

Preface
The respiratory immune response consists of multiple tiers of cellular responses that are engaged in a sequential manner in order to control infections. Stepwise engagement of effector functions with progressively increasing host fitness costs limits tissue damage. In addition, specific mechanisms are in place to promote disease tolerance in response to respiratory infections. Environmental factors, obesity and the ageing process can alter the efficiency and regulation of this tiered response, increasing pathology and mortality as a result. In this Review, we describe the cell types that coordinate pathogen clearance and tissue repair through serial secretion of cytokines, and discuss how the environment and comorbidity influence this response.
Introduction
The respiratory tract performs the crucial function of gas exchange that is necessary for life. The immune system that operates within the respiratory tract must therefore be compatible with this vital function in maintaining open airways at all times. However, the inhaled air is not always innocuous as it contains microbes and environmental particles, some of which can cause respiratory disease if they reach an appropriate niche. These inhalants need to be eliminated quickly by the immune system as failure to do so can lead to inflammatory responses that result in the swelling that closes the airways or infection that can lead to severe pneumonia.
To carry out pathogen containment, the immune response within the respiratory tract follows an ordered, stepwise program of engagement of distinct tiers of defense1. Local sensor cells first detect the invading microorganism. This detection event can trigger cell-intrinsic defense responses that contain the pathogen, lead to secretion of chemoattractants to recruit rapid responder cells such as neutrophils, and alert lung-resident lymphoid cells through the secretion of first order cytokines (Figure 1). The tissue-resident lymphocytes that respond to first order cytokines include innate lymphoid cells (ILCs), innate-like lymphocytes, natural killer (NK) cells and tissue-resident memory T (TRM) cells. These lymphocytes, in turn, transform first order cytokine signals into second order cytokines that recruit and enhance the activation of effector cells that can eliminate the pathogens or expel foreign particles. At each stage of the process, effector mechanisms are also activated that can potentially control the infection and thus prevent activation of subsequent immune responses, limiting the inflammatory damage. This stepwise program of immune defense thereby ensures that the minimum necessary response to a microbe is engaged. While the specific sensors and effector mechanisms vary, this core tiered response applies to both type 1 immune responses and type 2 immune responses as it represents an emerging pattern of immune regulation common to a wide range of pathogens.
Figure 1. A stepwise engagement of tiered responses following respiratory infection.
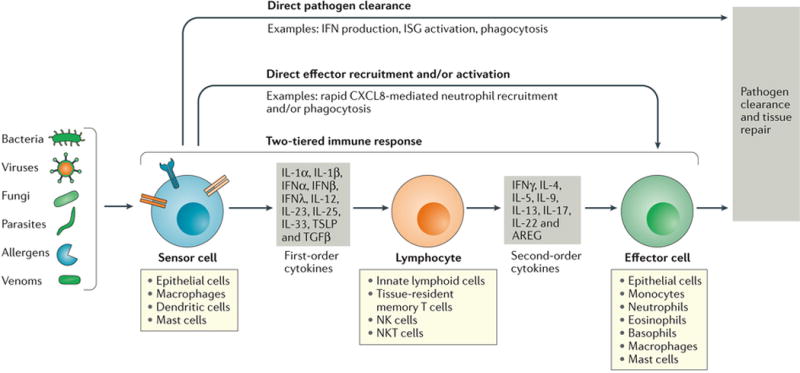
Pathogens and certain noxious compounds are detected by sensor cells located within the respiratory tract. Sensor cells immediately initiate innate immune responses that may be sufficient to clear localized infections. For example, sensor cells may secrete factors such as interferons (IFNs) that lead to pathogen clearance (direct pathogen clearance, top arrow). In some cases, first order cytokines directly recruit effector cells that clear pathogens; for example, CXCL8 mediate recruitment of neutrophils to clear bacteria (direct effector recruitment and/or activation, second arrow from the top). In addition, a two-tiered response can be engaged, in which sensor cells secrete first order cytokines that act on tissue-resident lymphoid cell populations, which integrate these signals and release appropriate second order cytokines. These cytokines in turn recruit and activate effector cells and effector functions specific to the pathogen type, which serve to promote pathogen clearance and tissue repair.
A variety of internal and external influences alter the activation or regulation of these tiers, often with pathological consequences. In particular, external environmental factors, including temperature and pollutants, can change the efficacy of antimicrobial responses, as can internal factors such as lung disease, ageing and obesity. Indeed, people at the extreme ends of the age spectrum as well as obese individuals are more susceptible to respiratory infections2,3, and understanding how these conditions alter immunological functions is vital for the design of therapies that improve clinical outcomes in these at-risk populations.
This Review will focus on a discussion of the different sensor and effector mechanisms of cellular responses that are responsible for antimicrobial host defenses in the conducting airways and lung immediately after a microbial infection. We have divided our discussions of type 1 and type 2 immune responses in order to better explore the shared features of these immune response paradigms. While respiratory immune responses often culminate in the priming of adaptive immune responses, we defer to other reviews on this topic4,5 and instead we focus on the early innate responses that occur within minutes to hours of respiratory challenge. We discuss how these responses are mediated by various cell types and cytokines, how internal and external factors disrupt the nature of these responses, and how pathology arises. Along the way, we highlight topics worthy of future investigation.
Constitutive airway defenses
Airway epithelial cells (AECs) contribute to keeping airways open by continually and actively defending against infection at its earliest stages, averting the need to recruit leukocytes for this task and preventing the accompanying inflammatory response. In addition to well-known roles in barrier defenses, mounting evidence shows that epithelial cells also suppress infection via cell-intrinsic innate immune responses. Further, airway cells serve as the key sensors of infection that inform the tissue-resident lymphocytes through the secretion of first order cytokines.
Airway diameter-dependent barrier defenses
The respiratory system consists of the conducting airways of the upper respiratory tract (nasal cavity, pharynx, larynx) and the lower respiratory tract (trachea, bronchi, bronchioles), and the respiratory zone (alveoli). Four major cell types produce a physical and chemical barrier to infection of the airways, including ciliated cells, mucus-secreting goblet cells, and club cells, which produce antimicrobial compounds and basal cells which, along with club cells, serve as regional progenitor cells to replenish the other cell types6,7. The proportion of each cell type, and the associated defense mechanisms, are compatible with the airway diameter (Figure 2). In the human respiratory tree, ciliated cells and mucus-secreting cells create the barrier defense in larger airways, whereas mucus-secreting cells become less frequent and secretory cells become more predominant in smaller airways. Within the alveoli, alveolar type 1 cells facilitate gas exchange whereas alveolar type 2 cells secrete pulmonary surfactant. Immune defenses are coordinated to airway size, as effective defenses of the large airways, such as a thick mucus layer, can be harmful rather than helpful in small airways. This is evident in diseases such as asthma and chronic obstructive pulmonary disease (COPD), in which mucus cell metaplasia in small airways contributes to airway obstruction8. The relationship between epithelial composition and airway diameter is a consideration when using animal models of respiratory infection; for example, in terms of epithelial composition, a mouse trachea is quite different from a human trachea but similar to a human bronchiole7,9 (Figure 2).
Figure 2. Composition of the airway epithelium varies with airway diameter.
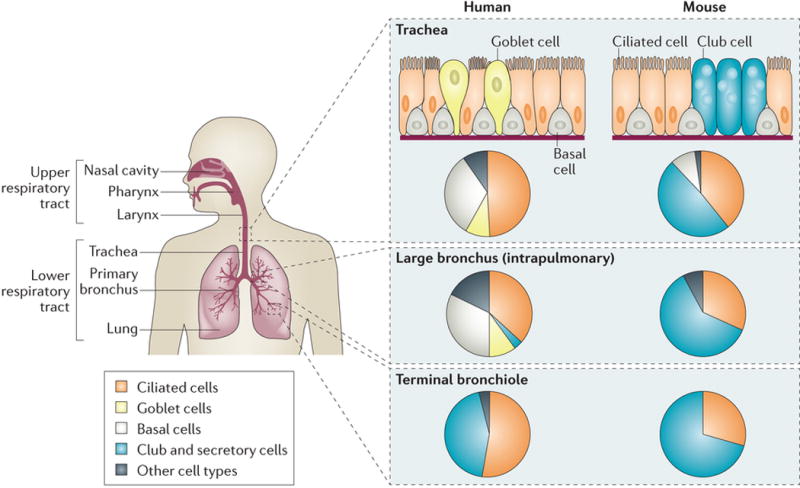
The conducting airways include the air passages from the nasal cavity to the terminal bronchioles. The same major cell types compose the airway epithelium lining in all these parts, but the relative proportion of each cell type varies with the airway diameter, which is consistent with differences in functional requirements. This is also evident when comparing the cellular composition of the human conducting airways with mouse airways. Mucous goblet cells are rare in the mouse airway epithelium beyond the upper airway; similarly, club cells are rare in human airways larger than the terminal bronchioles. The pie charts indicate approximate proportions of the major cell types at different locations of the respiratory system (compiled from references9,154).
Localized sensor responses
When a pathogen manages to enter the airway, its initial detection by sensor cells localized at the site of infection serves as the first tier of defense, promoting innate immune responses to clear limited infections and releasing first order cytokines that alert and arm local tissue-resident lymphocytes (Figures 1). In this section, we describe how local sensor cells detect and respond to infectious agents.
At steady state, the respiratory mucosa maintains its quiescence, in part through the function of alveolar macrophages. Alveolar macrophages reside inside the airway space of the alveoli, and within the mucus of the larger conducting airways. Recent work demonstrates that these tissue-resident macrophages arise from yolk sac progenitor cells distinct from hematopoietic stem cells, and that circulating monocytes contribute minimally to the alveolar macrophage compartment in young mice10,11. Alveolar macrophages constantly receive negative regulatory cues from AECs including CD200, transforming growth factor-β (TGFβ) and interleukin-10 (IL-10) which prevent their activation12. As such, alveolar macrophages function at steady state to clear particulates, apoptotic cells and cellular debris from the airways in order to maintain homeostatic tissue function (Figure 3A). A subset of mouse alveolar macrophages form connexin 43-mediated gap junctions with AECs, and even during LPS-induced inflammation these cells use Ca2+-dependent signaling to rapidly transmit immunosuppressive signals to local AECs and macrophages, helping to constrain consequent inflammation13.
Figure 3. Single and two-tiered responses in type 1 immunity. a.
