

Summary
The otherwise harmless skin inhabitant Staphylococcus epidermidis is a major cause of healthcare-associated medical device infections. The species' selective pathogenic potential depends on its production of surface adherent biofilms. Cell wall-anchored protein Aap promotes biofilm formation in S. epidermidis, independently from the polysaccharide intercellular adhesin PIA. Aap requires proteolytic cleavage to act as an intercellular adhesin. Whether and which staphylococcal proteases account for Aap processing is yet unknown. Here, evidence is provided that in PIA-negative S. epidermidis 1457Δica, the metalloprotease SepA is required for Aap-dependent S. epidermidis biofilm formation in static and dynamic biofilm models. qRT-PCR and protease activity assays demonstrated that under standard growth conditions, sepA is repressed by the global regulator SarA. Inactivation of sarA increased SepA production, and in turn augmented biofilm formation. Genetic and biochemical analyses demonstrated that SepA-related induction of biofilm accumulation resulted from enhanced Aap processing. Studies using recombinant proteins demonstrated that SepA is able to cleave the A domain of Aap at residue 335 and between the A and B domains at residue 601. This study identifies the mechanism behind Aap-mediated biofilm maturation, and also demonstrates a novel role for a secreted staphylococcal protease as a requirement for the development of a biofilm.
Graphical abstract
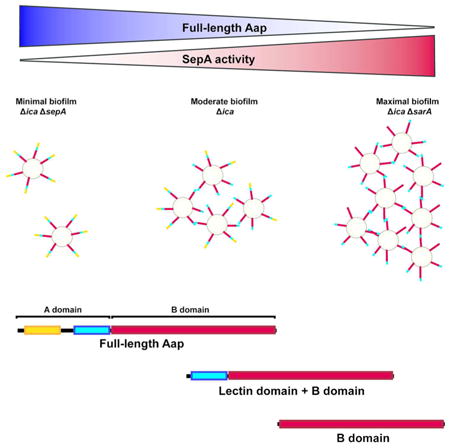
Cell wall-anchored adhesin Accumulation associated protein (Aap) significantly contributes to biofilm formation. Mature full length Aap prevents cell aggregation. High activity of metalloprotease SepA, such as in the absence of repressor SarA, results in increased cleavage of Aap and subsequent biofilm formation. Processing of Aap is primarily observed within the A domain at residue 335, removing the portion N-terminal to the lectin-like domain, but can also involve removal of the entire domain. Cleavage of Aap favors bacterial aggregation und biofilm formation. Thus, protease expression levels modulate adhesive S. epidermidis surface properties, biofilm formation and surface colonization.
Introduction
Staphylococcus epidermidis infections have emerged as a major problem in modern medicine. As an organism that is typically encountered as a commensal of human skin and mucous membranes (Otto, 2010, Grice & Segre, 2011), S. epidermidis is a prototypic opportunistic pathogen, causing infections almost exclusively in predisposed hosts. The risk factor most significantly associated with development of an infection is the use of implanted foreign materials, such as prosthetic joints, artificial heart valves, and central venous catheters (Darouiche, 2004). S. epidermidis is one of the most common organisms found in various types of medical device-associated infections (Donlan, 2001, Sievert et al., 2013) and the number one cause of healthcare associated-bloodstream infections, which are often secondary to medical device infections (Rupp, 2014). The coagulase-negative staphylococci (CoNS), of which S. epidermidis is the most abundant, are consistently reported within the top ten etiological agents in healthcare-associated infection (Sievert et al., 2013, Magill et al., 2014). There is clearly a need for effective treatment and prevention of S. epidermidis infections, which will require a detailed understanding of its pathogenesis.
The pathogenicity of S. epidermidis is linked to its ability to form adherent, multi-layered biofilms on artificial surfaces (Otto, 2014b, Mack et al., 2009). The biofilm mode of growth protects bacteria from host immune defences, as well as antibiotics, resulting in persistent difficult to treat infections (Morgenstern et al., 2016, Paharik & Horswill, 2016, Arciola et al., 2012). Biofilm maturation requires bacterial adhesins that mediate intercellular attachment and the eventual formation of an established biofilm structure. Research over the past two decades has identified several mechanisms that promote intercellular adhesion and biofilm accumulation in S. epidermidis. Polysaccharides, proteins, and extracellular DNA (eDNA) are the underlying functional molecules in the biofilm matrix. Despite sharing functional properties, these molecules differ with respect to their spatial organization within the biofilm. For example, the polysaccharide intercellular adhesin (PIA), synthesized by proteins encoded in the icaADBC operon, forms a sticky, glue-like extracellular matrix, while the cell wall-bound accumulation associated protein (Aap) strictly localizes to the bacterial cell surface (Buttner et al., 2015, Schommer et al., 2011). These apparent structural differences also result in distinct macroscopic and microscopic morphological phenotypes, and might reflect the ability of S. epidermidis to employ distinct biofilm forming modalities depending on the particular physical stresses present at different infection sites (Otto, 2014a). In support of this concept, a recent analysis of S. epidermidis clinical isolates found that the ica operon was more prevalent in strains that infected body sites with high shear stress, compared to those infecting lower-shear environments (Schaeffer et al., 2016). This suggests that PIA-independent, protein-dependent biofilms may be particularly relevant to infections in low-shear host niches.
Although PIA was previously considered to be essential for S. epidermidis biofilm development, multiple studies have reported that at least 30% of invasive isolates lack the icaADBC operon for PIA biosynthesis, with some infection types containing as many as 74% ica-negative isolates (Ziebuhr et al., 1997, Hellmark et al., 2013, Klug et al., 2003, Rohde et al., 2007). The cell wall-anchored adhesin Aap plays a critical role in biofilm formation in the absence of PIA (Hussain et al., 1997, Hennig et al., 2007, Rohde et al., 2005). In a rat central venous catheter model of S. epidermidis infection, an ica mutant in strain 1457 had no defect, while a single mutant lacking Aap was severely attenuated (Schaeffer et al., 2015), demonstrating that in certain infections, Aap-mediated surface colonization is favoured. Thus, understanding the molecular determinants promoting Aap-dependent biofilm formation is crucial for the development of novel approaches to combat S. epidermidis device-related infections.
Aap and its S. aureus orthologue SasG are sortase-anchored cell wall proteins that have been shown to promote both cell-surface and cell-cell adhesion. These proteins have two major domains, with the N-terminal A domain containing a predicted, 212 amino acid L-type lectin domain of unknown ligand specificity (Figure 1). The A domain has been shown to mediate primary attachment of bacteria to abiotic surfaces as well as to mammalian epithelial cell monolayers (Schaeffer et al., 2015, Roche et al., 2003, Macintosh et al., 2009, Conlon et al., 2014). The B domain is composed of several repeating G5 motifs with intervening E repeat spacers (Figure 1). Heterotypic interactions between B domain and Small basic protein (Sbp), a component of the biofilm matrix, contribute to Aap-dependent biofilm formation (Decker et al., 2015). In addition, extensive biochemical characterization has provided evidence for homotypic B domain interactions, leading to Zn2+-dependent assembly of a strong, elongated twisted rope-like structure that promotes intercellular accumulation (Conrady et al., 2013, Formosa-Dague et al., 2016, Corrigan et al., 2007, Gruszka et al., 2015, Gruszka et al., 2012, Geoghegan et al., 2010, Conrady et al., 2008, Kuroda et al., 2008). However, this process and its resulting biofilm maturation only occur once the A domain is proteolytically removed (Rohde et al., 2005). The necessity of proteolytic cleavage highlights the specific importance of proteases for Aap function as an intercellular adhesin, and also demonstrates that proteolytic processing is of fundamental importance for S. epidermidis to dynamically adjust functional cell surface properties. Strikingly, despite its obvious importance, the involvement of one or more specific S. epidermidis proteases in Aap processing and modulation of biofilm formation is unknown.
Figure 1.
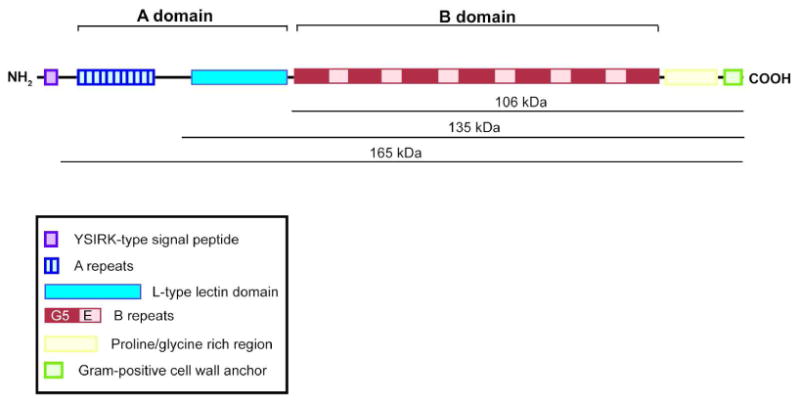
Diagram of S. epidermidis 1457 Accumulation-Associated Protein (Aap). Aap contains a YSIRK/GS-type signal peptide, an A domain consisting of A repeats and a lectin domain, and a B domain consisting of G5 domains with intervening spacer regions, referred to as E domains. The C terminal portion of the protein contains a proline/glycine-rich region and a cell wall anchor, including the LPXTG recognition site for Sortase A. The molecular weights shown correspond to the full-length protein and the cleavage products cut at amino acids 335 and 601. Molecular weights are predicted by ExPASY ProtParam tool and exclude the signal peptide N terminal to the AXA signal peptidase cleavage site.
