pathogen elimination directly via lysis of bacteria, and indirectly, via opsonization, while some molecular intermediates generated during these processes recruit immune and other cells to sites of inflammation [3]. The adjacent endothelial cells and the coagulation cascade are also activated and, apart from a hemostatic response, they serve to recruit additional immune cells as well as platelets and fibroblasts to promote the healing response.
Viewed from a systems dynamics perspective, inflammation may be simplified in a homeostatic circuit (Fig. 2) [4]. Disruption of normal cellular function by an ‘inducer’ (e.g. traumatic injury) is perceived by a ‘sensor’ (e.g. innate immune cells) as a deviation from the norm, or set point. For example, cellular stress causes release of damage-associated molecular patterns (DAMPs) that bind pattern-recognition receptors (PRRs) that activate innate immune cells [5, 6]. The sensor in turn will generate a ‘signal’ (e.g. pro-inflammatory cytokines) that will activate an ‘effector’ cell (e.g. lymphocytes), which in turn, will attempt to alleviate the inducer in order to restore the deranged variable within the normal range – for instance, by clearing off necrotic debris and repairing the injured tissue.
Figure 2. Simplified Homeostatic Circuit of the Inflammatory Response to Tissue Stress.
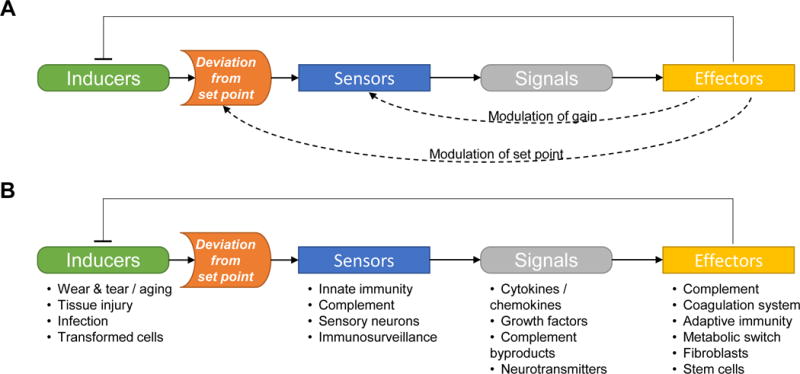
(a) The general composition of a homeostatic circuit (examples include control of blood glucose levels, tissue oxygenation, and core body temperature). The set point may be fixed at a certain range (which may vary from between tissues or organs), or may be adjustable depending on the context (e.g. the body temperature set point is raised during the febrile response). The sensitivity (or gain) of the sensors can be fine-tuned, making them more or less responsive to deviations from the set point.
(b) Inflammation has evolved to protect the organism from intrinsic and extrinsic challenges that, if not contained, can be detrimental to the involved tissue or even the entire body. It usually obeys the laws of homeostasis (although the system is in reality, more complex). Activators of the immune system (inducers, such as those described in Key Figure, Figure 1) are identified by the sensors as deviations from the set point, as a result of altered cell surface molecules and release of soluble mediators. The sensors initiate downstream cascades that attempt to alleviate the initial cause that deranged the system. In several disease states characterized by chronic inflammation (such as pancreatic cancer), the system is derailed secondary to imbalances between positive and negative regulators (e.g. additional activating signals elicited by cancer cells). These imbalances create new set points, modulate the threshold for sensor activation (gain) and alter the functions of effectors, such that instead of returning to baseline (i.e. homeostasis) the system is trapped in a self-perpetuating infinite loop that only benefits the cancer cells.
A very important aspect of all these processes is an ability to self- and cross-regulate the degree of activation for each component in order to prevent unnecessary propagation of inflammation and thus, result in collateral damage [4, 7, 8]. Almost every arm of the homeostatic circuit can set off negative feedback loops that either directly inhibit the effectors, or modulate the threshold of activation (‘gain’) of both sensors and effectors. The end result is the organized resolution of the inflammatory response and the minimization of harmful off-target effects. When these pathways malfunction and do not obey the laws of negative feedback and homeostasis, chronic inflammation ensues, and can be more detrimental than beneficial, leading to pathological consequences such as fibrosis, loss of organ function, and carcinogenesis (Fig. 1). The unrestrained propagation of cancer-associated fibro-inflammatory elements that originate in a host’s physiologic responses to injury, have led to the decades-old quip that tumors are “wounds that do not heal” [9, 10].
In the majority of cases, pancreatic cancer is characterized by a prominent inflammatory response [1, 11]. However, this type of inflammation presents considerable differences from the acute inflammation that accompanies wound healing: it is self-sustaining, unregulated, and permissive to cancer cell development [12, 13]. Thus, even though pancreatitis is a well-recognized risk factor for pancreatic cancer development, most patients with PDAC have no clinical history of overt pancreatitis. [14].
Numerous studies have established the importance of chronic inflammation in pancreatic carcinogenesis and progression [12]. The interplay between inflammation and cancer-initiating mutations is particularly interesting. Kras mutations are present in the majority of PDAC cases as well as in a significant percentage of pancreatic intraepithelial neoplasia (PanIN) lesions, even at early stages (over 30% of PanIN 1 harbor mutant Kras) [15]. Work done in well-validated genetically-engineered mouse models has revealed that mutant Kras not only initiates the stepwise progression from normal epithelium to acinar-to-ductal metaplasia (ADM), PanIN and invasive cancer, but also promotes immune cell infiltration through various mechanisms (reviewed in [13, 16]), including the release of pro-inflammatory cytokines IL-1α and IL-6 and chemoattractants such as granulocyte-monocyte colony stimulating factor (GM-CSF). In both rodents and humans, pancreatic inflammation also cooperates with Kras mutations to hyperactivate downstream pathways such as nuclear factor kappa B (NF-κB), signal transducer and activator of transcription 3 (STAT3), and cyclooxygenase-2 (COX2), which in turn, can accelerate carcinogenesis through an autocrine/paracrine feedback loops, release additional pro-inflammatory mediators, and further perpetuate inflammation (Figures 3 and 4) [13, 16]. We highlight several examples of homeostatic perturbations that sustain the pro-tumorigenic inflammatory response that accompanies PDAC.
Figure 3. Examples of Regulated and Dysregulated Inflammatory Reactions in the Pancreas.
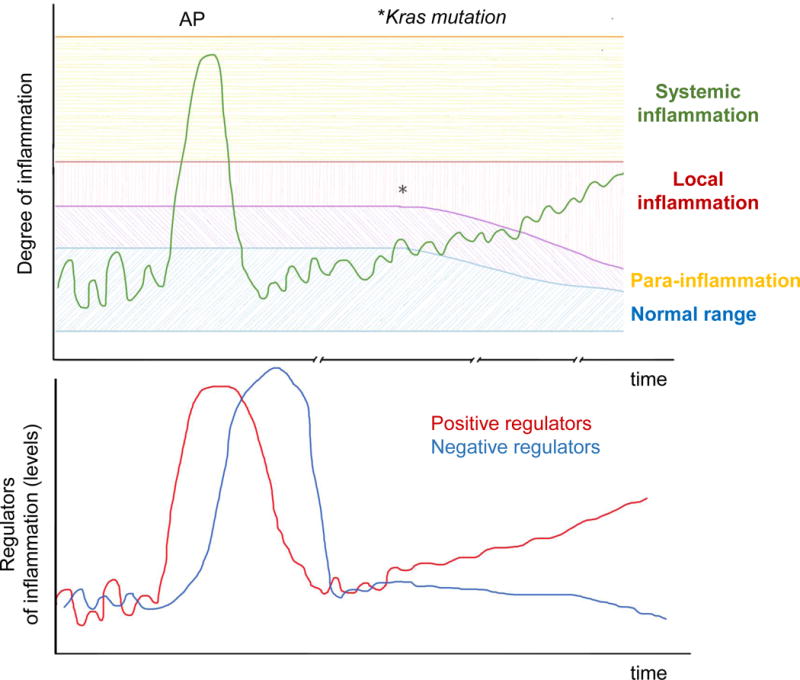
The output of a homeostatic system (e.g. degree of inflammation in the pancreas) can be plotted in a graph against time (top). In healthy tissues, pro-inflammatory mediators and downstream immune responses fluctuate within a narrow “normal range” that are not sufficient to trigger inflammation at the steady state. Cellular stress or injury can trigger an inflammatory reaction of variable magnitude, which can range from a minimal reaction to tissue stress (para-inflammation); to a more pronounced inflammatory reaction to overt cellular injury, as in mild acute pancreatitis (AP), characterized by local inflammation; to a full-blown systemic inflammatory response as seen in severe AP. The inflammatory response is driven by positive regulators (e.g. IL-6), but is eventually tuned down by negative regulators of the system (e.g. IL-10; bottom graph). Therefore, inflammation eventually subsides and the system returns back to normal (homeostasis).
When Kras is mutated, this balance is disturbed in two ways: (i) Kras mutant cells have a lower threshold for release of pro-inflammatory mediators; and (ii) the balance of positive and negative regulators of inflammation is tilted towards one that favors a sustained activation of immunosuppressive and pro-tumorigenic subsets.
Besides pro-inflammatory signals from transformed cells, the tumor-enabling inflammation is further perpetuated by (i) soluble mediators released by stressed non-transformed epithelial cells within the tumor microenvironment; and (ii) systemic promoters of low-grade inflammatory states such as smoking, obesity, and microbial dysbiosis.