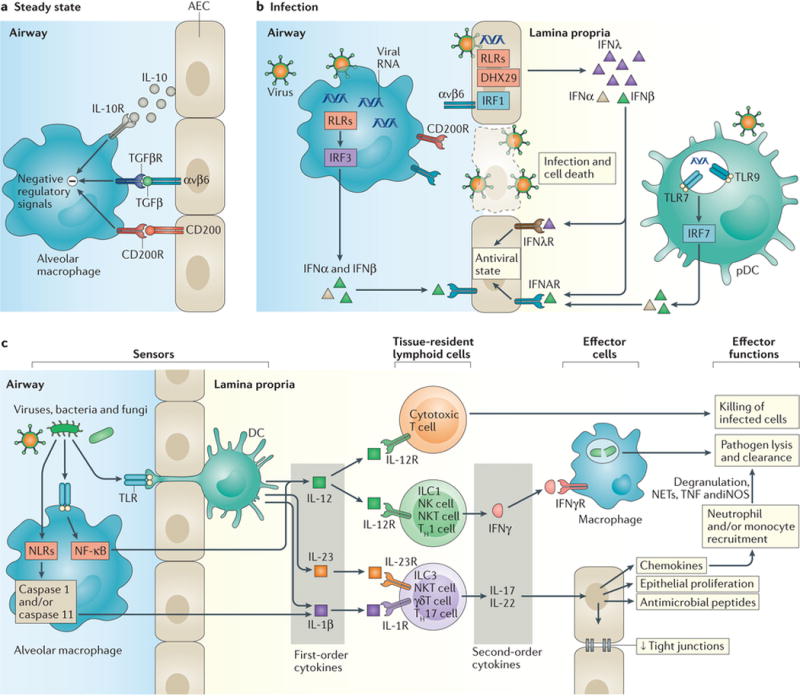
At steady state in the airways, alveolar macrophage activation is suppressed by negative regulatory signals in part delivered by CD200–CD200R and recognition of TGFβ presented by αvβ6 on airway epithelial cells (AECs). b |During infection, disruption of these interactions due to death of AECs enables activation of macrophages. Recognition of viruses by pattern recognition receptors expressed by airway epithelial cells (AECs) leads to secretion of interferon-λ (IFNλ), whereas recognition by endosomal Toll-like receptors (TLRs) in plasmacytoid DCs (pDCs) and cytosolic RIG-I-like receptors (RLRs) and DNA sensors in alveolar macrophages leads to IFNα/β production. These IFNs induce an antiviral state in proximal AECs, inducing IFN-stimulated genes that help constrain viral spread. c | TLRs expressed by alveolar macrophages and DCs that extend trans-epithelial processes enable the recognition of viral, fungal, and bacterial molecules, and bacterial pathogens in the airway leading to the production of first order cytokines including interleukin-12 (IL-12) and IL-23. Additional pathogen recognition via inflammasome activation leads to caspase-1-mediated activation and release of the first order cytokine IL-1β. These first order cytokines act on tissue-resident lymphoid populations of cytotoxic T lymphocytes to enhance direct killing of infected cells, and on innate lymphoid cells (ILCs), natural killer (NK) cells, NK T cells, and T cells to induce the production of appropriate second order cytokines including IFNγ, IL-17, and IL-22. These second order cytokines in turn act on AECs to induce chemokine production, antimicrobial peptide release, and increased proliferation and/or tight junction formation to enhance airway integrity and constrain pathogen spread. Local and chemokine-recruited phagocytes including neutrophils and monocytes are additionally activated by IFNγ, enhancing their phagocytic capabilities and leading to enhanced pathogen lysis and clearance.
Local sensor cells in type 1 immunity
Upon infection with viruses, bacteria, protozoa and fungi, type 1 immune responses are initiated by sensor cells including AECs, alveolar macrophages and dendritic cells (DCs) (Figure 3B). These sensors express pattern recognition receptors (PRRs) including Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), cytosolic DNA sensors and nucleotide oligomerization domain (NOD)-like receptors (NLRs). The specific combination of receptors, downstream signaling molecules, and anti-microbial effector pathways that predominate in AECs differs from those found in other immune cell populations. For example, during the antiviral response of macrophages, DCs, and plasmacytoid DCs (pDCs), secretion of type I interferons (IFNs) predominates. In AECs, type III interferons (lambda IFNs) are often secreted at higher levels than type I IFNs, and the ability to respond to type III IFNs is also epithelial-specific in both humans and mice14,15. In diverse cell types, engagement of RLRs at peroxisomes rather than mitochondrial membranes favors type III IFN induction16. Transcription factor requirements also differ for type III versus type I IFN induction downstream of RLRs. For example, interferon-regulatory factor 1 (IRF1) participates in a unique pathway regulating induction of type III but not type I IFNs16–18, and AECs use IRF1 for synthesis of type III IFNs18. AECs also have unique sensing requirements upstream of RLRs; for example, human AECs require the helicase DHX29 as a co-receptor to sense certain cytosolic nucleic acids, whereas monocyte-derived THP1 cells do not19. Thus, AECs have the ability to serve as specialized pathogen sensors, and they rely on specific regulatory co-factors in the initiation of IFN responses, which act in an autocrine and paracrine fashion to limit viral spread within the AECs.
In addition to AECs, innate leukocytes such as plasmacytoid DCs (pDCs) present in lungs respond to viruses20. Antibody depletion studies in mice show that pDCs are important for IFN production, viral clearance in the lung and limiting immunopathology21,22. The type I and type III IFNs released by pDCs, serve as first order cytokines, inducing the expression of interferon-stimulated genes (ISGs) with direct antiviral activities in proximal cells. IFNs additionally amplify subsequent local IFN production if an infection is not rapidly controlled by the local antiviral response. Alveolar macrophages express a wide range of TLRs and cytosolic sensors including retinoic acid inducible gene I (RIG-I), MDA5, and cyclic GMP–AMP synthetase (cGAS)23. Upon infection, the negative regulatory signals, including CD200 and TGFβ, that normally constrain alveolar macrophage activation are lost (by virtue of epithelial cell death, for example) and alveolar macrophages turn on their PRR signaling machinery12 (Figure 3B). Alveolar macrophages are a major source of type I IFNs during viral infections of the lung24. Thus, these cytokines mediate a single-tiered immune response which does not rely upon additional lymphoid cell amplification or effector cell recruitment in order to control the infection (Figure 3B).
In addition to the single-tiered response, two-tiered immune responses are deployed. Infections by viruses, bacteria, fungi, and protozoa are detected by sensor cells including alveolar macrophages, AECs and DCs (Figure 3C). IFNs secreted by these sensor cells promote the induction of chemokines including CC-chemokine ligand 2 (CCL2) from either the alveolar macrophages or from AECs in mice24, providing a means by which effector cells can be rapidly recruited to sites of infection. Alveolar macrophages additionally produce cytokines and chemokines including IL-6, tumour necrosis factor (TNF), IL-12, IL-23 and CCL3 during infections in response to specific PRR activation25 (Figure 3C).There are multiple DC subsets in the lung at steady state, and each subset appears to mediate distinct roles in innate and adaptive immunity1,26. Within the alveoli and to a lesser extent in the conducting airways, DCs extend trans-epithelial processes that sample antigens not phagocytosed by alveolar macrophages27,28. Upon pathogen detection, mouse lung DCs undergo TLR-dependent secretion of first order cytokines including IL-12 and IL-23 and migrate to draining lymph nodes to prime T cell responses29,30. This migration can depend on the complement anaphylatoxins C3a and C5a31 and NLRP3 inflammasome activation32 in addition to cytokine and chemokine signals. Inflammasome activation in response to virulence activity drives caspase-1-dependent release of IL-1β and/or IL-18 from PRR-activated macrophages and DCs33,34, providing additional first order cytokines signalling the presence of a virulent pathogen (Figure 3C).These first order cytokines act on tissue-resident lymphoid cells, which produce second order cytokines such as IFNγ, IL-17 and IL-22 (Figure 3C). The second order cytokines act on effector cells, which mediate elimination of pathogens.
Local sensor cells in type 2 immunity
Type 2 immunity is activated in response to inhaled allergens, venoms, or invasion by multicellular parasites. In contrast to type 1 immunity, during which PRRs mediate detection of pathogens, type 2 immunity is activated in response to recognition of tissue damage caused by parasites and other features associated with pathogen invasion such as the presence of proteases. Inhaled allergens, toxins, and venoms elicit similar type 2 immune responses. Certain allergens exhibit TLR4-dependent35 or β-glucan-dependent36 induction of first order cytokines in mouse models and in vitro analyses of human cells, respectively, indicating that PRRs may detect certain allergens. Sensors of allergens and helminths in the airways include AECs and mast cells that together coordinate the initial first order cytokines including thymic stromal lymphopoietin (TSLP), IL-25, IL-33 and IL-1β (Figure 4).
Figure 4. Single and two-tiered responses in type 2 immunity. a.
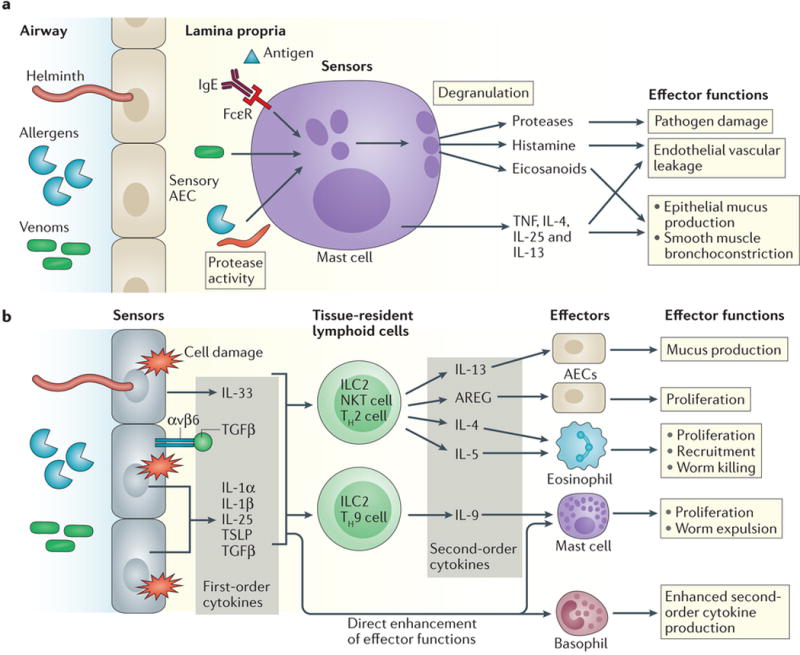
Mast cells can be activated directly in response to certain protease activities, venom proteins homologous to mammalian mast cell-activating proteins, or through antigen-specific IgE-mediated signaling through FcεR, whereupon they can form a single-tiered immune response to helminths and allergens. Activation leads to degranulation and release of proteases, histamine, and eicosanoids (including prostaglandins), as well as the production of certain effector cytokines. These compounds can directly initiate effector mechanisms that can promote worm expulsion but which are also associated with anaphylaxis in severe instances of allergy. b | Cell damage and protease activity from helminth infection or exposure to allergens and venoms leads to the secretion and release of the first order cytokines interleukin-25 (IL-25), TSLP, IL-33, IL-1β, and TGFβ (presented by αvβ6) from sensory airway epithelial cells (AECs). These cytokines in turn act on tissue-resident lymphoid cells including innate lymphoid cells (ILC2s), natural killer T cells, T helper 2 (Th2) cells and Th9 cells to drive secretion of appropriate second order cytokines, which act on mast cells, AECs, basophils, and eosinophils to initiate effector mechanisms aimed at worm expulsion and tissue repair. First order cytokines can also enhance basophil and mast cell recruitment and activation in order to appropriately calibrate the immune response.
Mast cells represent a unique class of sensor cell, in that they are long-lived resident cells that directly execute effector functions that eliminate helminths and allergens from the airway through a single-tiered immune response1 (Figure 4A). Mast cells are activated directly in response to protease activity and via IgE-mediated recognition of antigen, releasing effector molecules including proteases, histamine, and prostaglandins that promote vasodilation and smooth muscle peristalsis37. Although such single-tiered effector functions enable rapid expulsion of parasites from the airway, these functions can also lead to anaphylaxis and death in severe cases. In addition to these classical effector functions, mast cells can secrete first order cytokines including IL-25 (Ref.37), which enhanced by human epithelium-derived TSLP38, enabling these cells to directly amplify type 2 immune responses by acting on tissue-resident lymphocytes (Figure 4B).
Damage to the barrier integrity of the airway epithelium leads to release of alarmins. The best characterized airway alarmin is IL-33, an IL-1 family cytokine that is constitutively expressed and sequestered in the nuclei of alveolar epithelial cells39. In human airways, basal cells are the primary source of IL-33, whereas in mice alveolar type II cells are the predominate producers of IL-33 (Ref40), which underscores certain differences in airway cell function between these species (Figure 2). IL-33 is rapidly released from these cells upon exposure to allergens with protease activity41,42, during necrotic cell death43 or in response to mechanical stress44. In addition to the damage-mediated release of constitutively expressed alarmins, the expression and secretion of TSLP and IL-25 is increased in AECs during type 2 inflammatory responses. Human AECs express and secrete TSLP in response to protease activity, which is partly sensed by protease-activated receptor 2 (PAR-2)45. The expression and secretion of cytokine IL-25 is also induced in AECs in response to allergen exposure46,47, and together these three first order cytokines (IL-33, IL-25, and TSLP) make up the prototypical AEC response to allergens and parasitic infection. In mouse alveoli, alveolar type II epithelial cells are the primary source of IL-33 and TSLP during helminth infection, which is consistent with their role as cytokine producing cells48. More recently, secretion of TGFβ from AECs was shown to be essential during mouse allergic responses by acting on local ILC2s to enhance their proliferation and cytokine secretion49 (Figure 4B).
