

Abstract
In the pivotal TEMPO 3:4 trial, the arginine vasopressin V2‐receptor antagonist tolvaptan reduced the rate of kidney growth in patients with autosomal dominant polycystic kidney disease. Tolvaptan was initiated as daily morning/afternoon doses of 45/15 mg, and uptitrated weekly to 60/30 mg and 90/30 mg according to patient‐reported tolerability. The current report describes 3 phase 2 trials in adult autosomal dominant polycystic kidney disease subjects that were the basis for the titrated split‐dose regimen: a single ascending‐dose trial (tolvaptan 15 to 120 mg; n = 11), a multiple split‐dose trial (tolvaptan 15/15 mg, 30/0 mg, 30/15 mg, and 30/30 mg; n = 37), and an 8‐week open‐label safety and efficacy trial in 46 of the 48 subjects who participated in the prior 2 trials (tolvaptan 30/15 mg, 45/15 mg, 60/30 mg, and 90/30 mg). Urine osmolality (Uosm) was chosen as the biomarker of V2 receptor inhibition. Two tolvaptan doses per day were necessary to suppress Uosm to <300 mOsm/kg for 24 hours. The 45/15‐mg regimen was well tolerated and effective in suppressing Uosm in >50% of subjects. Therefore, this regimen was selected as the starting regimen for the TEMPO 3:4 trial. The 90/30‐mg regimen suppressed Uosm in 85% of subjects tested; however, only 28/46 subjects agreed to uptitrate to 90/30 mg due to tolerability. Higher concentrations of tolvaptan were less well tolerated, resulting in adverse events of pollakiuria, thirst, polyuria, nocturia, and a higher number of times out of bed to urinate. Subjects who agreed to uptitrate to 90/30 mg had lower eGFR than those who did not uptitrate.
Keywords: Tolvaptan, pharmacokinetics, pharmacodynamics, autosomal dominant polycystic kidney disease, urine osmolality, tolerability
Tolvaptan is an oral agent approved in the United States and Europe for the treatment of specific forms of hyponatremia.1, 2 Mechanistically, tolvaptan corrects serum sodium by inhibiting the binding of arginine vasopressin (AVP) to V2‐receptors in the distal nephron. This antagonism lowers the rate at which aquaporin‐2 channels are inserted into the membranes of renal collecting tubules, which in turn increases electrolyte‐free water excretion, or “aquaresis.” In the mammalian kidney and urine, 2 forms of aquaporin‐2 are detected: unglycosylated and glycosylated. Excretion of unglycosylated aquaporin‐2 has been shown to increase as concentrations of AVP increase or in response to administration of a V2‐receptor agonist.3 Excretion of unglycosylated aquaporin‐2 was hypothesized to decrease following tolvaptan administration.
Since the demonstration of its utility for clinically significant hyponatremia, tolvaptan has also been shown to have beneficial disease‐modifying effects in autosomal dominant polycystic kidney disease (ADPKD). In the pivotal TEMPO 3:4 (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes 3:4, NCT00428948) trial, subjects with a relatively early stage of ADPKD and a high likelihood of rapid disease progression (estimated creatinine clearance per Cockcroft‐Gault formula 4 ≥60 mL/min; total kidney volume ≥750 mL) were randomly assigned to receive tolvaptan (n = 961) or matching placebo (n = 484).5, 6 Over a 3‐year period, the tolvaptan group in TEMPO 3:4 exhibited a significantly lower rate of growth in total kidney volume, a lower rate of worsening kidney function, a lower rate of worsening kidney pain, and a slower decline in kidney function relative to the placebo group.6 The mechanisms underlying tolvaptan's beneficial effects in ADPKD have not been fully elucidated but likely involve downregulation of cAMP signaling, cell proliferation, and chloride‐driven fluid excretion.7, 8, 9, 10 Consequently, it was hypothesized that tolvaptan administration would decrease urinary excretion of cAMP.
In TEMPO 3:4, tolvaptan was initially dosed in daily morning and afternoon doses of 45 mg and 15 mg, respectively, with uptitration weekly to 60/30 mg and then to 90/30 mg according to patient‐reported tolerability. Patients remained on the highest tolerable dose for 36 months. This forced titration regimen was designed to accomplish 2 objectives. First, with splitting of the dose between 2 daily administrations, AVP receptor inhibition, as indicated by urine osmolality (Uosm) suppression to <300 mOsm/kg, could be maintained for 24 hours without excessive side effects. Second, by using a higher dose early in the day, followed by a lower dose 8 to 9 hours later, maximal inhibition could be produced on awakening, with a gradual falling off of effect during the night when frequent urination might lead to interruption of sleep. Since publication of the TEMPO 3:4 trial, there has been interest in better understanding how this dosing regimen was developed so that the rationale for uptitration to the 90/30‐mg dose regimen or to the highest tolerated dose are made in ADPKD patients on tolvaptan. With that in mind, we describe here 3 phase 2 trials that formed the basis of the tolvaptan dosing regimens used in TEMPO 3:4.
Methods
Ethics
The current report describes results from 3 phase 2 studies, referred to here as Trial 248, Trial 249, and Trial 250. Protocols for Trials 248 and 249 were approved by the Independent Institutional Review Board, Inc (Plantation, Florida); subjects were treated and monitored in the clinic (Orlando Clinical Research Center, Orlando, Florida). Trial 250 (NCT00413777) was conducted at 11 centers throughout the United States (Rogosin Institute, New York, New York; Northwest Renal Clinic, Inc, Portland, Oregon; Jacksonville Center for Clinical Research, Jacksonville, Florida; Davita Clinical Research, Minneapolis, Minnesota; Nephrology Clinical Research Center at the University of Virginia, Charlottesville, Virginia; Vanderbilt University Medical Center, Nashville, Tennessee; Johns Hopkins School of Medicine, Baltimore, Maryland; Mayo Medical Center, Rochester, Minnesota; University of Colorado Denver, Aurora, Colorado; University of Kansas Medical Center, Kansas City, Kansas; Center for Hypertension and Renal Disease Research, Atlanta, Georgia); ethics review was provided at each site. In all 3 studies, participants were required to provide informed consent prior to enrollment.
Trial Designs
Trial 248 was designed to assess the safety and pharmacokinetic (PK) and pharmacodynamic (PD) profiles of ascending single doses of tolvaptan in men and women 18 to 55 years of age who had ADPKD with preserved renal function. Confirmation of the ADPKD diagnosis via imaging was required. Exclusion criteria included serum creatinine at baseline >1.4 mg/dL for men and >1.2 mg/dL for women; serum creatinine >1.8 mg/dL if evidence of significant renal disease other than ADPKD; history of symptomatic kidney stones; proteinuria >2 g/24 h; absence of a kidney; hepatitis or HIV positive; and oral antibiotics within 30 days or any medications within 14 days of dosing, with the exception of angiotensin II receptor blockers and or angiotensin‐converting‐enzyme inhibitors. Eligible subjects (n = 11) were randomly assigned 2:1 to receive single oral doses of tolvaptan (n = 8) or matching placebo (n = 3) 72 hours apart on days 1, 4, 7, and 10. The 4 tested doses in the study were 15 mg, 30 mg, 60 mg, and 120 mg. After each dose the most appropriate next and higher dose was determined based on tolerability and expected vs observed PD response (in particular, Uosm suppression).
