

Abstract
A highly efficient and chemoselective method for the synthesis of diaryl disulfides is developed via a visible light-promoted coupling of readily accessible arenediazonium tetrafluoroborates and CS2. This practical and convenient protocol provides a direct pathway for the assembly of a series of disulfides in an environmentally friendly manner with good to excellent yields.
Keywords: arenediazonium tetrafluoroborates, carbon disulfide, chemoselectivity, diaryl disulfides, photocatalyst
Findings
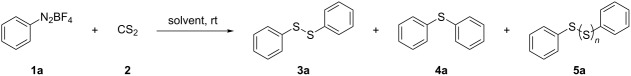
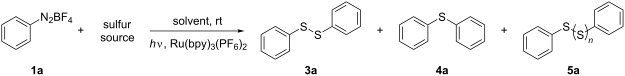
The development of methods for the functionalization of peptides and proteins under mild conditions is a current frontier in the fields of chemistry, biology and drug discovery [1–4]. Most of the pharmaceutically relevant proteins contain disulfide bonds, furthermore, the disulfide ligation and its established chemoselectivity is of great advantage for proteins’ functionalization [5]. In addition, disulfides also play valuable roles as versatile building blocks for industrial applications [6–8]. Thus, the development of methodologies for the synthesis of disulfides is rather desirable and many research groups have made great contributions to the synthesis of diaryl disulfides such as the Chandrasekaran group [9] and the Wacharasindhu group [10]. Indeed, the design of sustainable and useful transformations with applications in industry is considered of high practical value. In this context, carbon disulfide, a cheap and abundant chemical, has been widely used as reactant and solvent in both industry and materials science. For example, Batanero and co-workers reported an electrochemical transformation of carbon disulfide into diaryl disulfides [11]. Sunlight as abundant and almost infinitely available energy resource has been widely used for chemical transformations in the sense of cost, safety, availability, and environmental friendliness [12–15]. Herein, we report a visible light-mediated coupling of arenediazonium tetrafluoroborates and CS2 for the chemoselective assembly of diaryl disulfides as our continuing endeavor of utilizing arenediazonium tetrafluoroborates [16] for synthetic applications (Scheme 1).
Scheme 1.

Chemoselective assembly of diaryl disulfides.
We conducted our initial study with benzenediazonium tetrafluoroborate (1a) and CS2 (2) as model substrates to examine the feasibility of the formation of diphenyl disulfide (3a) (Table 1). Various solvents were screened and to our delight, it was found that the reaction of 1a and 2 in DMF and DMSO gave the desired product in a moderate yield of 54% and 53%, respectively (Table 1, entries 7 and 8). Unfortunately, under the applied conditions, the chemoselectivity of the reaction was poor, affording a mixture of unexpected diphenyl sulfide (4a) and diphenyl polysulfides (5a) as byproducts. Thus, a study to optimize the reaction conditions with regard to chemoselectivity and to minimize the formation of the byproducts was conducted.
Table 1.
Solvent screening for the coupling of benzenediazonium tetrafluoroborate (1a) and CS2 (2).a
 | ||||
| Entry | Solvents | Yield 3a (%)b | Yield 4a (%)b | Yield 5a (%)b |
| 1 | MeOH | n.d. | n.d. | n.d. |
| 2 | THF | 36 | 5 | n.d. |
| 3 | dioxane | n.d. | 14 | n.d. |
| 4 | acetone | n.d. | n.d. | n.d. |
| 5 | DCM | n.d. | n.d. | 19 |
| 6 | acetonitrile | n.d. | n.d. | n.d. |
| 7 | DMF | 54 | 3 | 31 |
| 8 | DMSO | 53 | 3 | 27 |
| 9 | hexane | n.d. | n.d. | 24 |
As recently surveyed, photoredox catalysts are widely employed for the generation of radicals for diverse radical reactions [19]. Further, the application of aryl radicals generated from aryldiazonium salts under visible light irradiation has also been studied [14–15] by taking advantage of visible light as abundant and environmentally friendly energy source for organic syntheses. The photochemistry of diazonium salts has been widely studied since the early 19th century, at which time, it was noticed that benzenediazonium nitrate turns red upon exposure to sunlight due to decomposition and formation of radical species [20]. Subsequently, the photodecomposition of diazonium salts by loss of nitrogen upon exposure to light has been utilized in organic synthesis for example to remove amino groups from anilines [21] or for arylation reactions [15,22].
