

Abstract
Monoclonal antibodies (mAbs) have emerged as a mainstream therapeutic option against cancer. mAbs mediate tumor cell-killing through several mechanisms including complement-dependent cytotoxicity (CDC). However, studies of mAb-mediated CDC against tumor cells remain largely dependent on in vitro systems. Previously developed and widely used NOD-scid IL2rγnull (NSG) mice support enhanced engraftment of many primary human tumors. However, NSG mice have a 2-bp deletion in the coding region of the hemolytic complement (Hc) gene, and it is not possible to evaluate CDC activity in NSG mice. To address this limitation, we generated a novel strain of NSG mice—NSG-Hc1—that have an intact complement system able to generate the membrane attack complex. Utilizing the Daudi Burkitt's human lymphoma cell line, and the anti-human CD20 mAb rituximab, we further demonstrated that the complement system in NSG-Hc1 mice is fully functional. NSG-Hc1 mice expressed CDC activity against Daudi cells in vivo following rituximab treatment and showed longer overall survival compared with rituximab-treated NSG mice that lack hemolytic complement. Our results validate the NSG-Hc1 mouse model as a platform for testing mechanisms underlying CDC in vivo and suggest its potential use to compare complement-dependent and complement-independent cytotoxic activity mediated by therapeutic mAbs.
Keywords: Immunotherapy, complement-dependent cytotoxicity (CDC), patient-derived xenograft (PDX), NSG mice, monoclonal antibody (mAb)
Introduction
Cancer cells, as well as stromal and vascular cells in the tumor microenvironment express novel antigens that distinguish them from their normal counterparts (Deckert, 2009; Nelson et al., 2010; Ravetch, 2010). With the continued advances in high–throughput sequencing tools, large-scale efforts to identify novel patient-specific tumor antigens are underway for personalized therapeutic interventions. As a result, monoclonal antibodies (mAbs), due to their specific reactivity to target antigens, are promising therapeutic agents for treating patients with malignancies (Scott et al., 2012).
The cytotoxic effects of mAbs are mediated through a number of mechanisms, including direct action of the antibody, receptor blockade activity, induction of apoptosis, delivery of a drug or cytotoxic agent, antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), regulation of T cell function, and specific effects on tumor vasculature and stroma (Scott et al., 2012). The function of the Fc region of antibodies, which is within the constant region, is particularly important for mediating tumor cell-killing through ADCC and CDC, as both of these cytotoxic mechanisms require recognition of the Fc constant region of cell-bound, aggregated mAbs, by either Fcγ receptors on effector cells (natural killer cells or macrophages), or activation of the complement system, respectively. Therefore, engineering the Fc region of mAbs to enhance their cytotoxic activity is currently an active area of investigation (Moore et al., 2010). However, experimental systems available to distinguish between complement-dependent and independent therapeutic benefit of these next-generation mAbs, especially in vivo, are limited.
Traditional in vitro mAb screening protocols using primary tumor cell lines are unreliable as predictors of efficacy responses in vivo because they do not represent the natural milieu of the human tumor microenvironment. Importantly, however, researchers can now use severely immunodeficient NOD-scid IL2rγnull (NSG) mice as a custom patient-derived xenograft (PDX) model to explore “personalized” therapy options (Brehm et al., 2016). NSG mice are widely used as hosts for growth of a variety of solid human tumors and hematological malignancies, i.e., acute myelogenous leukemia, (Ishikawa et al., 2007), acute lymphocytic leukemia, (Agliano et al., 2008), chronic lymphocytic leukemia (Wong et al., 2012), and chronic myelogenous leukemia (Zhang et al., 2010), to test experimental therapeutics. Others and we have shown that patient-derived tumors engrafted into NSG mice (NSG-PDX mice) closely recapitulate their corresponding primary neoplasm and can provide a virtually unlimited supply of tumor material, fostering multiple molecular and biological studies. One particular advantage of using NSG mice as PDX-based disease models is that they can also be engrafted with patient-derived immune systems to reconstitute a robust patient-specific immune system and to test immunotherapeutics (Tanaka et al., 2012).
The complement system includes >15 soluble proteins as well as numerous serosal proteins and receptors and functions in multiple biological processes including cytotoxic activity against cancer cells and killing of pathogens. The complement system is activated in three ways: via the classical pathway, which includes the proteins C1 through C4; the alternative pathway, with the participation of C3 and protein factors B, D, and P; and the lectin pathway, with the participation of mannose-binding lectin (MBL) and MBL-associated proteases (Ricklin et al., 2010; Tegla et al., 2011). All three pathways can lead to activation of the C5, C6, C7, C8, and C9 proteins, which initiate formation of the C5b-9 terminal complement complex on target cell membranes and this membrane attack complex causes a Ca2+ -dependent acute cell death (Tegla et al., 2011). The complement component C1q becomes activated when it recognizes the Fc portion of cell-bound mAbs, which can result in formation of pores on the target cell surface by the C5b-9 MAC, thus lysing the cell by CDC (Ricklin et al., 2010).
NSG mice have a key limitation preventing their use for testing complement-dependent cytotoxicity. NSG strains are congenic on the NOD/ShiLt strain background. NOD/ShiLt mice lack hemolytic complement due to a 2-bp deletion in the coding region of the hemolytic complement (Hc) gene, which encodes the C5 complement component and, the absence of a functional membrane attack complex prevents mAb-mediated CDC (Baxter and Cooke, 1993). In the present study we focused on the development of complement-sufficient NSG mice to test the CDC-mediated therapeutic efficacy of mAbs in vivo. To address this, we backcrossed the intact Hc gene from the NOD.CBALs-Hc1/Lt congenic strain to the NSG strain to generate a new strain of NSG mice that has an intact complement system: NOD.Cg-Hc1PrkdcscidIl2rgtm1Wjl/Sz, (NSG-Hc1) complement-sufficient mice. We then characterized NSG-Hc1 mice for their ability to mediate complement-independent versus complement-dependent cytotoxicity of rituximab anti-human CD20 antibody against Burkitt's B-cell lymphoma Daudi cells following engraftment into NSG and NSG-Hc1 mice. The increased survival of the Daudi cell-engrafted NSG-Hc1 mice following rituximab treatment indicate that the NSG-Hc1 mouse strain paired with the complement-deficient NSG strain will be a valuable platform for discriminating between complement-independent and CDC- therapeutic efficacy of mAbs in vivo.