Figure 4. Mediators of Tumor-Enabling Inflammation within the Pancreatic Cancer Microenvironment.
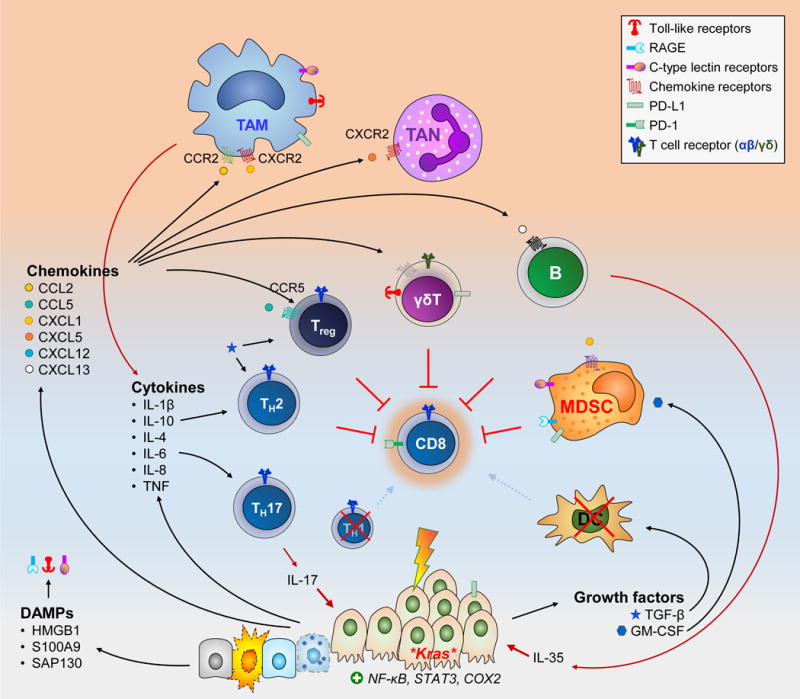
Cancer cells secrete a vast array of soluble factors that support an inflammatory infiltrate permissive of tumor growth. This is further fueled by similar mediators supplied by stressed non-transformed epithelial cells, particularly DAMPs that engage pattern recognition receptors (e.g. TLR-4, -7, and-9; CLR such as Mincle and Dectin-1; RAGE, etc)
The effects of each mediator are far more complex than shown above. Several of them can be secreted by more than one source and act on multiple cell types. For example TGF-β and IL-10 can be secreted by both the cancer cells and regulatory T cells (Treg). TGF-β can supports Treg differentiation as well as TH2 skewing of CD4+ T cells and disabling of CD8+ CTLs. The combination and relative ratios of different soluble mediators will ultimately dictate the outcome. Furthermore, cell surface molecules expressed on tumor and stromal cells (such as PD-L1) can also disable CTLs.
The end results of tumor-enabling inflammation can be conceptualized as (i) provision of cancer cells with growth signals (e.g. IL-6, IL-17); (ii) suppression of CTL anti-tumor activity; and (iii) activation of other non-immune stromal cells (shown in Figure 5).
HMGB1, high mobility group box 1; SAP130, Sin3-associated protein 130; TAN, tumor-associated neutrophils.
Imbalance of Soluble Mediators
Stressed cells, whether transformed or not, release DAMPs that bind PRRs and initiate or perpetuate inflammation. We and others have shown that the PDAC tumor microenvironment (TME) is rich in DAMPs, which bind Toll-like receptors (TLRs) and other PRRs such as the receptor for advanced glycation end-products (RAGE) in both mice and humans [13, 17–20]. In PDAC mouse models, ligation of several TLRs dramatically accelerates pancreatic oncogenesis through diverse mechanisms involving immune cell modulation as well as activation of pancreatic stellate cells (PSC) [17, 18, 21]. Other PRRs such as C-type lectin receptors (CLRs) also appear to have important roles. For instance, in both murine and human PDA, necroptotic epithelial cells release the histone deacetylase complex subunit SAP130 that acts as a DAMP by ligating Mincle – a CLR found on various myeloid cells – to promote suppression of antitumor immunity [19]. Disruption of the necroptotic pathway by genetic deletion or pharmacological inhibition of key signaling intermediates was shown to protect mice from pancreatic carcinogenesis, as did genetic ablation of Mincle[19].
Besides, PRRs can bind microbial byproducts that may originate from local or distant sites. Recent work has highlighted the role of oral microbial dysbiosis (i.e. imbalance of commensal flora and potentially pathogenic microorgansisms) in pancreatic carcinogenesis [13, 22, 23]. In a prospective human study featuring bacterial DNA profiling, overrepresentation of Neisseria elongata and Streptococcus mitis in saliva were strongly associated with PDAC [22]. Further, bacteria can directly access the pancreas. We found that orally administered Streptococcus mutans can reach the pancreas in mice, while a recent study identified Fusobacterium species in pancreatic tumor tissue and their presence correlated with higher mortality rates [18, 24]. Our ongoing investigations suggest that the intestinal microbiome is also deranged in PDAC patients and contributes to perturbations of the immune infiltrate, while germ-free mice are protected from pancreatic carcinogenesis secondary to immunogenic reprogramming of the TME (unpublished work).
Transformed pancreatic epithelial cells release a wide array of soluble factors, including cytokines (such as IL-1β, IL-6, IL-11, and TNF [18, 25, 26]), various chemokines (e.g. CCL2, CCL5, CXCL1, CXCL2, CXCL12 [19, 27–30]), and growth factors (such as GM–CSF and TGF-β [31, 32]), with profound effects on immune cell infiltration, activation status, as well as skewing towards cellular phenotypes that support tumor growth and immune escape (reviewed in the next section; Fig. 4). Some of the above are also released by non-transformed stressed cells [12]. The tumor-derived soluble factors often act in concert with each other, or with mediators derived from stromal cells, resulting in non-physiologic effects such as the generation of feed-forward loops that sustain the inflammatory reaction and prevent homeostasis [26, 33, 34]. For example, pancreatic epithelial cells with Kras mutations recruit myeloid cells that secrete high amounts of IL-6; the latter activates STAT3 on epithelial cells and upregulates pro-proliferative and anti-apoptotic molecules as well as extracellular matrix (ECM) modulating enzymes such as matrix metallopeptidase 7 (MMP7). This ultimately promotes PDAC progression and aggressiveness [26, 33].
The CXCL12–CXCR4 is one of the most well studied chemokine axes in multiple types of cancer [35, 36]. Both ligand and receptor are significantly upregulated on cancer cells as well as various other stromal cell types, and have important roles in bidirectional tumor-stroma communication in human PDAC, including promotion of tumor growth and invasion, enhancement of the cancer-associated fibroblast (CAF) compartment, and maintenance of intratumoral immunosuppression [36–38]. Intriguingly, it was recently implicated in neural invasion of PDAC: specifically, human peri-pancreatic Schwann cells were found to upregulate CXCR4 and CXCR7 in response to pancreatic cancer and associated hypoxia [30]. Cancer cells secrete CXCL12 which attracts peri-pancreatic nerves to infiltrate early PDAC lesions, rendering them less-responsive to pain [30]. Although this may be a defense mechanism to shield the patient from the intractable pain of pancreatic cancer-related neural invasion, it is clearly hijacked by cancer cells to promote loco-regional tumor dissemination [30].
Our understanding of the role of specific soluble mediators, particularly chemokines, is hampered by three important factors: First, several chemokines exhibit a great degree of redundancy, such that even if one is blocked, others can compensate for its absence. Second, chemokines exhibit promiscuity, acting on multiple receptors agonistically or antagonistically. Once again, targeting a single receptor might be insufficient to abrogate their effects. Lastly, even though many of the chemokines are conserved across mammalian species, some exhibit considerable structural and/or functional differences between humans and rodents, or may even be completely absent. One such example is CXCL8/IL-8, which is only present in humans and has important functions in recruitment of innate immune cells and angiogenesis, among others [39]. In conclusion, PDAC is characterized by profound imbalances of soluble mediators that have diverse and often overlapping functions. Therefore, investigations employing blockade of such promiscuous pathways should always take the above into consideration. Moreover, combination therapies against these mediators may be a better strategy for developing novel PDAC therapeutics.
Immune Cell Perturbations
The failure of the homeostasis of the immune system in the context of cancer development has been summarized in the concept of cancer immunoediting: Usually, the immune system successfully eradicates transformed cells (elimination). However, every once in a while cancer cells find ways to evade killing and persist in a “stealth mode” (equilibrium). As they accumulate additional mutations, the transformed cells that remain undetectable by cytotoxic immune effectors and can thrive in the pro-inflammatory tumor milieu are enriched and eventually prevail (escape). In pancreatic oncogenesis, the dynamics of immune cells are much more complex, with different cell types having multifaceted roles.
Myeloid cells such as neutrophils and macrophages have key roles in inflammatory diseases, including pancreatitis. Neutrophils – the predominant cell type involved in the early phases of acute inflammation – have been shown to contribute to pancreatic carcinogenesis and immunosuppression [11]. Chemokines such as CXCL5 and CXCL2 released by cancer cells and stromal cells, respectively, bind CXCR2 and attract neutrophils to the TME in human and murine PDAC [29]. Using murine PDAC cells, CXCL5 was shown to be upregulated in response to Kras mutations in a NF-κB-dependent manner in vitro. Moreover, genetic ablation of CXCR2 in a genetically-engineered mouse model of PDAC led to decreased neutrophil infiltration in tumors and augmented antitumor cytotoxic T cell immune responses, thus establishing a pro-tumorigenic role for tumor-associated neutrophils (TANs) in PDAC. [29].