Lymphocyte responses
The second tier of respiratory immune defenses is mediated by terminally differentiated tissue-resident lymphocytes including innate lymphoid cells (ILCs), NK cells, innate-like lymphocytes including NKT cells, mucosal-associated invariant T (MAIT) cells, epithelial γδ T cells, and tissue-resident memory T (TRM) cells. These cells serve as an intermediary controller50, integrating the cytokine signals from local sensor cells and producing effector cytokines that can recruit effector cell subsets and elicit appropriate responses to clear pathogens. These cells reside within the respiratory tract at the time of infection, and are thus distinct from recruited lymphocytes that arrive as a consequence of adaptive immunity several days later.
Tissue lymphocyte responses in type 1 immunity
ILCs are classified into three broad subsets based on their effector cytokine production and differentiation, and all three populations are found in the lungs of healthy humans51. Group 1 ILCs (ILC1s) are associated with the production of effector cytokines related to a Th1 response to viral and intracellular bacterial infections (Figure 3C). Group 3 ILCs (ILC3s) produce effector cytokines similar to those of Th17 cells in response to extracellular bacteria and fungi (Figure 3C). Pulmonary IL-17 production is dependent upon IL-23 signaling in mice25, and lung ILC3s are a main source of both IL-17 and IL-22 in response to IL-23 (Ref.52).
Natural killer (NK) cells are related to ILC1s and are rapidly recruited to the lung and activated upon influenza infection53. These cells are cytotoxic, and they recognize and lyse cells infected by viruses and intracellular bacteria. In addition to this cell-mediated killing, NK cells are a major source of cytokines during certain respiratory infections. NK cell activation by IL-12 leads to IFNγ production54, and in certain mouse models of infection NK cells also express IL-23R and are the dominant IL-22 producers55, and thereby help restoring the integrity of the epithelial barrier (Figure 3C).
Invariant NK T (iNKT) cells consist of multiple subsets of CD1d-restricted lipid responsive lymphocytes that serve as major producers of second order cytokines. Studies in other organ systems exhibit a highly diverse set of cytokine responses from NKT cell populations56, and these results extend to pulmonary NKT cells. For example, in mice iNKT cells express the IL-12 receptor and undergo IL-12-dependent activation and IFNγ secretion in the lungs during bacterial infection57, and they can secrete IL-22 and IL-17 in response to IL-1β and IL-23 during influenza infection58.
Studies in mice demonstrate that localized innate-like lymphocytes are also central to early and effective airway immunity. Lung γδ T cells secrete IL-17 in response to IL-1 and IL-23 signaling59, and they are an early and prominent source of IL-17 during Mycobacterium tuberculosis infection60. Conversely γδ T cells stimulated with IL-1 and IL-12 can produce IFNγ61, demonstrating that specific cytokine milieu elicits distinct effector cytokine responses from the same cell type.
During recall responses lung TRM cells protect against infection62, and memory Th1 and Th17 cells are major sources of IFNγ and IL-17/IL-22, respectively. Of note, adaptive T cells that make up the local TRM cell population represent a special case1 because these cells needed to go through an additional step of antigen-specific expansion and differentiation before being able to serve in their capacity to secrete second order cytokines in the tissue.
There are clear functional similarities between these different lymphoid cell populations in terms of the types of second order cytokines they can release. Even so, these cells retain non-redundant functions, such as the cytotoxic properties of NK cells, the ability of iNKT cells to respond to specific lipid species and the ability of TRM cells to respond to specific antigen peptides. Hence, although each lymphoid cell type can respond to first order cytokines, some of these cells can also respond to specific stimuli in order to mediate efficient antimicrobial defense.
Tissue lymphocyte responses in type 2 immunity
The first order cytokines released in response to helminth, venom or allergen detection direct the activation and function of tissue-resident lymphocytes, including NKT cells, ILC2s and TRM cells to produce appropriate second order cytokines, such as IL-4, IL-5, IL-13, amphiregulin (AREG) and IL-9 (Figure 4B). DCs also respond to second order cytokines (Box 1).
ILC2s are found in the lung at steady state in the absence of infection63. They proliferate and produce the canonical type 2 cytokines IL-5 and IL-13 during both helminth infections64 and allergic responses65 in mice. ILC2s constantly survey the airway epithelium and are highly responsive to AEC-derived cytokines including IL-33, IL-25, TSLP, and TGFβ49,64,65. While both IL-33 and IL-25 can promote ILC2 proliferation, studies in mouse models of airway hyperresponsiveness suggest that IL-33 is a more potent activator of these cells in vivo than IL-25 66, and that IL-25 activates a subset of innate lymphoid precursor cells that can be induced to differentiate into ILC2 or ILC3 cells67,68. TGFβ, but not IL-33, enhances ILC2 basal migration rates, priming ILC2s to respond to additional chemotactic stimuli in the airways49. Human ILC2s are also responsive to IL-1α/β, and IL-1 signaling enhances the expression of the receptors for IL-33, IL-25, and TSLP; thus, IL-1 is important for priming and amplifying the responsiveness of ILC2s to first order cytokines in the context of an unresolved infection69. ILC2s have also been implicated as a major source of IL-9 during lung stage helminth infections in mice as autocrine IL-9 signaling through IL-9R on ILC2s is necessary for sustained ILC2 function and epithelial repair48,70. Together these results suggest that the combination of AEC-derived first order cytokines in the lung specify ILC2 function (Figure 4B).
NKT cells are also responsive to first order cytokines, and thereby contribute to the coordination of type 2 effector responses. Mouse IL-17RB+ CD4+ NKT cells produce IL-13 in response to IL-25 (Ref. 71), and these cells produce both IL-4 and IL-13 in models of allergen-induced asthma72 and ozone-induced airway hyperresponsiveness73. TSLP and IL-33 similarly enhance NKT activation and cytokine secretion74,75. In the mouse lung, Vα14-bearing NKT cells reside in the intravascular space of the microvasculature, and respond to the lipid antigen α-galactosyl ceramide presented on CD1d-restricted to secrete IL-4 and recruit eosinophils76.
In the context of recurrent allergen exposure or infection, adaptive T cell populations in the airways have a role distinct from that of ILC2 and NKT cells. Memory Th2 cells can rapidly produce the effector cytokines IL-4, IL-5, and IL-13 in response to allergens and helminth infections, owing to their exquisite sensitivity to the small amount of antigens presented by local antigen-presenting cells (APCs). Th9 cells rapidly produce IL-9 in the lungs and have been implicated in allergic airway responses77 and IL-9 production is necessary for maximal allergen-induced mast cell accumulation and airway inflammation in mice78,79. Recently, a distinct CD4+ T cell subset secreting IL-21 was identified in the mouse lung in the context of allergic airway inflammation, and this IL-21 production supported development of eosinophilia80 (Figure 4B).
Adaptive T cells can also respond to first order cytokines independently of antigens. TSLP and IL-25 signaling enhances IL-9 production by Th9 cells78,81 and TSLP, IL-25, and IL-33 promote effector cytokine production by Th2 cells82,83, which demonstrates that first order cytokines from the AECs induce T cell-mediated cytokine responses. In addition to its role in stimulating mast cell proliferation, IL-9 has also been implicated in supporting ILC2 survival70, and IL-21 indirectly supports ILC2 cytokine secretion80 suggesting that paracrine regulation between different lymphocyte types helps to coordinate an optimal type 2 effector response (Figure 4B).
Effector responses
The signals from tissue-resident lymphoid cell populations serve to recruit and/or activate effector cell populations. These effector cells are the primary mode of pathogen clearance during serious infections when previous tiers of defenses have failed.
Effector responses in type 1 immunity
Some effector responses are directly initiated by sensors, such as during local control of viral infection within the airway epithelium by cell intrinsic defenses or localized interferon responses, and as described for pDCs (Figure 1). First order cytokines can also directly enhance effector responses; for example, cytotoxic T cells exhibit improved priming and cytotoxicity in response to DC-derived IL-12 (Ref84). Other responses require additional input from second order cytokines secreted by the tissue-resident lymphoid cells, which stimulate effector cell types in the tissue to promote pathogen clearance. IFNγ, IL-17 and IL-22 act on tissue macrophages to enhance phagocytosis of infected cells and infectious agents, and on AECs to produce antimicrobial peptides85, strengthen tight junctions, and induce proliferation and repair55,85 (Figure 3C). In addition to these responses, the recruitment of inflammatory cell types — particularly monocytes and neutrophils — is essential for the effective resolution of viral, bacterial and fungal respiratory infections.
Inflammatory monocytes are recruited to the lungs of infected mice in response to CCL7 and CCL2 (Ref.24,86) and play a central role in pathogen clearance. Recruited monocytes can serve as a major secondary source of cytokines including type I IFNs, TNF and IL-6, and their recruitment enhances subsequent neutrophil recruitment to the lung87,88. Once these inflammatory monocytes reach the site of infection they can differentiate either macrophages or DCs89, reinforcing sensor cell functions in the airway and increasing the local concentration of activated phagocytes and APCs86. Mouse monocytes can replenish alveolar macrophage populations by first differentiating into CD11c+ macrophages in the lung parenchyma and then migrating into the alveolar lumen90. Monocyte-derived macrophages and DCs also serve direct innate roles in pathogen elimination, for example monocyte-derived DCs produce TNF and/or iNOS89 and can directly engulf spores during respiratory fungal infection88 (Figure 3C).
Neutrophils arrive to the lung interstitium within minutes of high dose bacterial challenge, which is in some cases a response to the release of preexisting chemokine stores91. Pathogen sensing by AECs can also directly trigger AEC production of high levels of neutrophil chemoattractants. Thus, these cells can be directly recruited independent of lymphocyte-mediated signal amplification (Figure 1). In addition, AEC chemokine production can be amplified by the second order cytokines IL-17 and IFNγ92, allowing for additional neutrophil recruitment during later stages of infection. Movement of neutrophils into sites of infection is dependent upon their ability to secrete matrix metalloproteinase 9 (MMP9)93, which cleaves the collagen of the extracellular matrix (ECM). Cytokines such as TNF directly enhance MMP9 secretion from mouse neutrophils93, and thereby provide a regulatory checkpoint that prevents these cells from entering non-inflamed tissues.
Activated neutrophils clear fungi, bacteria and viruses through both phagocytosis of microbes and degranulation-mediated release of reactive oxygen species and antimicrobial peptides (Figure 3C). In addition, neutrophils release neutrophil extracellular traps (NETs)94, which contain DNA, histones, proteases and antimicrobial proteins. NET production is evident in the lungs of virally infected mice95 and is associated with optimal clearance of bacterial infections during septicaemia94 and fungal infections in lung tissue96. However, the toxic substances released by neutrophils can lead to irreparable damage and even death. Therefore, tight regulation of neutrophil responses is essential to prevent life-threatening immunopathology.
Effector responses in type 2 immunity
Type 2 immune effector responses can be mediated in either single- or two-tiered immune responses. As discussed above, resident mast cells can mediate a single-tiered response by secreting mediators that directly act on local effectors to induce parasite expulsion (Figure 4A). In the context of the two-tiered immune response, second order cytokine signals from innate-like lymphocytes initiate and amplify effector responses from AECs, basophils, eosinophils and macrophages in a coordinated effort to expel parasitic worms and restore damaged tissues (Figure 4B).