Materials and methods
Mice
The following stocks were obtained from research or Repository colonies at The Jackson Laboratory: NOD.CBALs-Hc1/LtJ, an NOD/ShiLt congenic mouse strain, carrying the Hc1 allele derived from CBALs mice (stock #004306); BALB/cByJ (stock #001026); and NOD-Prkdcscid Il2rgtmWjl/SzJ (NSG) mice (stock #005557). NOD.Cg-Hc1 PrkdcscidIl2rgtm1Wjl/SzJ, hereafter named NSG-Hc1, mouse colonies were maintained by sib matings. All mice were reared on NIH 31 M diet and acidified water ad libitum under modified barrier conditions at The Jackson Laboratory in a 12-hr dark/12-hr light cycle. The Institutional Animal Care and Use Committee of The Jackson Laboratory approved all animal procedures.
Sanger sequencing and genotyping
Total genomic DNA was prepared from 2-mm slices of tails of 4-6 week old mice using 50-mM NaOH heat step at 95°C followed by neutralization and centrifugation to pellet the debris. A 210-bp product containing exon 5 of the Hc gene was amplified using the following primer set: forward: CAATTAAAGCTTACTATAAGAAGGATTTTACAA and reverse: CAAGTTAGATCTAAGCACTAGCTACTCAAACAA. Bands of interest were excised and gel-extracted using the QIAquick Gel Extraction kit (Qiagen, Germantown, MD) and eluted in 30μl double-distilled water. If gel extraction was not necessary to separate multiple bands, PCR products were directly cleaned using ExoSAP-It (USB, Cleveland, OH). DNA samples were quantitated using the Nanodrop ND-1000 UV spectrophotometer (Nanodrop Technologies, Wilmington, DE). Sequencing reactions with gene-specific primers were carried out using BigDye® Terminator Cycle Sequencing chemistry and resolved on an AB3703xl (Applied Biosystems Life Technologies, Carlsbad, CA). cDNA was sequenced from both strands of NSG and NSG-Hc1 mice. Sequencher 4.9 (Gene Codes, Ann Arbor, MI) was used to assemble DNA sequences.
Cell culture, reagents, and antibodies
Sheep RBCs (SRBC) (Cat# 10H702) and rabbit antiserum to SRBC (Cat# 10K945) were purchased form Grainger (McAllen, Texas). Daudi Burkitt's lymphoma cells were purchased from the ATCC (CCL-213). SRBC were washed and maintained in HBSS medium. Daudi cells were thawed and cultured in RPMI-1640 medium (GIBCO# 21870) with 10% FBS, 2mM L-Glutamine, 100units/ml penicillin, and 100μg/ml streptomycin (GIBCO# 15140-122). Daudi cells were incubated at 37°C in a humidified atmosphere containing 5% CO2. RPMI growth medium was changed and cultures were split at a ratio of 1:10 every two to three days. Daudi cell viability was determined by Trypan Blue exclusion method. Rituximab (Cat# 680563) was purchased from Biogen Idec (Cambridge, MA).
Histology and immunohistochemistry
Mice were euthanized by CO2 asphyxiation followed by either cervical dislocation or thoracic puncture. Complete necropsy of the animals was performed and tissues were fixed in 10% neutral buffered formalin (NBF) solution, embedded in paraffin, and sectioned at 3-5 μm. Slides were stained with Mayer's hematoxylin and eosin (H&E), and anti-human CD45 antibody (clone 2B11+PD7/26, Dako Inc. Carpinteria, CA). Qualitative detection of CD45 antigens was done using the DISCOVERY DAB Map Detection Kit (RUO) (Ventana Medical Systems, Tucson, AZ) and photographs were taken with a photomicroscope (Olympus BX41).
Flow cytometry
Peripheral blood was collected from mice by puncture of the retro-orbital venous plexus directly into flow tubes containing FACS buffer (PBS containing 2% FBS and 0.01% NaN3). Mice were then euthanized by CO2 asphyxiation or cervical dislocation. Bone marrow was collected by flushing of the femurs. Spleen cells were collected following mechanical disaggregation. Single-cell suspensions prepared from blood, spleen, and bone marrow were subjected to RBC lysis using Ammonium-Chloride-Potassium (ACK) lysis buffer and washed twice before blocking in rabbit immunoglobulin commercially purchased from Sigma-Aldrich (St Louis, MO). Blocked RBC-depleted cells were stained using fluorescently conjugated antibodies to anti-human CD45 antibody (clone HI30) and anti-mouse CD45 antibody (clone A20), from BD Biosciences (San Jose, CA). Additionally, anti-human CD20 antibody (clone 2H7) from BioLegend (San Diego, CA) was used to confirm that the human Daudi cells were CD20 positive. Fixable viability dye eFluor® 780 from BD Biosciences (San Jose, CA) was used to stain dead cells to discriminate viable from non-viable cells during flow cytometric analysis. All phenotyping analyses were carried out by flow cytometry using a FACSCalibur (Becton Dickinson, San Jose, CA), and data were processed using FlowJo software.