Macrophages arrive to sites of inflammation after neutrophils and persist longer [40]; indeed, they are one of the main cell types involved in chronic inflammation [11, 41]. They infiltrate tumors in response to DAMPs and other danger signals are released secondary to tissue stress and hypoxia (which might be a consequence of chemo-/radio-therapy). They also mount responses to attempt to counteract danger signals and tissue stress; these include angiogenesis, remodeling of the ECM, and clearance of cellular debris [41]. Importantly, in mouse models of pancreatitis, they have been found to directly promote ADM via release of the pro-inflammatory cytokines RANTES and TNF, which activate NF-κB in acinar cells and upregulate several ADM-related proteins such as MMP9 [42]. Macrophages also act in concert with CD4+ T helper cells to direct adaptive immunity. Traditionally, they have been classified as M1 and M2 macrophages, depending on their mode of activation and cytokine profile, although this designation may be evolving. The imbalance in intratumoral macrophage-modulating factors usually leads to skewing of tumor-associated macrophages (TAMs) to an M2-like phenotype, as these tend to be pro-tumorigenic and T cell-suppressive [43]. By contrast, M1 macrophages usually support antitumor activity [43]. Since M1 polarization presumably correlates with the initiation of inflammatory responses, and M2 polarization with resolution, the perturbation observed in human and murine PDAC likely represents a maladaptive response with macrophages “locked” in an M2-like state that prevents antitumor immunity and supports tumor growth, invasion and metastasis as well as conferring resistance to chemoradiation [43–47]. An immunotherapeutic approach utilizing an agonistic CD40 antibody has shown promise in both murine and human PDAC through reprogramming of TAMs, which in turn became tumoricidal and facilitated depletion of tumor stroma [48]. TAM reprogramming was also achieved in another study in which mice with PDAC were subjected to inhibition of colony stimulating factor 1 (CSF1) or its receptor (CSF1R), resulting in reduced T cell-suppressive and augmented antigen presenting capacity [49].
CD8+ cytotoxic T lymphocytes (CTLs) are the main subset of antitumor effector cells; however they are usually relatively rare in PDAC-associated inflammatory infiltrates due to their exclusion by the associated stroma [38, 50, 51]. If present, they are suppressed by other immune and non-immune stromal cells via soluble factors such as IL-10 and TGF-β and inhibitory ligands such as PD-L1 [49, 52] (Figures 4 and 5). Notably, the aberrant expression of PD-L1 on tumor cells requires the presence of inflammation, once again indicating that homeostasis is deranged in the presence of oncogenic mutations [53, 54]. In addition, granulocytic and monocytic precursors of neutrophils and macrophages (and other myeloid cells), respectively, can give rise to subsets of myeloid-derived suppressor cells (MDSC), which, under the influence of tumor-derived factors such as GM-CSF, can directly inhibit antitumor immunity [31, 32, 55, 56]. Of note, most immunotherapeutic approaches aim to augment CTL effector responses.
Figure 5. Dysregulated Fibroblast Function in PDAC Leads to Desmoplasia where Cancer Cells Thrive.
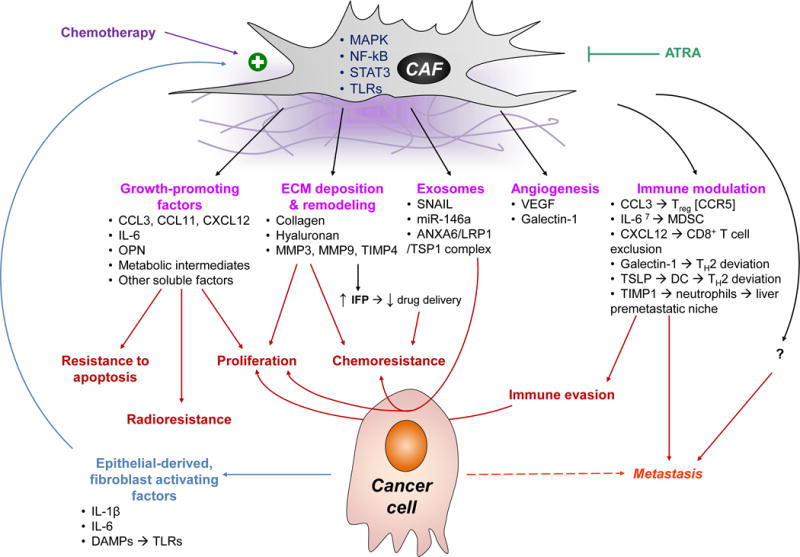
CAFs have a bidirectional communication with the epithelial compartment of the tumor. Epithelial-derived factors such as cytokines and chemokines, as well as DAMPs that ligate PRRs promote their activation via the MAPK, NF-κB, and STAT3 pathways [17, 18, 71, 104]. CAFs in turn provide, pro-proliferative and anti-apoptotic signals [18, 71, 105, 106]; promote angiogenesis [71, 104, 107]; modulate the ECM; and contribute to chemo- and radio-resistance [105, 106]. Importantly, CAFs also engage in crosstalk with immune cells and promote tumor tolerance: they recruit immunosuppressive cells such as Treg and MDSC [18, 104]; they skew helper T cells to TH2 deviation [25, 107, 108]; they sequester cytotoxic CD8+ T cells away from cancer cells [38, 51]; and recruit neutrophils to distant organs such as the liver, generating pre-metastatic niches [98, 99]. A newly appreciated mode of intercellular communication is exosomes, which are upregulated on CAFs under stressful conditions, such as hypoxia and exposure to chemotherapy [106, 109]. All the aforementioned mechanisms make the TME more hospitable to cancer cells, and contribute to tumor growth, immune escape, and metastasis. Agents such as all-trans retinoic acid (ATRA) may have a role in blocking their activation and disrupting their pro-tumorigenic function [110].
IFP, interstitial fluid pressure; MMP, matrix metalloprotease; OPN, osteoprotegerin; TSLP; thymic stromal lymphopoietin; VEGF, vascular endothelial growth factor.
CD4+ T cells have been reported to harbor either anti- or pro-tumorigenic effects, depending on their sub-differentiation. For instance, on the one hand, T helper 1 (TH1) cells have been shown to provide support to CTLs and to enable tumor rejection; however they are usually under-represented among intrapancreatic CD4+ T cells [57]. TH2 cells on the other hand, can promote pancreatitis and support pancreatic oncogenesis by enabling immune escape [17, 58, 59]. IL-17-producing CD4+ T cells (TH17) are also present in the transformed pancreas and have been found to provide growth support to cancer cells overexpressing the IL-17 receptor under the influence of mutant Kras in a PDAC mouse model [60]. Lastly, regulatory T cells (Treg), one of the most potent CTL-suppressive immune cell types, are recruited in the PDAC milieu via tumor-derived chemokines acting on the chemokine receptor CCR5 (such as CCL3 and CCL5), and their presence correlates with adverse patient outcomes [18, 28, 61]. Not surprisingly, ablation of CD4+ T cells in mice dramatically protects from pancreatitis and significantly delays PDAC development in the setting of mutant Kras [58, 59]. This exemplifies the inappropriate skewing of T helper cells in favor of pro-inflammatory and pro-tumorigenic subsets (i.e. TH2, TH17 and Treg) in pancreatic cancer, constituting in turn, a typical example of failed defense mechanisms and deregulated homeostasis in the host.
An often neglected function of immune cells is their contribution to tissue regeneration. Accordingly, Rag1−/− mice exhibit impaired pancreas regeneration following pancreatitis [62]. Although not well-studied in the context of pancreatic injury, IL-17 has an established role in the regeneration of other organs such as the liver [63]. It is likely that the aforementioned IL-17-producing T cells are also part of a pro-regenerative program in response to bystander tissue injury in the pancreas; however the presence of oncogenic Kras mutations leads to a completely different outcome, as explained above [60]. Moreover, IL-22, a cytokine of the IL-10 family typically produced by CD4+ T cells (often together with IL-17), has been found to be secreted in response to Aryl hydrocarbon receptor (AhR) signaling and to promote tissue repair following acute pancreatitis in mice [64]. By contrast, chronic intraperitoneal administration of AhR ligands in mice with chronic pancreatitis has been shown to lead to high IL-22 production by CD4+ T cells, as well as PSC activation, in addition to exacerbated inflammation and fibrosis [65]. Interestingly, previous work examining Th17 cells in pancreatic oncogenesis also found elevated levels of IL-22 in PDAC-infiltrating CD4+ T cells, suggesting that IL-22 may be contributing to PDAC fibro-inflammatory reaction and might constitute a link between smoking and other activators of AhR signaling, chronic pancreatitis, and PDAC [60].