S. epidermidis secretes 3 major proteases: a metalloprotease SepA, a cysteine protease Ecp, and a serine protease Esp (Teufel & Gotz, 1993, Moon et al., 2001, Oleksy et al., 2004). Research on the role of proteases in staphylococcal biofilms has focused on the dispersal phase of the biofilm life cycle, in which the biofilm matrix is degraded and bacterial cells return to a free, planktonic state (Tsang et al., 2008, Mootz et al., 2013, Boles & Horswill, 2008). The secreted S. epidermidis serine protease Esp is well known as an inhibitor of S. aureus biofilm formation and colonization (Sugimoto et al., 2013). Esp can degrade existing S. aureus biofilms by cleaving specific proteins in the biofilm matrix (Sugimoto et al., 2013, Iwase et al., 2010), and can prevent S. aureus biofilm formation by degrading the autolysin Atl, thereby eliminating eDNA production in the biofilm matrix (Chen et al., 2013). The functions of Ecp and SepA in the biofilm life cycle are less well characterized.
In this report, we show that PIA-independent biofilm formation in S. epidermidis requires the secreted protease SepA. Aap processing is dependent on SepA, and studies with recombinant Aap showed that SepA is able to cleave Aap at two residues: Leu 335 and Leu 601. The global regulator SarA negatively regulates sepA and therefore inhibits SepA-mediated biofilm formation. The present study gives novel insights into the molecular mechanisms used by S. epidermidis to regulate biofilm formation by posttranslational mechanisms.
Results
SepA is required for PIA-independent S. epidermidis biofilm formation
In the absence of PIA, S. epidermidis forms a protein-dependent biofilm that is primarily mediated by the cell wall-anchored adhesin Aap, which requires processing by an unknown staphylococcal protease (Rohde et al., 2005, Schaeffer et al., 2015). To characterize the mechanism of protein-dependent biofilm formation, we used an allelic replacement mutant of the icaADBC operon in the S. epidermidis central venous catheter infection isolate 1457 (Mack et al., 1992). 1457Δica was used as the reference strain in all biofilm experiments in this study. As expected, mutation of aap in 1457Δica completely abolished biofilm formation in a microtiter plate assay (Fig. 2A). To investigate the importance of secreted proteases in this Aap-dependent biofilm, we tested mutants of the secreted metalloprotease SepA and cysteine protease Ecp. We found that biofilm formation in a microtiter plate assay was greatly reduced in the 1457ΔicaΔsepA mutant, to a similar extent as the 1457ΔicaΔaap mutant (Fig. 2A). The 1457ΔicaΔecp mutant showed a modest defect in biofilm formation as well, although complementation on a high-copy plasmid did not restore biofilm formation (data not shown). To more accurately model the in vivo conditions of a S. epidermidis biofilm infection, we tested the 1457Δica and 1457ΔicaΔsepA strain pair in a flow cell biofilm (Fig. 2C-F). Biomass was greatly reduced in 1457ΔicaΔsepA as assessed by confocal laser scanning microscopy (Fig. 2E,F vs. 2C,D) and as demonstrated by quantification of the biomass across several images (Fig. 2B).
Figure 2.
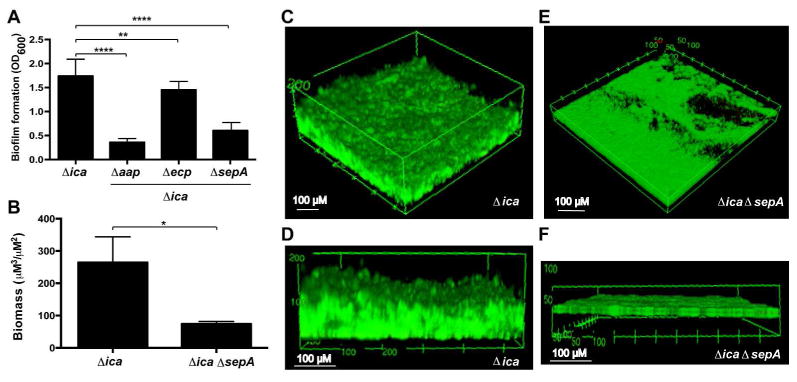
Mutation of the sepA metalloprotease results in decreased PIA-independent biofilm formation. A. Microtiter plate biofilm formation. Strains were grown statically for 20 hours in microtiter plates and the biofilm biomass was stained with crystal violet. Biofilm formation is shown as the absorbance at 600 nm of the solubilized crystal violet. Results are pooled from three experiments with three replicates each. One-way ANOVA with multiple comparisons (Bonferroni correction) was performed. ** indicates p < 0.01; **** p < 0.0001. B. COMSTAT2 results quantifying total biomass in flow cell images. Biomass in live and dead channels was quantified from three images per strain and combined. Total biomass is reported as volume per square μM. * indicates p < 0.05. C-F. Flow cell biofilm formation. Strains were grown under flowed media for 40 hours and stained with the BacLight live:dead kit. Angled (C) and side (D) views of representative images from 1457Δica at 20× magnification are shown. Angled (E) and side (F) views of representative images from 1457ΔicaΔsepA at 20 × magnification are shown. 3D projections were created using FIJI.
The level of SepA activity correlates with Aap-dependent biofilm formation
To further examine the role of SepA, a complementing sepA plasmid (psepA) was constructed using a staphylococcal vector backbone (Pang et al., 2010). psepA completely restored biofilm capacity to the 1457ΔicaΔsepA mutant compared to a vector control (Fig. 3A). To confirm this result, we tested biofilm formation of 1457ΔicaΔsepA treated with exogenously added protease. Mature SepA shares 79.3% amino acid identity with its S. aureus orthologue Aureolysin (Aur) and both are able to degrade casein (Teufel & Gotz, 1993, Arvidson, 1973). We therefore hypothesized that Aur could substitute for SepA activity. The addition of purified Aur enhanced biofilm formation of 1457ΔicaΔsepA (Fig. 3B). A dose-dependent increase in biofilm formation in 1457Δica treated with Aur was also observed (Fig. 3C), even though this strain secretes SepA. This observation suggests that endogenous SepA activity is not high enough to maximally promote biofilm formation in our WT strain.
Figure 3.
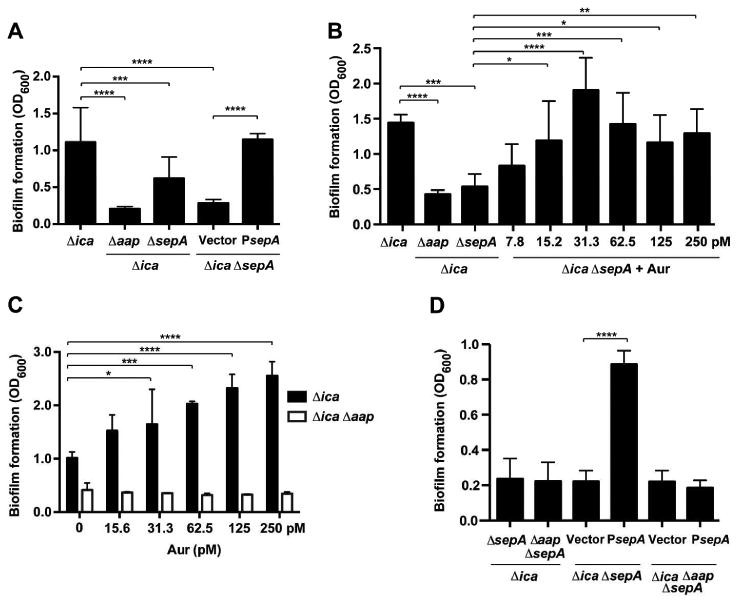
Complementation of sepA mutant by plasmid and exogenous protease. Strains were grown statically for 20 hours in 48-well microtiter plates. With the exception of (C), oneway ANOVA with multiple comparisons (Bonferroni correction) was performed. * indicates p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001. A. Microtiter biofilm assay using strains complemented with psepA. Results are pooled from three experiments with three replicates each. B. Microtiter biofilm assay with Aureolysin (Aur) added to complement 1457ΔicaΔsepA. Results are pooled from two experiments with three replicates each. C. Microtiter biofilm assay with Aur added to 1457Δica and 1457ΔicaΔaap. One representative experiment is shown. Two-way ANOVA with multiple comparisons (Bonferroni correction) was performed. D. Microtiter biofilm assay using strains complemented with psepA. Results are pooled form three experiments with three replicates each.
To assess the Aap dependence of these biofilm phenotypes, 1457Δica and 1457ΔicaΔaap were treated with either purified Aur or transformed with psepA plasmid. No increase in biofilm formation was observed with the addition of Aur to 1457ΔicaΔaap (Fig. 3C). Similarly, the psepA complementation plasmid did not enhance biofilm formation in the 1457ΔicaΔaapΔsepA mutant (Fig. 3D), further indicating the Aap dependence of the biofilm. Altogether, these results suggest that SepA/Aur enhance S. epidermidis biofilm formation and that this mechanism requires Aap.