In vitro complement-dependent cytotoxicity assay
Whole mouse blood was collected directly into Becton Dickinson microtainer tubes with serum separator and allowed to clot by leaving undisturbed for 15 minutes at room temperature. The clots were removed by centrifugation and the sera were removed using sterile pipettes. Complement activity in mouse sera was determined by measuring the lysis of antibody-coated SRBC in various serum dilutions. Briefly, SRBC cells incubated with anti-SRBC antibody (EA cells) were prepared by mixing an equal volume of RBCs (1 × 109 cells/ml) and 1:10 diluted rabbit anti-SRBC Ab in HBSS and incubating at 37°C for 30 min. The complement activity was determined by mixing 25μl of EA cells at 109/ml in HBSS medium with 175μl of 1:5 diluted mouse sera in a V-bottom 96-well plate. EA cells incubated either with ACK lysis buffer or only HBSS medium were used as positive and negative controls, respectively. EA cells and sera were incubated for 1 hour at 37 °C and centrifuged for 10 min at 2,500xg, and the OD 405 of the supernatant measured to assess EA cell lysis. Percentage complement-dependent lysis in mouse sera was calculated relative to cells lysed in ACK lysis buffer (100% lysis) and cells incubated with HBSS only (0% lysis).
In vivo complement-dependent cytotoxicity assay
On day 0 age-matched female NSG and NSG-Hc1 mice were divided into five groups (5-7 mice/group) and injected with 1 × 105 viable Daudi cells via the tail vein. Mice that were not injected with tumor cells were used as additional controls. At 10 days post-engraftment, cohorts of mice were injected intraperitoneally (IP) with either with rituximab (25μg/g) in 200μL of PBS or with 200 μL of PBS only. Tumor-engrafted mice were monitored three times per week and euthanized by CO2 asphyxiation followed by thoracic puncture when they exhibited hind leg paralysis or showed >20% weight loss. Tumor burden and tumor invasiveness in mice were evaluated via flow cytometry, histology, and immunohistochemistry. Statistical differences in survival between the treated and control groups were analyzed by Kaplan-Meier plots using Prism statistical software. Statistical analysis was performed using the Log-Rank test, and differences were considered significant at a P-value of <0.05.
Results
Generation of NSG-Hc1 mice
Absence of hemolytic complement lytic activity in NOD mice results from a 2-base pair deletion in the Hc gene, which encodes the C5 complement component. The termination codon UGA is present 4bp downstream from the deletion that causes the lack of C5 protein expression (Fig 1A,B), (Baxter and Cooke, 1993). The defective Hc0 allele expressed by NOD/ShiLt mice (Hc locus, Chr 2) was replaced by a wild-type Hc1 allele from CBA by outcrossing a CBA/Ls female to an NOD/ShiLt male mouse, followed by ten backcrosses to NOD/ShiLt mice and fixation of sex chromosomes from NOD/ShiLt mice (Chen et al., 2005). We obtained the homozygous NOD-Hc1 congenic mouse strain, carrying Chr 2 alleles derived from CBALs, including the Hc1 allele, i.e., NOD.CBALs-Hc1/Lt (stock #004306), from The Jackson Laboratory Repository. NOD.CBALs-Hc1/Lt males were mated with female NSG mice. cDNA from F1 offspring mice were subjected to targeted Sanger sequencing capturing up to a 0.21-kb region on exon 5 of the Hc gene. The F1 mice were heterozygous for the wild-type Hc gene (Hc0/Hc1), the Prkdcscid mutation, and the Il2rgtm1Wj l allele. These heterozygous F1 mice were then intercrossed to produce NSG mice homozygous for the Prkdcscid, IL2rgtm1Wjl, and Hc1 alleles, i.e., NSG-Hc1 mice (Fig. 1A,B). The colony is maintained by sib matings of NSG-Hc1 mice.
Figure 1.
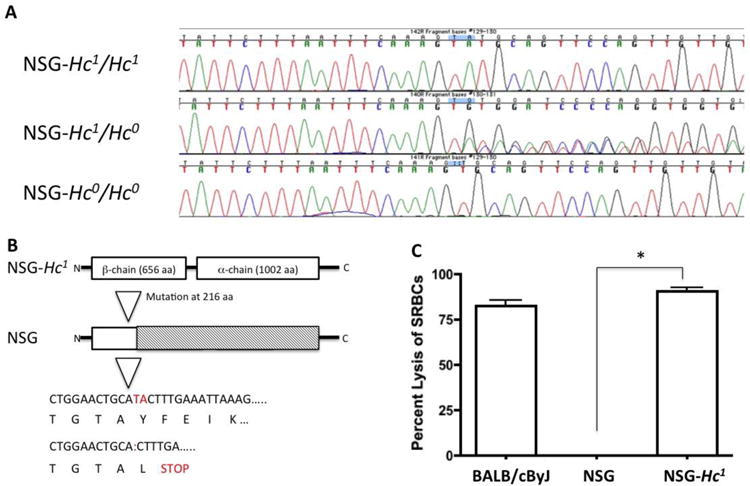
Representative image of PCR- and sequencing-based analysis of (A) the Hc gene from NSG and NSG-Hc1 mice demonstrating a 2 bp deletion in NSG mice. (B) Protein structure showing a 2-bp deletion leading to a stop codon and elimination of the α and β chains in NSG mice. (C) Representative image demonstrating level of lysis of antibody-coated SRBC following incubation with 1:5 diluted sera from age- and sex-matched BALB/cByJ, NSG, or NSG-Hc1 mice. Sera from BALB/cByJ mice was used as positive complement control. Data are reported as the mean ± SD. p < 0.0001.
In vitro characterization of complement-dependent cytotoxicity in NSG-Hc1 mice
Sera from 9-10-month-old male BALB/cByJ, NSG, and NSG-Hc1 mice were collected and the level of complement activity in mouse sera was determined by examining the capacity of 1:5 diluted mouse sera to lyse antibody-coated SRBC, i.e., sensitized cells (EA cells). Sera from BALB/cBy mice, which carry the wild-type Hc1 allele, was used as a positive control. Sera from NSG-Hc1 mice produced similar levels of lysis of EA cells as sera from BALB/cBy mice (Fig. 1C). However, <0.1% of EA-cell lysis was observed when incubated with sera from NSG mice (Fig. 1C). These results confirm that the congenic Hc1 allele in NSG-Hc1 mice transcribes a functional C5 protein.