In addition to their roles as primary sensors of infection, AECs mediate effector functions that contribute to resolution of disease (Figure 4B). Second order cytokines from lymphoid cells promote type 2 effector responses including airway cell replication and remodeling. IL-13 signaling in AECs has several effects, such as induced production of CCL11 in mouse alveolar and bronchial epithelial cells97, and enhanced mucus production from bronchial cells98. Repair of the airway epithelium is important for effective resolution of anti-helminth responses, and ILC2s have a central role in this process by producing IL-5, IL-13, and AREG which promote epithelial regrowth48,70,99.
Eosinophils are largely absent from the lung at baseline, and are recruited in response to IL-5 derived from ILC2, Th2 cells and NKT cells, and CCL11 derived from AECs100 (Figure 4B). Human eosinophils express the TSLP receptor, and TSLP signaling enhances their chemotaxis and survival, supporting eosinophilic responses in the face of sustained tissue damage101. Eosinophils can thus be recruited in response to either sensor- or lymphoid cell-derived signals, and once recruited are responsive to additional signals produced by local sensors, which serves as a checkpoint to minimize their prolonged activation. Eosinophil degranulation results in the release of compounds toxic to helminths including major basic protein (MBP) and eosinophil cationic protein (ECP), as well as additional release of IL-4 (Ref.102) to induce and enhance Th2 cell and B cell responses to infection.
Basophils are recruited to the airways during allergen exposure and helminth infections, where they produce IL-4 and interact with local memory T cells to enhance Th2 cell-mediated production of IL-5 and IL-13 and thereby potentiate airway inflammation103,104 (Figure 4B). In addition, basophils can be stimulated by the first order cytokines IL-33 and TSLP directly and by IgE105. Both human and mouse basophils proliferate and increase expression of receptors including IL-33R in response to TSLP106, and IL-33 signaling enhances basophil cytokine production and consequent airway inflammation75,104, and thereby provide a direct pathway for damaged AECs to mediate basophil responses. T cell-derived IL-3 can also promote basophil activation and proliferation, but even in the absence of IL-3 signaling TSLP is sufficient to activate basophils directly106,107. Basophils play a protective role in the context of recurrent N. brasiliensis infections in mice108, and basophil-derived IL-4 can also act on ILC2s, enhancing the production of IL-9, IL-13, and CCL11 to increase mast cell recruitment, eosinophilia, and inflammation104.
Mast cell-derived chymase plays a role in the degradation of alarmins including IL-33 in vivo in mice109 allowing for the rapid termination of type 2 responses. In addition, mast cells can constrain airway inflammation in response to papain in mice owing to their ability to produce IL-2 and enhance Treg cell proliferation110. Interestingly, mast cell-derived proteases can also cleave IL-33 into more potent forms to enhance ILC2 activation111, and whether these cells amplify or constrain the magnitude of the immune response is probably context dependent. Thus, both effector cytokines and cell types can amplify the immune response as needed, but can also initiate negative feedback mechanisms that help to terminate immune responses when they are no longer required.
Disease tolerance in the airway
Disease tolerance is an important host defense pathway that centers on improving the ability of the host to tolerate tissue damage, and is independent from the resistance mechanisms that target pathogen elimination112. This concept is distinct from the classical immunological paradigms of central and peripheral immunological tolerance. Disease tolerance is an essential survival strategy against respiratory infections, and the mechanisms by which it is mediated are only beginning to be understood. Enhancing disease tolerance, in addition to antimicrobial host defenses, represents an important alternative strategy for future therapeutic interventions.
The mechanisms of disease tolerance are not well understood, but recent studies highlight emerging principles. ILC2s restore airway epithelial integrity after severe influenza A virus (IAV) infection via AREG secretion without altering viral load99 (Figure 4B). Administering AREG to mice suffering from a lethal IAV bacterial coinfection can promote host survival without affecting pathogen burden113, which demonstrates that host resistance to infection and survival can be uncoupled by tolerance mechanisms. IL-22 is similarly protective against IAV-associated epithelial cell death and damage without altering viral burden both in vitro and in vivo55,58 (Figure 3C).
Tissue repair responses are not the only pathway to disease tolerance. For example, reducing the magnitude of inflammatory responses can improve survival without compromising antimicrobial host defenses. In mice lacking the TLR and RLR signaling machinery necessary for IAV detection, disruption of caspase-1/11 activation can reverse IAV-associated lung tissue damage and mortality without altering pathogen burden95. Caspase-1 is also deleterious during later stages of pneumonic plague without offering an advantage in bacterial clearance114, and is responsible for causing CD4+ T cell loss in HIV-1 infection without affecting viral load115. In mice disease tolerance against IAV is additionally bolstered by glucose consumption during infection through a mechanism involving prevention of cell death downstream of IFN signaling in the hind brain116. Future identification of other disease tolerance mechanisms is needed for the design of novel anti-inflammatory agents.
Modifiers of respiratory immunity
The stepwise engagement of the immune response is the template for respiratory immunity, but many internal and external factors can disrupt this otherwise ordered process (Figure 5). Environmental and metabolic factors as well as the ageing process can change the way cells in the airways are activated or respond to stimuli, often to a deleterious end. The IFN response in particular is susceptible to many such influences, and given its important effector and regulatory roles in respiratory immunity this disruption probably has substantial consequences although these are not yet fully understood. For example, asthma is associated with a diminished IFN response to rhinovirus infection but the precise relationship between this finding and disease pathogenesis is not yet known (Box 2).
Figure 5. Internal and external factors increase respiratory disease susceptibility.
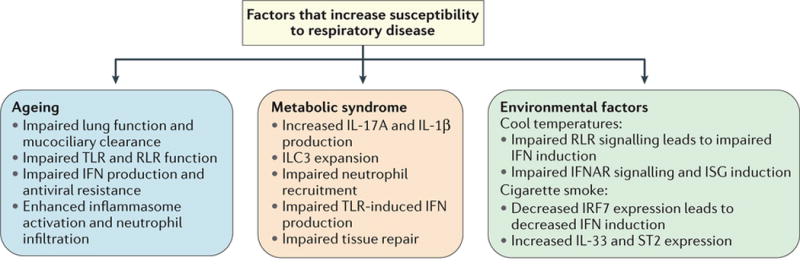
Both internal and external influences can alter early respiratory immune responses. External factors like cold temperatures and cigarette smoke can both impair sensor cell functionality and antiviral responses. Chronic conditions such as ageing and metabolic syndrome can also have profound effects, altering the functions of sensors, lymphoid cells, and effector responses, thus disrupting various stages of the tiered respiratory immune response. Together these factors contribute to the increases in respiratory morbidity and mortality observed in populations affected by these factors.
External factors that alter respiratory immune responses
The mucosal surface of the respiratory epithelium is exposed to the external environment with every breath, which occur approximately 20 times per minute. Therefore the characteristics of the inhaled air, including its temperature, can potentially impact the biology of the surface epithelium. Changing the ambient temperature of AECs profoundly impacts their antiviral defense responses117,118. Rhinovirus, which causes the common cold, is known to replicate more robustly at nasal cavity temperatures of 33–35°C than at core body temperature of 37°C. Studies in mouse primary airway cells showed that recognition of rhinovirus via the RLR pathway and signaling downstream of the type I IFN receptor are more effective at 37°C compared with at 33°C117. This phenomenon is largely responsible for the temperature-dependence of rhinovirus replication in mouse AECs. By comparison, human bronchial epithelial cells have relatively attenuated IFN responses during rhinovirus infection. However, other double-stranded RNA-dependent defense responses, including apoptosis and RNAseL activity, can control rhinovirus replication in human epithelial cells, and these responses are also diminished at nasal cavity temperature compared with body temperature118. These findings suggest that cooler temperatures in the nasal cavity — which is further cooled when the inhaled air temperature is low — create a niche in which rhinovirus, and perhaps other respiratory viruses, can evade natural antiviral defense mechanisms.
In addition to temperature, evidence indicates that inhaled pollutants impact the airway innate immune responses to viral infection, with many studies focusing specifically on the effects of cigarette smoke. In a study of nasal epithelial cells from human smokers, the IFN response to IAV was dampened, and this correlated with diminished expression of the IFN-inducing transcription factor IRF7 and hypermethylation of the IRF7 promoter119. In vitro studies of AECs have also shown that exposure to chemicals in cigarette smoke diminishes levels of IRF7 and other key signaling molecules that mediate IFN responses120. These effects may partially underlie the enhanced susceptibility of smokers to viral respiratory infections. A recent study in mice also points to a possible role for IL-33 in exacerbation of respiratory viral infection in smokers. Exposure of mice to cigarette smoke led to increased expression of IL-33 in bronchial and alveolar epithelial cells, and increased expression of the IL-33 receptor ST2 on macrophages and NK cells, thereby setting the stage for an exaggerated inflammatory response following a viral challenge121. These findings highlight how environmental factors can alter the regulation of the tiered structure of the respiratory immune response to pathological effect.
Impact of ageing on lung immunity
The ageing process has a wide range of disruptive effects on both adaptive and innate immunity, and older adults (over the age of 65) are more susceptible to respiratory pathogens, which account for ~90% of the mortality associated with influenza and pneumonia in the United States each year3. Older individuals exhibit marked changes in lung physiology including reduced respiratory muscle functionality and impaired mucociliary clearance, which reduce the ability to eliminate pathogens from the airway122 (Figure 5). Ageing also has broad effects on innate immunity, with older individuals exhibiting impairments in signaling through many TLRs and a skewing in hematopoiesis that favours myeloid cells123. On the other hand, ageing is also generally associated with increased baseline levels of inflammation.
In aged mice, increased prostaglandin D2 expression in the lung suppresses DC migration to draining lymph nodes, which leads to impaired development of an effective immune response124. Lung NK cell cytotoxicity and IFNγ secretion are decreased in mouse models of ageing and influenza125. Upon LPS stimulation aged mice exhibit increased pulmonary CXCL1 and CXCL2 levels and neutrophil infiltration, which may be linked to increased IL-1β production126. In addition, ageing impairs the ability of human peripheral blood monocytes to produce IFNs upon IAV infection or RIG-I stimulation95, and thereby impairing host resistance mechanisms within these cells. The combination of impaired antiviral resistance (due to IFN defects) combined with enhanced inflammasome responses and neutrophil activation results in lethal consequences following IAV infection95,127.
Impact of metabolic disease on lung immunity
Metabolic syndrome increases the risk for respiratory diseases; morbidly obese humans are more likely to require hospitalization2 and have an increased risk of developing asthma after IAV infection128 (Figure 5). In general obesity is associated with increased levels of inflammatory M1 macrophages, CD8+ T cells, and decreased levels of anti-inflammatory M2 macrophages or Treg cells. Furthermore, in the mouse lungs a high fat diet is associated with increased numbers of monocytes and CD3+ lymphocytes even in the absence of infection129. Mice on a high fat diet exhibit increased airway hyperactivity due to an expansion of ILC3-like cells that produce IL-17A in the lung in response to IL-1β released from M1 macrophages130. Mice with obesity show a reduced ability to control bacterial pneumonia131, and impaired antimicrobial host defenses may be a consequence of reduced neutrophil recruitment into the alveoli, which is also reduced during airway injury and LPS challenge in obese mice129. Interestingly, during mouse H1N1 IAV infections, obesity is associated with increased lung pathology and impaired tissue repair without altered viral titers, suggesting that disease tolerance may be additionally compromised in obese individuals132. Peripheral blood cells from obese humans exhibit impaired IFN responses to TLR stimulation, equal to old individuals133, and this impairment may have similarly broad implications for loss of antiviral resistance and feedback regulation of respiratory immunity. Metabolic syndrome thus alters the frequency and responsiveness of respiratory cell populations and disease tolerance mechanisms, and thereby disrupts the normal tiered structure of the immune response and increases the risk of infection-related immunopathology and reduced pathogen control.