Biofilm formation and SepA activity are repressed by SarA
SarA is a global transcriptional regulator that has been demonstrated to strongly repress expression of the secreted proteases in S. aureus (Cassat et al., 2006, Shaw et al., 2004). Although casein zymography has previously suggested increased protease activity in S. epidermidis sarA mutants (Lai et al., 2007), we sought to test SarA regulation of sepA by quantitative RT-PCR as well as a SepA-specific activity assay. Quantitative RT-PCR was used to compare expression of sepA, ecp and esp in S. epidermidis 1457Δica and 1457ΔicaΔsarA. After six hours of growth, sepA expression was 22.3-fold up-regulated in 1457ΔsarA (Table 1; p<0.001; CI 19.1 – 26.1). In contrast, no significant regulation was observed for ecp or esp (Table 1), respectively. sepA up-regulation was still evident after 16 hours of growth (7.8-fold up-regulation; CI 2.2 – 27.2; p<0.05). Still, no regulation of ecp became apparent, while there was a slight but significant up-regulation of esp (6.3-fold up-regulation; CI: 2.4 – 16.9; p<0.01).
Table 1. qPCR analysis of protease expression in S. epidermidis.
| 6 h | 18 h | |||
|---|---|---|---|---|
|
| ||||
| Fold regulationa (CI) | p-valueb | Fold regulationa (CI) | p-valueb | |
| Δ ica versus ΔicaΔsarA | ||||
| sepA | 22.3 (19.1 – 26.1) | <0.0001 | 7.8 (2.2 – 27.2) | 0.01 |
| ecp | -2.2 (-11.1 – 2.2) | 0.24 | 2.7 (-1,4 – 10.2) | 0.1 |
| esp | 1.9 (-1.1 – 4.1) | 0.07 | 6.3 (2.4 – 16.9) | 0.006 |
|
| ||||
| ΔicaΔsarA versus ΔicaΔsarAΔecp | ||||
| sepA | 1.0 (-1.9 – 2.0) | 0,99 | 1.6 (-1.1 – 2.9) | 2.3 |
|
| ||||
| ΔicaΔsarA versus Δ icaΔsarAΔesp | ||||
| sepA | -1.39 (-2.6 – 1.3) | 0.22 | 1.3 (-4.9 – 8.8) | 0.69 |
calculated using the 2ΔΔCT method (Livak & Schmittgen, 2001)
two-tailed Student's t-test
To test SepA activity in S. epidermidis, we used a fluorescein-labelled peptide that is cleaved by Aur between the Asn and Ile residues (Kavanaugh et al., 2007). We hypothesized that SepA would cleave this substrate at the same site (Fig. 4A). In S. epidermidis, low-level SepA activity was observed at the expected cleavage site (Fig. 4B, note faint band). This activity was diminished in 1457ΔicaΔsepA. In accordance with transcriptional data, the fluorescein-labelled peptide assay revealed greatly increased SepA activity in 1457ΔicaΔsarA. This activity was decreased in the 1457ΔicaΔsarAΔsepA mutant, confirming that enhanced protease activity in 1457ΔicaΔsarA is SepA-mediated (Fig. 4B). Although SarA did not affect ecp transcription, we tested whether Ecp activity was altered in a ΔsarA mutant using a FRET substrate based on the CXCR2 neutrophil receptor protein, which is cleaved by the S. aureus protease ScpA (Fig. 4C) (Olson et al., 2014, Laarman et al., 2012). We found that activity was enhanced approximately three-fold in 1457ΔicaΔsarA compared to 1457Δica, indicating that SarA represses Ecp at a post-transcriptional level (Fig. 4D). Due to the lack of an Esp-specific substrate, SarA regulation of Esp activity was not tested.
Figure 4.
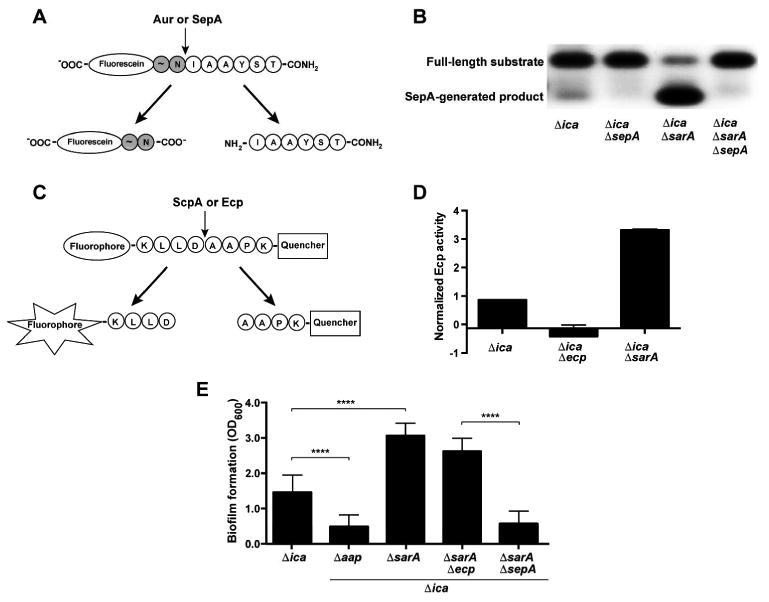
SarA negatively regulates SepA activity, Ecp activity, and SepA-dependent biofilm formation. A. Fluorescein-labelled peptide for activity of S. aureus Aur and S. epidermidis SepA. The cleavage leaves a negatively-charged, fluorescein-labelled species that can be observed by running the reaction on an agarose gel. B. Activity assay for SepA. Cell-free spent media from S. epidermidis strains was incubated with the fluorescein-labeled substrate. Reactions were run on an agarose gel and visualized under UV light. The negative image of the gel is shown. C. FRET (fluorescence resonance energy transfer)-based substrate for S. aureus ScpA and S. epidermidis Ecp. The peptide has an N-terminal fluorophore and C-terminal quencher linked to the terminal Lys residues. Cleavage allows fluorescence by separating the fluorophore from the quencher. D. Activity assay for Ecp. Cell-free spent media from S. epidermidis strains were incubated with the FRET substrate. Reaction rates (fluorescence/time) were averaged from two experiments with three replicates each and normalized to wild type 1457. E. Microtiter biofilm assay. Results are pooled from three experiments with three replicates each. One-way ANOVA with multiple comparisons (Bonferroni correction) was performed. **** indicates p < 0.0001.
Since we have observed enhanced biofilm formation in conditions with increased SepA activity, we tested the impact of SarA on PIA-independent biofilm formation. As expected, the 1457ΔicaΔsarA mutant had markedly enhanced biofilm formation in this background, but the 1457ΔicaΔsarAΔsepA displayed a biofilm phenotype similar to that of 1457ΔicaΔsepA (Fig. 4E). These observations indicated that the SarA-mediated enhancement in biofilm formation is due to increased SepA activity. Since SarA also represses Ecp activity, we tested whether Ecp contributed to the biofilm phenotype of the ΔsarA mutant. However, biofilm formation in 1457ΔicaΔsarAΔecp was similar to 1457ΔicaΔsarA, suggesting that Ecp is not required for SarA-mediated biofilm enhancement (Fig. 4D). This indicates that even with enhanced Ecp activity, this protease does not detectably contribute to biofilm formation under these conditions.
SepA promotes Aap processing in S. epidermidis
Multiple previous studies have shown that the B domain of Aap and its S. aureus orthologue SasG facilitate intercellular adhesion and biofilm maturation by forming a Zn2+-dependent twisted rope structure (Geoghegan et al., 2010, Conrady et al., 2013). In Aap, proteolytic removal of the A domain is required to allow B domain-mediated intercellular adhesion to occur (Rohde et al., 2005). We hypothesized that Aap-mediated biofilm enhancement by SepA occurs through processing of Aap. To investigate this question, Western blots of cell wall preparations were performed using an antibody to the B domain of Aap. The cell wall of 1457Δica revealed two prominent bands running above and below 250 kDa standard (Fig. 5A), corresponding to full-length and processed Aap (Fig. 1). Multiple faint bands at lower molecular weights were also present, and all of these bands were absent in a 1457ΔicaΔaap mutant control. Although full-length cell wall-anchored Aap is predicted to be 165 kDa, we observed the protein running larger than 250 kDa, which has previously been observed and is possibly due to remaining cell wall fragments from the isolation procedure (Schaeffer et al., 2015). Processed Aap containing only the B domain is expected to run at ∼200 kDa in these samples (Rohde et al., 2005) and surprisingly, we observed very little of this form of the protein. The observed intermediate band (just below 250 kDa) is likely Aap that is processed within the A domain (Fig. 5A). In testing protease knockout strains, the 1457ΔicaΔecp mutant showed no change in Aap processing from the 1457Δica background. In contrast, Aap in 1457ΔicaΔsepA shifted almost completely to the full-length form, while in 1457ΔicaΔsepA complemented by psepA the full length-form is absent, with the amount of processed Aap increased.
Figure 5. Aap processing by S. epidermidis secreted proteases.