In vivo characterization of complement-dependent cytotoxicity in NSG-Hc1 mice
CDC of rituximab against Daudi cells was used to evaluate the utility of NSG-Hc1 mice as a platform for testing the CDC-dependent therapeutic efficacy of mAb therapy against human tumors in vivo. Age- and sex-matched NSG and NSG-Hc1 mice were injected intravenously (IV) with 1×105 Daudi cells per mouse. At 10 days post-engraftment, mice were injected IP with either PBS or rituximab. At 38 days post-injection of Daudi cells, we observed significant weight loss and hind limb paralysis in PBS-treated NSG and NSG-Hc1 mice when compared to rituximab-treated mice (Fig. 2A). However, no significant difference in overall survival was observed between PBS-treated NSG mice (median survival=36 days) and PBS-treated NSG-Hc1 mice (median survival=38 days) (Fig. 2B). All mice in the PBS-treated cohorts exhibited either >20% weight loss or hind limb paralysis and were subsequently euthanized by 42 days post-engraftment. Rituximab treatment resulted in a significant complement-independent increase in the overall survival of NSG mice associated with a delayed loss of body weight and hind limb paralysis (median survival=46 days) when compared to the PBS-treated cohorts, (p<0.05) (Fig. 2A, B). The rituximab-treated NSG-Hc1 mice survived throughout the 52-day observation period. No loss of body weight was observed in rituximab-treated NSG-Hc1 mice throughout the observation period, suggesting that the functional complement system in NSG-Hc1 mice treated with rituximab mice significantly improved mouse survival by mediating effective CDC activity against human tumor cells (Fig. 2A, B).
Figure 2.
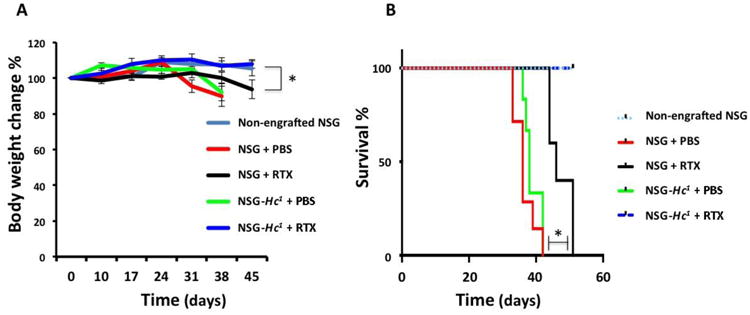
(A) Change in body weight over time in different cohorts of age- and sex-matched NSG and NSG-Hc1 mice engrafted with Daudi cells. (B) Kaplan-Meier survival plot demonstrate percent of survival over time in different cohorts of age- and sex-matched NSG and NSG-Hc1 mice engrafted with Daudi cells. Non-engrafted NSG mice were used as negative controls. Data are reported as percentage of the mean ± SE. PBS, phosphate buffer saline; RTX, rituximab; p < 0.05.
Flow cytometry analysis of circulating human CD45+ cells in peripheral blood at 38 days post-engraftment confirmed the CDC-mediated antitumor activity in NSG-Hc1 mice, as we observed significantly lower numbers of human CD45+ Daudi cells in peripheral blood of rituximab-treated NSG-Hc1 mice when compared to rituximab-treated NSG mice (p<0.05) (Fig. 3A). Interestingly, we observed a significant reduction in the number of human CD45+ cells in the bone marrow of rituximab-treated NSG mice (˜3% of viable cells) when compared to PBS treated NSG mice (˜63% of viable cells), suggesting a complement independent inhibitory effect of rituximab on bone marrow metastasis of tumor cells (p<0.05) (Fig. 3B). Other than complement mediated lytic activity, Rituximab is known to induce tumor-cell killing through direct apoptosis and/or ADCC (Scott et al., 2012). Although NSG mice lack natural killer cells to mediate NK cell dependent ADCC, there could be possible involvement of other mouse immune cells (e.g., macrophage and neutrophil) mediated ADCC in NSG mice. However, further investigation will be needed for clarification of the underlying mechanism. On the other hand, rituximab treatment greatly reduced bone marrow infiltration of human CD45+ in NSG-Hc1 mice (0.2% of viable cells) when compared to PBS-treated NSG-Hc1 mice (˜51% of viable cells), suggesting a more pronounced inhibitory effect of rituximab on bone marrow metastasis in the presence of complement, (p<0.05) (Fig. 3B). In addition, numbers of double negative cells of non-hematopoietic origin were significantly reduced in mice showing higher human CD45+ engraftment rates, suggesting an effective replacement of cells of nonhematopoietic origin as well by outgrowth of human tumor cells, (Fig. 3A, B).
Figure 3.
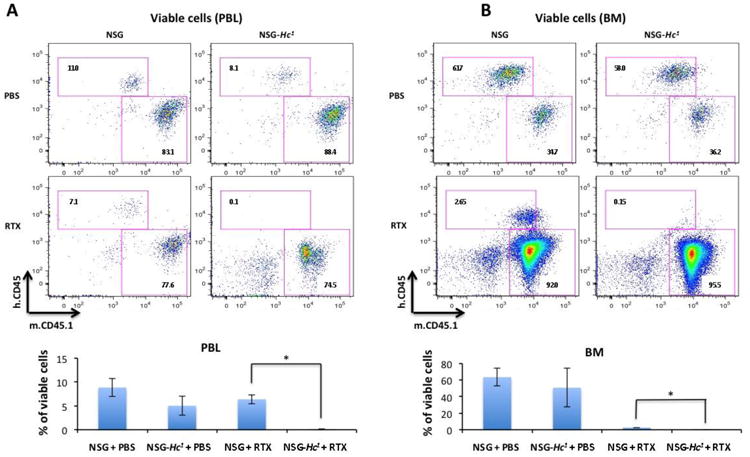
Representative flow cytometry analysis of human CD45+ cells in (A) peripheral blood and (B) bone marrow 38 days post-engraftment demonstrating percentages of viable cells in different cohorts of age- and sex-matched NSG and NSG-Hc1 mice displaying levels of Daudi cell engraftment. Non-engrafted NSG mice were used as negative controls (data not shown). Data are reported as percentage of the mean ± SD. PBS, phosphate buffer saline; RTX, rituximab; p < 0.05.