Trial 249 was designed to assess the safety, PK, and PD profiles of split‐dose tolvaptan in subjects who had ADPKD with preserved renal function. Inclusion and exclusion criteria were identical to those of Trial 248, with the exception that subjects could be 18 to 60 years of age. Eligible subjects were randomized 1:1:1:1 to receive the following split‐dose regimens on 5 consecutive days: tolvaptan 15 mg am + tolvaptan 15 mg pm (15/15 mg; n = 9); tolvaptan 30 mg am + placebo pm (30 mg daily [QD]; n = 9); tolvaptan 30 mg am + tolvaptan 15 mg pm (30/15 mg; n = 9); and tolvaptan 30 mg am + tolvaptan 30 mg pm (30/30 mg; n = 10). Safety, PK, and PD profiles were evaluated on day 1 and day 5.
Trial 250 (TEMPO 2:4) was an open‐label, 2‐part trial designed to assess the safety and efficacy of split‐dose tolvaptan regimens in 46 subjects who were previously enrolled in Trials 248 and 249. Additional exclusion criteria in Trial 250 included an estimated creatinine clearance per Cockcroft‐Gault formula of at least 30 mL/min on day 1; anticipated renal‐replacement therapy within 1 year of trial entry; diuretic use for 1 week prior to enrollment or anticipated need for diuretics prior to the month‐2 visit. Eligible subjects (n = 46) underwent forced titration of split‐dose oral tolvaptan regimens over a 4‐week period according to the following pattern: 30/15 mg → 45/15 mg → 60/30 mg → 90/30 mg. Dosing was then sustained at the highest tolerated dose until 8 weeks after the start of trial. Tolerability was self‐assessed outside of the clinic.
In all 3 trials, subjects were administered a calcium (500 mg) + vitamin D supplement twice daily to stabilize parathyroid hormone levels, which would be expected to blunt the variability of cAMP concentrations in blood and urine. Fluids were unrestricted.
Sample Collection
Trial 248
Blood samples (5 mL with sodium heparin as anticoagulant) were collected for PK analyses at predose and 1, 2, 3, 4, 8, 12, 16, 24, 30, 36, and 48 hours postdose on days 1, 4, 7, and 10. Plasma was evenly pipetted into 2 aliquots and stored at –20°C or colder until analyzed for tolvaptan.
Baseline PD blood and urine samples collected on day –1 were taken relative to the expected dosing time of tolvaptan on following days. PD blood samples were taken on day –1 and after each dose administered on days 1, 4, 7, and 10. Ten mL for determination of serum Na+, K+, and osmolality were collected at predose and 4, 8, 12, 16, and 24 hours postdose.
Urine samples were collected for PD analyses on days 1, 4, 7, and 10 across the following intervals: 0 to 4, 4 to 8, 8 to 12, 12 to 16, 16 to 24, 24 to 28, 28 to 32, 32 to 36, 36 to 40, and 40 to 48 hours postdose. Subjects voided within 30 minutes prior to dosing and prior to the end of the collection interval. For each interval the volume of pooled voids over the interval was recorded, and aliquots were taken for the determination of sodium, osmolality, cAMP, and unglycosylated aquaporin‐2.
Fluid intake was recorded for the same intervals as urine collection so that fluid balance could be determined for each interval. Calculation of fluid intake included water used for dosing, as well as foods with high water content (eg, soup, gelatine). The number of times out of bed (to drink or urinate) during the sleep period (ie, between 16 and 24 hours postdose) on days –1 and 1, 4, 7, and 10 were recorded and included the subject's first void after arising for the day's activities.
Trial 249
Trial 249 had the same pre‐ and postdose sampling times (relative to the morning tolvaptan dose) and the same analytes as Trial 248. PK blood samples were collected through 24 hours on day 1 and through 48 hours on day 5. PD blood samples were collected through 24 hours on days –1, 1, and 5. Urine samples were collected through 24 hours on day 1 and through 48 hours on day 5. Fluid intake was recorded for the same intervals as urine collection. Number of times out of bed (to drink or urinate) during the sleep period (ie, between 16 and 24 hours postdose) on days –1 to 6 were recorded.
Trial 250
Urine osmolality was assessed from spot urine samples obtained on the day and morning prior to the first day of dosing (baseline) and then after at least 4 days of dosing on the day and morning prior to the weekly clinic visit during the titration period. Samples were collected 3 times during the day: (1) as part of the second morning void after awakening, prior to consuming any liquids or food and prior to the morning (first) dose of tolvaptan, (2) about 4 pm on the baseline day or prior to the afternoon (second) dose of tolvaptan, and (3) prior to bedtime.
Pharmacokinetic Analyses
Tolvaptan plasma concentrations were quantified by validated assay using high‐performance liquid chromatography with tandem mass spectrophotometric detection.11 Plasma concentrations of tolvaptan were analyzed, and standard PK assessments were performed by noncompartmental methods using WinNonlin Pro, version 4 (Pharsight Corporation, Mountain View, California). Analysis was performed using actual times postdose.
The primary tolvaptan PK endpoints in Trial 248 were maximum plasma concentration (Cmax), time to maximum plasma concentration (tmax), area under the curve from time 0 to infinity (AUC∞), apparent clearance from plasma after extravascular administration (CL/F), and terminal‐phase elimination half‐life (t1/2,z). The primary tolvaptan PK endpoints in Trial 249 were day 1 and day 5 Cmax, tmax, and area under the curve from time 0 to 24 hours postdose (AUC0‐24h). For the 30‐mg QD dose at day 5, Cmax at steady state and CL/F at steady state were determined. Secondary endpoints were t1/2,z and the day 5/day 1 ratios of Cmax and AUC0‐24h.
Pharmacodynamic Analyses
Urine osmolality and creatinine were determined at the study sites’ clinical chemistry laboratories according to standard operating procedures.
Plasma AVP concentrations were determined by radioimmunoassay at the Department of Laboratory Medicine and Pathology, Mayo Clinic (Rochester, Minnesota) according to published methodologies.12
Urine cAMP concentrations were determined following isolation from urine samples using a standard anion‐exchange column (Dowex‐1 in formate form; Dow Water Processing Solutions, Edina, Minnesota). An internal standard (8‐methylamino cAMP) was used to correct for recovery losses. Once eluted from the column, the cAMP was allowed to react under acidic conditions with chloracetaldehyde to give a fluorescent 1,N(6)‐ethano‐derivative of cAMP. The modified nucleotides were excited at 316 nm, and emission was measured at 410 nm in a fluorometric detector. Quantitation was by peak height measurement against a calibration standard. Analysis was performed in Dr Torres's laboratory, Mayo Translational PKD Center (Rochester, Minnesota).
Unglycosylated aquaporin‐2 concentrations in urine samples were determined by denaturation of protein in a concentrated sample, followed by electrophoresis, visualization by chemiluminescent aquaporin‐2 antibody, and quantitation using image‐analysis software. Analysis was performed in Dr Torres's laboratory, Mayo Translational PKD Center.
In both Trials 248 and 249, the primary PD endpoint was Uosm. Secondary endpoints included duration and percentage of subjects with Uosm <300 mOsm/kg on day 5 (duration determined as the end time of the last consecutive collection interval with a Uosm <300 mOsm/kg); urinary excretion of cAMP (adjusted for urinary creatinine) and unglycosylated aquaporin‐2 were determined by multiplying the concentration by urine volume for each collection interval and adding all the intervals in the 24‐hour period (if urine volume was not recorded for an interval, 24‐hour excretion was not determined); urine volume and 24‐hour fluid balance (fluid intake minus urine volume); and number of times out of bed to urinate during the sleep period. In Trial 250 the primary PD endpoints were spot Uosm in samples obtained prior to the first dose and percentage of subjects with Uosm <300 mOsm/kg for this sample.
Safety Assessments
In Trials 248 and 249, safety was monitored by adverse event (AE) reporting, vital signs (heart rate, blood pressure after sitting ≥5 minutes, and weight), electrocardiogram, physical examination, and clinical laboratory tests. For 7 days after the last dose of tolvpatan, assessments were made of any ongoing AEs, new serious AEs, and the use of concomitant medications.