Other immune cell types are also being identified to be co-opted by cancer cells and to contribute to pancreatic oncogenesis. B cells were recently found to infiltrate PDAC in both humans and mouse models [66, 67]. Stromal cell-derived CXCL13 was identified as one of the main chemoattractants for B cells, which in turn promoted tumor progression directly, via secretion of IL-35, supporting cancer cell proliferation; and indirectly, via promotion of immunosuppressive TAMs and subsequent disabling of CTLs [66, 67]. Further, we have shown that γδ T cells infiltrate the chronically-inflamed pancreas, as well as early pre-neoplastic lesions and invasive PDAC in mice and humans; this in turn, prevents CD4+ and CD8+ αβ T cells from mounting an antitumor immune response [52]. Mechanistically, the immunosuppressive effect of γδ T cells was found to be mediated by T cell inhibitory ligands such as PD-L1 and Galectin-9 [52]. In mice, depletion of γδ T cells or deletion of chemokine receptors involved in their recruitment (e.g. CCR2, CCR5, or CCR6) led to a significant increase in the infiltration of CD4+ and CD8+ αβ T cells, and afforded protection from the development and progression of PDAC lesions [52]. Of note, γδ T cells have also been reported to be an intrapancreatic source of IL-17; however their pro-tumorigenic effects appear to be independent of IL-17 production since IL-17 levels are not significantly affected upon γδ T cell depletion [52, 60]. In these human correlative studies, the majority of intrapancreatic γδ T cells were found to be Vδ1+, and this particular subset has been previously reported to have a protumorigenic role; by contrast, Vδ2+ T cells harbor tumoricidal capacity and have been exploited for immunotherapy against PDAC and other malignancies [68].
Cancer-associated Fibrosis
Tissue fibrosis has its roots in the normal wound healing process, particularly at sites where adequate regeneration cannot occur. Resident tissue fibroblasts are normally activated by acute inflammation and engage in functions that promote epithelial regeneration, including angiogenesis, modulation of the inflammatory response, provision of growth signals, as well as synthesis of ECM proteins that can act as a cellular scaffold for epithelial cells, among other things [10]. Unlike the physiologic state where fibroblasts either revert back to quiescence or die once tissue integrity is restored, chronic inflammatory states (such as chronic pancreatitis and PDAC) are characterized by persistent activation of fibroblasts and other stromal cells, leading to the formation of an abnormal ECM that contributes to a state of desmoplasia.
Although desmoplasia was identified as a hallmark of PDAC several decades ago, its pathophysiologic significance is not well understood [69, 70]. The predominant effector cell is the CAF, which originates from activated resident fibroblasts (such as pancreatic stellate cells). CAFs exhibit bi-directional communication with both epithelial cells and immune cells [38, 71]. They are activated by DAMPs, cytokines, and various other agents (including chemotherapeutics) and contribute to PDAC progression through a myriad of mechanisms (Fig. 5). Although attempts to deplete CAFs have yielded contradictory results, they remain a very attractive target for therapeutic intervention since they represent a central hub coordinating aberrant stromal homeostasis [69, 72–74].
The fibro-inflammatory response of PDAC is visible very early on in the process of carcinogenesis, even at the stage of ADM, and is then progressively intensified [69]. The end result is the generation of a dense, gel-like extracellular matrix with unique physicochemical properties [75]. Several macromolecules of the gel-like ECM, such as hyaluronic acid (HA), absorb and retain significant amounts of water, thus increasing the interstitial fluid pressure (IFP) of the tumor; this dramatically elevated IFP is evident by the collapsed blood vessels in areas of desmoplasia, and appears to be an important hurdle to adequate delivery of drugs in the tumor milieu, due to both inadequate perfusion and opposition to their extravasation [75].
Interestingly, similar stromal modifications have been found not only in primary tumors, but also at sites of future metastases. For example, in both murine and human PDAC, the metastatic liver has been shown to exhibit significant alterations in its microarchitecture that are evident even with isolated tumor cells [70, 76]; these include the deposition of various ECM proteins (such as HA, collagen I and fibronectin), and activation of myofibroblast-like cells, as well as infiltrating immune cells [70, 76]. These changes are remarkable in that they have been shown to promote the engraftment of pancreatic cancer cells in mouse models and thus, constitute a defining characteristic of the hepatic premetastatic niche [77, 78]. Successful treatment of liver metastases in mice leads to regression of metastasis-associated ECM changes, attesting to the addiction of pancreatic cancer cells to desmoplasia [76].
Additional Local and Systemic Imbalances
Autophagy, Endoplasmic Reticulum Stress, and Metabolic Perturbations
Stressed pancreatic epithelial cells accumulate metabolic intermediates that damage vital macromolecules and intracellular organelles. One of their cardinal defense mechanisms is the process of autophagy, which allows the removal of cellular debris that propagate cellular injury; limits the accumulation of DAMPs and the resultant inflammatory reaction; and recycles essential metabolic intermediates to facilitate cellular repair (reviewed in [12]). Disruption of autophagy via genetic deletion of its key mediator autophagy related 5 (ATG5) in mice led to increased levels of reactive oxygen species, augmented endoplasmic reticulum (ER) stress, and development of spontaneous chronic atrophic pancreatitis [79]. Conceivably, autophagy appears to limit ER stress and pro-tumorigenic mediators, and protect during incipient pancreatic oncogenesis [80, 81].
The coin is flipped in established PDAC: both mutant Kras and microenvironmental stressors, such as intratumoral hypoxia and nutrient deprivation, significantly increase autophagic flux, which benefits cancer cells by protecting them from toxic intermediates, including chemotherapeutics, and by supporting tumor metabolism [82, 83]. Cancer cells have a very high turnover of proteins, lipids, and other molecules, and therefore rely on a “personalized menu” of metabolic intermediates, as well as on raw material derived through autophagy [12, 15]. Metabolic reprogramming is considered a hallmark of cancer, and likely originates from homeostatic circuits that attempt to mobilize readily usable intermediates to support stressed cells [83]. However, its effects not only nurture the cancer cells but also affect the TME and propagate tumor-enabling inflammation. Further, epithelial and stromal cell crosstalk leads to changes in the metabolic intermediates in the tumor milieu that support the cancer cells [84]. One study recently showed that human cancer cells can coerce PSC to activate autophagy in vitro so as to provide non-essential amino acids such as alanine that fuel the cancer cells’ tricarboxylic acid cycle and support lipid and protein synthesis [85]. The above were verified in vivo using xenograft mouse models, although the cancer cell-derived mediators responsible for this process remain unidentified.
Conditions that further deregulate metabolism, such as obesity, can also fuel inflammation and accelerate pancreatic carcinogenesis [86]. For example, a recent study in an orthotopic PDAC mouse model found that obesity leads to adipocyte activation, which can in turn recruit neutrophils to the pancreas via IL-1β signaling [87]. The latter can then enhance PSC activation, thus supporting tumor growth and augmented desmoplasia, promoting chemoresistance [87]. Adipocytes have also been shown to promote proliferation of murine pancreatic cancer cells in vitro by providing glutamine, especially in nutrient-poor conditions [88]. On the other extreme of metabolic perturbations is cancer cachexia, a dreaded complication of PDAC which leads to wasting, and occurs with advanced disease. In mouse models of PDAC-associated cachexia, several tumor-derived pro-inflammatory factors (e.g. IL-6 and TGF-β) have been shown to modulate the metabolism at a systemic level, as evidenced by altered levels of circulating glucose and ketones, [89, 90]. The metabolic stress leads to elevated glucocorticoid levels that contribute to immune suppression and tumor tolerance [89].
In summary, it is now becoming evident that several homeostatic processes that were traditionally viewed at the cancer cell level, such as the response to cellular stress and the regulation of metabolism, need to be revisited and considered in conjunction with the tumor’s surrounding stromal cells.
Coagulation
Another consequence of deregulated homeostasis at a systemic level is the unbalanced activation of the coagulation system. Thromboembolic disease occurs in up to 57% of PDAC patients and in extreme cases it may manifest as migratory thrombophlebitis (Trousseau’s syndrome) or disseminated intravascular coagulation [91, 92]. Although initially theorized to be related to tumor cell emboli and mucins secreted by tumor cells into the circulation, the hypercoagulability associated with cancer is now believed to be a result of multiple overlapping mechanisms involving imbalances in positive and negative regulators of the coagulation pathway; secretory products of cancer cells and activated immune cells; systemic changes in endothelial cells of both venous and arterial conduits; as well as widespread platelet activation [92]. Importantly, several intermediates of the coagulation cascade including fibrinogen may act as mitogenic signals for fibroblasts that perpetuate chronic inflammation [93, 94].
Metastasis
An important evolving phenomenon in our understanding of metastasis is the contribution of the immune system to this process [76, 95]. Metastasis has been conceptualized as a sequential process whereby cancer cells detach from their surroundings, migrate through the extracellular matrix, gain access to blood vessels or lymphatics, travel to distant sites, and exit these conduits at distant organs, remaining dormant or forming metastatic foci [96]. The immune system has been shown to contribute to all these steps through multiple complementary mechanisms (reviewed in [95]). In PDAC, one recent study showed that macrophages were recruited to the liver of mice at sites seeded by isolated cancer cells [47]. There, they secreted granulin, a factor that in turn activated hepatic stellate cells to synthesize a dense ECM, and supported tumor cell proliferation in the liver in a periostin-dependent manner, thus promoting metastasis. The above is reminiscent of a foreign body reaction, or a granuloma formation in response to infectious agents; however, a main difference here is that cancer cells can exploit it to their advantage.