Based on the above research results, we envisioned that a radical pathway may facilitate the formation of diaryl disulfides. Therefore the photocatalyst Ru(bpy)3(PF6)2 (bpy = 2,2’-bipyridine) [23] and a 20 W blue-light LED were chosen as catalyst and the source of visible light, respectively for our model reaction (Table 2). A variety of solvents was evaluated and eventually, it was found that the coupling of benzenediazonium tetrafluoroborate (1a) and CS2 (2) in ethanol as the solvent gave the desired product diphenyl disulfide (3a) in 77% yield accompanied by only 8% of the undesired diphenyl polysulfides (Table 2, entry 4). Switching to DMSO as the solvent for the reaction afforded exclusively the desired product 3a in excellent yield (88%, Table 2, entry 6). Next, other sulfur sources were also examined, such as S8, NaSH, Na2S, Na2S2O3, Na2S2O4 and K2S2O8, however, none of them provided the desired product in an acceptable yield (Table 2, entries 7–13).
Table 2.
Screening of the solvents and sulfur sources for the visible light-mediated coupling of benzenediazonium tetrafluoroborate (1a) and CS2 (2) and other sulfur sources in the presence of Ru(bpy)3(PF6)2 as the photocatalyst.a
 | |||||
| Entry | Solvent | Sulfur source | Yield 3a (%)b | Yield 4a (%)b | Yield 5a (%)b |
| 1 | MeOH | CS2 | 88 | <1 | 10 |
| 2 | H2O | CS2 | 47 | 5 | 20 |
| 3 | THF | CS2 | 87 | <1 | 8 |
| 4 | EtOH | CS2 | 77 | n.d. | 8 |
| 5 | acetone | CS2 | 79 | 7 | 3 |
| 6 | DMSO | CS2 | 88 | n.d. | <1 |
| 7 | DMSO | S8 | 14 | 9 | 67 |
| 8 | DMSO | Na2S | n.d. | 43 | 11 |
| 9 | DMSO | Na2S2O3 | n.d. | n.d. | n.d. |
| 10 | DMSO | Na2S2O4 | n.d. | n.d. | n.d. |
| 11 | DMSO | K2S2O8 | n.d. | n.d. | 4 |
| 12 | DMSO | NaSH | 22 | 28 | 14 |
| 13 | DMSO | (NH4)2S2O8 | n.d. | n.d. | 4 |
aReaction conditions: 1a (0.1 mmol), sulfur sources (0.2 mmol), Ru(bpy)3(PF6)2 (0.001 mmol), blue light (20 W), solvents (2 mL), rt, 6 h; byields were determined by HPLC using 3a and 4a as the external standards, the yield of 5a is based on the integration of the corresponding HPLC peaks [17–18]; n.d. = not determined.
In order to maximize the yields, varying amounts of CS2 (2) were also tested (Table 3) and it was found that the CS2 loading had a considerable influence on the reaction. By decreasing the loading of CS2 from 2 equiv to 0.5 equiv, the yield of the product 3a dropped to 42%, whereas increasing amounts of CS2 did not significantly increase the yield of the product. Subsequently, different photocatalysts were investigated and it turned out that the choice of catalyst also had a significant impact on our model reaction. Ru(bpy)3Cl2 catalyzed this coupling to afford the desired product 3a in a moderate yield of 65% (Table 3, entry 8). However, when the iridium-based photocatalysts Ir(ppy)3 [24], [Ir(ppy)2(bpy)]PF6 and [Ir(ppy)2(dtbbpy)]PF6 (bpy = 2,2’-bipyridine, ppy = 2-phenylpyridine, dtbbpy = 4,4’-di-tert-butyl-2,2’-bipyridine) [25–26] were used, the product yield of diphenyl disulfide (3a) was much lower compared to reactions performed in the presence of ruthenium catalysts (Table 3, entries 9–11).
Table 3.