In Trial 250, safety was assessed by regular monitoring of AEs, directed physical examinations and vital signs, clinical laboratory tests, and electrocardiograms. Information on comorbidities commonly or rarely associated with ADPKD was solicited. A secondary endpoint of tolerability used descriptive assessment of subject self‐reported tolerance of each dose regimen of tolvaptan during the first 4 weeks of trial treatment. Responses to the question, “Could you tolerate taking this dose of tolvaptan for the rest of your life? (‘Yes’ or ‘No’)” were collected and summarized.
Statistical Analyses
In Trials 248 and 249, plasma concentration data of tolvaptan were summarized by treatment and time point. PK parameters of tolvaptan and PD endpoints, absolute values, and changes from baseline were summarized by treatment group and treatment or trial day using descriptive statistics. (SAS [version 8.2, SAS Institute, Cary, North Carolina] and S‐Plus [version 6.1, TIBCO Corp, Palo Alto, California]). In Trial 250, Uosm in samples prior to the first dose, prior to the second dose, and prior to bedtime were summarized by treatment using descriptive statistics. Number (%) of subjects with Uosm <300 mOsm/kg at each time point and dose, and a “Yes” response to the tolerability question at each dose were also reported (SAS, version 8.2).
Results
Subject Disposition and Baseline Characteristics
Eleven subjects with ADPKD enrolled in Trial 248, of whom 8 received 4 ascending doses of tolvaptan and 3 received 4 placebo doses. Thirty‐seven subjects enrolled in Trial 249 and received either tolvaptan 15/15 mg (n = 9), 30 mg QD (n = 9), 30/15 mg (n = 9), or 30/30 mg (n = 10). All 48 subjects in Trials 248 and 249 completed the trials. Of these, 47 were screened, and 46 enrolled in the tolvaptan dose‐titration portion of Trial 250 (Figure 1). The intended starting dose in Trial 250 was tolvaptan 30/15 mg, but 1 subject started on a tolvaptan dose of 15/15 mg and titrated upward after day 8. Sixteen subjects downtitrated from the 60/30‐mg dose due to tolerability. Of the 28 subjects who titrated to the 90/30‐mg dose, 7 answered “No” to the question of lifetime tolerability (see Safety Assessments [Methods]) at week 4 and downtitrated; all of the latter subjects remained on the downtitrated dose of tolvaptan until completing week 8 of the trial.
Figure 1.
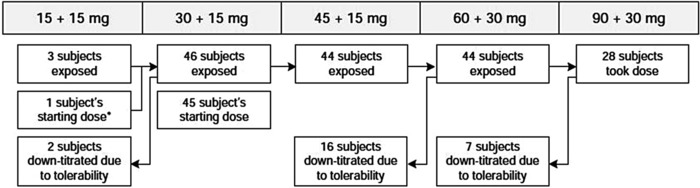
Subject disposition flow chart in Trial 250.
Baseline characteristics of subjects in each trial are presented in Table 1. In Trial 248, 72.7% of subjects (6/8 [75.0%] in the tolvaptan group and 2/3 [66.7%] in the placebo group) received concomitant medications during the trial for treatment of hypertension, including benazepril, captopril, fosinopril, irbesartan, lisinopril, losartan, and olmesartan. In Trial 249, 75.7% of subjects (28/37) received concomitant medications during the trial for the treatment of hypertension, including candesartan, enalapril, fosinopril, irbesartan, lisinopril, olmesartan ramipril, valsartan, and valsartan hydrochlorothiazide.
Table 1.
Baseline Characteristics of Enrolled Subjects in Trials 248, 249, and 250
| Trial 248 | Trial 249 | Trial 250 | ||
|---|---|---|---|---|
| Characteristic | Tolvaptan (n = 8) | Placebo (n = 3) | All Subjects (N = 37) | All Subjects (N = 46) |
| Age, years | 39 (9) | 31 (4) | 42 (8) | 42 (8) |
| Weight, kg | 73 (21) | 90 (10) | 78 (21) | 80 (21) |
| Sex, n (%) | ||||
| Male | 2 (25) | 2 (67) | 8 (22) | 12 (26) |
| Female | 6 (75) | 1 (33) | 29 (78) | 34 (74) |
| Race, n (%) | ||||
| White | 7 (88) | 3 (100) | 35 (95) | 45 (98) |
| Black | 0 (0) | 0 (0) | 1 (3) | 1 (2) |
| Other | 1 (13) | 0 (0) | 1 (3) | 0 (0) |
| Serum creatinine, mg/dL | 1.04 (0.27) | 1.27 (0.32) | 1.03 (0.31) | 1.28 (0.38) |
Values are mean (standard deviation) except where noted.
Pharmacokinetic Profiles
PK parameters for Trial 248 and Trial 249 on day 5 are presented in Table 2. PK parameters for day 1 in Trial 249 are not presented as the accumulation ratios of tolvaptan were approximately 1, indicating no evidence of tolvaptan accumulation or differing pharmacokinetics following multiple administrations.
Table 2.
Mean (SD) Plasma Pharmacokinetic Parameters for Tolvaptan Following Single (Trial 248) or Multiple (Trial 249) Dosing in Subjects With ADPKD
| Trial 248 | Trial 249 (Day 5) | |||||||
|---|---|---|---|---|---|---|---|---|
| 15 mg | 30 mg | 60 mg | 120 mg | 15/15 mg | 30 mg QD | 30/15 mg | 30/30 mg | |
| (n = 8) | (n = 8) | (n = 8) | (n = 8) | (n = 9) | (n = 9) | (n = 9) | (n = 10) | |
| Cmax, ng/mL | 146 | 263 | 481 | 917 | 190 | 330 | 269 | 295 |
| (35) | (75) | (177) | (237) | (61) | (230) | (69) | (122) | |
| tmax, hoursa | 1.00 | 1.00 | 1.50 | 1.50 | 9.00 | 1.98 | 0.98 | 5.47 |
| (1.00‐2.00) | (1.00‐2.00) | (1.00‐3.00) | (1.00‐3.00) | (0.95‐9.98) | (0.98‐2.98) | (0.97‐9.95) | (0.93‐12.02) | |
| AUC∞, ng⋅h/mL | 880 | 1430 | 4150 | 7740 | … | … | … | … |
| (318)b | (615)c | (1140)d | (3100)d | |||||
| AUC0‐24h, ng⋅h/mL | … | … | … | … | 1890 | 2140 | 2770 | 2990 |
| (1070) | (1620) | (2020) | (1640) | |||||
| t1/2,z, hours | 4.5 | 4.3 | 5.1 | 5.6 | 6.2 | 4.3 | 6.4 | 4.7 |
| (2.7) b | (1.3) c | (1.0) d | (2.0) d | (3.3) | (1.2) e | (3.7) d | (1.8) f | |
| CL/F, mL/min/kg | 3.78 | 6.03 | 3.99 | 4.45 | ND | 5.38 | ND | ND |
| (1.69) b | (2.30) c | (1.93) d | (2.66) d | (4.88) | ||||
| Rac, Cmax, ratio | … | … | … | … | 1.04 | 1.03 | 1.04 | 0.91 |
| (0.45) | (0.18) | (0.26) | (0.22) | |||||
| Rac, AUC, ratio | … | … | … | … | 1.16 | 1.09 | 1.21 | 1.02 |
| (0.38) | (0.22) | (0.24) | (0.13) | |||||
In Trial 248, subjects were administered each dose separated by a 72‐hour washout. In Trial 249, split doses were administered 8 hours apart. A placebo tablet was administered in the afternoon for the 30‐mg QD regimen.