Even more strikingly, deranged immune responses can create premetastatic niches – altered microenvironments in distant organs that are conducive to future colonization by circulating tumor cells. For example, in an orthotopic mouse model of PDAC as well as in intraperitoneally disseminated colon cancer mouse model, tumor cells were found to promote the expansion of intrahepatic MDSC through the release of soluble factors such as KC/CXCL1, creating an immune-privileged hepatic microenvironment that favors [97]. Moreover, Kruger’s laboratory recently showed that stromal cells in human and murine PDAC and other tumors could release tissue inhibitor of metalloproteinases-1 (TIMP-1), which acts in the premetastatic liver to induce the secretion of ECM proteins such as fibronectin, and chemokines such CXCL12 [98, 99]. ; the interaction of TIMP-1 with CD63 on hepatic stellate cells mediates some of these effects and leads to neutrophil recruitment in a CXCL12/CXCR4-dependent manner, supporting liver homing of cancer cells [98, 99]. Another study reported that murine PDAC-derived exosomes constitute an important source of immunomodulating factors such as macrophage migration inhibitory factor (MIF); these are taken up by hepatic Kupffer cells to induce a TGF-β-mediated signaling cascade that involves ECM remodeling and recruitment of additional bone marrow-derived cells, ultimately promoting murine PDAC metastasis to the liver [77].
Collectively, these current findings exemplify how primary tumors can globally affect other biological processes at an organismal level that allow them to survive hostile microenvironments, evade destruction by the immune system, grow and spread to distant organs. Additional mechanisms that are likely to underlie PDAC metastasis include the modulation of distant organ microvessel architecture (e.g. in response to DAMPs and low-grade inflammation) to facilitate disseminated tumor cell (DTC) homing and extravasation; modulation of metabolism to support DTC growth; and altered angiogenesis in response to hypoxia, as has been shown for other cancer types [100].
Concluding Remarks
It is now clear that the tumor microenvironment is of vital importance in influencing the behavior of tumor cells, their ability to evolve and spread, as well as their response to chemo-immune-therapeutic treatments. It must be appreciated that the relationship between deregulated homeostatic responses and cancer progression is most likely not a deterministic phenomenon. Instead, it is a consequence of natural selection whereby cancer cells that are able to thrive in conditions of chronic inflammation and fibrosis are selectively, the ones that survive. The presence of multiple clones of cancer cells further complicates this picture, as these malignant clones might exploit host physiology by benefiting from chronic inflammation in different ways and/or using redundant pathways. As a result, devising anti-cancer therapies to attack a single physiological process is frequently ineffective. Several novel discoveries in PDAC have revealed new possibilities but have also generated several questions that need to be answered (see Outstanding Questions). By thinking of the pathophysiology of the tumor microenvironment as a whole and in the context of deranged homeostatic responses, me may better understand the process of cancer development and its resistance to treatment, which may inform novel ways to profile (Box 1) and treat such a devastating disease (Box 2). As an analogy, one might consider the management of septic shock, where antibiotics alone are not enough to treat it, given that an important part of its pathophysiology originates from a derailed immune response; thus, approaches to treating pancreatic cancer may be viewed in a parallel fashion, where it is important to consider the tumor in relation to the host’s physiology, and vice versa. Outcomes may improve only when interventions targeting the perturbed immune homeostasis are instituted: it is time to think outside the box!
Box 1. Tumor Profiling Should Include the Microenvironment.
The traditional approach to most solid malignancies has been surgical removal of the tumors when feasible, accompanied by high-dose chemotherapeutic agents postoperatively, depending on clinicopathological parameters that dictate the risk of locoregional and or distant recurrence. If resection is not feasible due to size or involvement of critical surrounding structures (e.g. major blood vessels), the patient may be treated with chemotherapy and/or radiation therapy first, in an attempt to “shrink” the tumor and convert it to resectable. If the tumor is unresectable to begin with, the patient is usually treated with high doses of chemotherapy. Even though most of these treatments have the potential to prolong survival, they often come with serious side effects that compromise a patient’s quality of life. Ultimately, the tumors recur and metastatic disease with its associated complications/death ensues.
This remarkable ability of cancer to recur originates from is its inherent potential to mutate and adapt to its environment. The selection pressure imposed by chemotherapeutic agents will promote the emergence of a minority of resistant clones that may find shelter under the protective veil of the co-opted fibro-inflammatory stroma. They remain dormant until they gain strength and eventually form new colonies. This phenomenon of resistance is not entirely cell-autonomous, but it also depends on the microenvironment that may supply a favorable scaffold, soluble mediators, nutrients, and signals that shield the cancer cell from chemotherapeutic agents.
In the past decade, several novel types of biomarkers have been introduced to clinical practice that can either enhance the accuracy of prognosis or predict response to certain types of adjuvant therapies, and have gained clinical utility in breast cancer. Although useful, such approaches of mutational analysis entirely neglect the stroma and the systemic perturbations of homeostatic responses. Not surprisingly, they haven’t been successful in PDAC, which relies so heavily on fibro-inflammation. We therefore need to capture the status of the tumor microenvironment in order to better identify opportunities for treatment.
A theoretical example to better profiling would be a combination of:
-
-
Histopathological and immunohistochemical analysis of tumor and peritumoral tissue (reaction to tumor), including immune infiltrates, lymphovascular and neural structures, fibrosis etc
-
-
Mutational analysis of cancer cells
-
-
Microdissection of tumor stroma and subsequent gene expression profiling
-
-
Analysis of upregulated circulating cytokines, chemokines
-
-
Circulating exosome profiling
-
-
Intestinal microbiome profiling
-
-
Analysis of a host’s gene polymorphisms that may be potentially implicated in homeostatic response outcomes and pathophysiology.
Box 2. Clinician’s Corner: A Novel Approach to Cancer Treatment.
Imagine if we could gather extensive intelligence (comprehensive profiling, such as the theoretical example in Box 1) before attacking the enemy (the cancer cells). Once we know what is happening behind the enemy lines, we first take down the enemy’s supply line, destroy the supportive infrastructure, disable the defenses, and isolate the enemy before mounting our heads-on attack. The enemy will have nowhere to hide and would be entirely vulnerable to our weapons (e.g. chemotherapy). What if we could approach cancer in this way, i.e. restore the aberrant homeostatic responses back to normal (to the extent that it is possible) and then deal with the cancer cells, while continuing to prevent aberrant physiology. With appropriate intelligence we could also predict the stromal responses after treatment and manage them pre-emptively (e.g. by depleting DAMPs, or using neutralizing antibodies for chemokines or TLRs).
Ultimately, the goal should be not only to eradicate the transformed cells, but also to restore the lost equilibria and re-establish homeostasis. To this end, it must be kept in mind that non-targeted therapies such as traditional chemotherapeutics and radiation therapy can injure the uninvolved parenchymal cells and the surrounding supportive tissue – particularly when delivered at maximal tolerated doses – thus propagating the vicious cycle of inflammation and fibrosis. On the contrary, lowering the treatment doses and combining multiple drugs with targeted agents and other modalities might be the key to delivering a more accurate hit to cancer cells while sparing the stroma and uninvolved epithelial cells. This is an area that certainly deserves further exploration.
Outstanding Questions Box.
Can cancer screening be performed by monitoring the changing tumor microenvironment (TME)? It is now well established that the generation of the TME begins very early during carcinogenesis, even at the pre-malignant stages. Theoretically, if we can detect those changes, we should be able to develop novel biomarkers for the early detection of cancer as well as for monitoring their progression and response to treatment.
Can we improve our understanding of the biologic behavior of each patient’s pancreatic tumor by better examining the TME? The optimization of methods that allow propagation of patient-derived cancer cells and subsequent genomic analyses (e.g. using organoids and xenografts) has enabled profiling of the mutational landscape of individual patient’s tumors and has the potential to identify suitable personalized targeted therapies. However such approaches are usually inadequate to account for the composition of the stromal compartment and the respective active pathways. We speculate that the negating effect of the stromal reaction is one of the reasons of why many seemingly appropriate targeted therapies fail in the clinic.
Can combination treatments against cancer cells and the stromal reaction improve the efficacy of targeted therapies? Does the order of administration matter? Even though studying the TME has yielded several critical mediators that are attractive as therapeutic targets (e.g. T cell checkpoint molecules), interventions against them have not been effective as monotherapy. For example, neutralizing antibodies against PD-1/PD-L1 are largely ineffective in PDAC when given alone; however when other aspects of the TME are modulated (e.g. CAFs, TAMs), such agents become efficacious. Further, the majority of patients who receive these agents (mostly in the context of clinical trials) are heavily pre-treated with chemotherapeutics that have unpredictable consequences on the TME. Many novel drugs tested as monotherapies in small scale phase 1 and 2 trials are abandoned as a consequence of this very reason. In contrast, administering TME-modulating agents before chemo- or radiation therapy may be constitute a more promising strategy and should be considered for future trials.
Trends Box.
Persistent low-grade inflammation is much more common during carcinogenesis than extremes of inflammatory response. The latter can result from exposure to environmental insults (e.g. smoking-related substances); from states that perpetuate chronic injury, such as chronic pancreatitis; or from internal imbalances that constantly release pro-infammatory mediators, such as obesity and microbial dysbiosis.
Tumor-enabling inflammation is one of the hallmarks of cancer and is especially prominent in PDAC. It usually starts as imbalance of positive and negative regulators (such as soluble mediators and immune cell polarization) and co-operates with oncogenic mutations (e.g. Kras) to create feed-forward loops that perpetuate the state.