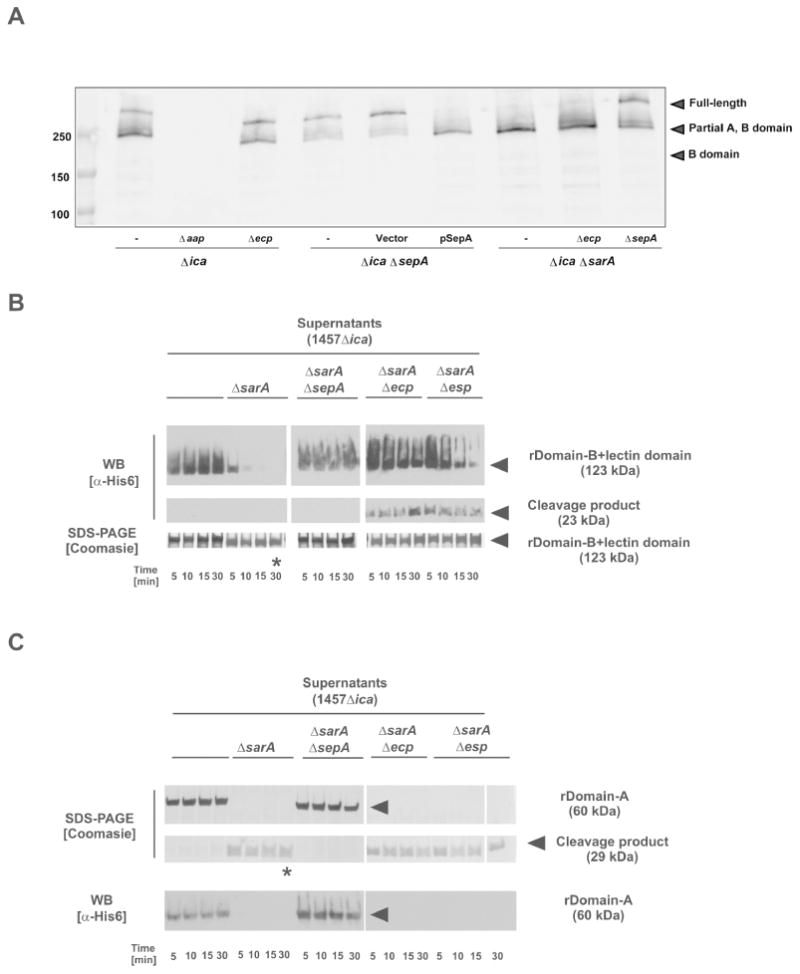
A. Western blot of cell wall preparations from S. epidermidis 1457Δica background strains. Samples were blotted and analyzed using a rabbit anti-rDomain B antiserum. Bound antibodies were detected using anti-rabbit IgG coupled to IR800 (Licor). B. Analysis of rDomain-B_LLD cleavage by incubation with concentrated cell-free supernatants protease overexpressing strain 1457ΔicaΔsarA and corresponding protease knock-out mutants incubated with rDomain-B_LLD. After incubation at 37 °C reactions were loaded onto 4-15 % gradient gels. After separation proteins were stained using Coomassie blue or blotted onto PVDF membrane. Proteins were detected using a mouse anti-His6 IgG and an anti-mouse IgG coupled to peroxidase. * indicates band for which the N-terminal sequence was determined. C. Analysis of rDomain-A cleavage by incubation with concentrated cell-free supernatants protease overexpressing strain 1457ΔicaΔsarA and corresponding protease knock-out mutants incubated with rDomain-B+212. After incubation at 37 °C reactions were loaded onto 4-15 % gradient gels. After separation proteins were stained using Coomassie blue or blotted onto PVDF membrane. Proteins were detected using a mouse anti-His6 IgG and an anti-mouse IgG coupled to peroxidase. Results shown in panel B and C were obtained using supernatants from S. epidermidis 1457-M10ΔsarA and corresponding protease knock-out mutants.
We also assessed the contribution of the SarA repressor to Aap processing (Fig. 5A). In 1457ΔicaΔsarA, Aap was mostly processed to the intermediate form, as observed with 1457ΔicaΔsepA × psepA, both conditions that have high levels of SepA activity. The 1457ΔicaΔsarAΔsepA mutant regained full-length Aap, confirming that SepA mediates SarA-increased Aap processing (Fig. 5A). The 1457ΔicaΔsarAΔecp mutant displayed the same Aap processing pattern as observed in the 1457ΔicaΔsarA mutant. These results suggest that SepA proteolytically cleaves Aap, and Ecp does not have detectable impact under these conditions. Further, these results mirror the biofilm findings demonstrating that increased SepA, but not Ecp, activity enhances biofilm formation. Since a small amount of processed Aap remains in 1457ΔicaΔsepA, another protease such as Esp may also cleave Aap in this region, or the protein may undergo spontaneous degradation, as has been observed in S. aureus SasG (Geoghegan et al., 2010).
Mapping of Aap cleavage sites
Next, a series of experiments was performed with the goal of defining the SepA cleavage sites within Aap, as well as further test the involvement of the other major secreted proteases, Ecp and Esp, in Aap processing. Due to the instability of recombinant full-length Aap, recombinant Aap fragments carrying an N-terminal His6 tag were expressed in E. coli and purified by nickel affinity chromatography. These fragments correspond to the A domain (rDomain-A; amino acids 54 - 614) and the B domain along with the 212 amino acid lectin-like domain portion of the A domain (rDomain-B_LLD; aa 398 - 1507), thus including the anticipated cleavage site between the A and B domains (Fig. 1) (Rohde et al., 2005).
Incubation of rDomain-B_LLD with supernatants of 1457Δica for 30 minutes at 37°C did not result in a detectable degradation of the recombinant 123 kDa protein, as investigated by SDS-PAGE and Western blots using anti-His6 antibodies. In contrast, incubation of rDomain-B_LLD with 1457ΔicaΔsarA supernatants resulted in a rapid decrease of immunoreactivity of the 123 kDa band in anti-His6 Western blots (Fig. 5B), while SDS PAGE analysis only documented a slight shift of the protein towards a lower molecular weight. Thus evidence was provided that the Aap degrading activity of the 1457ΔicaΔsarA supernatants leads to an N-terminal degradation of rDomain-B_LLD. Indeed, N-terminal sequencing of non-anti-His6 reactive protein after 30 minutes of incubation (Figure 5B) identified amino acids mapping to positions 601 – 608 of full length Aap, confirming N-terminal cleavage between the 212 amino acid lectin-like domain and the first G5 domain of the Aap B domain (Fig. 1). The anticipated, N-terminal tagged 23 kDa cleavage product only became apparent in Western blot analysis after 5 minutes of incubation on ice, but also disappeared thereafter (data not shown), suggesting that the lectin-like domain undergoes further degradation, resulting in smaller protein species undetectable by the methods applied here.
Incubation of rDomain-A with supernatants from 1457ΔicaΔsarA, but not from 1457Δica, also led to a rapid degradation of the recombinant protein, associated with the appearance of a 29 kDa protein species detectable by SDS-PAGE (Fig. 5C). The 29 kDa protein, unlike the mature recombinant protein, was undetectable by anti-His6 Western blot analysis. Thus, rDomain-A is processed, resulting in removal of its His6-tagged N-terminus. N-terminal sequencing of the 29 kDa protein (Figure 5C) identified amino acids mapping to 335 – 343 of the mature Aap protein (Fig. 1). Taken together, these data suggest that Aap contains at least two independent cleavage sites N-terminal to the positions of amino acid 335 and amino acid 601.
Since we observed enhanced Aap processing in 1457ΔicaΔsarA and 1457ΔicaΔsarAΔecp, but not 1457ΔicaΔsarAΔsepA (Fig. 5A), we predicted that SepA was the primary protease involved in the observed degradation of recombinant Aap sub-domains. Indeed, incubation of rDomain-B_LLD or rDomain-A with concentrated culture supernatants from 1457ΔicaΔsarAΔsepA did not result in any degradation of the recombinant protein as detected by SDS-PAGE or anti-His6 Western blot analysis. On the contrary, supernatants of 1457ΔicaΔsarAΔecp or 1457ΔicaΔsarAΔesp retained full rDomain-A processing activity. However, a differential pattern was found when rDomain-B_LLD was incubated with supernatants from 1457ΔicaΔsarAΔecp or 1457ΔicaΔsarAΔesp. Unlike the findings made using 1457ΔicaΔsarA supernatants, even after 30 minutes of incubation, unprocessed recombinant protein was detectable with almost unchanged (ΔicaΔsarAΔecp) or lowered (ΔicaΔsarAΔesp) signal intensity (Fig. 5B). Furthermore, a 29 kDa, anti-His6-reactive band appeared at increasing intensities over time. Thus, despite unchanged sepA expression in 1457ΔicaΔsarAΔecp and 1457ΔicaΔsarAΔesp as compared to 1457ΔicaΔsarA (Table 1), rDomain-B_LLD turn-over is slowed down in the absence of Ecp or Esp. Taken together, these results indicate that SepA is necessary for processing of rDomain-A and rDomain-B_LLD, while Ecp and Esp contribute to processing of rDomain-B_LLD.
Discussion
Staphylococcus epidermidis is a prevalent cause of medical device-associated infections, which are mediated by biofilm formation and are a significant burden to the healthcare system. Although there are many published studies on S. aureus biofilms, there are fewer mechanistic investigations of biofilm formation in the coagulase-negative staphylococci. Previous work has shown that S. epidermidis can produce a protein-dependent biofilm that is primarily mediated by the cell wall adhesin Aap. In this study, we found that SepA is required for Aap-dependent biofilm formation, and SepA functions by removing the N-terminal domain of Aap and promoting the homophilic interactions of this protein necessary for biofilm accumulation. This is the first report of a staphylococcal secreted protease that enhances biofilm capacity. These findings indicate that coagulase-negative staphylococci exhibit distinct biofilm development mechanisms that warrant further investigation.