ADPKD, autosomal dominant polycystic kidney disease; AUC, area under the curve; CL/F, total body clearance; Cmax, peak plasma concentration; ND, not determined; QD, daily; Rac, ratio of accumulation day 5/day 1; SD, standard deviation; t1/2,z, terminal phase elimination half‐life.
Values are median (min‐max).
n = 4.
n = 7.
n = 5.
n = 8.
n = 9.
In Trial 248 the increase in tolvaptan exposure (AUC∞) with increasing single oral doses of 15‒120 mg was dose proportional (Table 2). In Trial 249 a dose‐related increase in AUC following increased split doses was also observed. Median plasma concentration‐time profiles for tolvaptan in Trial 248 and on day 5 in Trial 249 are shown in Figure 2. Median tolvaptan plasma concentrations following a 30‐mg QD dose in Trial 249 were ∼25% higher compared to the value in the 248 trial. As a CYP3A4‐sensitive substrate, intersubject variability for this compound is high, and the PK parameters for this dose are within the ranges of previously reported values in healthy subjects.13, 14, 15
Figure 2.
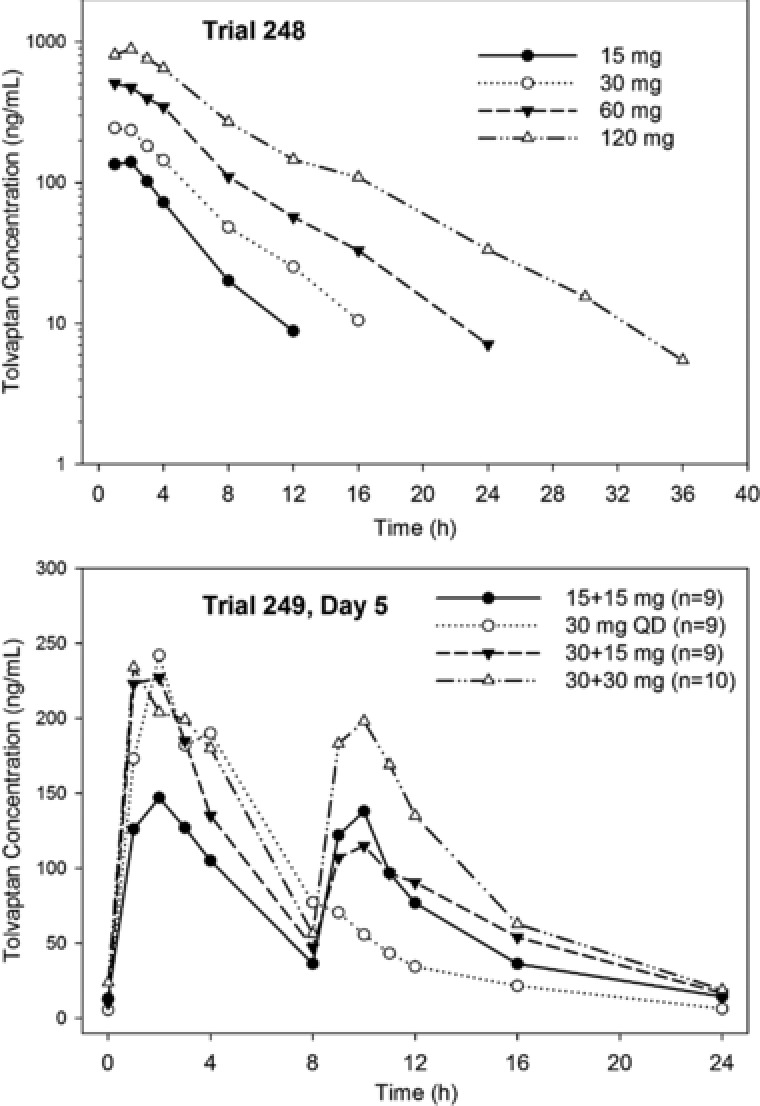
Median tolvaptan plasma concentrations in subjects with ADPKD following single doses (Trial 248) or on day 5 following split doses (Trial 249).
Pharmacodynamic Effects
Animal studies and human PK/PD trials in which free water clearance was determined indicated that Uosm is a good surrogate measure of AVP/aquaporin‐2 action and a biomarker of inhibition at the V2‐receptor.11, 13, 16 In Trial 248 increasing doses of tolvaptan produced greater decreases in Uosm (Figure 3) and increased the percentage of subjects in whom Uosm was suppressed below 300 mOsm/kg at 16 to 24 hours postdose (Table 3). Figure 4 shows the correlation between Uosm and urine excretion rate for urine samples collected from 8 subjects for 0 to 4, 4 to 8, 8 to 12, 12 to 16, 16 to 24 hours at baseline and following 120 mg of tolvaptan. The asymptote for Uosm appeared to be around 50 mOsm/kg; increasing the urine excretion rate beyond 6 mL/min did not produce further reductions in osmolality.
Figure 3.
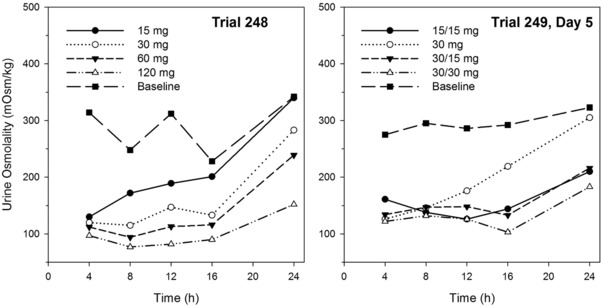
Mean urine osmolality (mOsm/kg) at the end of the collection interval following tolvaptan single doses (Trial 248) or multiple doses for 5 days (Trial 249) in subjects with ADPKD.
Table 3.