Screening of photocatalysts for the visible light-mediated coupling of benzenediazonium tetrafluoroborate (1a) and CS2 (2).a
 | ||||
| Entry | 2 (equiv) | photocatalyst | solvent | Yield 3a (%)b |
| 1 | 0.5 | Ru(bpy)3(PF6)2 | DMSO | 42 |
| 2 | 1 | Ru(bpy)3(PF6)2 | DMSO | 47 |
| 3 | 1.5 | Ru(bpy)3(PF6)2 | DMSO | 53 |
| 4 | 2 | Ru(bpy)3(PF6)2 | DMSO | 88 |
| 5 | 2.5 | Ru(bpy)3(PF6)2 | DMSO | 55 |
| 6 | 3 | Ru(bpy)3(PF6)2 | DMSO | 57 |
| 7 | – | Ru(bpy)3(PF6)2 | CS2 | n.d. |
| 8 | 2 | Ru(bpy)3Cl2 | DMSO | 65 |
| 9 | 2 | Ir(ppy)3 | DMSO | 57 |
| 10 | 2 | Ir(ppy)2(bpy)(PF6) | DMSO | 73 |
| 11 | 2 | Ir(ppy)2(dtbbpy)(PF6) | DMSO | 8 |
| 12 | 2 | none | DMSO | 53 |
aReaction conditions: 1a (0.1 mmol), photocatalyst (0.001 mmol), blue light (20 W), solvent (2 mL), rt, 6 h; byields were determined by HPLC using 3a as the external standard.
A plausible reaction mechanism has been proposed and is depicted in Scheme 2. We envision that the phenyl radical I was initially generated under visible light irradiation [14–15]. Subsequently, the radical I attacked the sulfur atom of carbon disulfide to provide the intermediate II which can be converted to radical intermediate III through the cleavage of the carbon–sulfur bond accompanied with the release of a carbon sulfide [11]. The active radical intermediate III can transform into three types of products through different pathways. Firstly, diaryl disulfide 3 is obtained through a dimerization of radical intermediates III, whereas the reaction of radical III with phenyl radical I is leading to byproduct 4. Finally, radical III can react with various equivalents of CS2 with release of carbon sulfide to generate aryl-polythio radicals IV and V. The combination of the latter intermediates with radical I then finally affords polysulfides 5.
Scheme 2.

A plausible reaction mechanism.
To demonstrate the scope of the reaction, a series of arenediazonium tetrafluoroborates was utilized in the reaction with CS2 to generate the corresponding diaryl disulfides (Table 4). Arenediazonium tetrafluoroborates 1b–p with both, electron-withdrawing and donating groups successfully underwent transformation, affording the corresponding coupling products 3b–p in good to excellent yields (42–99%). Also sterically demanding substrates gave the desired products in good yields (3d, 3f, 3g, 3i, 3m and 3n) and functional groups such as chloro, bromo, ester, methyl, nitro, and phenyl groups were also compatible with the reaction conditions.
Table 4.
Reaction scope of the visible light-mediated coupling of arenediazonium tetrafluoroborates 1 with CS2 (2).