Early studies on S. epidermidis biofilms focused on PIA as the critical adhesin, biofilm matrix component, and virulence factor in S. epidermidis (Otto, 2009, Otto, 2013). Other mechanisms of biofilm growth were mostly overlooked because PIA-negative strains are generally poor biofilm formers in microtiter biofilm assays. However, in a flow cell assay as well as in vivo conditions, a 1457Δaap single mutant shows a marked defect in biofilm formation (Schaeffer et al., 2015) These differences are not easily distinguishable in the typical microtiter biofilm assay because PIA can likely compensate for the lack of other adhesins. To focus on PIA-independent, Aap-dependent mechanisms of biofilm formation, we optimized the biofilm assay for a S. epidermidis strain that does not produce PIA (1457Δica). Our results corroborated previous findings that Aap is critical for biofilm formation in the absence of PIA (Schaeffer et al., 2015). Under these conditions, both Aap and SepA are required for biofilm formation (Fig. 2A). We also found that SepA and its S. aureus orthologue Aur enhanced PIA-negative biofilm formation (Fig. 3A-3C), and that this enhancement required Aap (Fig. 3C, D).
A previous study found that exogenous addition of trypsin or elastase enhanced PIA-negative biofilm formation in the clinical S. epidermidis strain 5189 by cleaving Aap at the juncture between its A and B domains (Rohde et al., 2005). However, a native staphylococcal protease that processed Aap had not been identified. There has been extensive research describing the structure of the B domain in Aap and SasG, as well as its ability to dimerize. The homodimerization of adjacent Aap proteins forms a twisted rope structure that is able to withstand significant shear force, allowing strong intercellular association (Formosa-Dague et al., 2016). This process requires Zn2+, which is coordinated by residues within each G5/E repeat of the B domain (Conrady et al., 2013, Conrady et al., 2008). Interestingly, SepA is a metalloprotease that also requires divalent cations for activity, with a preference for Zn2+ (Teufel & Gotz, 1993). Therefore, our work shows that Aap-mediated intercellular adhesion requires Zn2+ at two steps, corroborated by previous findings that Zn2+ chelation inhibits SasG-mediated biofilm formation(Geoghegan et al., 2010).
Based on the biofilm phenotypes of the sepA mutant (Fig. 2), we hypothesized that SepA processed Aap to enhance biofilm formation. Western blots revealed that Aap on the S. epidermidis cell wall is processed to multiple forms, with the most prominent processed band appearing just below 250 kDa, as well as multiple faint bands of lower molecular weight. The samples consistently run at a higher molecular weight than predicted Aap, which has been observed in previous studies of Aap and SasG (Geoghegan et al., 2010, Schaeffer et al., 2015, Rohde et al., 2005), and may be partially explained by remaining cell wall fragments. Another explanation for the increase in apparent molecular weight is the presence of acidic amino acids (Asp and Glu) in Aap. Multiple studies have reported slowed SDS-PAGE migration of proteins with greater than 11% acidic amino acids (Whitfield et al., 1995, Guan et al., 2015). According to the ExPASy ProtParam tool, full-length Aap contains 16.5% Asp and Glu residues and has a predicted pI of 4.65. We predict that the most abundant processed form (∼250 kDa) corresponds to the cleavage product within the A domain at amino acid 335. We expect that the smaller bands represent the product of cleavage at amino acid 601, as well as other potential sites that are present in the repeats of the B domain. Overexpression of sepA, either by a plasmid or in a ΔsarA mutant, results in a loss of the full-length protein and increased presence of the processed forms (Fig. 5A). Experiments with recombinant truncations of Aap incubated with S. epidermidis supernatants demonstrated that SepA cleaves Aap at two sites, Leu 335 and Leu 601. While the recombinant experiments showed that Ecp, as well as Esp, have some Aap cleavage activity (Fig. 5D), this was not observed with the cell wall Western blot, potentially due to the inability to detect these lower abundant cleavage products. Although both the 1457ΔicaΔsepA and 1457ΔicaΔsarAΔsepA mutants retain some processed Aap, especially the form that is processed within the A domain (Fig. 5A), their biofilm formation is greatly decreased relative to 1457Δica (Fig.1, Fig. 2) or 1457ΔicaΔsarA (Fig. 4D).
In terms of the effect on biofilm formation, the results of the Aap Western blot compared with our biofilm studies suggest a model in which the N terminus of the A domain is inhibitory to biofilm formation. Rather than biofilm formation occurring with the presence of processed Aap, biofilm formation seems to be de-repressed when the full-length form of Aap is lost. Although a previous study showed that the full A domain alone of SasG inhibited SasG-mediated intercellular accumulation (Geoghegan et al., 2010), it is not known whether it is the entire A domain that is responsible. Our results suggest that the portion N terminal to the lectin domain specifically inhibits biofilm formation. The absence of a cleavage product corresponding to a total loss of the A domain suggests that Aap can mediate intercellular adhesion even when the lectin domain is present. Further, the function of free A domain in the biofilm matrix is not known, nor is the specific ligand bound by the lectin domain of Aap. Clearly, much remains to be clarified regarding the structure and function of Aap.
In S. aureus, the SepA homolog Aureolysin has been reported to be positively regulated by the agr quorum-sensing system (Shaw et al., 2004). A previous report also showed decreased S. epidermidis extracellular protease activity in an Δagr mutant based on casein zymography, which is generally accepted to indicate Aur or SepA metalloprotease activity (Lai et al., 2007). However, the S. epidermidis ΔrnaIII mutant does not show differential regulation of sepA compared to wildtype in a microarray (Olson et al., 2014). Additionally, the ΔrnaIII mutant did not demonstrate a loss of SepA activity in our fluorescein-labelled peptide assay (data not shown). Collectively, these findings indicate that sepA is not agr quorum-sensing regulated in S. epidermidis.
SarA has previously been shown to down-regulate extracellular protease activity in S. epidermidis based on a casein zymography assay (Lai et al., 2007). We found that SarA negatively regulates SepA and Ecp activity using substrates specific to these enzymes (Fig. 4B-4C), and that a ΔsarA mutant had markedly enhanced biofilm formation and Aap processing relative to 1457Δica (Fig. 4D), both phenotypes being dependent on SepA. This demonstrates that in an Aap-mediated biofilm, SarA is a strong negative regulator of biofilm formation. Similar observations have recently been made for Embp-induced biofilms, which are also negatively regulated by SarA (Christner et al., 2012). On the contrary, in ica-positive S. epidermidis, ΔsarA mutants have decreased ica expression and lower biofilm capacity (Tormo et al., 2005, Handke et al., 2007). Our results highlight the varying effects of this global regulatory system depending on the particular biofilm morphology.
SepA has been shown to enhance S. epidermidis biofilm formation via increased processing of the autolysin AtlE to its active form (Christner et al., 2012). This phenotype occurs due to strengthening the biofilm matrix with eDNA, since active AtlE induces autolysis. However, the conditions in our study seem to favour a protein-dependent, rather than eDNA-dependent biofilm. Our findings show that SepA enhancement of biofilm formation requires the presence of Aap (Fig. 2), rather than any other SepA substrate. Further, the previous study would suggest that a sepA mutant should have less eDNA in the biofilm. However, an assay to detect eDNA in the biofilm matrix showed no difference between wild type S. epidermidis 1457 and a sepA mutant in our biofilm growth conditions (data not shown). Therefore, we observe a protein-dependent biofilm that requires Aap and its processing by SepA. Our work shows that SepA has another mechanism to promote biofilm formation in addition to enhancing autolysis.
This finding also supports the possibility of multispecies biofilm formation of S. aureus and S. epidermidis. We found that both SepA and S. aureus Aur can process Aap to its biofilm-inducing form. Although one might predict that both enzymes could also process S. aureus SasG, a previous study did not find that Aur or other secreted S. aureus proteases were required for SasG cleavage (Geoghegan et al., 2010). Interestingly, the Leu 335 and Leu 601 SepA cleavage residues identified in the present study are absent in all annotated forms of SasG, which suggests that SasG processing may occur via another mechanism. Recent work has also demonstrated that SasG and Aap B domains are able to form heterodimers, providing a mechanism for intercellular adhesion between S. aureus and S. epidermidis in a biofilm (Formosa-Dague et al., 2016). Because S. aureus Aur can promote S. epidermidis PIA-independent biofilm formation (Fig. 3), these collective findings suggest that S. epidermidis might have the ability to co-opt S. aureus protease activity to promote biofilm formation, which could be beneficial in particular shared host niches.
Overall, this work demonstrates that the secreted protease SepA is critical to S. epidermidis protein-dependent biofilm formation by processing Aap. Our findings add to previous work showing that SepA helps to defend against the host immune system by cleaving the antimicrobial peptide dermicidin (Lai et al., 2007) and promoting S. epidermidis survival following phagocytosis by human neutrophils (Cheung et al., 2010). Therefore, SepA has multiple roles as a virulence factor, both promoting the biofilm lifestyle and providing defences against the human innate immune system. Strategies to interfere with S. epidermidis proteolytic activity or Aap processing could open new avenues to treat certain types of biofilm infections.
Experimental procedures
Strains and plasmids
Strains, plasmids, and bacteriophages are listed in Table S1. Unless otherwise noted, strains were cultured at 37 °C with 200 RPM shaking. E. coli was grown in lysogeny broth (LB) or on LB agar plates containing 100 μg/mL ampicillin to maintain plasmids when necessary. S. aureus was grown in tryptic soy broth (TSB) or on TSB agar plates, and S. epidermidis was grown in TSB, on TSB agar plates, or in brain-heart infusion broth (BHI). Staphylococcal plasmids were maintained with growth in 10 μg/mL chloramphenicol. Chromosomal tetM transductants were selected by growth on TSB agar plates containing 2.5 μg/mL tetracycline, and dhfr transductants were selected by growth on TSB agar plates containing 10 μg/mL trimethoprim.