Pharmacodynamic Parameters for Tolvaptan Following Single (Trial 248) or Multiple (Trial 249) Dosing in Subjects With ADPKD
| Trial 248 | Trial 249 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 15 mg | 30 mg | 60 mg | 120 mg | 15/15 mg | 30 mg QD | 30/15 mg | 30/30 mg | ||
| Parameter | Endpoint | (n = 8) | (n = 8) | (n = 8) | (n = 8) | (n = 9) | (n = 9) | (n = 9) | (n = 10) |
| Urine osmolality <300 mOsm/kg, 16‐24 hours postdose, n (%) | Day –1 | 0 (0) | 5 (55.6) | 2 (22.2) | 1 (11.1) | 5 (50.0) | |||
| Treatmenta | 3 | 7 | 7 | 8 | 8 | 4 | 7 | 9 | |
| (37.5) | (87.5) | (87.5) | (100) | (88.9) | (44.4) | (77.8) | (90.0) | ||
| Number of times out of bed to urinate, 16‐24 hours postdose | Day –1 | 1.38 (0.52) | 2.78 (0.83) | 1.78 (0.67) | 1.89 (0.60) | 1.50 (0.53) | |||
| Change on day 1 | ‐0.13 | 0.88 | 0.88 | 2.38 | 0.11 | 0.44 | 0.44 | 0.80 | |
| (0.64) | (1.89) | (0.164) | (1.85) | (1.27) | (0.88) | (1.24) | (1.14) | ||
| Change on day 5 | … | … | … | … | 0.0 | 0.33 | 0.44 | 0.70 | |
| (1.58) | (0.71) | (1.01) | (1.06) | ||||||
| 24‐hour fluid balance, mL | Day –1 | 241 | 182 | 525 | 212 | –114 | |||
| (753) | (663) | (461) | (429) | (479) | |||||
| Change on day 1 | –156 | –707 | –659 | –1325 | –707 | –901 | –896 | –754 | |
| (962) | (1090) | (1350) | (848) | (1790) | (1060) | (685) | (1110) | ||
| Change on day 5 | … | … | … | … | 558 | 243 | –36 | –99 | |
| (1040) | (1513) | (770) | (782) | ||||||
| Serum sodium, mEq/L | Predose b | 139 | 138 | 139 | 138 | 138 | 138 | 140 | 139 |
| (1.60) | (1.96) | (2.19) | (3.42) | (2.00) | (162) | (1.72) | (1.75) | ||
| Change to 24 hours postdose, day 1 | –0.63 (1.92) | 1.38 (1.19) | 1.88 (1.55) | 3.00 (3.21) | 3.56 (2.24) | 2.89 (1.17) | 1.22 (1.92) | 3.30 (2.67) | |
| Change to 24 hours postdose, day 5 | … | … | … | … | 1.89 (1.69) | 1.78 (1.79) | 0.11 (1.90) | 2.00 (2.16) | |
| Serum arginine vasopressin, pg/mL | Predoseb | 1.84 (1.27) | 1.65 (1.18) | 1.63 (1.50) | 1.34 (0.95) | 0.82 (0.40) | 1.11 (1.09) | 0.54 (0.11) | 1.34 (0.77) |
| Change to 24 hours postdose, day 1 | 1.16 (1.00) | 2.29 (1.83) | 2.25 (1.56) | 2.75 (1.40) | 1.54 (1.34) | 0.37 (1.03) | 0.91 (0.90) | 2.63 (2.34) | |
| Change to 24 hours postdose, day 5 | … | … | … | … | 0.68 | 0.03 | 0.70 | 0.43 | |
| (0.68) | (0.93) | (0.62) | (0.74) | ||||||
| 24‐hour cAMP urine excretion, nmol/mg creatinine | Day –1 | 1.47 (0.41) | 1.84 (0.30) | 1.47 (0.17) | 1.86 (0.52) | 1.56 (0.47) | |||
| Change on day 1 | 0.12 | 0.43 | 0.26 | 0.19 | –0.18 | –0.19 | –0.07 | –0.01 | |
| (0.24) | (0.28) | (0.27) | (0.23) | (0.17) | (0.29) | (0.20) | (0.10) | ||
| Change on day 5 | … | … | … | … | –0.16 | –0.03 | –0.04 | –0.11 | |
| (0.35) | (0.24) | (0.26) | (0.31) | ||||||
| 24‐hour unglycosylated aquaporin‐2 urine excretion, μg | Day –1 | 168 (61) | 131 (109) | 159 (115) | 101 (50) | 73 (64) | |||
| Change on day 1 | –67 | 60 | ND | ND | –80 | –41 | –32 | –29 | |
| (48) | (247) | (87) | (90) | (78) | (40) | ||||
| Change on day 5 | … | … | … | … | –87 | –72 | –50 | –56 | |
| (80) | (74) | (42) | (67) | ||||||
In Trial 248, subjects were administered each dose separated by a 72‐hour washout. In Trial 249, split doses were administered 8 hours apart. A placebo tablet was administered in the afternoon for the 30‐mg QD regimen. Values are mean (standard deviation) except where noted.
ADPKD, autosomal dominant polycystic kidney disease; ND, not determined, urine too dilute; QD, daily.
Day 5 values for Trial 249.
In Trial 248, value is prior to administration of each dose. In Trial 249, value is prior to dosing on day 1.
Figure 4.
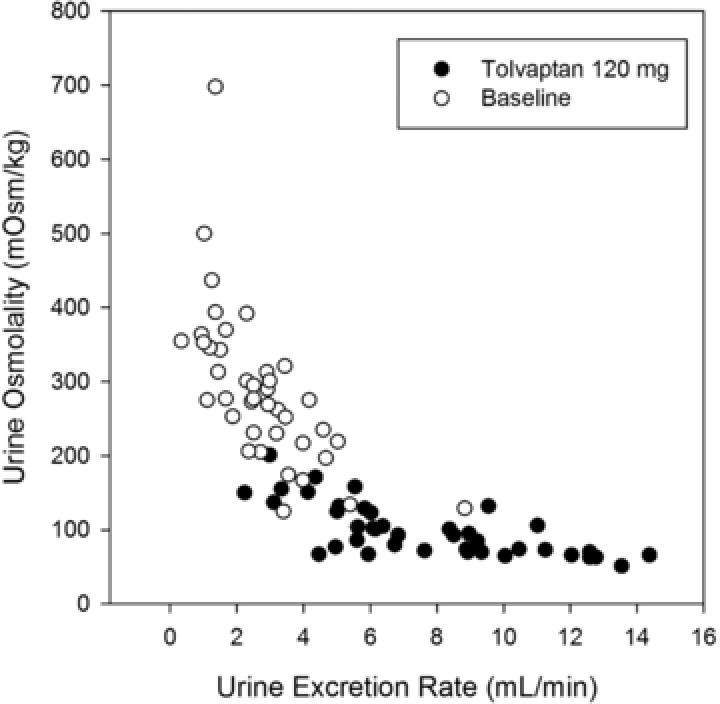
Urine osmolality vs urine excretion rate in 8 ADPKD subjects at baseline or following 120 mg tolvaptan.
In Trial 249 split‐dose regimens combining 15‐ and 30‐mg doses suppressed Uosm similarly to single doses of 60 and 120 mg (Figure 3), but the peak plasma concentrations of tolvaptan were lower following split doses (Figure 2). The variability in baseline Uosm was large, with coefficients of variation for mean values above 50%. Variability in subject responsiveness to tolvaptan was also large, as the coefficient of variation values for changes from baseline in Uosm were also approximately 50%.
The previous analyses from Trials 248 and 249 assessed Uosm over the collection interval and, thus, did not address whether Uosm at 24 hours postdose, the end of the 16‐ to 24‐hour collection interval, may have been higher when tolvaptan concentrations are at their lowest. The spot urine collections in Trial 250 addressed this question. Results showed that Uosm was indeed least suppressed immediately prior to the first dose of tolvaptan, whereas greater suppression was achieved prior to the second and bedtime doses (Figure 5 ). Higher doses not only produced lower mean Uosm at every dose tested but also reduced the percentage of subjects with clinically significant breakthrough; for the highest dose (90/30 mg), 100% of subjects had Uosm <300 mOsm/kg prior to the second dose and prior to bedtime, and 85% were suppressed prior to the first dose (Table 4).
Figure 5.
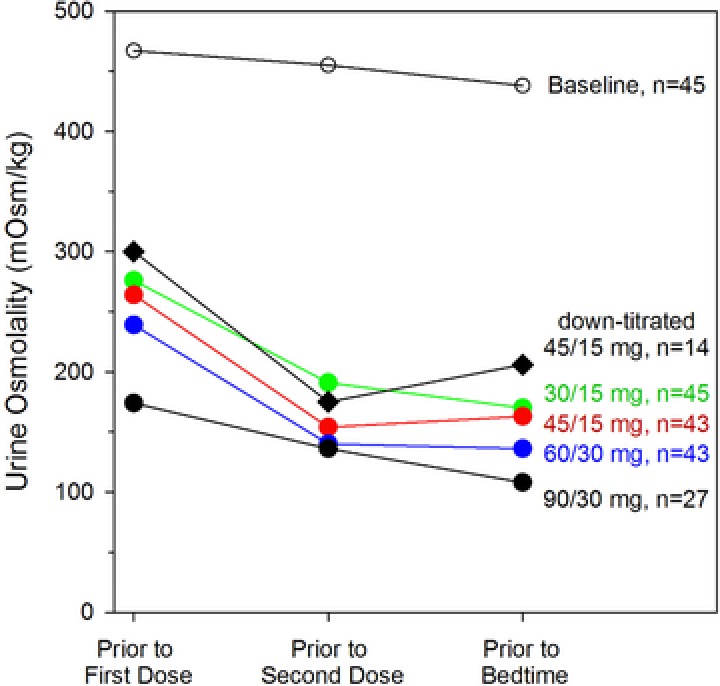
Mean spot urine osmolality concentrations following multiple doses of tolvaptan in subjects with ADPKD (Trial 250).