Cancer-associated fibroblasts likely originate from maladaptive tissue regeneration programs. They are a vital component of the PDAC microenvironment, supporting cancer cells in various ways such as provision of pro-proliferative and anti-apoptotic cues, metabolic supplementation, angiogenesis, and generation of local immunosuppression.
Imbalances in homeostatic responses contribute to resistance to various therapeutic approaches. Conversely, some treatment modalities themselves (e.g. radiotherapy and some chemotherapeutic drugs) may inadvertently fuel the tumor-enabling inflammation secondary to bystander homeostatic responses (e.g. activation immune and other pro-tumorigenic stromal cells). We must therefore develop strategies that will attempt to tackle such perturbations.
Acknowledgments
CPZ is supported by the Center for Metastasis Research Fellowship (Memorial Sloan Kettering Cancer Center, NY). GM is supported by NIH grants CA168611, CA155649, and CA206105; the Department of Defense Peer Reviewed Medical Research Program; the Lustgarten Foundation; AACR-PanCan; and the Hirshberg Foundation for Pancreatic Cancer Research.
Glossary
- Acinar-to-ductal metaplasia (ADM)
process of de-differentiation of pancreatic acinar cells and assumption of immature ductal epithelial cell features in response to cellular stress and injury. It is necessary for regeneration and is reversible once the noxious stimulus is removed; however in the presence of Kras mutations it becomes irreversible and can proceed to PanIN formation and PDAC development.
- Angiogenesis
the process of forming new blood vessels. It occurs physiologically during development, as well as transiently during wound healing in adults. Cancers cells release various factors that stimulate angiogenesis in a persistent and haphazard fashion, in order to augment oxygen and nutrient supply and sustain their heightened proliferation and metabolism
- Aryl hydrocarbon receptor (AhR)
a cytosolic ligand-activated transcription factor with important roles in T cell differentiation and xenobiotic response to toxic metabolites. It is activated by binding various naturally occurring compounds as well as synthetic polycyclic aromatic hydrocarbons and various other toxic intermediates
- Autophagy
the process of recycling damaged intracellular organelles and macromolecular structures via sequestration in double-membrane vesicles (termed ‘autophagosomes’) that eventually fuse with lysosomes leading to degradation of their contents. It is upregulated under conditions of starvation as a homeostatic response to nutrient deprivation
- Bystander effects of homeostatic responses
whenever a tissue is exposed to stress (e.g. radiation injury), it activates protective responses to shield it from the injurious insult. In the case of cancer, malignant cells may benefit from mediators (e.g. chemokines) released by non-malignannt cells as part of these protective responses
- Cachexia
A multifactorial clinical syndrome associated with advanced malignancy, characterized by weight loss, skeletal muscle wasting, poor performance status, and partial refractoriness to nutritional supplementation. It is especially prominent in PDAC and correlates with decreased survival
- Cancer-associated fibroblasts (CAFs)
mesenchymal cells originating from tissue-resident fibroblasts that, under the influence of tumor-derived cues, assume an activated secretory phenotype providing nutrients and growth signals to cancer cells; supporting the propagation of cancer-associated inflammation; and contributing to resistance to chemotherapy, radiation therapy and immunotherapy. There is no universal specific marker for their detection, although CD140a (PDGF-Rα) and fibroblast activation protein (FAP) have been proposed in separate studies
- Coagulation cascade
The main defense system responsible for hemostasis. Upon exposure of subendothelial collagen, tissue factor, and other molecules (e.g. due to endothelial injury), platelets adhere to the site and become activated, initiating the sequential activation of coagulation factors. The end result is the generation of a fibrin polymer that stabilizes the adherent platelets and contributes to the inflammatory response
- Complement system
A set of soluble factors that continuously probe for invading pathogens as well as damaged cells and supplement the immune system’s function in eliminating the former. It is activated rapidly and results in generation of multiple intermediates that contribute not only to immune clearance but also to inflammation, angiogenesis, tissue repair, as well as various pathogenic conditions
- Damage-associated molecular patterns (DAMPs)
molecules released by injured or dying cells (e.g. S100 proteins, heat shock proteins, HMGB1, fibronectin, etc), which bind to and activate pattern-recognition receptors (e.g. Toll-like receptors)
- Desmoplasia [GR: δέσμη (bundle) + πλάθω (to make)]
the intense fibrotic reaction that accompanies neoplastic and non-neoplastic processes and is result of deranged remodeling of the ECM
- Extracellular matrix (ECM)
a dense, complex substance that fills the interstitial space of organs. It is composed of various fibrillary (such as collagen) and globular proteins, as well as glycosaminoglycans and proteoglycans – the main one being hyaluronic acid (HA). It functions as a supportive mesh for epithelial cells, providing contact signals via cell surface molecules (e.g. integrins). It is usually formed by fibroblasts and serves as a reservoir for growth factors (e.g. TGF-β), chemokines and other molecules which are embedded in the matrix and are released during the remodeling phase
- Exosomes
microvesicles (40–150 nm in diameter) originating from inward budding of the plasma membrane and subsequent processing in multivesicular bodies, with eventual release in the surroundings; they carry proteins and nucleic acids (mRNA, miRNA, DNA) and are emerging as key players in intercellular communication in health and disease
- Gamma delta (γδ) T cells
T cells bearing the γδ T cell receptor (TCR). They are not antigen-restricted and recognize antigens on pathogens as well on stressed cells via their TCRs and other receptors (e.g. PRRs). They are found in the peripheral blood as well as in various organs that interface with the external environment (e.g. skin, IEL compartment, liver etc) and are among the first responders to tissue stress and infection. Recent studies have found conflicting roles for γδ T cells in carcinogenesis. Based on the δ chain of their TCR, two main subsets have been identified in humans - Vδ1 and Vδ2, although Vδ3 and other much less studied subsets exist
- Granulin
glycoprotein involved in wound healing. It is associated with fibroblast migration and has variable effects on epithelial cell growth
- Intraepithelial lymphocytes (IELs)
immune cells of lymphocyte lineage that reside within the epithelial lining of the mucosa of various organs (such as the intestine). Unlike naïve T cells (that have not been previously exposed to antigens), they do not require priming and are able to be activated rapidly, thus serving as a first line of defense against invading pathogens as well as stressed epithelial cells
- Kupffer cells
liver-resident macrophages located within the lumen of hepatic sinusoids, adherent to their walls, and having important roles in sampling and clearance of damaged cells and microbes in health and disease. Upon activation, they release several soluble mediators that can affect immune responses locally within the liver as well as systemically (e.g. in the bone marrow)
- M1 and M2 macrophages
conceptual framework of macrophage polarization, initially described for infectious diseases and imperfectly applied to tumor-associated macrophages. M1 (‘classical’) are induced by interferon-γ and LPS and promote TH1 responses, while M2 (‘alternative’) macrophages are induced by IL-4, IL-10, IL-13, and FcRγ signaling and support TH2 responses
- Metabolic reprogramming
alterations in the cancer cell’s metabolism in response to cellular stress, increased proliferation and hypovascularity that lead to relative nutrient deprivation and hypoxia. A classic example is the augmented uptake of glucose and preferential funneling into glycolysis, even under normoxic conditions, termed the ‘Warburg effect’ after Otto Warburg
- Necroptosis
a recently identified process of cell death with morphological features of necrosis but a more organized mode of initiation and propagation (resembling apoptosis), via engagement of death receptors or several PRRs, and activation of receptor interaction protein kinase 1 (RIP1) and RIP3 downstream: ‘programmed necrosis’
- Opsonization
the process of coating a cell (endogenous, such as apoptotic cell; or exogenous, such as a microbe) with “opsonins” – molecules that promote complement activation on the surface of that cell. Such molecules include antibodies (especially IgM), complement proteins (e.g. C3b), and other circulating proteins (e.g. ficolins)
- Pancreatic Intraepithelial Neoplasia (PanIN)
premalignant alterations of the pancreatic ductal epithelium. According to the overall cellular morphology as well as the nuclear architecture and atypia, they are subdivided into PanIN-1, -2, and -3 – the latter representing carcinoma in situ. They correlate with progressive accumulation of mutations and increasing risk for progression to PDAC. PanIN-1 and -2 are quite common in patients over 40 years
- Pancreatic stellate cells (PSCs)
fibroblast-like cells that reside in the pancreas in a quiescent form, characterized by expression desmin and storage of vitamin A. When activated (e.g. by cellular injury/stress, cytokines, chemokines, DAMPs), they assume a myofibroblast phenotype, lose vitamin A cytoplasmic droplets, and express α-smooth muscle actin (α-SMA). They are the primary source of ECM molecules in health and disease, and contribute to pancreatic fibrosis, regeneration, inflammation and tumorigenesis. They are likely the primary source of CAFs in the context of pancreatic cancer although this is an area of ongoing investigation and debate
- Pattern recognition receptors (PRRs)
a broad class of receptors important in the innate immune system. They sense evolutionarily conserved molecular motifs originating from microbes (e.g. lipopolysaccharide, LPS) as well as from stressed cells (DAMPs) and contribute to various immune-related processes. They comprise four families: Toll-like receptors (TLRs); C-type lectin receptors (CLRs; e.g. Dectin-1); NOD-like receptors (NLRs); and RIG-like receptors (RLRs).