Concluding remarks
Specialized defense mechanisms are in place to protect the respiratory system against infections while minimizing tissue damage. Pathogens that manage to penetrate the mucus layer and enter the AECs trigger cell intrinsic defenses and the release of first order cytokines that inform tissue-resident lymphocytes. In addition, resident macrophages, DC and mast cells serve as the primary sensors of incoming pathogens that breach the epithelial barrier. Various innate and memory lymphocytes resident in the tissue interpret the combination of first order cytokines to produce second order cytokines that in turn act on the effector cells to eliminate pathogens and noxious substances. While these responses are optimized for immune defense in the lungs, various external and internal factors, including ambient temperature, pollutants, ageing and obesity can alter the efficiency with which the tiers of defense are engaged. In addition, the highly regulated nature of the respiratory immune system may be conducive to tumor metastasis and growth (Box 3).
Early immune responses in the lung are still riddled with many unanswered questions. For example, what are the molecular pathways that enforce disease tolerance? Can we leverage such knowledge for therapeutic treatment of acute and chronic inflammatory diseases? Another area of interest is to determine the functional consequences of triggering iNKT cells or TRM cells with cytokine versus specific antigens. In addition to magnitude of response, are there fundamentally different immunological outcome of stimulating these lymphocytes with first order cytokines versus antigens? Further areas of research might investigate whether prior exposure to a given pathogen or allergen dictates future responses to unrelated antigens. For instance, does exposure to virus X establish different tissue-resident lymphoid cell composition, and if so, does subsequent challenge by allergen or bacteria induce distinct outcomes? Can this explain susceptibility of individuals to co-infections or asthma?
Finally, both ageing and obesity increase human susceptibility to a range of airway infections, either by impairing host resistance mechanisms and/or by impairing disease tolerance. Reducing pathological inflammation in these populations may provide an alternative means of limiting morbidity and mortality from infectious diseases. Thus, understanding how engagement of tiered immune responses integrates with disease tolerance mechanisms, and how ageing and obesity alter this underlying tiered structure, will offer further insights for better treatment of respiratory diseases.
Box 1 | Dendritic cells sense pathogens and cytokines in type 2 immunity.
Dendritic cells (DCs) in the lung are highly responsive to first order cytokines derived from airway epithelial cells (AECs) and second order cytokines derived from lymphocytes. AEC-derived chemokines, such as CCL20, promote immature DC migration to the airway36. Interleukin-33 (IL-33) directly promotes airway DC activation and migration to lymph nodes in the context of allergic airway inflammation134, and skin models of allergy suggest that epithelium-derived TSLP can similarly activate DCs135. Airway DCs are also responsive to proximal signals from group 2 innate lymphoid cells (ILC2s), and ILC2-derived IL-13 induces DC migration to draining lymph nodes136. Migration of CD11 b+ DCs and CD301b+ DCs from the airways is essential for T helper 2 (Th2) cell differentiation and type 2 immunity in the context of allergic inflammation137 and lung helminth infections138, respectively. Whether AEC-derived cytokines are necessary or sufficient to initiate or enhance these DC responses remains to be determined, however TSLP is dispensable for Nippostrongylus brasiliensis lung infections in vivo139, and helminth products may directly stimulate DC activation in this model. DCs constantly take up antigens from the airway, and certain common allergens interact with DC receptors such as Toll-like receptor 4 and DC-SIGN140,141. Thus, DC activation in the context of type 2 immunity can occur as a consequence of both direct pathogen sensing and indirect activation by first order and second order cytokines.
Box 2 | Asthma and the impaired respiratory innate defense.
Asthma is a chronic lung disease affecting ~10% of the population and is most often characterized by asymptomatic periods punctuated with episodes of wheezing and respiratory distress. During the past decade, nucleic-acid based virus detection methods have demonstrated that the most common trigger for these disease exacerbations is infection with a respiratory virus and most frequently the rhinovirus — ~90% of childhood asthma attacks are virus-related142. Early work on bronchial epithelial cells collected from patients demonstrated a defect in rhinovirus-triggered induction of both type I and type III interferons (IFNs) in cells from asthmatics compared with those from healthy controls143,144. Subsequent studies have largely corroborated these findings but with mixed results145–147. Although this is an area of active investigation, it remains unclear whish exact molecular mechanism accounts for the IFN defect in asthmatics, or whether this defect is a cause or an effect of asthma pathogenesis. Nevertheless, the observation that epithelial IFN responses are diminished in asthma is intriguing as it could potentially explain why rhinovirus infections have severe respiratory consequences in patients with asthma but not in healthy individuals. Recent work suggests that the bacterial microbiota may also impact respiratory antiviral defense and asthma pathogenesis. As part of a longitudinal study of children at risk for asthma, investigators could retrospectively examine the early life microbiota of children who subsequently developed asthma. This study revealed links between the presence of certain bacteria in the nasopharynx during viral respiratory infection in infancy, febrile respiratory infection in infancy, and subsequent development of asthma148. Together, these studies suggest that aberrant immune responses during respiratory virus infection may lie at the heart of asthma pathogenesis.
Box 3 | Early responses to lung metastases.
How the immune system recognizes and responds to metastatic cancer cells that migrate to the lung is an active area of investigation. Cytotoxic natural killer (NK) cells and CD8+ T cells are important for tumor lysis and elimination, and the mechanisms that mediate these responses against tumor cells in the lung are not fully understood. In mice, circulating tumor cells that arrive in the lung are first engulfed by neutrophils, monocytes, and non-alveolar macrophages and/or dendritic cells (DCs) in the lung vasculature in a stepwise fashion149. CCL2-driven monocyte recruitment is important for tumor cell establishment and survival in the lung, in part due to the production of VEGF which supports tumor endothelial extravasation150. Neutrophils similarly enable tumor extravasation and survival through the production of both interleukin-1β (IL-1β) and MMP-9 (Refs.151,152), which act on the endothelium and alter the local microenvironment in a manner that ultimately supports tumor proliferation151. Neutrophils may additionally suppress systemic and local NK cell activation, further enabling the survival of circulating tumor cells151. CD103+ lung resident DCs, in contrast to these other myeloid cell populations, are important for the development of an effective anti-tumor immune response. These cells phagocytose tumor cells and migrate to the mesenteric lymph node within 72 hours of tumor cell instillation, enhancing T cell proliferation149. Alveolar macrophages suppress this DC proliferation and subsequent T cell response in part through TGFβ secretion in an effort to maintain lung homeostasis in the absence of activation of pattern recognition receptors and thereby enabling metastatic proliferation153. Thus, a plausible model of the role of immune cells in lung metastasis is that local and recruited myeloid cells attempt to maintain a homeostatic state in the lung tissue due to a lack of pathogen-associated signals and in doing so they suppress the development of an otherwise effective anti-tumor response.
Online summary.
The respiratory system consists of the upper respiratory tract — the nasal cavity, pharynx and larynx; and the lower respiratory tract — the conducting airway (trachea, bronchi) and the respiratory zone (alveoli). The composition of the epithelial cell types and the resident myeloid and lymphoid cell types depends on the diameter of the airway.
Airway cells are the first to respond to invading pathogens. They serve multiple functions in their capacity to provide barrier function, act as innate sensors to secrete first order cytokines, and serve as effector of antimicrobial defence.
Following respiratory infection by viral, bacterial, fungal and protozoan pathogens, type 1 immune responses are engaged. These infections are recognized through pattern recognition receptors in sensor cells including airway epithelial cells, macrophages, dendritic cells and plasmacytoid dendritic cells.
Infection by helminths or inhalation of allergens result in the engagement of the type 2 immune responses. Epithelial cells and mast cells detect the activities of the helminths and allergens to secrete cytokines that stimulate the next tier of immune response.
Sensor cells secrete discrete first order cytokines in response to pathogens, and activate tissue-resident lymphocytes to secrete second order cytokines. These cytokines in turn activate effector cell types to initiate pathogen elimination and tissue repair.
Various internal and external factors can alter the effectiveness of the signals mediated by the sensors and effectors, leading to tipping of the balance away from antimicrobial host defenses and towards pathological inflammation. Strategies to improve disease tolerance may be needed to combat infectious diseases in such vulnerable populations.
Acknowledgments
The authors would like to thank the Howard Hughes Medical Institute and the US National Institutes of Health (NIH) for their support of research in the laboratory (AI054359, HHSN272201100019C). E.F.F. was supported by funding from the NIH (T32 HL007974-11 and K08 AI119139-01). R.D.M. was supported by funding from the NIH (T32 AI007019-38 and T32 AI055403) and the Francis Trudeau Trainee Fellowship.
Glossary terms
- First Order Cytokines
A group of cytokines released from the cells that initially sense the presence of a pathogen. These cytokines primarily serve to alert tissue-resident lymphoid cell populations in order to coordinate an appropriate immune response to the pathogen
- Second Order Cytokines
A group of cytokines released from tissue lymphoid cells in response to signals from first order cytokines. These cytokines serve to recruit effector cells and activate effector and tissue repair functions in order to help resolve infections
- Type 1 Immune Responses
A group of related immune responses to viruses, bacteria, fungi, and protozoa that trigger immune responses characterized by cytokines IFN-γ, TNF-α, IL-17 and IL-22. These second order cytokines are secreted by ILC1, ILC3, NK, NKT, innate-like lymphocytes, Th1 and Th17 cells. Effectors also include cytotoxic T cell to kill infected cells
- Type 2 Immune Responses
A group of related immune responses to macroparasites, allergens, and certain venoms that trigger immune responses defined by the cytokines IL-4, IL-5, IL-9 and IL-13. These second order cytokines are secreted by ILC2, NKT, Th2 and Th9 cells
- Ciliated Cells
The predominant cell type in the surface epithelium of the conducting airways. Cilia at the apical surface of these cells beat continuously in a coordinated manner to propel airway mucus towards the mouth and nose, where mucus (and entrapped particles) can be removed via coughing or swallowing
- Goblet Cells
These cells produce airway mucins, the key components of the protective barrier impeding pathogen entry into the airway epithelium
- Club Cells
These secretory cells (formerly known as Clara cells) produce detoxifying and antimicrobial compounds that contribute to defense of the airway mucosa. Club cells have also been reported to serve as progenitor cells with the ability to replicate and/or differentiate into ciliated cells
- Basal Cells
These cells serve as regional progenitor cells of the airway epithelium, with the ability to proliferate in response to damage and differentiate into other surface epithelial cell types
- Pattern Recognition Receptors (PRRs)
Germ-line encoded receptors that recognize conserved pathogen-associated molecular patterns and activate signalling cascades that initiate an immune response
- RIG-I-like Receptors (RLRs)
A group of cytosolic PRRs consisting of RIG-I, MDA5 and LGP2 that recognize viral RNA in infected cells and initiate downstream inflammatory and interferon responses by signalling through the mitochondrial adapter protein MAVS
- Type I Interferons
Mammalian type I IFNs consist of IFN-α (13 subtypes in humans), IFN-β, IFN-к, IFN-δ, IFN-ε, IFN-τ, IFN-ω, and IFN-ζ. They all bind to and signal through the receptor consisting of IFNAR1/IFNAR2 dimer and trigger transcription of genes involved in antiviral defense (a.k.a “interferon stimulated genes.”)
- Type III Interferons
Type III IFNs consist of IFN-λ1, IFN-λ2 and IFN-λ3, and are potent antiviral cytokines secreted by diverse cell types following PRR detection of viral infection. Upon binding to the IFNλ receptor (IL28RA/IL10RB dimer) these interferons trigger transcription of genes involved in antiviral defense (a.k.a “interferon stimulated genes.”)