Moreover, as expected, histological analysis of various organs, including ovary, kidney, liver, spleen, lungs and lymph nodes, of mice at 38 days post-engraftment demonstrated reduced numbers of Daudi cells in tissues from rituximab-treated NSG-Hc1 mice compared to the other groups of mice (Fig. 4A, B).
Figure 4.
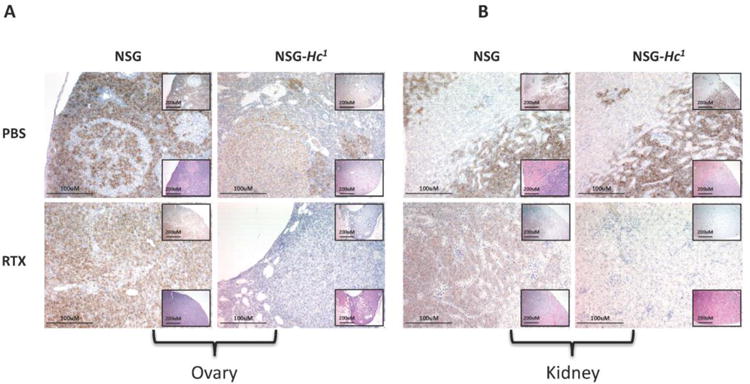
Representative images of the (A) ovary and (B) kidney 38 days post-engraftment displaying level of Daudi cell engraftment (brown stained cells) in different cohorts of age- and sex-matched rituximab-treated NSG and NSG-Hc1 mice. Low-magnification images at top right showing anti-CD45 staining IHC, at bottom right showing H&E staining, (×10, scale bar = 200um) showing; high-magnification image (×20, scale bar = 100um) showing anti-human CD45 IHC. PBS, phosphate buffer saline; RTX, rituximab. There were also increased numbers of Daudi cells in the livers, spleens, lungs, and lymph nodes of NSG mice (data not shown).
Discussion
mAb therapeutics recognize tumor-associated antigens and guide the immune system to destroy tumor cells while sparing normal cells. Primary mechanisms contributing to cytotoxic mAb-induced tumor-cell killing include apoptosis, CDC, and ADCC (Scott et al., 2012). Rituximab is a chimeric human-mouse mAb specific for human CD20 antigen, and is used in the treatment of B-cell malignancies (Maloney, 1999; McLaughlin, 2001; Johnson and Glennie, 2003) and rheumatoid arthritis (Silverman and Weisman, 2003; Edwards and Cambridge, 2006). Currently, efforts are underway to enhance both CDC-and/or ADCC-mediated cytotoxic activity of therapeutic antibodies, including rituximab, by engineering amino acid sequences of the Fc regions of these mAbs (Moore et al., 2010). However, because existing human tumor xenograft mouse models have significant limitations for comparing in vivo evaluation of mAb-mediated CDC versus complement-independent cytotoxicity against human patient-derived tumors, few Fc-engineering approaches for enhancing CDC activity have been systematically tested in these platforms. In this report, we have generated and characterized a complement-sufficient NSG-Hc1 mouse model that can be used in concert with complement-deficient NSG mice to specifically test mAb-mediated CDC against tumors as well as infectious microorganisms in vivo. The C5-deficient NSG mouse supports analysis of complement-independent mAb-induced apoptosis while the NSG-Hc1 mouse model supports both mAb-induced apoptosis and CDC without any contribution from mouse or human NK cells. Using the NSG-Hc1 mouse model, we demonstrated the contribution of hemolytic complement to the effective therapeutic activity of rituximab, given only as a single dose in a human B-lymphoma xenograft model (Fig 2-3). In a separate experiment, two successive doses of rituximab following Daudi cell injection in NSG-Hc1 mice were able to completely clear engrafted tumor cells and no sign of disease was observed during the 6 months of observation period (data not shown).
Traditionally, to demonstrate the ability of mAbs to function via CDC, researchers have relied upon rapid in vitro screening protocols, using primary tumor cell lines and serum as a source of complement. However, in vitro systems do not represent the natural milieu of the human tumor microenvironment, and substantial discrepancies between the CDC potency of mAbs in vitro and in vivo exist, in part because of the upregulation of membrane-bound complement regulators (CD46, CD55, and CD59) (Gelderman et al., 2004; Watson et al., 2006; Macor et al., 2007), and/or the loss of target antigen when tumor cells invade the liver or spleen (Williams et al., 2006; Beum et al., 2008). The NSG-Hc1 mouse model provides not only a severely immunodeficient environment that successfully supports in vivo growth of a variety of human tumors, as evidenced by a number of earlier studies (Ishikawa et al., 2007; Agliano et al., 2008; Zhang et al., 2010; Tanaka et al., 2012; Wong et al., 2012; Shultz et al., 2014; Brehm et al., 2016), but also a fully functional complement system enabling testing of CDC in vivo.
The NSG-Hc1 model addresses limitations of existing immunodeficient mouse strains that restrict their use for discriminating between mAb-induced apoptosis and mAb-mediated CDC activity. Recently, the in vivo CDC activity of a rituximab-derived CDC-optimized mAb was evaluated by injecting human serum in combination with rituximab into complement-deficient NOG mice (Sato et al., 2010). Although injection of human serum provided a measure of activity of the mAb-mediated CDC for a short period of time, insufficient replenishment of the consumed serum-derived complement may have been insufficient for supporting prolonged CDC and may require additional exogenous manipulation or supplementation. Moreover, high levels of human complement are toxic to immunodeficient mice, as this can induce intravascular hemolysis of mouse erythrocytes and other human complement-induced disorders (Tateno et al., 2004).