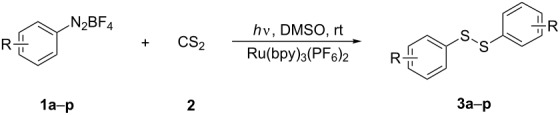 | |||
| Substrate 1a | Product 3, yieldb | Substrate 1a | Product 3, yieldb |
 1a |
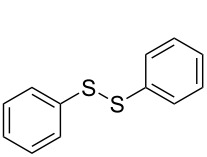 3a, 80%, 50%c |
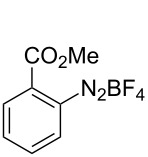 1i |
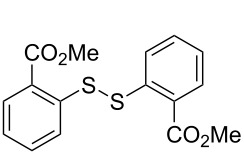 3i, 94%, 82%c |
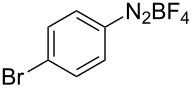 1b |
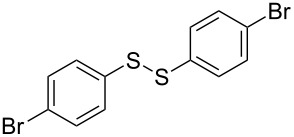 3b, 81%, 78%c |
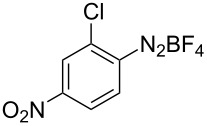 1j |
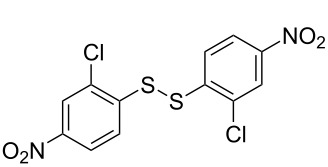 3j, 99%, 85%c |
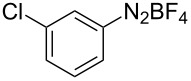 1c |
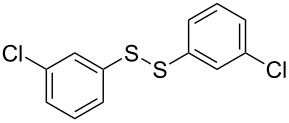 3c, 85%, 72%c |
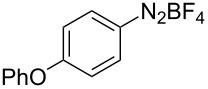 1k |
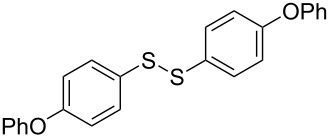 3k, 70% |
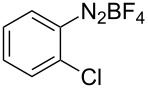 1d |
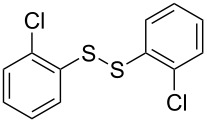 3d, 94% |
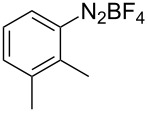 1l |
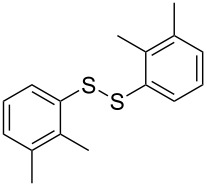 3l, 76% |
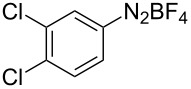 1e |
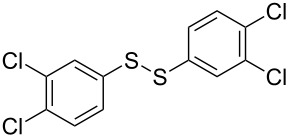 3e, 90% |
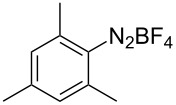 1m |
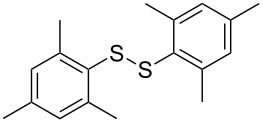 3m, 56% |
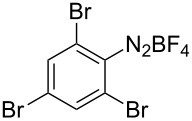 1f |
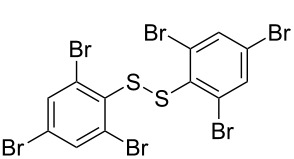 3f, 88% |
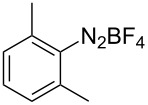 1n |
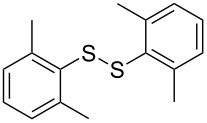 3n, 42% |
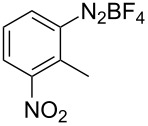 1g |
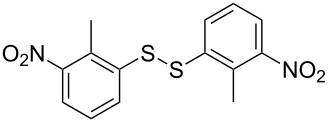 3g, 88% |
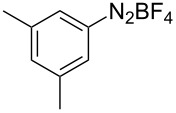 1o |
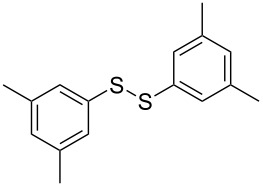 3o, 56% |
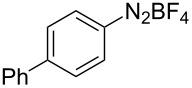 1h |
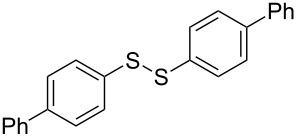 3h, 76% |
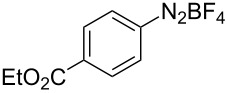 1p |
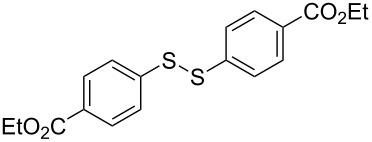 3p, 90%c, 92%c,d |
aReaction conditions: 1 (0.1 mmol), CS2 (0.2 mmol), Ru(bpy)3(PF6)2 (0.001 mmol), blue light (20 W), DMSO (2 mL), rt, 6 h; bisolated yields after chromatography on silica gel; cthe reactions were carried out with the diazonium salts 1 at a 5 mmol scale; dacetone was used as the solvent.
Conclusion
In conclusion, we have developed an efficient method for the synthesis of diaryl disulfides through the coupling of arenediazonium tetrafluoroborates and CS2. This straightforward visible light-promoted process proceeds under mild reaction conditions and is applicable for the assembly of a wide range of diaryl disulfides. Further studies to clearly understand the reaction mechanism and the synthetic applications are ongoing in our laboratory.
Supporting Information
Experimental procedures, characterization data and copies of 1H and 13C NMR spectra for final compounds.
Acknowledgments
This work was supported by the Wuhan University of Technology.