Table 4.
Percentage of Subjects With Urine Osmolality <300 mOsm/kg and Percentage of Subjects Who Tolerated Titrated Split‐Dose Regimens of Tolvaptan in Subjects With ADPKD in Trial 250
| Week of Treatment and Dose | ||||||
|---|---|---|---|---|---|---|
| Day 0 | Week1 | Week 2 | Week 3 | Week 4 | ||
| Baseline | 30/15 mg | 45/15 mg | 60/30 mg | 45/15 mg | 90/30 mg | |
| (n = 45)a | (n = 45)a | (n = 43)a | (n = 43)a | (n = 14)a | (n = 27)a | |
| Time of day | ||||||
| Prior to first dose | 24 | 64 | 70 | 77 | 42 | 85 |
| Prior to second dose | 33 | 84 | 97.7 | 97.7 | 92.9 | 100 |
| Prior to bedtime | 38 | 91.1 | 93 | 97.7 | 91.7 | 100 |
| Subjects tolerating, % | ||||||
| Dose after 1 week | – | 96 (44/46) | 100 (44/44) | 64 (28/44) | – | 75 (21/28) |
Subjects tolerating is based on the proportion of subjects having a “yes” response to the following query on their tolerability of the tolvaptan dose: “Could you tolerate taking this dose of tolvaptan for the rest of your life, please answer only yes or no?”
Number of subjects with urine osmoality sample at any time of day.
In Trial 248 the mean change from baseline in 24‐hour urine volume increased dose‐proportionally across the 30‐, 60‐, and 120‐mg single doses (Figure 6), consistent with the increased and longer duration of suppression of Uosm. In Trial 249 the mean change from baseline in 24‐hour urine volume also increased following all split‐dose regimens, but there was no evidence of a dose effect, again consistent with the Uosm result. Following multiple doses, urine volume was approximately 20% lower on day 5 than on day 1, consistent with observations in healthy subjects17 and in patients with heart failure.18
Figure 6.
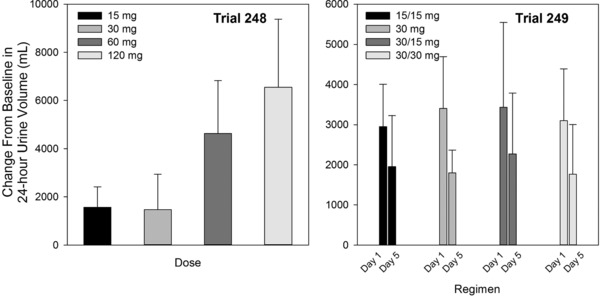
Mean (SD) change from baseline 24‐hour urine volume following tolvaptan single doses (Trial 248) or multiple doses for 5 days (Trial 249) in subjects with ADPKD.
Tolerability to the continued increase in urine output during the nighttime hours was assessed by the change in the number of times subjects got out of bed to urinate (Table 3). Increases in the number of times out of bed to urinate was highest for the highest doses (120 mg QD, 60 mg total dose) in each trial.
In Trial 248 mean 24‐hour fluid balance was similar for the 30‐ and 60‐mg doses, but an overall dose‐related decrease was observed (Table 3) despite the fact that subjects had ad libitum water freely available. Although fluid balance decreased on day 1 across all split‐dose regimens in Trial 249, there was no evidence of a dose relationship (Table 3), consistent with all regimens producing the same change in urine volume. Fluid balance became more positive by day 5 as fluid intake increased (data not shown) and urine volume decreased.
As expected with negative fluid balances on day 1, serum sodium increases were observed at 24 hours postdose (Table 3). In Trial 248 the 120‐mg dose with the largest negative fluid balance had the greatest mean change in serum sodium (3 mmol/L). In Trial 249 increases in serum sodium were lower on day 5 than on day 1, as subjects’ fluid balances were closer to 0.
Although tolvaptan was effectively increasing urine output and increasing serum sodium, there was no dose‐ or exposure‐related increase in serum AVP concentrations following single ascending doses or 5 days of dosing (Table 3). In Trial 249, mean increases in AVP on day 5 were smaller than the mean changes on day 1.
As markers of tolvaptan action, urinary excretion of cAMP, adjusted by urine creatinine, was not decreased following the tolvaptan doses administered in Trials 248 and 249 (Table 3). The change in excretion was highly variable with standard deviations greater than the absolute mean changes for most treatments. However, the mean changes in the urinary excretion of unglycosylated aquaporin 2 were negative on both day 1 and day 5 of Trial 249 with decreases of about 50%. The changes were not statistically significant due to the large variability in response. Following 60‐ and 120‐mg single doses of tolvaptan in Trial 248, the urine became too dilute for determination of aquaporin 2 concentrations.
Safety and Tolerability
Table S1 and Table S2 summarize the most commonly reported treatment‐emergent adverse events (TEAEs) in Trials 248, 249, and during the first 8 weeks of Trial 250. In Trial 248, 2 of the 3 placebo‐treated subjects reported 5 TEAEs. More subjects receiving 15 mg tolvaptan experienced TEAEs than with any other dose: 4 of 8 (50.0%) vs 2 of 8 (25.0%) with 120 mg and 1 of 8 (12.5%) with 30 and 60 mg. In Trial 249, 21 of 37 subjects had TEAEs, with no clear dose‐ or regimen‐related trends observed; dry mouth (29.7%) and fatigue (13.5%) were the most frequently reported TEAEs. In Trials 248 and 249 most treatment‐related AEs were considered mild in intensity, and none were considered severe. There were no deaths during the trials, no serious AEs, and no discontinuations due to AEs. No clinically meaningful changes were observed by electrocardiogram, vital signs, or clinical laboratory tests.
In Trial 250, all subjects reported at least 1 TEAE during the first 8 weeks; most were mild to moderate in intensity. No AEs led to discontinuation of trial medication in the first 8 weeks, and no deaths occurred. The incidence rates of severe and serious AEs were 30.4% (n = 14) and 2.2% (n = 1), respectively. One subject experienced 4 serious TEAEs (abdominal pain [n = 2], pelvic pain, and ruptured ovarian cyst). The 4 most frequently reported TEAEs (Table S1) were pollakiuria (47.8%), thirst (41.3%), nocturia (23.9%), and polyuria (21.7%), all related to the aquaretic side effects of tolvaptan. No signs or symptoms of liver toxicity were observed in any subject.
Trial 250 also had subjects self‐report the tolerance of each tolvaptan dose regimen during the first 4 weeks of treatment (“Could you tolerate taking this dose of tolvaptan for the rest of your life? [Yes or No]”). At week 4, a “No” meant the subject was downtitrated to the 60/30‐mg dose, whereas a “Yes” meant that the subject remained on 90/30 mg until week 8. As presented in Figure 1 and Table 4, tolerability of the 30/15‐mg regimen was good, with 44 of 46 subjects uptitrating to 45/15 mg. All of these 44 subjects then uptitrated to the 60/30‐mg dose. However, following the 60/30‐mg dose, only 28 subjects were able to uptitrate to the 90/30‐mg dose, and 7 of these then downtitrated to 60/30 mg. Consequently, only 21 of 48 subjects tolerated the highest dose regimen of 90/30 mg.