Several studies indicate that, despite differences between murine and human complement, murine complement is effective for studies using xenograft mouse models to assess mAb-mediated CDC activity against human cancers. Murine complement interacts with human tumor cells differently than does human complement, due to the species specificity of some complement inhibitory proteins, particularly CD59 (Zhao et al., 1998). In addition, mouse complement activity has been found to be lower than that of human (Ong and Mattes, 1989); however, earlier studies have shown that complement from one species can interact effectively with antibodies from phylogenetically distant species to trigger CDC (Alexander and Steiner, 1980). These interactions are possible because surface residues, i.e., Glu318, Lys320, and Lys322, present at the CH2 domain of the Fc region, where complement protein, e.g., C1q, binds to trigger subsequent CDC, are highly conserved throughout evolution in all human IgGs; rat IgG2b and IgG2c; mouse IgG2a, IgG2b, and IgG3; guinea pig IgG1; and rabbit IgGs (Duncan and Winter, 1988). Although three isotype residues present at the CH2 domain differ from their highly conserved counterparts, i.e., mouse IgG1 (Arg322), rat IgG1 (Thr318 and Arg320), and rat IgG2a (Thr318), it has been shown that these differences do not affect either C1q binding or complement-dependent lysis (Duncan and Winter, 1988), suggesting that antibodies containing the CH2 domain from murine, rat, guinea, or human, can be fixed effectively by murine complement. In fact, in the present study, rituximab, an antibody containing a human Fc region, was able to interact effectively with murine complement to trigger effective CDC. Because most mAbs used in the clinic contain either mouse-, rabbit-, or human-derived Fc regions, we are therefore confident that the NSG-Hc1 complement-sufficient model system will be ideally -suited for testing the CDC activity of a wide variety of these mAbs. Moreover, the lack of interference from human complement-induced disorders, and the long-term endogenous complement production, will enable prolonged experiments in our NSG-Hc1 model system, which is an important advantage over the complement-deficient NSG and NOG models.
Recent reports demonstrating interplay between mAb-mediated ADCC and CDC are contradictory, with some reports suggesting that both pathways act synergistically, resulting in greater killing than either mechanism alone (Pfeiffer et al., 2005; van Meerten et al., 2006), and others suggesting antagonistic mechanisms of action (Kennedy et al., 2004; Lefebvre et al., 2006). Therefore, the interaction of ADCC and CDC in mediating mAb-dependent effects warrants further investigations. One particular advantage of NSG-Hc1 mice is that they can either be engrafted with natural killer (NK) cells isolated from healthy donors, or NK cells can be induced from engrafted CD34+ HSCs in the presence of certain human cytokines (Tanaka et al., 2012). Other strains of mice that express NOD congenically derived signal regulatory protein α (Sirpa), including B6. CgRag2tm1FwaIL2rgtmiWjl SirpaNOD/ShiLt (BRGS) mice, engraft at heightened levels following human HSC injection and have an intact complement system (Yamauchi et al., 2013). However, unlike B6.CgRag2tm1FwaIL2rgtmWjl and BRGS mice, the NSG and NSG-Hc1 strains offer a matched set of strains that differ only in the presence or absence of complement activity. Engraftment of these two strains with the same human tumor followed by mAb treatment should precisely distinguish between the apoptotic affect of the mAb and the CDC.
Complement anaphylatoxins, i.e. C5a and C3a, are powerful immune modulators and can either propagate or suppress tumor progression by changing the composition and function of immune cells, including monocytes, macrophages, dendritic cells, neutrophils, basophils, eosinophils, mast cells, T-cells, myeloid derived suppressor cells (MDSC) within the tumor microenvironment through their receptors, i.e. C5a receptor (C5aR) and C3a receptor (C3aR), respectively (Sayegh et al., 2014). It has also been reported that murine C5a can activate human C5aR in a dose-dependent manner to the same level as human C5a (Schatz-Jakobsen et al., 2014). Moreover, recent reports have indicated that generation of the most effective therapeutic strategies requires not only targeting tumor or stroma cells, but also methods to overcome blockade of anti-tumor immune responses presented by lymphoid immune suppressor cells such as regulatory T cells (Treg), tumor associated macrophages (TAM) and distinct populations of myeloid cells such as myeloid derived suppressor cells (MDSC). In a preliminary experiment, we observed higher percentages of human myeloid cell development in the blood of NSG-Hc1 mice transplanted with human HSCs at 12 weeks post engraftment as compared to NSG mice, suggesting possible complement dependent immune modulation of composition and function of human immune cell engraftment. Therefore, humanized NSG-Hc1 mice, may also serve as a PDX based cancer-model system to evaluate C5a dependent immune modulation in tumor microenvironment as well as a platform for testing novel immunotherapies. In addition, humanized NSG-Hc1 mice model may represent an advanced, clinically relevant model to conduct mechanistic studies for C5a dependent inflammatory diseases, i.e. sepsis, anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis, etc., and complement-targeted therapy.
In summary, in this study we have developed a complement-sufficient NSG mouse model for measuring mAb-mediated CDC activity in vivo. This model has significant potential to not only to foster novel insights into the molecular mechanisms underlying direct interactions between mAb-mediated apoptosis and CDC, but to also promote efficacy studies of the next generation Fc-engineered mAbs, which ultimately can be translated to the clinic for treatment of patients.
Acknowledgments
The authors thank Allison Ingalls, Michelle Farley and Bruce Gott for skillfully managing mouse colonies.
Finalcial support for the authors: This work was supported in part by National Institutes of Health grant 1R24 OD018259, and UC4DK104218 (DLG, MAB, and LDS), CA034196 (LDS and MKV), CA171983 (LDS), the Myeloma Foundation (MKV), a grant from the Maine Cancer Foundation (LDS), and support from the Raymond and Beverly Sackler Foundation (LDS). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflict of Interest: MAB and DLG are consultants for and have research grant support from The Jackson Laboratory. The other authors have no conflicts to declare.
Disclosure of Potential Conflict of Interest: MAB and DLG are consultants for and have research grant support from The Jackson Laboratory. The other authors have no conflicts to declare.
Author's Contributions: Conception and design: MKV and LDS.
Development of Methodology: MKV and LDS.
Acquisition of Data: MKV, JC and LB.
Analysis and interpretation of data: MKV, LB and LDS.