References
- 1.Bode J W. Curr Opin Drug Discovery Dev. 2006;9:765–775. [PubMed] [Google Scholar]
- 2.Pratt M R, Bertozzi C R. Chem Soc Rev. 2005;34:58–68. doi: 10.1039/b400593g. [DOI] [PubMed] [Google Scholar]
- 3.Prescher J A, Dube D H, Bertozzi C R. Nature. 2004;430:873–877. doi: 10.1038/nature02791. [DOI] [PubMed] [Google Scholar]
- 4.Kolb H C, Finn M G, Sharpless K B. Angew Chem, Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 5.Woycechowsky K J, Raines R T. Curr Opin Chem Biol. 2000;4:533–539. doi: 10.1016/S1367-5931(00)00128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oae S. Organic Sulfur Chemistry, Structure and Mechanism. Boca Raton, FL: CRC Press; 1991. [Google Scholar]
- 7.Cremlyn R J. An Introduction to Organosulfur Chemistry. New York: John Wiley and Sons; 1996. [Google Scholar]
- 8.Jocelyn D C. Biochemistry of the Thiol Groups. New York: Academic Press; 1992. [Google Scholar]
- 9.Bhar D, Chandrasekaran S. Synthesis. 1994:785–786. doi: 10.1055/s-1994-25573. [DOI] [Google Scholar]
- 10.Tankam T, Poochampa K, Vilaivan T, Sukwattanasinitt M, Wacharasindhu S. Tetrahedron. 2016;72:788–793. doi: 10.1016/j.tet.2015.12.036. [DOI] [Google Scholar]
- 11.Barba F, Ranz F, Batanero B. Tetrahedron Lett. 2009;50:6798–6799. doi: 10.1016/j.tetlet.2009.09.102. [DOI] [Google Scholar]
- 12.Ravelli D, Fagnoni M, Albini A. Chem Soc Rev. 2013;42:97–113. doi: 10.1039/C2CS35250H. [DOI] [PubMed] [Google Scholar]
- 13.Lang X, Chen X, Zhao J. Chem Soc Rev. 2014;43:473–486. doi: 10.1039/C3CS60188A. [DOI] [PubMed] [Google Scholar]
- 14.Hari D P, König B. Angew Chem, Int Ed. 2013;52:4734–4743. doi: 10.1002/anie.201210276. [DOI] [PubMed] [Google Scholar]
- 15.Hofmann J, Heinrich M R. Tetrahedron Lett. 2016;57:4334–4340. doi: 10.1016/j.tetlet.2016.08.034. [DOI] [Google Scholar]
- 16.Qin H-L, Zheng Q, Bare G A L, Wu P, Sharpless K B. Angew Chem, Int Ed. 2016;55:14155–14158. doi: 10.1002/anie.201608807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Arisawa M, Tanaka K, Yamaguchi M. Tetrahedron Lett. 2005;46:4797–4800. doi: 10.1016/j.tetlet.2005.05.024. [DOI] [Google Scholar]
- 18.Zysman-Colman E, Harpp D N. J Org Chem. 2003;68:2487–2489. doi: 10.1021/jo0265481. [DOI] [PubMed] [Google Scholar]
- 19.Prier C K, Rankic D A, MacMillan D W C. Chem Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horspool W M, Lenci F. CRC Handbook of Organic Photochemistry and Photobiology. 2nd ed. 1 & 2. Boca Raton: CRC press; 2003. [Google Scholar]
- 21.He L, Qiu G, Gao Y, Wu J. Org Biomol Chem. 2014;12:6965–6971. doi: 10.1039/C4OB01286K. [DOI] [PubMed] [Google Scholar]
- 22.Xue D, Jia Z-H, Zhao C-J, Zhang Y-Y, Wang C, Xiao J. Chem – Eur J. 2014;20:2960–2965. doi: 10.1002/chem.201304120. [DOI] [PubMed] [Google Scholar]
- 23.Dedeian K, Djurovich P I, Garces F O, Carlson G, Watts R J. Inorg Chem. 1991;30:1685–1687. doi: 10.1021/ic00008a003. [DOI] [Google Scholar]
- 24.Slinker J D, Gorodetsky A A, Lowry M S, Wang J, Parker S, Rohl R, Bernhard S, Malliaras G G. J Am Chem Soc. 2004;126:2763–2767. doi: 10.1021/ja0345221. [DOI] [PubMed] [Google Scholar]
- 25.Schank K, Leider R, Lick C, Glock R. Helv Chim Acta. 2004;87:869–924. doi: 10.1002/hlca.200490085. [DOI] [Google Scholar]
- 26.Carnell A J, Johnstone R A W, Parsy C C, Sanderson W R. Tetrahedron Lett. 1999;40:8029–8032. doi: 10.1016/S0040-4039(99)01610-X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures, characterization data and copies of 1H and 13C NMR spectra for final compounds.