- Plasmacytoid DCs (pDCs)
Specialized sensory cells that express TLR7 and TLR9 and rapidly produce large amounts of type I interferons in response to viral infection
- Amphiregulin (AREG)
An epidermal growth factor (EGF)-like protein that promotes epithelial cell growth and tissue repair through EGFR signalling. AREG is important for promoting disease tolerance in lung infection models
- Alarmins
Constitutively expressed molecules that are released upon cell membrane rupture that alert the immune system
- Innate Lymphoid Cells (ILCs)
A group of lymphoid cells that lack B or T cell receptors and play that are resident in a variety of different organs. They are a major source of second order cytokines that maintain tissue homeostasis and promoting effective microbial clearance
- Airway Hyperresponsiveness (AHR)
A pathological state in which narrowing of the conducting airways can be easily triggered. AHR is a diagnostic feature of asthma and contributes to airway obstruction
- Neutrophil Extracellular Traps (NETs)
An extracellular mesh of chromatin that contains histones, neutrophil-derived proteases, and antimicrobial molecules. NETs are released from neutrophils in certain inflammatory scenarios and are important for ensnaring and trapping extracellular pathogens
- Chymase
A family of serine proteases primarily expressed by mast cells and released upon degranulation that can initiate proteolytic degradation of numerous substrates including IL-33 and the extracellular matrix
- Disease Tolerance
A host strategy for improving disease outcomes by improving tissue repair or reducing the detrimental impact of inflammatory signals
- Metabolic Syndrome
A set of risk factors including obesity, elevated blood pressure, and insulin resistance that are associated with an increased risk of heart disease, diabetes, and other negative clinical outcomes
Biography
Ryan Molony is a Ph.D. candidate at Yale University (USA) in the laboratory of Akiko Iwasaki in the Department of Immunobiology. His research focuses on the link between human ageing and increased susceptibility to influenza viral infection. Ryan previously completed B.S. and M.S. degrees at the University of Connecticut (USA), where he studied the role of metalloregulatory proteins in transcription factor activation in the laboratory of Michael Lynes in the Department of Molecular & Cell Biology.
Ellen Foxman is a physician-scientist and an assistant professor in the Department of Laboratory Medicine at Yale University. Her research focuses on the anti-viral defense mechanisms of the airway epithelium. During her post-doctoral training with Akiko Iwasaki, she investigated the impact of ambient temperature on innate immune defense against rhinovirus, the common cold virus. She also studied host defense mechanisms during her M.D.-Ph.D. training at Stanford University, where she worked with Eugene Butcher to understand how neutrophils navigate to sites of inflammation in response to chemotactic signals.
Home page: http://medicine.yale.edu/labmed/people/ellen_foxman.profile
Akiko Iwasaki received her Ph.D. from the University of Toronto (Canada), and her postdoctoral training from the National Institutes of Health (USA). She joined Yale University (USA) as a faculty in 2000, and currently is an Investigator of the HHMI and a Waldemar von Zedtwitz Professor of Immunobiology, and of Molecular Cellular and Developmental Biology. Akiko Iwasaki’s research focuses on the mechanisms of immune defense against viruses at the mucosal surfaces. Her laboratory is interested in how innate recognition of viral infections lead to the generation of adaptive immunity and ultimately to design better vaccines.
Footnotes
Competing interests statement
The authors declare no competing interests.
References
- 1.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morgan OW, et al. Morbid obesity as a risk factor for hospitalization and death due to 2009 pandemic influenza A (H1N1) disease. PloS one. 2010;5:e9694. doi: 10.1371/journal.pone.0009694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thompson WW, Shay DK, Weintraub E, et al. Mortality associated with influenza and respiratory syncytial virus in the united states. JAMA. 2003;289:179–186. doi: 10.1001/jama.289.2.179. [DOI] [PubMed] [Google Scholar]
- 4.Braciale TJ, Sun J, Kim TS. Regulating the adaptive immune response to respiratory virus infection. Nat Rev Immunol. 2012;12:295–305. doi: 10.1038/nri3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol. 2015;16:343–353. doi: 10.1038/ni.3123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kotton DN, Morrisey EE. Lung regeneration: mechanisms, applications and emerging stem cell populations. Nat Med. 2014;20:822–832. doi: 10.1038/nm.3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rock JR, Randell SH, Hogan BL. Airway basal stem cells: a perspective on their roles in epithelial homeostasis and remodeling. Dis Model Mech. 2010;3:545–556. doi: 10.1242/dmm.006031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curran DR, Cohn L. Advances in mucous cell metaplasia: a plug for mucus as a therapeutic focus in chronic airway disease. Am J Respir Cell Mol Biol. 2010;42:268–275. doi: 10.1165/rcmb.2009-0151TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Treuting, P.a.D., SM. Comparative Anatomy and Histology: A Mouse and Human Atlas. Elsevier; 2012. [Google Scholar]
- 10.Gomez Perdiguero E. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518:547–551. doi: 10.1038/nature13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guilliams M, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210:1977–1992. doi: 10.1084/jem.20131199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. 2014;14:81–93. doi: 10.1038/nri3600. [DOI] [PubMed] [Google Scholar]
- 13.Westphalen K, et al. Sessile alveolar macrophages communicate with alveolar epithelium to modulate immunity. Nature. 2014;506:503–506. doi: 10.1038/nature12902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mordstein M, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol. 2010;84:5670–5677. doi: 10.1128/JVI.00272-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khaitov MR, et al. Respiratory virus induction of alpha-, beta- and lambda-interferons in bronchial epithelial cells and peripheral blood mononuclear cells. Allergy. 2009;64:375–386. doi: 10.1111/j.1398-9995.2008.01826.x. [DOI] [PubMed] [Google Scholar]
- 16.Odendall C, et al. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat Immunol. 2014;15:717–726. doi: 10.1038/ni.2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Odendall C, Kagan JC. The unique regulation and functions of type III interferons in antiviral immunity. Curr Opin Virol. 2015;12:47–52. doi: 10.1016/j.coviro.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ueki IF, et al. Respiratory virus-induced EGFR activation suppresses IRF1-dependent interferon lambda and antiviral defense in airway epithelium. J Exp Med. 2013;210:1929–1936. doi: 10.1084/jem.20121401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sugimoto N, Mitoma H, Kim T, Hanabuchi S, Liu YJ. Helicase proteins DHX29 and RIG-I cosense cytosolic nucleic acids in the human airway system. Proc Natl Acad Sci U S A. 2014;111:7747–7752. doi: 10.1073/pnas.1400139111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu Rev Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 21.Wang H, Peters N, Schwarze J. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J Immunol. 2006;177:6263–6270. doi: 10.4049/jimmunol.177.9.6263. [DOI] [PubMed] [Google Scholar]
- 22.Kaminski MM, Ohnemus A, Cornitescu M, Staeheli P. Plasmacytoid dendritic cells and Toll-like receptor 7-dependent signalling promote efficient protection of mice against highly virulent influenza A virus. J Gen Virol. 2012;93:555–559. doi: 10.1099/vir.0.039065-0. [DOI] [PubMed] [Google Scholar]
- 23.Juarez E, et al. Differential expression of Toll-like receptors on human alveolar macrophages and autologous peripheral monocytes. Respir Res. 2010;11:2. doi: 10.1186/1465-9921-11-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goritzka M, et al. Alveolar macrophage-derived type I interferons orchestrate innate immunity to RSV through recruitment of antiviral monocytes. J Exp Med. 2015;212:699–714. doi: 10.1084/jem.20140825. This study uses a mouse RSV system to show that type I inteferons released from alveolar macrophages induce CCL2 expression necessary for inflammatory monocyte recruitment and viral clearance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Happel KI, et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J Exp Med. 2005;202:761–769. doi: 10.1084/jem.20050193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neyt K, Lambrecht BN. The role of lung dendritic cell subsets in immunity to respiratory viruses. Immunol Rev. 2013;255:57–67. doi: 10.1111/imr.12100. [DOI] [PubMed] [Google Scholar]
- 27.Thornton EE, et al. Spatiotemporally separated antigen uptake by alveolar dendritic cells and airway presentation to T cells in the lung. J Exp Med. 2012;209:1183–1199. doi: 10.1084/jem.20112667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vermaelen KY, Carro-Muino I, Lambrecht BN, Pauwels RA. Specific migratory dendritic cells rapidly transport antigen from the airways to the thoracic lymph nodes. J Exp Med. 2001;193:51–60. doi: 10.1084/jem.193.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Persson EK, et al. IRF4 transcription-factor-dependent CD103+ CD11b+ dendritic cells drive mucosal T helper 17 cell differentiation. Immunity. 2013;38:958–969. doi: 10.1016/j.immuni.2013.03.009. [DOI] [PubMed] [Google Scholar]
- 30.Bafica A, et al. TLR9 regulates Th1 responses and cooperates with TLR2 in mediating optimal resistance to Mycobacterium tuberculosis. J Exp Med. 2005;202:1715–1724. doi: 10.1084/jem.20051782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kandasamy M, et al. Complement mediated signaling on pulmonary CD103+ dendritic cells is critical for their migratory function in response to influenza infection. PLoS Pathog. 2013;9:e1003115. doi: 10.1371/journal.ppat.1003115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ichinohe T, et al. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci USA. 2011;108:5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen IY, Ichinohe T. Response of host inflammasomes to viral infection. Trends Microbiol. 2015;23:55–63. doi: 10.1016/j.tim.2014.09.007. [DOI] [PubMed] [Google Scholar]
- 34.von Moltke J, Ayres JS, Kofoed EM, Chavarria-Smith J, Vance RE. Recognition of bacteria by inflammasomes. Annu Rev Immunol. 2013;31:73–106. doi: 10.1146/annurev-immunol-032712-095944. [DOI] [PubMed] [Google Scholar]
- 35.Hammad H, et al. House dust mite allergen induces asthma via Toll-like receptor 4 triggering of airway structural cells. Nat Med. 2009;15:410–416. doi: 10.1038/nm.1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nathan AT, Peterson EA, Chakir J, Wills-Karp M. Innate immune responses of airway epithelium to house dust mite are mediated through beta-glucan-dependent pathways. J Allergy Clin Immunol. 2009;123:612–618. doi: 10.1016/j.jaci.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Voehringer D. Protective and pathological roles of mast cells and basophils. Nat Rev Immunol. 2013;13:362–375. doi: 10.1038/nri3427. [DOI] [PubMed] [Google Scholar]
- 38.Allakhverdi Z, et al. Thymic stromal lymphopoietin is released by human epithelial cells in response to microbes, trauma, or inflammation and potently activates mast cells. J Exp Med. 2007;204:253–258. doi: 10.1084/jem.20062211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moussion C, Ortega N, Girard JP. The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PloS one. 2008;3:e3331. doi: 10.1371/journal.pone.0003331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Byers DE, et al. Long-term IL-33–producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest. 2013;123:3967–3982. doi: 10.1172/JCI65570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Haenuki Y, et al. A critical role of IL-33 in experimental allergic rhinitis. J Allergy Clin Immunol. 2012;130:184–194. e111. doi: 10.1016/j.jaci.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Snelgrove RJ, et al. Alternaria-derived serine protease activity drives IL-33–mediated asthma exacerbations. J Allergy Clin Immunol. 2014;134:583–592. e586. doi: 10.1016/j.jaci.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lüthi AU, et al. Suppression of interleukin-33 bioactivity through proteolysis by apoptotic caspases. Immunity. 2009;31:84–98. doi: 10.1016/j.immuni.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 44.Kakkar R, Hei H, Dobner S, Lee RT. Interleukin 33 as a mechanically responsive cytokine secreted by living cells. J Biol Chem. 2012;287:6941–6948. doi: 10.1074/jbc.M111.298703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kouzaki H, O’Grady SM, Lawrence CB, Kita H. Proteases induce production of thymic stromal lymphopoietin by airway epithelial cells through protease-activated receptor-2. J Immunol. 2009;183:1427–1434. doi: 10.4049/jimmunol.0900904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barlow JL, et al. IL-33 is more potent than IL-25 in provoking IL-13–producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allergy Clin Immunol. 2013;132:933–941. doi: 10.1016/j.jaci.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 47.Tamachi T, et al. IL-25 enhances allergic airway inflammation by amplifying a T H 2 cell–dependent pathway in mice. J Allergy Clin Immunol. 2006;118:606–614. doi: 10.1016/j.jaci.2006.04.051. [DOI] [PubMed] [Google Scholar]