Writing, review, and/or revision of manuscript: MKV, SBS, MAB, DLG and LDS.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agliano A, Martin-Padura I, Mancuso P, Marighetti P, Rabascio C, Pruneri G, Shultz LD, Bertolini F. Human acute leukemia cells injected in NOD/LtSz-scid/IL-2Rgamma null mice generate a faster and more efficient disease compared to other NOD/scid-related strains. International journal of cancer. Journal international du cancer. 2008;123:2222–7. doi: 10.1002/ijc.23772. [DOI] [PubMed] [Google Scholar]
- Alexander RJ, Steiner LA. Fixation of frog and guinea pig complement by antibodies from the bullfrog, Rana catesbeiana. Molecular immunology. 1980;17:1263–73. doi: 10.1016/0161-5890(80)90023-1. [DOI] [PubMed] [Google Scholar]
- Baxter AG, Cooke A. Complement lytic activity has no role in the pathogenesis of autoimmune diabetes in NOD mice. Diabetes. 1993;42:1574–8. doi: 10.2337/diab.42.11.1574. [DOI] [PubMed] [Google Scholar]
- Beum PV, Lindorfer MA, Taylor RP. Within peripheral blood mononuclear cells, antibody-dependent cellular cytotoxicity of rituximab-opsonized Daudi cells is promoted by NK cells and inhibited by monocytes due to shaving. J Immunol. 2008;181:2916–24. doi: 10.4049/jimmunol.181.4.2916. [DOI] [PubMed] [Google Scholar]
- Brehm MA, Bortell R, Verma M, Shultz LD, Greiner DL. Chapter 11 - Humanized Mice in Translational Immunology A2 - Tan, Seng-Lai In: Translational Immunology. Academic Press; Boston: 2016. pp. 285–326. [Google Scholar]
- Chen J, Reifsnyder PC, Scheuplein F, Schott WH, Mileikovsky M, Soodeen-Karamath S, Nagy A, Dosch MH, Ellis J, Koch-Nolte F, Leiter EH. “Agouti NOD”: identification of a CBA-derived Idd locus on Chromosome 7 and its use for chimera production with NOD embryonic stem cells. Mamm Genome. 2005;16:775–83. doi: 10.1007/s00335-005-0007-1. [DOI] [PubMed] [Google Scholar]
- Deckert PM. Current constructs and targets in clinical development for antibody-based cancer therapy. Curr Drug Targets. 2009;10:158–75. doi: 10.2174/138945009787354502. [DOI] [PubMed] [Google Scholar]
- Duncan AR, Winter G. The binding site for C1q on IgG. Nature. 1988;332:738–740. doi: 10.1038/332738a0. [DOI] [PubMed] [Google Scholar]
- Edwards JC, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nature reviews. Immunology. 2006;6:394–403. doi: 10.1038/nri1838. [DOI] [PubMed] [Google Scholar]
- Gelderman KA, Kuppen PJ, Okada N, Fleuren GJ, Gorter A. Tumor-specific inhibition of membrane-bound complement regulatory protein Crry with bispecific monoclonal antibodies prevents tumor outgrowth in a rat colorectal cancer lung metastases model. Cancer research. 2004;64:4366–72. doi: 10.1158/0008-5472.CAN-03-2131. [DOI] [PubMed] [Google Scholar]
- Ishikawa F, Yoshida S, Saito Y, Hijikata A, Kitamura H, Tanaka S, Nakamura R, Tanaka T, Tomiyama H, Saito N, Fukata M, Miyamoto T, Lyons B, Ohshima K, Uchida N, Taniguchi S, Ohara O, Akashi K, Harada M, Shultz LD. Chemotherapy-resistant human AML stem cells home to and engraft within the bone-marrow endosteal region. Nature biotechnology. 2007;25:1315–21. doi: 10.1038/nbt1350. [DOI] [PubMed] [Google Scholar]
- Johnson P, Glennie M. The mechanisms of action of rituximab in the elimination of tumor cells. Seminars in oncology. 2003;30:3–8. doi: 10.1053/sonc.2003.50025. [DOI] [PubMed] [Google Scholar]
- Kennedy AD, Beum PV, Solga MD, DiLillo DJ, Lindorfer MA, Hess CE, Densmore JJ, Williams ME, Taylor RP. Rituximab infusion promotes rapid complement depletion and acute CD20 loss in chronic lymphocytic leukemia. J Immunol. 2004;172:3280–8. doi: 10.4049/jimmunol.172.5.3280. [DOI] [PubMed] [Google Scholar]
- Lefebvre ML, Krause SW, Salcedo M, Nardin A. Ex vivo-activated human macrophages kill chronic lymphocytic leukemia cells in the presence of rituximab: mechanism of antibody-dependent cellular cytotoxicity and impact of human serum. Journal of immunotherapy (Hagerstown, Md: 1997) 2006;29:388–97. doi: 10.1097/01.cji.0000203081.43235.d7. [DOI] [PubMed] [Google Scholar]
- Macor P, Tripodo C, Zorzet S, Piovan E, Bossi F, Marzari R, Amadori A, Tedesco F. In vivo targeting of human neutralizing antibodies against CD55 and CD59 to lymphoma cells increases the antitumor activity of rituximab. Cancer research. 2007;67:10556–63. doi: 10.1158/0008-5472.CAN-07-1811. [DOI] [PubMed] [Google Scholar]
- Maloney DG. Preclinical and phase I and II trials of rituximab. Seminars in oncology. 1999;26:74–8. [PubMed] [Google Scholar]
- McLaughlin P. Rituximab: perspective on single agent experience, and future directions in combination trials. Critical reviews in oncology/hematology. 2001;40:3–16. doi: 10.1016/s1040-8428(01)00130-5. [DOI] [PubMed] [Google Scholar]
- Moore GL, Chen H, Karki S, Lazar GA. Engineered Fc variant antibodies with enhanced ability to recruit complement and mediate effector functions. MAbs. 2010;2:181–9. doi: 10.4161/mabs.2.2.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–74. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- Ong GL, Mattes MJ. Mouse strains with typical mammalian levels of complement activity. Journal of immunological methods. 1989;125:147–58. doi: 10.1016/0022-1759(89)90088-4. [DOI] [PubMed] [Google Scholar]