The sepA complementation plasmid psepA was generated by cloning sepA and its promoter region from S. epidermidis 1457 chromosomal DNA using primers E1 and E2 (listed in Table S2) into a TOPO vector, then digesting with NheI and XhoI and ligating into pCM29 (Pang et al., 2010).
Generation of S. epidermidis mutants
An esp allelic replacement mutant was constructed by first cloning 5′ and 3′ regions of esp (amplified using primers 2304/2305 and 2306/2307) into the EcoRI-BamHI (5” region) and SalI-PstI sites of pUC19 (Yanisch-Perron et al., 1985). dhfr, encoding trimethoprim resistance (Handke et al., 2007), and pROJ6448, containing a temperature sensitive gram positive replicon (Projan & Archer, 1989), were subsequently ligated into the SalI and PstI sites, respectively, to generate pNF281. Allelic replacement experiments with S. epidermidis 1457 were performed as previously described (Schaeffer et al., 2015). Transfer of plasmids psepA and pCM28 from E. coli into S. epidermidis was accomplished by electroporation or phage transduction with phage 187. Electroporation of S. epidermidis was performed using plasmids isolated from E. coli strain DC10B (Maliszewski & Nuxoll, 2014, Monk et al., 2012). Transduction by phage 187 was performed according to Winstel et al., with S. aureus PS187ΔhsdRΔSauUS1 carrying the respective plasmid as the donor strain (Winstel et al., 2015, Winstel et al., 2016). S. epidermidis double and triple mutants were created by phage crosses with phage 71 (Olson & Horswill, 2014, Dean et al., 1973) or phage A6C (Rohde et al., 2005).
Microtiter biofilm assay
Strains were picked from TSB agar plates and grown overnight in BHI, then diluted 1:200 in 50% BHI. 10 μg/mL chloramphenicol was added at both steps to cultures where necessary to maintain plasmids. 1 mL/well of the 1:200 subculture was added to a 48-well tissue culture-treated plate. Plates were incubated without shaking at 37 °C for 18-20 hours, then media was aspirated and biomass was washed once by pipetting gently with 750 μL per well of sterile dH2O. Adherent biomass was stained with 1 mL per well 0.1% crystal violet for 5 minutes, then washed once with 1 mL sterile dH2O by gently pipetting. Final stained biomass was solubilized in 1 mL per well isopropanol and transferred to a 96-well plate with 200 μL per well to measure absorbance at 600 nm on a Tecan plate reader. For Aureolysin-treated biofilms, purified S. aureus Aureolysin (BioCentrum) suspended in PBS was added to biofilms at time 0.
Flow cell biofilm assay
Flow cell apparatus was set up as previously described (Boles & Horswill, 2008). Briefly, flow cell channels were created by gluing acid-etched coverslips to polycarbonate chambers. Overnight cultures grown in BHI were diluted 1:100 in BHI and injected into chambers, where bacteria were allowed to attach for 1 hour at room temperature before flow was initiated. The biofilms were grown for 40-45 hours in a 37 °C room, with media flow of 2% BHI at a pump speed of 3.25 RPM. After growth, the chambers were injected with BacLight live/dead stain (Thermo Fisher Scientific) according to the manufacturer's protocol and imaged by confocal microscopy. 3D images from the confocal microscopy were generated with ImageJ Fiji software (Schindelin et al., 2012) and biomass was quantified using Comstat 2.1 (Heydorn et al., 2000, Vorregaard, 2008).
Aap western blot of cell wall samples
To isolate cell walls, strains were grown statically in 5 mL of 50% BHI for 16.5 hours to mimic biofilm assay conditions. Cells were harvested by centrifugation at 4,000 × g for 5 minutes at 4 °C, then washed in 2 mL cold 50 mM Tris and 1mM EDTA (TE) pH 8.0 with 1× protease inhibitor cocktail (Sigma). After a second centrifugation, cell pellets were resuspended to a total volume of 100 μL in TE pH 8.0 with 20% sucrose and 1× protease inhibitor cocktail. 10 μg lysostaphin was added to each sample, and samples were incubated at 37 °C for 15 minutes. Samples were centrifuged at 13,000 × g for 10 minutes and supernatants were collected and quantified by the BioRad Bradford assay using BSA as a standard. Samples were diluted to equal protein concentrations, loaded at 1 μg per well on a 4-20% gradient SDS-PAGE gel, and blotted to 0.2 μM pore size PVDF using a Turbo transfer blot apparatus.
For the Aap Western blot, the membrane was incubated for one hour in blocking buffer of 5% milk in PBS, overnight in primary rabbit anti-Aap B domain antibody (Rohde et al., 2005, Rohde et al., 2007) diluted 1:100,000 in 5% milk in PBS + 0.1% Tween 20, and for one hour in secondary IR800-labeled goat-anti rabbit (Licor) diluted 1:20,000 in 5% milk in PBS + 0.1% Tween 20. Between steps, the membrane was washed with PBS + 0.1% Tween 20. Before imaging, the membrane was washed with PBS to remove Tween. Imaging was performed on a Licor Odyssey scanner.
Expression of recombinant Aap domains
Expression and purification of recombinant, His6-tagged Aap domain A (amino acids 54 – 614) was carried out essentially as described (Rohde et al., 2005). For expression of a N-terminally His6-tagged protein spanning the predicted 212 amino acid lectin-like domain and the repetitive B domain (amino acids 398 - 1507), the corresponding coding region was amplified using primers aap1165_for and aap1785_rev and genomic DNA from S. epidermidis 5179 as a template. Purified amplicons were cloned into pENTR/D-TOPO and subsequently subcloned into DEST17, giving pDESTdomain B+212. Correctness of clones was validated using restriction digestion and sequencing of vector – insert junctions. rDomain-B_LLD and His6-tagged recombinant domain A were expressed and purified essentially as described (Rohde et al., 2005).
Aap proteolysis assay and N-terminal sequencing
Supernatants were obtained from cultures of S. epidermidis 1457-M10, 1457-M10ΔsarA and corresponding mutants defective in expression of sepA, esp, or ecp. Supernatants from 50 ml overnight cultures were cleared by centrifugation, and subsequently 10-fold concentrated using centrifuge cartridges (molecular weight cut-off 10.000 Da; Merck Millipore, Darmstadt, Germany). Recombinant rDomain A (10 μg) or rDomain B+212 (10 μg) were incubated with 5 μl concentrated culture supernatants at 37 °C. Samples were heated at 70 °C in LDS buffer for 7 min, and then separated by SDS-PAGE using 4 – 15 % Bis-Tris gradient gels (Invitogen, Karlsruhe, Germany). Proteins were made visible using Coomassie staining. In some experiments, proteins were detected by Western blot using anti-His6 IgG (1:10,000) and peroxidase-coupled anti-mouse IgG (1:10,000). N-terminal sequencing by Edman degradation and mass spectrometry has been described elsewhere (Sturenburg et al., 2002).
Quantification of sepA, ecp and esp expression
For the analysis of differential gene expression, triplicates of overnight cultures of strains 1457-M10 and 1457-M10ΔsarA were diluted 1:100 and grown for 6 hours and 18 hours in 20 ml of tryptic soy broth (TSB), respectively. RNA was isolated as described previously (Franke et al., 2007). Briefly, bacteria were immersed in RNAprotect Bacteria reagent (Qiagen, Hilden, Germany) and lysed. RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany) and a sample volume containing 1μg of RNA was digested twice by use of DNA-free DNase (Ambion, Life Technologies, Darmstadt, Germany). A volume of 5 μl of DNase-digested RNA was reverse-transcribed, using the iScript cDNA Synthesis Kit (BioRad, Hercules, CA, U.S.A.). The resulting cDNA was frozen immediately at -80°C until further use. Relative quantification of gene expression was performed on a Light Cycler 480 (Roche Applied Science, Mannheim, Germany). The staphylococcal housekeeping genes gyrB, rho and tpiA, coding for DNA gyrase subunit B, transcription termination factor rho and triosephosphate isomerase tpiA respectively, were used as internal reference genes (Sihto et al., 2014, Crawford et al., 2014, Theis et al., 2007). The TaqMan Fast Advanced Master Mix 2× (Thermo Fisher Scientific) according to the manufacturer's instructions, duplicates of the reaction were set up manually in a reaction volume of 15 μl.
All primers and probes were designed using Primer3 (Untergasser et al., 2012, Koressaar & Remm, 2007) and are listed in table S2. The hydrolysis probes contain FAM as fluorophore and BHQ1 as quencher. All primers and probes were synthesized by MWG Eurofins, Germany. Cycling and subsequent analysis of qPCR data was performed as described previously (Weiser et al., 2016). Values were assumed to be normally distributed and the two-tailed Student's t-test was performed with the significance level set to 0.017 to account for multiple testing.
SepA activity assay
Overnight cultures grown in TSB were diluted to an OD600 of 0.2 in TSB and allowed to grow for 3 hours. Cultures were filtered through Spin-X tubes and the filtrates were tested for SepA activity as previously described (Kavanaugh et al., 2007). 20 μL reactions were set up containing 15 μL of spent media with fluorescein-labeled substrate at a final concentration of 150 μM. Reactions were buffered to 20 mM Tris-HCl pH 8.0. EDTA was added at 1 mM for SepA inhibition reactions. Reactions were incubated at 37°C for 3 hours, then 20 μL of 50% glycerol was added to each reaction to enable loading in an agarose gel. 20 μL of each reaction was loaded on an agarose gel and imaged using a GelDoc camera.