- Periostin
ECM protein normally found in periosteum of bones (hence the name). It is upregulated in several cancer types (e.g. ovarian and pancreatic cancer) and interacts with multiple receptors, such as integrins, on cancer cells to facilitate their survival, invasion, and metastasis, among others.
- Rag1−/− mice
genetically-engineered mice harboring an inactivating mutation of the recombination activation gene Rag-1, resulting in defective V(D)J recombination of B and T cell receptors and hence lack of B and T cells due to early developmental arrest
- Schwann cells
glial cells that are wrapped around nerve fibers of peripheral nerves. They synthesize the insulating myelin sheath and provide structural support to peripheral neurons
- Stroma
the supportive (i.e. non-parenchymal) part of an organ. It can be divided into an acellular component (the ECM) and a cellular component. The latter is composed of a variety of cells such as mesenchymal stem cells, fibroblasts, vascular and lymphatic endothelial cells, neurons and Schwann cells (or other nerve supportive cells), adipocytes, etc
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
None declared.
References
- 1.Greer JB, Whitcomb DC. Inflammation and pancreatic cancer: an evidence-based review. Curr Opin Pharmacol. 2009;9:411–418. doi: 10.1016/j.coph.2009.06.011. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, et al. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 3.Ricklin D, et al. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–827. doi: 10.1016/j.cell.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 7.Cao X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat Rev Immunol. 2016;16:35–50. doi: 10.1038/nri.2015.8. [DOI] [PubMed] [Google Scholar]
- 8.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dvorak HF. Tumors: Wounds That Do Not Heal. N Engl J Med. 1986;315:1650–1659. doi: 10.1056/NEJM198612253152606. [DOI] [PubMed] [Google Scholar]
- 10.Dvorak HF. Tumors: wounds that do not heal-redux. Cancer Immunol Res. 2015;3:1–11. doi: 10.1158/2326-6066.CIR-14-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zheng L, et al. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology. 2013;144:1230–1240. doi: 10.1053/j.gastro.2012.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gukovsky I, et al. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1199–1209 e1194. doi: 10.1053/j.gastro.2013.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zambirinis CP, et al. Pancreatic cancer, inflammation, and microbiome. Cancer J. 2014;20:195–202. doi: 10.1097/PPO.0000000000000045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeo TP, Lowenfels AB. Demographics and epidemiology of pancreatic cancer. Cancer J. 2012;18:477–484. doi: 10.1097/PPO.0b013e3182756803. [DOI] [PubMed] [Google Scholar]
- 15.Kleeff J, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. doi: 10.1038/nrdp.2016.22. [DOI] [PubMed] [Google Scholar]
- 16.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144:1220–1229. doi: 10.1053/j.gastro.2013.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ochi A, et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. The Journal of experimental medicine. 2012;209:1671–1687. doi: 10.1084/jem.20111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zambirinis CP, et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J Exp Med. 2015;212:2077–2094. doi: 10.1084/jem.20142162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seifert L, et al. The necrosome promotes pancreatic oncogenesis via CXCL1 and Mincle-induced immune suppression. Nature. 2016;532:245–249. doi: 10.1038/nature17403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kang R, et al. The receptor for advanced glycation end products (RAGE) sustains autophagy and limits apoptosis, promoting pancreatic tumor cell survival. Cell Death Differ. 2010;17:666–676. doi: 10.1038/cdd.2009.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ochi A, et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J Clin Invest. 2012;122:4118–4129. doi: 10.1172/JCI63606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farrell JJ, et al. Variations of oral microbiota are associated with pancreatic diseases including pancreatic cancer. Gut. 2012;61:582–588. doi: 10.1136/gutjnl-2011-300784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fan X, et al. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut. 2016 doi: 10.1136/gutjnl-2016-312580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mitsuhashi K, et al. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget. 2015;6:7209–7220. doi: 10.18632/oncotarget.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.De Monte L, et al. Intratumor T helper type 2 cell infiltrate correlates with cancer-associated fibroblast thymic stromal lymphopoietin production and reduced survival in pancreatic cancer. The Journal of experimental medicine. 2011;208:469–478. doi: 10.1084/jem.20101876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukuda A, et al. Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell. 2011;19:441–455. doi: 10.1016/j.ccr.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanford DE, et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: a role for targeting the CCL2/CCR2 axis. Clin Cancer Res. 2013;19:3404–3415. doi: 10.1158/1078-0432.CCR-13-0525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tan MC, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182:1746–1755. doi: 10.4049/jimmunol.182.3.1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chao T, et al. CXCR2-Dependent Accumulation of Tumor-Associated Neutrophils Regulates T-cell Immunity in Pancreatic Ductal Adenocarcinoma. Cancer Immunol Res. 2016;4:968–982. doi: 10.1158/2326-6066.CIR-16-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Demir IE, et al. Early pancreatic cancer lesions suppress pain through CXCL12-mediated chemoattraction of Schwann cells. Proc Natl Acad Sci U S A. 2017;114:E85–E94. doi: 10.1073/pnas.1606909114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bayne LJ, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor regulates myeloid inflammation and T cell immunity in pancreatic cancer. Cancer Cell. 2012;21:822–835. doi: 10.1016/j.ccr.2012.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pylayeva-Gupta Y, et al. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell. 2012;21:836–847. doi: 10.1016/j.ccr.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lesina M, et al. Stat3/Socs3 activation by IL-6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456–469. doi: 10.1016/j.ccr.2011.03.009. [DOI] [PubMed] [Google Scholar]
- 34.Daniluk J, et al. An NF-kappaB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J Clin Invest. 2012;122:1519–1528. doi: 10.1172/JCI59743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun X, et al. CXCL12/CXCR4/CXCR7 chemokine axis and cancer progression. Cancer Metastasis Rev. 2010;29:709–722. doi: 10.1007/s10555-010-9256-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shakir M, et al. The chemokine receptors CXCR4/CXCR7 and their primary heterodimeric ligands CXCL12 and CXCL12/high mobility group box 1 in pancreatic cancer growth and development: finding flow. Pancreas. 2015;44:528–534. doi: 10.1097/MPA.0000000000000298. [DOI] [PubMed] [Google Scholar]
- 37.Singh AP, et al. CXCL12/CXCR4 protein signaling axis induces sonic hedgehog expression in pancreatic cancer cells via extracellular regulated kinase- and Akt kinase-mediated activation of nuclear factor kappaB: implications for bidirectional tumor-stromal interactions. J Biol Chem. 2012;287:39115–39124. doi: 10.1074/jbc.M112.409581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feig C, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110:20212–20217. doi: 10.1073/pnas.1320318110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705–716. doi: 10.1016/j.immuni.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Varol C, et al. Macrophages: development and tissue specialization. Annu Rev Immunol. 2015;33:643–675. doi: 10.1146/annurev-immunol-032414-112220. [DOI] [PubMed] [Google Scholar]
- 41.Chang JH, et al. Role of immune cells in pancreatic cancer from bench to clinical application: An updated review. Medicine (Baltimore) 2016;95:e5541. doi: 10.1097/MD.0000000000005541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liou GY, et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J Cell Biol. 2013;202:563–577. doi: 10.1083/jcb.201301001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ruffell B, et al. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012;33:119–126. doi: 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ireland L, et al. Chemoresistance in Pancreatic Cancer Is Driven by Stroma-Derived Insulin-Like Growth Factors. Cancer Res. 2016;76:6851–6863. doi: 10.1158/0008-5472.CAN-16-1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weizman N, et al. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene. 2014;33:3812–3819. doi: 10.1038/onc.2013.357. [DOI] [PubMed] [Google Scholar]
- 46.Seifert L, et al. Radiation Therapy Induces Macrophages to Suppress T-Cell Responses Against Pancreatic Tumors in Mice. Gastroenterology. 2016;150:1659–1672 e1655. doi: 10.1053/j.gastro.2016.02.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nielsen SR, et al. Macrophage-secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol. 2016;18:549–560. doi: 10.1038/ncb3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beatty GL, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu Y, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74:5057–5069. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clark CE, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67:9518–9527. doi: 10.1158/0008-5472.CAN-07-0175. [DOI] [PubMed] [Google Scholar]
- 51.Ene-Obong A, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145:1121–1132. doi: 10.1053/j.gastro.2013.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Daley D, et al. gammadelta T Cells Support Pancreatic Oncogenesis by Restraining alphabeta T Cell Activation. Cell. 2016;166:1485–1499 e1415. doi: 10.1016/j.cell.2016.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut. 2017;66:124–136. doi: 10.1136/gutjnl-2016-312078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim SO, et al. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell. 2016;30:925–939. doi: 10.1016/j.ccell.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pergamo M, Miller G. Myeloid-derived suppressor cells and their role in pancreatic cancer. Cancer Gene Ther. 2016 doi: 10.1038/cgt.2016.65. [DOI] [PubMed] [Google Scholar]