- Pfeiffer M, Stanojevic S, Feuchtinger T, Greil J, Handgretinger R, Barbin K, Jung G, Martin D, Niethammer D, Lang P. Rituximab mediates in vitro antileukemic activity in pediatric patients after allogeneic transplantation. Bone marrow transplantation. 2005;36:91–7. doi: 10.1038/sj.bmt.1705014. [DOI] [PubMed] [Google Scholar]
- Ravetch J. In vivo veritas: the surprising roles of Fc receptors in immunity. Nat Immunol. 2010;11:183–5. doi: 10.1038/ni0310-183. [DOI] [PubMed] [Google Scholar]
- Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785–97. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato F, Ito A, Ishida T, Mori F, Takino H, Inagaki A, Ri M, Kusumoto S, Komatsu H, Iida S, Okada N, Inagaki H, Ueda R. A complement-dependent cytotoxicity-enhancing anti-CD20 antibody mediating potent antitumor activity in the humanized NOD/Shi-scid, IL-2Rgamma(null) mouse lymphoma model. Cancer immunology, immunotherapy: CII. 2010;59:1791–800. doi: 10.1007/s00262-010-0905-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayegh ET, Bloch O, Parsa AT. Complement anaphylatoxins as immune regulators in cancer. Cancer medicine. 2014;3:747–58. doi: 10.1002/cam4.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz-Jakobsen JA, Yatime L, Larsen C, Petersen SV, Klos A, Andersen GR. Structural and functional characterization of human and murine C5a anaphylatoxins. Acta crystallographica. Section D, Biological crystallography. 2014;70:1704–17. doi: 10.1107/S139900471400844X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott AM, Allison JP, Wolchok JD. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012;12:14. [PMC free article] [PubMed] [Google Scholar]
- Shultz LD, Goodwin N, Ishikawa F, Hosur V, Lyons BL, Greiner DL. Human cancer growth and therapy in immunodeficient mouse models. Cold Spring Harbor protocols. 2014;2014:694–708. doi: 10.1101/pdb.top073585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman GJ, Weisman S. Rituximab therapy and autoimmune disorders: prospects for anti-B cell therapy. Arthritis and rheumatism. 2003;48:1484–92. doi: 10.1002/art.10947. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Saito Y, Kunisawa J, Kurashima Y, Wake T, Suzuki N, Shultz LD, Kiyono H, Ishikawa F. Development of mature and functional human myeloid subsets in hematopoietic stem cell-engrafted NOD/SCID/IL2rgammaKO mice. J Immunol. 2012;188:6145–55. doi: 10.4049/jimmunol.1103660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateno C, Yoshizane Y, Saito N, Kataoka M, Utoh R, Yamasaki C, Tachibana A, Soeno Y, Asahina K, Hino H, Asahara T, Yokoi T, Furukawa T, Yoshizato K. Near completely humanized liver in mice shows human-type metabolic responses to drugs. The American journal of pathology. 2004;165:901–12. doi: 10.1016/S0002-9440(10)63352-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegla CA, Cudrici C, Patel S, Trippe R, 3rd, Rus V, Niculescu F, Rus H. Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunologic research. 2011;51:45–60. doi: 10.1007/s12026-011-8239-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meerten T, van Rijn RS, Hol S, Hagenbeek A, Ebeling SB. Complement-induced cell death by rituximab depends on CD20 expression level and acts complementary to antibody-dependent cellular cytotoxicity. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:4027–35. doi: 10.1158/1078-0432.CCR-06-0066. [DOI] [PubMed] [Google Scholar]
- Watson NF, Durrant LG, Madjd Z, Ellis IO, Scholefield JH, Spendlove I. Expression of the membrane complement regulatory protein CD59 (protectin) is associated with reduced survival in colorectal cancer patients. Cancer immunology, immunotherapy: CII. 2006;55:973–80. doi: 10.1007/s00262-005-0055-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams ME, Densmore JJ, Pawluczkowycz AW, Beum PV, Kennedy AD, Lindorfer MA, Hamil SH, Eggleton JC, Taylor RP. Thrice-weekly low-dose rituximab decreases CD20 loss via shaving and promotes enhanced targeting in chronic lymphocytic leukemia. J Immunol. 2006;177:7435–43. doi: 10.4049/jimmunol.177.10.7435. [DOI] [PubMed] [Google Scholar]
- Wong KK, Brenneman F, Chesney A, Spaner DE, Gorczynski RM. Soluble CD200 is critical to engraft chronic lymphocytic leukemia cells in immunocompromised mice. Cancer research. 2012;72:4931–43. doi: 10.1158/0008-5472.CAN-12-1390. [DOI] [PubMed] [Google Scholar]
- Yamauchi T, Takenaka K, Urata S, Shima T, Kikushige Y, Tokuyama T, Iwamoto C, Nishihara M, Iwasaki H, Miyamoto T, Honma N, Nakao M, Matozaki T, Akashi K. Polymorphic Sirpa is the genetic determinant for NOD-based mouse lines to achieve efficient human cell engraftment. Blood. 2013;121:1316–25. doi: 10.1182/blood-2012-06-440354. [DOI] [PubMed] [Google Scholar]
- Zhang B, Strauss AC, Chu S, Li M, Ho Y, Shiang KD, Snyder DS, Huettner CS, Shultz L, Holyoake T, Bhatia R. Effective targeting of quiescent chronic myelogenous leukemia stem cells by histone deacetylase inhibitors in combination with imatinib mesylate. Cancer cell. 2010;17:427–42. doi: 10.1016/j.ccr.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XJ, Zhao J, Zhou Q, Sims PJ. Identity of the residues responsible for the species-restricted complement inhibitory function of human CD59. J Biol Chem. 1998;273:10665–71. doi: 10.1074/jbc.273.17.10665. [DOI] [PubMed] [Google Scholar]