Ecp activity assay
Overnight cultures were diluted to an OD600 of 0.1 and grown in 25 mL TSB in 125 mL flasks. At each time point, 500 μL was removed from each culture and filtered through Spin-X tubes. The resulting spent media were tested for Ecp activity using the FRET substrate based on CXCR2, with the sequence (5-FAM)-Lys-Leu-Leu-Asp-Ala-Ala-Pro-Lys-(QXL520)-OH (AnaSpec, Fremont, CA) (Olson et al., 2014). Spent media from each culture was buffered by mixing 2 μL of 2M Tris-HCl pH 7.4 with 198 μL spent media. In a black 96 well microtiter plate, 10 μL of 50 μM FRET substrate in dH2O was mixed with 90 μL buffered spent media and incubated in a TECAN plate reader at 37 °C for 30 minutes, with fluorescence (Excitation 490, Emission 520) measured every minute with the gain set to 100. Activity is calculated as the slope of the fluorescence over time.
Acknowledgments
We thank Dr. Carolyn Schaeffer Koziol for advice on S. epidermidis genetic manipulation, Dr. Eric Ransom for assistance with cloning of sepA, Friedrich Buck for help with N-terminal sequencing, and Gesche Kroll for excellent technical assistance. Michael Otto kindly provided 1457ΔsepA. S. aureus PS187ΔhsdRΔSauUS1 and bacteriophage 187 were obtained from Andreas Peschel. AEP is funded by an American Heart Association Predoctoral Fellowship, award number 14PRE19910005. ARH and PDF were supported by projects 3 and 2, respectively, of NIH grant AI083211.
Footnotes
Author contributions: Alexandra E. Paharik performed experiments, interpreted data, and wrote the paper.
Marta Kotasinska performed experiments and interpreted data.
Anna Both performed experiments, interpreted data, and wrote the paper.
Tra-my Hoang performed experiments and interpreted data.
Henning Büttner performed experiments and interpreted data.
Paroma Roy performed experiments and interpreted data.
Paul D. Fey designed the study, interpreted data, and wrote the paper.
Alexander R. Horswill designed the study, interpreted data, and wrote the paper.
Holger Rohde designed the study, interpreted data, and wrote the paper.
References
- Arciola CR, Campoccia D, Speziale P, Montanaro L, Costerton JW. Biofilm formation in Staphylococcus implant infections. A review of molecular mechanisms and implications for biofilm-resistant materials. Biomaterials. 2012;33:5967–5982. doi: 10.1016/j.biomaterials.2012.05.031. [DOI] [PubMed] [Google Scholar]
- Arvidson S. Studies on extracellular proteolytic enzymes from Staphylococcus aureus. II. Isolation and characterization of an EDTA-sensitive protease. Biochimica et biophysica acta. 1973;302:149–157. doi: 10.1016/0005-2744(73)90017-x. [DOI] [PubMed] [Google Scholar]
- Boles BR, Horswill AR. Agr-mediated dispersal of Staphylococcus aureus biofilms. PLoS pathogens. 2008;4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttner H, Mack D, Rohde H. Structural basis of Staphylococcus epidermidis biofilm formation: mechanisms and molecular interactions. Frontiers in cellular and infection microbiology. 2015;5:14. doi: 10.3389/fcimb.2015.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassat J, Dunman PM, Murphy E, Projan SJ, Beenken KE, Palm KJ, Yang SJ, Rice KC, Bayles KW, Smeltzer MS. Transcriptional profiling of a Staphylococcus aureus clinical isolate and its isogenic agr and sarA mutants reveals global differences in comparison to the laboratory strain RN6390. Microbiology. 2006;152:3075–3090. doi: 10.1099/mic.0.29033-0. [DOI] [PubMed] [Google Scholar]
- Chen C, Krishnan V, Macon K, Manne K, Narayana SV, Schneewind O. Secreted proteases control autolysin-mediated biofilm growth of Staphylococcus aureus. The Journal of biological chemistry. 2013;288:29440–29452. doi: 10.1074/jbc.M113.502039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung GY, Rigby K, Wang R, Queck SY, Braughton KR, Whitney AR, Teintze M, DeLeo FR, Otto M. Staphylococcus epidermidis strategies to avoid killing by human neutrophils. PLoS pathogens. 2010;6:e1001133. doi: 10.1371/journal.ppat.1001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christner M, Heinze C, Busch M, Franke G, Hentschke M, Bayard Duhring S, Buttner H, Kotasinska M, Wischnewski V, Kroll G, Buck F, Molin S, Otto M, Rohde H. sarA negatively regulates Staphylococcus epidermidis biofilm formation by modulating expression of 1 MDa extracellular matrix binding protein and autolysis-dependent release of eDNA. Molecular microbiology. 2012;86:394–410. doi: 10.1111/j.1365-2958.2012.08203.x. [DOI] [PubMed] [Google Scholar]
- Conlon BP, Geoghegan JA, Waters EM, McCarthy H, Rowe SE, Davies JR, Schaeffer CR, Foster TJ, Fey PD, O'Gara JP. Role for the A domain of unprocessed accumulation-associated protein (Aap) in the attachment phase of the Staphylococcus epidermidis biofilm phenotype. Journal of bacteriology. 2014;196:4268–4275. doi: 10.1128/JB.01946-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrady DG, Brescia CC, Horii K, Weiss AA, Hassett DJ, Herr AB. A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:19456–19461. doi: 10.1073/pnas.0807717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrady DG, Wilson JJ, Herr AB. Structural basis for Zn2+-dependent intercellular adhesion in staphylococcal biofilms. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E202–211. doi: 10.1073/pnas.1208134110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigan RM, Rigby D, Handley P, Foster TJ. The role of Staphylococcus aureus surface protein SasG in adherence and biofilm formation. Microbiology. 2007;153:2435–2446. doi: 10.1099/mic.0.2007/006676-0. [DOI] [PubMed] [Google Scholar]
- Crawford EC, Singh A, Metcalf D, Gibson TW, Weese SJ. Identification of appropriate reference genes for qPCR studies in Staphylococcus pseudintermedius and preliminary assessment of icaA gene expression in biofilm-embedded bacteria. BMC research notes. 2014;7:451. doi: 10.1186/1756-0500-7-451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darouiche RO. Treatment of infections associated with surgical implants. The New England journal of medicine. 2004;350:1422–1429. doi: 10.1056/NEJMra035415. [DOI] [PubMed] [Google Scholar]
- Dean BA, Williams RE, Hall F, Corse J. Phage typing of coagulase-negative staphylococci and micrococci. The Journal of hygiene. 1973;71:261–270. doi: 10.1017/s0022172400022737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker R, Burdelski C, Zobiak M, Buttner H, Franke G, Christner M, Sass K, Zobiak B, Henke HA, Horswill AR, Bischoff M, Bur S, Hartmann T, Schaeffer CR, Fey PD, Rohde H. An 18 kDa scaffold protein is critical for Staphylococcus epidermidis biofilm formation. PLoS pathogens. 2015;11:e1004735. doi: 10.1371/journal.ppat.1004735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donlan RM. Biofilms and device-associated infections. Emerging infectious diseases. 2001;7:277–281. doi: 10.3201/eid0702.010226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa-Dague C, Speziale P, Foster TJ, Geoghegan JA, Dufrene YF. Zinc-dependent mechanical properties of Staphylococcus aureus biofilm-forming surface protein SasG. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:410–415. doi: 10.1073/pnas.1519265113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke GC, Dobinsky S, Mack D, Wang CJ, Sobottka I, Christner M, Knobloch JK, Horstkotte MA, Aepfelbacher M, Rohde H. Expression and functional characterization of gfpmut3.1 and its unstable variants in Staphylococcus epidermidis. Journal of microbiological methods. 2007;71:123–132. doi: 10.1016/j.mimet.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Geoghegan JA, Corrigan RM, Gruszka DT, Speziale P, O'Gara JP, Potts JR, Foster TJ. Role of surface protein SasG in biofilm formation by Staphylococcus aureus. Journal of bacteriology. 2010;192:5663–5673. doi: 10.1128/JB.00628-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice EA, Segre JA. The skin microbiome. Nature reviews Microbiology. 2011;9:244–253. doi: 10.1038/nrmicro2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruszka DT, Whelan F, Farrance OE, Fung HK, Paci E, Jeffries CM, Svergun DI, Baldock C, Baumann CG, Brockwell DJ, Potts JR, Clarke J. Cooperative folding of intrinsically disordered domains drives assembly of a strong elongated protein. Nature communications. 2015;6:7271. doi: 10.1038/ncomms8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruszka DT, Wojdyla JA, Bingham RJ, Turkenburg JP, Manfield IW, Steward A, Leech AP, Geoghegan JA, Foster TJ, Clarke J, Potts JR. Staphylococcal biofilm-forming protein has a contiguous rod-like structure. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E1011–1018. doi: 10.1073/pnas.1119456109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Y, Zhu Q, Huang D, Zhao S, Jan Lo L, Peng J. An equation to estimate the difference between theoretically predicted and SDS PAGE-displayed molecular weights for an acidic peptide. Scientific reports. 2015;5:13370. doi: 10.1038/srep13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handke LD, Slater SR, Conlon KM, O'Donnell ST, Olson ME, Bryant KA, Rupp ME, O'Gara JP, Fey PD. SigmaB and SarA independently regulate polysaccharide intercellular adhesin production in Staphylococcus epidermidis. Canadian journal of microbiology. 2007;53:82–91. doi: 10.1139/w06-108. [DOI] [PubMed] [Google Scholar]