- 56.Ugel S, et al. Tumor-induced myeloid deviation: when myeloid-derived suppressor cells meet tumor-associated macrophages. J Clin Invest. 2015;125:3365–3376. doi: 10.1172/JCI80006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tassi E, et al. Carcinoembryonic antigen-specific but not antiviral CD4+ T cell immunity is impaired in pancreatic carcinoma patients. J Immunol. 2008;181:6595–6603. doi: 10.4049/jimmunol.181.9.6595. [DOI] [PubMed] [Google Scholar]
- 58.Demols A, et al. CD4(+)T cells play an important role in acute experimental pancreatitis in mice. Gastroenterology. 2000;118:582–590. doi: 10.1016/s0016-5085(00)70265-4. [DOI] [PubMed] [Google Scholar]
- 59.Zhang Y, et al. CD4+ T lymphocyte ablation prevents pancreatic carcinogenesis in mice. Cancer Immunol Res. 2014;2:423–435. doi: 10.1158/2326-6066.CIR-14-0016-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McAllister F, et al. Oncogenic Kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell. 2014;25:621–637. doi: 10.1016/j.ccr.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang Y, et al. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS One. 2014;9:e91551. doi: 10.1371/journal.pone.0091551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Folias AE, et al. Aberrant innate immune activation following tissue injury impairs pancreatic regeneration. PLoS One. 2014;9:e102125. doi: 10.1371/journal.pone.0102125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rao R, et al. Interleukin 17-producing gammadeltaT cells promote hepatic regeneration in mice. Gastroenterology. 2014;147:473–484 e472. doi: 10.1053/j.gastro.2014.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xue J, et al. Aryl hydrocarbon receptor regulates pancreatic IL-22 production and protects mice from acute pancreatitis. Gastroenterology. 2012;143:1670–1680. doi: 10.1053/j.gastro.2012.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xue J, et al. Aryl Hydrocarbon Receptor Ligands in Cigarette Smoke Induce Production of Interleukin-22 to Promote Pancreatic Fibrosis in Models of Chronic Pancreatitis. Gastroenterology. 2016;151:1206–1217. doi: 10.1053/j.gastro.2016.09.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pylayeva-Gupta Y, et al. IL35-Producing B Cells Promote the Development of Pancreatic Neoplasia. Cancer Discov. 2016;6:247–255. doi: 10.1158/2159-8290.CD-15-0843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gunderson AJ, et al. Bruton Tyrosine Kinase-Dependent Immune Cell Cross-talk Drives Pancreas Cancer. Cancer Discov. 2016;6:270–285. doi: 10.1158/2159-8290.CD-15-0827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oberg HH, et al. Novel bispecific antibodies increase gammadelta T-cell cytotoxicity against pancreatic cancer cells. Cancer Res. 2014;74:1349–1360. doi: 10.1158/0008-5472.CAN-13-0675. [DOI] [PubMed] [Google Scholar]
- 69.Neesse A, et al. Stromal biology and therapy in pancreatic cancer: a changing paradigm. Gut. 2015;64:1476–1484. doi: 10.1136/gutjnl-2015-309304. [DOI] [PubMed] [Google Scholar]
- 70.Whatcott CJ, et al. Desmoplasia in Primary Tumors and Metastatic Lesions of Pancreatic Cancer. Clin Cancer Res. 2015;21:3561–3568. doi: 10.1158/1078-0432.CCR-14-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Erez N, et al. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17:135–147. doi: 10.1016/j.ccr.2009.12.041. [DOI] [PubMed] [Google Scholar]
- 72.Neesse A, et al. Stromal biology and therapy in pancreatic cancer. Gut. 2011;60:861–868. doi: 10.1136/gut.2010.226092. [DOI] [PubMed] [Google Scholar]
- 73.Rhim AD, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–747. doi: 10.1016/j.ccr.2014.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sherman MH, et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80–93. doi: 10.1016/j.cell.2014.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.DuFort CC, et al. Mounting Pressure in the Microenvironment: Fluids, Solids, and Cells in Pancreatic Ductal Adenocarcinoma. Gastroenterology. 2016;150:1545–1557 e1542. doi: 10.1053/j.gastro.2016.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aiello NM, et al. Metastatic progression is associated with dynamic changes in the local microenvironment. Nat Commun. 2016;7:12819. doi: 10.1038/ncomms12819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Costa-Silva B, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–826. doi: 10.1038/ncb3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kruger A. Premetastatic niche formation in the liver: emerging mechanisms and mouse models. J Mol Med (Berl) 2015;93:1193–1201. doi: 10.1007/s00109-015-1342-7. [DOI] [PubMed] [Google Scholar]
- 79.Diakopoulos KN, et al. Impaired autophagy induces chronic atrophic pancreatitis in mice via sex- and nutrition-dependent processes. Gastroenterology. 2015;148:626–638 e617. doi: 10.1053/j.gastro.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 80.Antonucci L, et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci U S A. 2015;112:E6166–6174. doi: 10.1073/pnas.1519384112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dumartin L, et al. ER stress protein AGR2 precedes and is involved in the regulation of pancreatic cancer initiation. Oncogene. 2016 doi: 10.1038/onc.2016.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Guo JY, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ying H, et al. Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell. 2012;149:656–670. doi: 10.1016/j.cell.2012.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tape CJ, et al. Oncogenic KRAS Regulates Tumor Cell Signaling via Stromal Reciprocation. Cell. 2016;165:910–920. doi: 10.1016/j.cell.2016.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sousa CM, et al. Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature. 2016;536:479–483. doi: 10.1038/nature19084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Philip B, et al. A high-fat diet activates oncogenic Kras and COX2 to induce development of pancreatic ductal adenocarcinoma in mice. Gastroenterology. 2013;145:1449–1458. doi: 10.1053/j.gastro.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Incio J, et al. Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discov. 2016;6:852–869. doi: 10.1158/2159-8290.CD-15-1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Meyer KA, et al. Adipocytes promote pancreatic cancer cell proliferation via glutamine transfer. Biochem Biophys Rep. 2016;7:144–149. doi: 10.1016/j.bbrep.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Flint TR, et al. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Antitumor Immunity. Cell Metab. 2016;24:672–684. doi: 10.1016/j.cmet.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Greco SH, et al. TGF-beta Blockade Reduces Mortality and Metabolic Changes in a Validated Murine Model of Pancreatic Cancer Cachexia. PLoS One. 2015;10:e0132786. doi: 10.1371/journal.pone.0132786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Khorana AA, Fine RL. Pancreatic cancer and thromboembolic disease. Lancet Oncol. 2004;5:655–663. doi: 10.1016/S1470-2045(04)01606-7. [DOI] [PubMed] [Google Scholar]
- 92.Varki A. Trousseau’s syndrome: multiple definitions and multiple mechanisms. Blood. 2007;110:1723–1729. doi: 10.1182/blood-2006-10-053736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Qi Q, et al. Hyperfibrinogen Is Associated With the Systemic Inflammatory Response and Predicts Poor Prognosis in Advanced Pancreatic Cancer. Pancreas. 2015;44:977–982. doi: 10.1097/MPA.0000000000000353. [DOI] [PubMed] [Google Scholar]
- 94.Davalos D, Akassoglou K. Fibrinogen as a key regulator of inflammation in disease. Semin Immunopathol. 2012;34:43–62. doi: 10.1007/s00281-011-0290-8. [DOI] [PubMed] [Google Scholar]
- 95.Kitamura T, et al. Immune cell promotion of metastasis. Nat Rev Immunol. 2015;15:73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Connolly MK, et al. Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. J Leukoc Biol. 2010;87:713–725. doi: 10.1189/jlb.0909607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Seubert B, et al. Tissue inhibitor of metalloproteinases (TIMP)-1 creates a premetastatic niche in the liver through SDF-1/CXCR4-dependent neutrophil recruitment in mice. Hepatology. 2015;61:238–248. doi: 10.1002/hep.27378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Grunwald B, et al. Pancreatic Premalignant Lesions Secrete Tissue Inhibitor of Metalloproteinases-1, Which Activates Hepatic Stellate Cells Via CD63 Signaling to Create a Premetastatic Niche in the Liver. Gastroenterology. 2016;151:1011–1024 e1017. doi: 10.1053/j.gastro.2016.07.043. [DOI] [PubMed] [Google Scholar]
- 100.McAllister SS, Weinberg RA. The tumour-induced systemic environment as a critical regulator of cancer progression and metastasis. Nat Cell Biol. 2014;16:717–727. doi: 10.1038/ncb3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Linkermann A, et al. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14:759–767. doi: 10.1038/nri3743. [DOI] [PubMed] [Google Scholar]
- 103.Vesely MD, et al. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 104.Mace TA, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73:3007–3018. doi: 10.1158/0008-5472.CAN-12-4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hwang RF, et al. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008;68:918–926. doi: 10.1158/0008-5472.CAN-07-5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Richards KE, et al. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene. 2016 doi: 10.1038/onc.2016.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Martinez-Bosch N, et al. Galectin-1 drives pancreatic carcinogenesis through stroma remodeling and Hedgehog signaling activation. Cancer Res. 2014;74:3512–3524. doi: 10.1158/0008-5472.CAN-13-3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tang D, et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int J Cancer. 2012;130:2337–2348. doi: 10.1002/ijc.26290. [DOI] [PubMed] [Google Scholar]
- 109.Leca J, et al. Cancer-associated fibroblast-derived annexin A6+ extracellular vesicles support pancreatic cancer aggressiveness. J Clin Invest. 2016;126:4140–4156. doi: 10.1172/JCI87734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Froeling FE, et al. Retinoic acid-induced pancreatic stellate cell quiescence reduces paracrine Wnt-beta-catenin signaling to slow tumor progression. Gastroenterology. 2011;141:1486–1497. 1497 e1481–1414. doi: 10.1053/j.gastro.2011.06.047. [DOI] [PubMed] [Google Scholar]