Discussion
In PCK rats, an animal model of cystic renal disease, constant inhibition of the V2 receptor was shown to reduce the rate of disease progression,19 and in other animal models, suppression of Uosm and renal homogenate cAMP were shown to be markers of AVP inhibition.16 It was therefore hypothesized that keeping Uosm <300 mOsm/kg, hypotonic compared to plasma osmolality, would produce an effective treatment for human ADPKD. A previously published report indicated that a single tolvaptan dose of at least 120 mg per day was required to maintain Uosm at <300 mOsm/kg for 24 hours in healthy subjects.11 As it was unclear how the progressive renal dysfunction characterizing ADPKD might impact tolvaptan PK and PD, 3 phase 2 studies on tolvaptan in this population were conducted.
Subjects in the first trial, Trial 248, initiated single‐dose tolvaptan at a relatively low level to maximize safety, ie, 15 mg. Subsequent dose escalation to 120 mg QD was based on tolerability, with a particular focus on aquaretic AEs (thirst, nocturia, etc). Results indicated that tolvaptan PK and PD in ADPKD subjects with well‐preserved renal function (serum creatinine <1.4 mg/dL for men and <1.2 mg/dL for women) were similar to those in healthy subjects. Tolvaptan increased urine excretion rate when plasma concentrations were greater than 25 ng/mL and produced maximal increases in urine excretion rate when concentrations reached 150 ng/mL. The dose‐related suppression of Uosm in Trial 248 (Figure 3) and dose‐related increase in the percentage of subjects with Uosm <300 mOsm/kg at 16 to 24 hours postdose (Table 3) were associated with increasing 24‐hour urine volumes (Figure 6), as concentrations of tolvaptan remained in the effective range for a longer period of time. The increase in urine volume was associated with decreased tolerability to the drug, as subjects showed a greater number of times out of bed to urinate as dose increased (Table 3).
Trial 249 explored the ability of split‐dose regimens to suppress Uosm with the possibility of reduced 24‐hour urine volume but also evaluated 30 mg QD as 7 of 8 subjects at this dose in Trial 248 did have Uosm <300 mOsm/kg from 16 to 24 hours postdose. All the split‐dose regimens had lower mean urine osmolality and more subjects with Uosm <300 mOsm/kg in the 16‐ to 24‐hour period, validating the benefit of split dosing. The 15/15‐mg, 30/15‐mg, and 30/30‐mg regimens produced similar reductions in Uosm (Figure 3) with similar changes in 24‐hour urine volume (Figure 6), likely reflecting the fact that all split tolvaptan dose regimens suppressed Uosm to a maximal extent for at least some period of time. Additionally, there was a large intersubject variability in the change in urine output in response to tolvaptan, with percentage coefficients of variation around 50% (Figure 6). The suppression of Uosm for the split‐dose regimens was similar to that for the single 60‐mg dose, but with a smaller increase in total daily urine volume. There was a difference in nighttime tolerabilities of the dose regimens, as the mean number of times out of bed to urinate did increase as total daily dose increased from 30 to 45 to 60 mg (Table 3). Consistent with results seen following single tolvaptan doses of 60 to 480 mg to healthy subjects,11 mean AVP concentrations in ADPKD subjects did not show large, <3 pg/mL, or dose‐dependent increases following tolvaptan administration.
Elevated cAMP plays a central role in ADPKD pathogenesis via a network of signaling pathways in cystic tissue, including those involved not only in fluid excretion, but also epithelial tubulogenesis, cell proliferation, extracellular matrix interactions, interstitial inflammation, and fibrosis.9 For the single‐ and multiple‐tolvaptan‐dose regimens tested in ADPKD subjects, no change was seen in urinary excretion of cAMP. Although animal models showed reduced cAMP concentrations in renal tissue following treatment with V2‐receptor antagonists,19 it may be that cellular changes in cAMP are not reflected in urine, as excretion of unglycosylated aquaporin 2 did appear to be decreased, consistent with V2‐receptor inhibition.
AVP is the most powerful agonist for cAMP generation in freshly isolated collecting ducts,20 and it was therefore hypothesized that the highest level of AVP antagonism commensurate with good safety and tolerability might generate the most significant clinical benefit in subjects with ADPKD. Consequently, subjects in Trial 250 underwent a forced‐titration protocol with higher tolvaptan doses than those used in Trial 249 (30/15 mg → 45/15 mg → 60/30 mg → 90/30 mg over 4 weeks), and dosing was sustained at the highest tolerated level until 8 weeks after the start of the trial. As anticipated, increasing doses increased the suppression of Uosm and the duration of time that concentrations remained <300 mOsm/kg. However, even the 90/30‐mg regimen was not sufficient to suppress early morning (prior to the first dose) Uosm in 15% of the 27 subjects with Uosm measurements.
Sixteen subjects treated with the 60/30‐mg regimen indicated that they could not tolerate that dose and downtitrated to 45/15 mg at week 4. The mean spot Uosm values for this group were about 25 mOsm/kg higher when compared to the mean values for all subjects treated with this regimen (Figure 5), and fewer than half of these subjects had Uosm suppressed in the early morning (Table 4).
Because the hypothesis for ADPKD treatment is that greater/longer suppression of Uosm (greater inhibition of AVP) would produce better efficacy, it was of interest to explore reasons why some subjects could not uptitrate. Tolerability to dosing was associated with the adverse events related to tolvaptan's action in promoting free water clearance, as the most common treatment‐emergent AEs in Trial 250 were pollakiuria, thirst, nocturia, and polyuria. Given the limitations imposed on tolvaptan therapy by aquaretic AEs, the identification of biomarkers associated with renal function was explored first. Subjects who were able to titrate to the 90/30‐mg dose in Trial 250 had lower baseline spot Uosm values on awakening, less change from baseline following a 30/15‐ or 45/15‐mg tolvaptan dose, and lower baseline estimated glomerular filtration rate (eGFR) than those subjects who were unable to titrate beyond the 60/30‐mg split dose (Table 5). These results suggest that ADPKD subjects with more compromised kidney function, as reflected in lower ability to concentrate urine and lower eGFR, were less susceptible to or better able to tolerate the aquaretic side effects of tolvaptan. This was most likely due to the fact that subjects with lower GFR were shown to have smaller increases in urine excretion rate and total daily urine volume following tolvaptan treatment when compared to subjects with higher GFR.21
Table 5.
Mean (SD) Baseline eGFR and Spot Urine Osmolality on Awakening or Prior to First Dose in Trial 250
| Urine Osmolality | ||||
|---|---|---|---|---|
| Group | Baseline | Change From Baseline Following 30/15 mg | Change From Baseline Following 45/15 mg | Baseline eGFRMDRD |
| Did not uptitrate past 60/30 mg (n = 17) | 558 (226) | –230 (197) | –278 (239) | 66.5 (18.0) |
| Titrated to 90/30 mg (n = 28) | 411 (212) | –156 (220) | –158 (181) | 53.5 (17.3) |
Units: Uosm, mOsm/kg; eGFR, mL/(min·1.73 m2).
eGFR, estimated glomerular filtration rate; SD, standard deviation.