- 48.Mohapatra A, et al. Group 2 innate lymphoid cells utilize the IRF4-IL-9 module to coordinate epithelial cell maintenance of lung homeostasis. Mucosal Immunol. 2016;9:275–286. doi: 10.1038/mi.2015.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Denney L, et al. Pulmonary Epithelial Cell-Derived Cytokine TGF-beta1 Is a Critical Cofactor for Enhanced Innate Lymphoid Cell Function. Immunity. 2015;43:945–958. doi: 10.1016/j.immuni.2015.10.012. The authors of this study show that TGFβ is released from airway epithelial cells upon allergen exposure, and is necessary for ILC2 proliferation and cytokine production, and for subsequent type 2 immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kotas ME, Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–827. doi: 10.1016/j.cell.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Grove KC, et al. Characterization and Quantification of Innate Lymphoid Cell Subsets in Human Lung. PloS one. 2016;11 doi: 10.1371/journal.pone.0145961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Van Maele L, et al. Activation of Type 3 innate lymphoid cells and interleukin 22 secretion in the lungs during Streptococcus pneumoniae infection. J Infect Dis. 2014;210:493–503. doi: 10.1093/infdis/jiu106. [DOI] [PubMed] [Google Scholar]
- 53.Pribul PK, et al. Alveolar macrophages are a major determinant of early responses to viral lung infection but do not influence subsequent disease development. J Virol. 2008;82:4441–4448. doi: 10.1128/JVI.02541-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou G, Juang SWW, Kane KP. NK cells exacerbate the pathology of influenza virus infection in mice. Eur J Immunol. 2013;43:929–938. doi: 10.1002/eji.201242620. [DOI] [PubMed] [Google Scholar]
- 55.Kumar P, Thakar MS, Ouyang W, Malarkannan S. IL-22 from conventional NK cells is epithelial regenerative and inflammation protective during influenza infection. Mucosal Immunol. 2013;6:69–82. doi: 10.1038/mi.2012.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coquet JM, et al. Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4-NK1.1- NKT cell population. Proc Natl Acad Sci U S A. 2008;105:11287–11292. doi: 10.1073/pnas.0801631105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brigl M, et al. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med. 2011;208:1163–1177. doi: 10.1084/jem.20102555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paget C, et al. Interleukin-22 is produced by invariant natural killer T lymphocytes during influenza A virus infection: potential role in protection against lung epithelial damages. J Biol Chem. 2012;287:8816–8829. doi: 10.1074/jbc.M111.304758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sutton CE, et al. Interleukin-1 and IL-23 Induce Innate IL-17 Production from γδ T Cells, Amplifying Th17 Responses and Autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 60.Lockhart E, Green AM, Flynn JL. IL-17 production is dominated by gammadelta T cells rather than CD4 T cells during Mycobacterium tuberculosis infection. J Immunol. 2006;177:4662–4669. doi: 10.4049/jimmunol.177.7.4662. References 59 and 60 show that δγ T cells are a main source of the level 2 cytokine IL-17, which is induced in response to the level 1 cytokines IL-1 and IL-23. [DOI] [PubMed] [Google Scholar]
- 61.Skeen MJ, Ziegler HK. Activation of gamma delta T cells for production of IFN-gamma is mediated by bacteria via macrophage-derived cytokines IL-1 and IL-12. J Immunol. 1995;154:5832–5841. [PubMed] [Google Scholar]
- 62.Teijaro JR, et al. Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J Immunol. 2011;187:5510–5514. doi: 10.4049/jimmunol.1102243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mjösberg JM, et al. Human IL-25-and IL-33-responsive type 2 innate lymphoid cells are defined by expression of CRTH2 and CD161. Nat Immunol. 2011;12:1055–1062. doi: 10.1038/ni.2104. [DOI] [PubMed] [Google Scholar]
- 64.Neill DR, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–1370. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Halim TY, Krauß RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 2012;36:451–463. doi: 10.1016/j.immuni.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 66.Barlow JL, et al. IL-33 is more potent than IL-25 in provoking IL-13–producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J Allerg Clin Immunol. 2013;132:933–941. doi: 10.1016/j.jaci.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 67.Huang Y, et al. IL-25-responsive, lineage-negative KLRG1hi cells are multipotential/inflammatory/′type 2 innate lymphoid cells. Nat Immunol. 2015;16:161–169. doi: 10.1038/ni.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Saenz SA, et al. IL-25 simultaneously elicits distinct populations of innate lymphoid cells and multipotent progenitor type 2 (MPPtype2) cells. J Exp Med. 2013;210:1823–1837. doi: 10.1084/jem.20122332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ohne Y, et al. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat Immunol. 2016;17:646–655. doi: 10.1038/ni.3447. [DOI] [PubMed] [Google Scholar]
- 70.Turner JE, et al. IL-9–mediated survival of type 2 innate lymphoid cells promotes damage control in helminth-induced lung inflammation. J Exp Med. 2013;210:2951–2965. doi: 10.1084/jem.20130071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Terashima A, et al. A novel subset of mouse NKT cells bearing the IL-17 receptor B responds to IL-25 and contributes to airway hyperreactivity. J Exp Med. 2008;205:2727–2733. doi: 10.1084/jem.20080698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Akbari O, et al. Essential role of NKT cells producing IL-4 and IL-13 in the development of allergen-induced airway hyperreactivity. Nat Med. 2003;9:582–588. doi: 10.1038/nm851. [DOI] [PubMed] [Google Scholar]
- 73.Pichavant M, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med. 2008;205:385–393. doi: 10.1084/jem.20071507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Nagata Y, Kamijuku H, Taniguchi M, Ziegler S, Seino KI. Differential role of thymic stromal lymphopoietin in the induction of airway hyperreactivity and Th2 immune response in antigen-induced asthma with respect to natural killer T cell function. Int Arch Allergy Immunol. 2007;144:305–314. doi: 10.1159/000106319. [DOI] [PubMed] [Google Scholar]
- 75.Smithgall MD, et al. IL-33 amplifies both Th1-and Th2-type responses through its activity on human basophils, allergen-reactive Th2 cells, iNKT and NK cells. Int Immunol. 2008;20:1019–1030. doi: 10.1093/intimm/dxn060. [DOI] [PubMed] [Google Scholar]
- 76.Scanlon ST, et al. Airborne lipid antigens mobilize resident intravascular NKT cells to induce allergic airway inflammation. J Exp Med. 2011;208:2113–2124. doi: 10.1084/jem.20110522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaplan MH, Hufford MM, Olson MR. The development and in vivo function of T helper 9 cells. Nat Rev Immunol. 2015;15:295–307. doi: 10.1038/nri3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yao W, et al. Interleukin-9 is required for allergic airway inflammation mediated by the cytokine TSLP. Immunity. 2013;38:360–372. doi: 10.1016/j.immuni.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kearley J, et al. IL-9 governs allergen-induced mast cell numbers in the lung and chronic remodeling of the airways. Am J Respir Crit Care Med. 2011;183:865–875. doi: 10.1164/rccm.200909-1462OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Coquet JM, et al. Interleukin-21-Producing CD4(+) T Cells Promote Type 2 Immunity to House Dust Mites. Immunity. 2015;43:318–330. doi: 10.1016/j.immuni.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 81.Angkasekwinai P, Chang SH, Thapa M, Watarai H, Dong C. Regulation of IL-9 expression by IL-25 signaling. Nat Immunol. 2010;11:250–256. doi: 10.1038/ni.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Omori M, Ziegler S. Induction of IL-4 expression in CD4+ T cells by thymic stromal lymphopoietin. J Immunol. 2007;178:1396–1404. doi: 10.4049/jimmunol.178.3.1396. [DOI] [PubMed] [Google Scholar]
- 83.Schmitz J, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–490. doi: 10.1016/j.immuni.2005.09.015. This study established IL-33 as an IL-1 family member and demonstrated its signaling paradigm and its role in the eventual induction of level 2 cytokines and type 2 immunity. [DOI] [PubMed] [Google Scholar]
- 84.Nizzoli G, et al. Human CD1c+ dendritic cells secrete high levels of IL-12 and potently prime cytotoxic T-cell responses. Blood. 2013;122:932–942. doi: 10.1182/blood-2013-04-495424. [DOI] [PubMed] [Google Scholar]
- 85.Aujla SJ, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aldridge JR, Jr, et al. Proc Natl Acad Sci U S A. 2009;106:5306–5311. doi: 10.1073/pnas.0900655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Maus UA, et al. Monocytes are potent facilitators of alveolar neutrophil emigration during lung inflammation: role of the CCL2-CCR2 axis. J Immunol. 2003;170:3273–3278. doi: 10.4049/jimmunol.170.6.3273. [DOI] [PubMed] [Google Scholar]
- 88.Espinosa V, et al. Inflammatory monocytes orchestrate innate antifungal immunity in the lung. PLoS Pathog. 2014;10:e1003940. doi: 10.1371/journal.ppat.1003940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lin KL, Suzuki Y, Nakano H, Ramsburg E, Gunn MD. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J Immunol. 2008;180:2562–2572. doi: 10.4049/jimmunol.180.4.2562. [DOI] [PubMed] [Google Scholar]
- 90.Landsman L, Jung S. Lung macrophages serve as obligatory intermediate between blood monocytes and alveolar macrophages. J Immunol. 2007;179:3488–3494. doi: 10.4049/jimmunol.179.6.3488. [DOI] [PubMed] [Google Scholar]
- 91.Kreisel D, et al. In vivo two-photon imaging reveals monocyte-dependent neutrophil extravasation during pulmonary inflammation. Proc Natl Acad Sci U S A. 2010;107:18073–18078. doi: 10.1073/pnas.1008737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sun K, Salmon SL, Lotz SA, Metzger DW. Interleukin-12 promotes gamma interferon-dependent neutrophil recruitment in the lung and improves protection against respiratory Streptococcus pneumoniae infection. Infect Immun. 2007;75:1196–1202. doi: 10.1128/IAI.01403-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bradley LM, Douglass MF, Chatterjee D, Akira S, Baaten BJ. Matrix metalloprotease 9 mediates neutrophil migration into the airways in response to influenza virus-induced toll-like receptor signaling. PLoS Pathog. 2012;8:e1002641. doi: 10.1371/journal.ppat.1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122:2784–2794. doi: 10.1182/blood-2013-04-457671. [DOI] [PubMed] [Google Scholar]
- 95.Pillai PS, et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science. 2016;352:463–466. doi: 10.1126/science.aaf3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bruns S, et al. Production of Extracellular Traps against Aspergillus fumigatus In Vitro and in Infected Lung Tissue Is Dependent on Invading Neutrophils and Influenced by Hydrophobin RodA. PLoS Pathog. 2010;6:e1000873. doi: 10.1371/journal.ppat.1000873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li L, et al. Effects of Th2 cytokines on chemokine expression in the lung: IL-13 potently induces eotaxin expression by airway epithelial cells. J Immunol. 1999;162:2477–2487. [PubMed] [Google Scholar]
- 98.Kuperman DA, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8:885–889. doi: 10.1038/nm734. This study demonstrates that STAT6-mediated IL-13 signaling in epithelial cells is critical for effector functions associated with airway hyperreactivity, including enhanced mucus production. [DOI] [PubMed] [Google Scholar]