The split‐dose regimens were designed to use a higher dose early in the day and a lower dose 8 to 9 hours later to produce maximal AVP inhibition on awakening, with a gradual falling off of effect during the night when frequent urination might lead to interruption of sleep. As predicted from Trial 249, all split‐dose regimens in Trial 250 resulted in good suppression of Uosm, although even the 90/30‐mg regimen exhibited 15% clinical breakthrough on awakening, as measured by Uosm >300 mOsm/kg. Tolerability of the 30/15‐ and 45/15‐mg doses was good but decreased for the 60/30‐ and 90/30‐mg doses. Accordingly, patients in the pivotal TEMPO 3:4 trial initiated tolvaptan treatment at a split dose of 45/15 mg, based on the evidence that this was the highest split dose that was most tolerable in ADPKD patients, followed by a forced titration to 60/30 mg and 90/30 mg according to subject‐reported tolerability.6
Conclusion
Tolvaptan PK and PD in ADPKD subjects with well‐preserved renal function are similar to those observed in healthy subjects. Trials 248, 249, and 250 demonstrated that oral tolvaptan provided effective inhibition of AVP, as measured by suppression of Uosm. Overall, split dosing of tolvaptan offered the best balance between suppression for 24 hours and tolerability. Most subjects could tolerate the 45/15‐mg dose, a dose that effectively inhibited the fluid balance effects of AVP, but tolerability declined at higher doses (60/30 and 90/30 mg), with fewer than half of subjects on the 90/30‐mg regimen reporting that they could tolerate that dose for the rest of their lives. Subjects with less compromised kidney function or greater suppression of Uosm in response to tolvaptan therapy may have had greater sensitivity to aquaretic AEs, suggesting that added attention on the part of the clinician may be required in this population during tolvaptan titration. Overall, the forced‐titration protocol used in TEMPO 3:4 was designed to provide the most constant and complete inhibition of the vasopressin V2‐receptor while being tolerated during chronic treatment.
Disclosures
A.B.C. and V.E.T. are members of the TEMPO 3:4 and 4:4 Steering Committee. S.E.S., J.O., and F.S.C. are employees of Otsuka Pharmaceutical Development & Commercialization, Inc (Rockville, Maryland). A.B.C. and V.E.T. have received research funding from Otsuka Pharmaceutical Development & Commercialization, Inc. A.B.C. has received consultancy fees from Otsuka Pharmaceutical Development & Commercialization, Inc. David Norris of Ecosse provided editorial assistance for the methods section and formatted text and references to comply with journal requirements with funding by Otsuka.
Supporting information
Table S1. Most Commonly Reported Treatment‐Emergent Adverse Events. Events are included if they occurred in ≥ 2 subjects in Trial 248 and at ≥ 5% incidence in Trial 249 and Trial 250 (first 8 weeks).
Table S2. Most Commonly Reported Treatment‐Emergent Adverse Events in Placebo‐treated Subjects in Trial 248.
References
- 1. Berl T, Quittnat‐Pelletier F, Verbalis JG, et al. Oral tolvaptan is safe and effective in chronic hyponatremia. J Am Soc Nephrol. 2010;21(4):705–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Schrier RW, Gross P, Gheorghiade M, et al. Tolvaptan, a selective oral vasopressin V2‐receptor antagonist, for hyponatremia. N Engl J Med. 2006;355(20):2099–2112. [DOI] [PubMed] [Google Scholar]
- 3. Baumgarten R, van de Pol MH, Deen PM, van Os CH, Wetzels JF. Dissociation between urine osmolality and urinary excretion of aquaporin‐2 in healthy volunteers. Nephrol Dial Transplant. 2000;15(8):1155–1161. [DOI] [PubMed] [Google Scholar]
- 4. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. [DOI] [PubMed] [Google Scholar]
- 5. Torres VE, Meijer E, Bae KT, et al. Rationale and design of the TEMPO (Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes) 3‐4 Study. Am J Kidney Dis. 2011;57(5):692–699. [DOI] [PubMed] [Google Scholar]
- 6. Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367(25):2407–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamaguchi T, Nagao S, Wallace DP, et al. Cyclic AMP activates B‐Raf and ERK in cyst epithelial cells from autosomal‐dominant polycystic kidneys. Kidney Int. 2003;63(6):1983–1994. [DOI] [PubMed] [Google Scholar]
- 8. Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11(7):1179–1187. [DOI] [PubMed] [Google Scholar]
- 9. Torres VE, Harris PC. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J Am Soc Nephrol. 2014;25(1):18–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reif GA, Yamaguchi T, Nivens E, Fujiki H, Pinto CS, Wallace DP. Tolvaptan inhibits ERK‐dependent cell proliferation, Cl(–) secretion, and in vitro cyst growth of human ADPKD cells stimulated by vasopressin. Am J Physiol Renal Physiol. 2011;301(5):F1005–F1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shoaf SE, Wang Z, Bricmont P, Mallikaarjun S. Pharmacokinetics, pharmacodynamics, and safety of tolvaptan, a nonpeptide AVP antagonist, during ascending single‐dose studies in healthy subjects. J Clin Pharmacol. 2007;47(12):1498–1507. [DOI] [PubMed] [Google Scholar]
- 12. Skowsky WR, Rosenbloom AA, Fisher DA. Radioimmunoassay measurement of arginine vasopressin in serum: development and application. J Clin Endocrinol Metab. 1974;38(2):278–287. [DOI] [PubMed] [Google Scholar]
- 13. Shoaf SE, Bramer SL, Bricmont P, Zimmer CA. Pharmacokinetic and pharmacodynamic interaction between tolvaptan, a non‐peptide AVP antagonist, and furosemide or hydrochlorothiazide. J Cardiovasc Pharmacol. 2007;50(2):213–222. [DOI] [PubMed] [Google Scholar]
- 14. Shoaf SE, Bricmont P, Mallikaarjun S. Absolute bioavailability of tolvaptan and determination of minimally effective concentrations in healthy subjects. Int J Clin Pharmacol Ther. 2012;50(2):150–156. [DOI] [PubMed] [Google Scholar]
- 15. Shoaf SE, Kim SR, Bricmont P, Mallikaarjun S. Pharmacokinetics and pharmacodynamics of single‐dose oral tolvaptan in fasted and non‐fasted states in healthy Caucasian and Japanese male subjects. Eur J Clin Pharmacol. 2012;68(12):1595–1603. [DOI] [PubMed] [Google Scholar]
- 16. Wang X, Wu Y, Ward CJ, Harris PC, Torres VE. Vasopressin directly regulates cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2008;19(1):102–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shoaf SE, Ohzone Y, Ninomiya S, et al. In vitro P‐glycoprotein interactions and steady‐state pharmacokinetic interactions between tolvaptan and digoxin in healthy subjects. J Clin Pharmacol. 2011;51(5):761–769. [DOI] [PubMed] [Google Scholar]
- 18. Hauptman PJ, Zimmer C, Udelson J, et al. Comparison of two doses and dosing regimens of tolvaptan in congestive heart failure. J Cardiovasc Pharmacol. 2005;46(5):609–614. [DOI] [PubMed] [Google Scholar]
- 19. Wang X, Gattone V 2nd, Harris PC, Torres VE. Effectiveness of vasopressin V2 receptor antagonists OPC‐31260 and OPC‐41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005;16(4):846–851. [DOI] [PubMed] [Google Scholar]
- 20. Yasuda G, Jeffries WB. Regulation of cAMP production in initial and terminal inner medullary collecting ducts. Kidney Int. 1998;54(1):80–86. [DOI] [PubMed] [Google Scholar]
- 21. Shoaf SE, Bricmont P, Mallikaarjun S. Pharmacokinetics and pharmacodynamics of oral tolvaptan in patients with varying degrees of renal function. Kidney Int. 2014;85(4):953–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Most Commonly Reported Treatment‐Emergent Adverse Events. Events are included if they occurred in ≥ 2 subjects in Trial 248 and at ≥ 5% incidence in Trial 249 and Trial 250 (first 8 weeks).
Table S2. Most Commonly Reported Treatment‐Emergent Adverse Events in Placebo‐treated Subjects in Trial 248.