- 99.Monticelli LA, et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12:1045–1054. doi: 10.1031/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pope SM, et al. IL-13 induces eosinophil recruitment into the lung by an IL-5- and eotaxin-dependent mechanism. J Allergy Clin Immunol. 2001;108:594–601. doi: 10.1067/mai.2001.118600. [DOI] [PubMed] [Google Scholar]
- 101.Wong CK, Hu S, Cheung PFY, Lam CWK. Thymic Stromal Lymphopoietin Induces Chemotactic and Prosurvival Effects in Eosinophils. Am J Respir Cell Mol Biol. 2010;43:305–315. doi: 10.1165/rcmb.2009-0168OC. [DOI] [PubMed] [Google Scholar]
- 102.Rosenberg HF, Dyer KD, Foster PS. Eosinophils: changing perspectives in health and disease. Nat Rev Immunol. 2013;13:9–22. doi: 10.1038/nri3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wakahara K, et al. Basophils are recruited to inflamed lungs and exacerbate memory Th2 responses in mice and humans. Allergy. 2013;68:180–189. doi: 10.1111/all.12072. [DOI] [PubMed] [Google Scholar]
- 104.Motomura Y, et al. Basophil-derived interleukin-4 controls the function of natural helper cells, a member of ILC2s, in lung inflammation. Immunity. 2014;40:758–771. doi: 10.1016/j.immuni.2014.04.013. [DOI] [PubMed] [Google Scholar]
- 105.Sharma M, Bayry J. Autoimmunity: Basophils in autoimmune and inflammatory diseases. Nat Rev Rheumatol. 2015;11:129–131. doi: 10.1038/nrrheum.2014.199. [DOI] [PubMed] [Google Scholar]
- 106.Siracusa MC, et al. TSLP promotes interleukin-3-independent basophil haematopoiesis and type 2 inflammation. Nature. 2011;477:229–233. doi: 10.1038/nature10329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lantz CS, et al. Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature. 1998;392:90–93. doi: 10.1038/32190. [DOI] [PubMed] [Google Scholar]
- 108.Ohnmacht C, Voehringer D. Basophils protect against reinfection with hookworms independently of mast cells and memory Th2 cells. J Immunol. 2010;184:344–350. doi: 10.4049/jimmunol.0901841. [DOI] [PubMed] [Google Scholar]
- 109.Roy A, et al. Mast cell chymase degrades the alarmins heat shock protein 70, biglycan, HMGB1, and interleukin-33 (IL-33) and limits danger-induced inflammation. J Biol Chem. 2014;289:237–250. doi: 10.1074/jbc.M112.435156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Morita H, et al. An interleukin-33-mast cell-interleukin-2 axis suppresses papain-induced allergic inflammation by promoting regulatory T cell numbers. Immunity. 2015;43:175–186. doi: 10.1016/j.immuni.2015.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lefrançais E, et al. Central domain of IL-33 is cleaved by mast cell proteases for potent activation of group-2 innate lymphoid cells. Proc Natl Acad Sci U S A. 2014;111:15502–15507. doi: 10.1073/pnas.1410700111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. 2012;335:936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jamieson AM, et al. Role of tissue protection in lethal respiratory viral-bacterial coinfection. Science. 2013;340:1230–1234. doi: 10.1126/science.1233632. This study uses a influenza/bacteria coinfection model system to demonstrate that disease lethality can occur in the absence of changes in effective pathogen burden or control due to impaired disease tolerance mechanisms. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sivaraman V, et al. Yersinia pestis Activates Both IL-1B and IL-1 Receptor Antagonist to Modulate Lung Inflammation during Pneumonic Plague. PLoS Pathog. 2015;11:e1004688. doi: 10.1371/journal.ppat.1004688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Doitsh G, et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505:509–514. doi: 10.1038/nature12940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang A, et al. Opposing Effects of Fasting Metabolism on Tissue Tolerance in Bacterial and Viral Inflammation. Cell. 2016;166:1512–1525. e1512. doi: 10.1016/j.cell.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Foxman EF, et al. Temperature-dependent innate defense against the common cold virus limits viral replication at warm temperature in mouse airway cells. Proc Natl Acad Sci U S A. 2015;112:827–832. doi: 10.1073/pnas.1411030112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Foxman EF, Storer JA, Vanaja K, Levchenko A, Iwasaki A. Two interferon-independent double-stranded RNA-induced host defense strategies suppress the common cold virus at warm temperature. Proc Natl Acad Sci U S A. 2016;113:8496–8501. doi: 10.1073/pnas.1601942113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jaspers I, et al. Reduced expression of IRF7 in nasal epithelial cells from smokers after infection with influenza. Am J Respir Cell Mol Biol. 2010;43:368–375. doi: 10.1165/rcmb.2009-0254OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bauer CM, et al. Cigarette smoke suppresses type I interferon-mediated antiviral immunity in lung fibroblast and epithelial cells. J Interferon Cytokine Res. 2008;28:167–179. doi: 10.1089/jir.2007.0054. [DOI] [PubMed] [Google Scholar]
- 121.Kearley J, et al. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity. 2015;42:566–579. doi: 10.1016/j.immuni.2015.02.011. This study illustrates how cigarette smoke alters IL-33 signaling by promoting increased epithelial IL-33 expression, as well as decreased ST2 expression on ILCs and increased ST2 expression on NK cells and macrophages, predisposing the lung to more readily engage inflammatory effector functions. [DOI] [PubMed] [Google Scholar]
- 122.Lowery EM, Brubaker AL, Kuhlmann E, Kovacs EJ. The aging lung. Clin Interv Aging. 2013;8 doi: 10.2147/CIA.S51152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shaw AC, Goldstein DR, Montgomery RR. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 2013;13:875–887. doi: 10.1038/nri3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhao J, Zhao J, Legge K, Perlman S. Age-related increases in PGD 2 expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J Clin Invest. 2011;121:4921–4930. doi: 10.1172/JCI59777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Beli E, et al. Natural killer cell function is altered during the primary response of aged mice to influenza infection. Mech Ageing Dev. 2011;132:503–510. doi: 10.1016/j.mad.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Gomez CR, et al. Advanced age exacerbates the pulmonary inflammatory response after lipopolysaccharide exposure. Crit Care Med. 2007;35:246–251. doi: 10.1097/01.CCM.0000251639.05135.E0. [DOI] [PubMed] [Google Scholar]
- 127.Brandes M, Klauschen F, Kuchen S, Germain RN. A systems analysis identifies a feedforward inflammatory circuit leading to lethal influenza infection. Cell. 2013;154:197–212. doi: 10.1016/j.cell.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Camargo CA, Weiss ST, Zhang S, Willett WC, Speizer FE. Prospective study of body mass index, weight change, and risk of adult-onset asthma in women. Arch Intern Med. 1999;159:2582–2588. doi: 10.1001/archinte.159.21.2582. [DOI] [PubMed] [Google Scholar]
- 129.Manicone AM, Gong K, Johnston LK, Giannandrea M. Diet-induced obesity alters myeloid cell populations in naïve and injured lung. Respir Res. 2016;17:1–11. doi: 10.1186/s12931-016-0341-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kim HY, et al. Interleukin-17-producing innate lymphoid cells and the LRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med. 2014;20:54–61. doi: 10.1038/nm.3423. This study demonstrates that an increase in airway hyperreactivity in obese mice occurs as a consequence of NLRP3-mediated IL-1β production, which in turn induces increased IL-17 production by ILC3s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ubags ND, et al. A comparative study of lung host defense in murine obesity models: insights into neutrophil function. Am J Respir Cell Mol Biol. 2016 doi: 10.1165/rcmb.2016-0042OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.O’Brien KB, et al. Impaired wound healing predisposes obese mice to severe influenza virus infection. J Infect Dis. 2012;205:252–261. doi: 10.1093/infdis/jir729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Teran-Cabanillas E, Montalvo-Corral M, Caire-Juvera G, Moya-Camarena SY, Hernández J. Decreased interferon-α and interferon-β production in obesity and expression of suppressor of cytokine signaling. Nutrition. 2013;29:207–212. doi: 10.1016/j.nut.2012.04.019. [DOI] [PubMed] [Google Scholar]
- 134.Besnard AG, et al. IL-33-activated dendritic cells are critical for allergic airway inflammation. Eur J Immunol. 2011;41:1675–1686. doi: 10.1002/eji.201041033. [DOI] [PubMed] [Google Scholar]
- 135.Soumelis V, et al. Human epithelial cells trigger dendritic cell–mediated allergic inflammation by producing TSLP. Nat Immunol. 2002;3:673–680. doi: 10.1038/ni805. [DOI] [PubMed] [Google Scholar]
- 136.Halim Timotheus YF, et al. Group 2 Innate Lymphoid Cells Are Critical for the Initiation of Adaptive T Helper 2 Cell-Mediated Allergic Lung Inflammation. Immunity. 2014;40:425–435. doi: 10.1016/j.immuni.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Plantinga M, et al. Conventional and monocyte-derived CD11b+ dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity. 2013;38:322–335. doi: 10.1016/j.immuni.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 138.Kumamoto Y, et al. CD301b(+) dermal dendritic cells drive T helper 2 cell-mediated immunity. Immunity. 2013;39:733–743. doi: 10.1016/j.immuni.2013.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Massacand JC, et al. Helminth products bypass the need for TSLP in Th2 immune responses by directly modulating dendritic cell function. Proc Natl Acad Sci U S A. 2009;106:13968–13973. doi: 10.1073/pnas.0906367106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Trompette A, et al. Allergenicity resulting from functional mimicry of a Toll-like receptor complex protein. Nature. 2009;457:585–588. doi: 10.1038/nature07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shreffler WG, et al. The major glycoprotein allergen from Arachis hypogaea, Ara h 1, is a ligand of dendritic cell-specific ICAM-grabbing nonintegrin and acts as a Th2 adjuvant in vitro. J Immunol. 2006;177:3677–3685. doi: 10.4049/jimmunol.177.6.3677. [DOI] [PubMed] [Google Scholar]
- 142.Gern JE. The ABCs of rhinoviruses, wheezing, and asthma. J Virol. 2010;84:7418–7426. doi: 10.1128/JVI.02290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wark PA, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med. 2005;201:937–947. doi: 10.1084/jem.20041901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Contoli M, et al. Role of deficient type III interferon-lambda production in asthma exacerbations. Nat Med. 2006;12:1023–1026. doi: 10.1038/nm1462. [DOI] [PubMed] [Google Scholar]
- 145.Sykes A, et al. Rhinovirus-induced interferon production is not deficient in well controlled asthma. Thorax. 2014;69:240–246. doi: 10.1136/thoraxjnl-2012-202909. [DOI] [PubMed] [Google Scholar]
- 146.Edwards MR, et al. Impaired innate interferon induction in severe therapy resistant atopic asthmatic children. Mucosal Immunol. 2013;6:797–806. doi: 10.1038/mi.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Baraldo S, et al. Deficient antiviral immune responses in childhood: distinct roles of atopy and asthma. J Allergy Clin Immunol. 2012;130:1307–1314. doi: 10.1016/j.jaci.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 148.Teo SM, et al. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe. 2015;17:704–715. doi: 10.1016/j.chom.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Headley MB, et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 2016;531:513–517. doi: 10.1038/nature16985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Qian BZ, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–225. doi: 10.1038/nature10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Spiegel A, et al. Neutrophils suppress intraluminal NK-mediated tumor cell clearance and enhance extravasation of disseminated carcinoma cells. Cancer Discov. 2016:CD-15-1157. doi: 10.1158/2159-8290.CD-15-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Acuff HB, Carter KJ, Fingleton B, Gorden DL, Matrisian LM. Matrix metalloproteinase-9 from bone marrow-derived cells contributes to survival but not growth of tumor cells in the lung microenvironment. Cancer Res. 2006;66:259–266. doi: 10.1158/0008-5472.CAN-05-2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Sharma SK, et al. Pulmonary alveolar macrophages contribute to the premetastatic niche by suppressing antitumor T cell responses in the lungs. J Immunol. 2015;194:5529–5538. doi: 10.4049/jimmunol.1403215. [DOI] [PubMed] [Google Scholar]
- 154.Boers JE, Ambergen AW, Thunnissen FB. Number and proliferation of clara cells in normal human airway epithelium. Am J Respir Crit Care Med. 1999;159:1585–1591. doi: 10.1164/ajrccm.159.5.9806044. [DOI] [PubMed] [Google Scholar]