- Hellmark B, Soderquist B, Unemo M, Nilsdotter-Augustinsson A. Comparison of Staphylococcus epidermidis isolated from prosthetic joint infections and commensal isolates in regard to antibiotic susceptibility, agr type, biofilm production, and epidemiology. International journal of medical microbiology : IJMM. 2013;303:32–39. doi: 10.1016/j.ijmm.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Hennig S, Nyunt Wai S, Ziebuhr W. Spontaneous switch to PIA-independent biofilm formation in an ica-positive Staphylococcus epidermidis isolate. International journal of medical microbiology : IJMM. 2007;297:117–122. doi: 10.1016/j.ijmm.2006.12.001. [DOI] [PubMed] [Google Scholar]
- Heydorn A, Nielsen AT, Hentzer M, Sternberg C, Givskov M, Ersboll BK, Molin S. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology. 2000;146(Pt 10):2395–2407. doi: 10.1099/00221287-146-10-2395. [DOI] [PubMed] [Google Scholar]
- Hussain M, Herrmann M, von Eiff C, Perdreau-Remington F, Peters G. A 140-kilodalton extracellular protein is essential for the accumulation of Staphylococcus epidermidis strains on surfaces. Infection and immunity. 1997;65:519–524. doi: 10.1128/iai.65.2.519-524.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase T, Uehara Y, Shinji H, Tajima A, Seo H, Takada K, Agata T, Mizunoe Y. Staphylococcus epidermidis Esp inhibits Staphylococcus aureus biofilm formation and nasal colonization. Nature. 2010;465:346–349. doi: 10.1038/nature09074. [DOI] [PubMed] [Google Scholar]
- Kavanaugh JS, Thoendel M, Horswill AR. A role for type I signal peptidase in Staphylococcus aureus quorum sensing. Molecular microbiology. 2007;65:780–798. doi: 10.1111/j.1365-2958.2007.05830.x. [DOI] [PubMed] [Google Scholar]
- Klug D, Wallet F, Kacet S, Courcol RJ. Involvement of adherence and adhesion Staphylococcus epidermidis genes in pacemaker lead-associated infections. Journal of clinical microbiology. 2003;41:3348–3350. doi: 10.1128/JCM.41.7.3348-3350.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koressaar T, Remm M. Enhancements and modifications of primer design program Primer3. Bioinformatics. 2007;23:1289–1291. doi: 10.1093/bioinformatics/btm091. [DOI] [PubMed] [Google Scholar]
- Kuroda M, Ito R, Tanaka Y, Yao M, Matoba K, Saito S, Tanaka I, Ohta T. Staphylococcus aureus surface protein SasG contributes to intercellular autoaggregation of Staphylococcus aureus. Biochemical and biophysical research communications. 2008;377:1102–1106. doi: 10.1016/j.bbrc.2008.10.134. [DOI] [PubMed] [Google Scholar]
- Laarman AJ, Mijnheer G, Mootz JM, van Rooijen WJ, Ruyken M, Malone CL, Heezius EC, Ward R, Milligan G, van Strijp JA, de Haas CJ, Horswill AR, van Kessel KP, Rooijakkers SH. Staphylococcus aureus Staphopain A inhibits CXCR2-dependent neutrophil activation and chemotaxis. The EMBO journal. 2012;31:3607–3619. doi: 10.1038/emboj.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Villaruz AE, Li M, Cha DJ, Sturdevant DE, Otto M. The human anionic antimicrobial peptide dermcidin induces proteolytic defence mechanisms in staphylococci. Molecular microbiology. 2007;63:497–506. doi: 10.1111/j.1365-2958.2006.05540.x. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Macintosh RL, Brittan JL, Bhattacharya R, Jenkinson HF, Derrick J, Upton M, Handley PS. The terminal A domain of the fibrillar accumulation-associated protein (Aap) of Staphylococcus epidermidis mediates adhesion to human corneocytes. Journal of bacteriology. 2009;191:7007–7016. doi: 10.1128/JB.00764-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack D, Davies AP, Harris LG, Knobloch JK, Rohde H. Staphylococcus epidermidis Biofilms: Functional Molecules, Relation to Virulence, and Vaccine Potential. Topics in current chemistry. 2009;288:157–182. doi: 10.1007/128_2008_19. [DOI] [PubMed] [Google Scholar]
- Mack D, Siemssen N, Laufs R. Parallel induction by glucose of adherence and a polysaccharide antigen specific for plastic-adherent Staphylococcus epidermidis: evidence for functional relation to intercellular adhesion. Infection and immunity. 1992;60:2048–2057. doi: 10.1128/iai.60.5.2048-2057.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill SS, Edwards JR, Bamberg W, Beldavs ZG, Dumyati G, Kainer MA, Lynfield R, Maloney M, McAllister-Hollod L, Nadle J, Ray SM, Thompson DL, Wilson LE, Fridkin SK. Multistate point-prevalence survey of health care-associated infections. The New England journal of medicine. 2014;370:1198–1208. doi: 10.1056/NEJMoa1306801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maliszewski KL, Nuxoll AS. Use of electroporation and conjugative mobilization for genetic manipulation of Staphylococcus epidermidis. Methods Mol Biol. 2014;1106:125–134. doi: 10.1007/978-1-62703-736-5_11. [DOI] [PubMed] [Google Scholar]
- Monk IR, Shah IM, Xu M, Tan MW, Foster TJ. Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. mBio. 2012;3 doi: 10.1128/mBio.00277-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon JL, Banbula A, Oleksy A, Mayo JA, Travis J. Isolation and characterization of a highly specific serine endopeptidase from an oral strain of Staphylococcus epidermidis. Biological chemistry. 2001;382:1095–1099. doi: 10.1515/BC.2001.138. [DOI] [PubMed] [Google Scholar]
- Mootz JM, Malone CL, Shaw LN, Horswill AR. Staphopains modulate Staphylococcus aureus biofilm integrity. Infection and immunity. 2013 doi: 10.1128/IAI.00377-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgenstern M, Post V, Erichsen C, Hungerer S, Buhren V, Militz M, Richards G, Moriarty F. Biofilm formation increases treatment failure in Staphylococcus epidermidis device-related osteomyelitis of the lower extremity in human patients. Journal of orthopaedic research : official publication of the Orthopaedic Research Society. 2016 doi: 10.1002/jor.23218. [DOI] [PubMed] [Google Scholar]
- Oleksy A, Golonka E, Banbula A, Szmyd G, Moon J, Kubica M, Greenbaum D, Bogyo M, Foster TJ, Travis J, Potempa J. Growth phase-dependent production of a cell wall-associated elastinolytic cysteine proteinase by Staphylococcus epidermidis. Biological chemistry. 2004;385:525–535. doi: 10.1515/BC.2004.062. [DOI] [PubMed] [Google Scholar]
- Olson ME, Horswill AR. Bacteriophage Transduction in Staphylococcus epidermidis. Methods Mol Biol. 2014;1106:167–172. doi: 10.1007/978-1-62703-736-5_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson ME, Todd DA, Schaeffer CR, Paharik AE, Van Dyke MJ, Buttner H, Dunman PM, Rohde H, Cech NB, Fey PD, Horswill AR. Staphylococcus epidermidis agr quorum-sensing system: signal identification, cross talk, and importance in colonization. Journal of bacteriology. 2014;196:3482–3493. doi: 10.1128/JB.01882-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcus epidermidis--the ‘accidental’ pathogen. Nature reviews Microbiology. 2009;7:555–567. doi: 10.1038/nrmicro2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcus colonization of the skin and antimicrobial peptides. Expert review of dermatology. 2010;5:183–195. doi: 10.1586/edm.10.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcal infections: mechanisms of biofilm maturation and detachment as critical determinants of pathogenicity. Annual review of medicine. 2013;64:175–188. doi: 10.1146/annurev-med-042711-140023. [DOI] [PubMed] [Google Scholar]
- Otto M. Physical stress and bacterial colonization. FEMS microbiology reviews. 2014a;38:1250–1270. doi: 10.1111/1574-6976.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto M. Staphylococcus epidermidis pathogenesis. Methods Mol Biol. 2014b;1106:17–31. doi: 10.1007/978-1-62703-736-5_2. [DOI] [PubMed] [Google Scholar]
- Paharik AE, Horswill AR. The Staphylococcal biofilm: adhesins, regulation, and host response. Microbiol Spectrum. 2016;4 doi: 10.1128/microbiolspec.VMBF-0022-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang YY, Schwartz J, Thoendel M, Ackermann LW, Horswill AR, Nauseef WM. agr-Dependent interactions of Staphylococcus aureus USA300 with human polymorphonuclear neutrophils. Journal of innate immunity. 2010;2:546–559. doi: 10.1159/000319855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Projan SJ, Archer GL. Mobilization of the relaxable Staphylococcus aureus plasmid pC221 by the conjugative plasmid pGO1 involves three pC221 loci. Journal of bacteriology. 1989;171:1841–1845. doi: 10.1128/jb.171.4.1841-1845.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche FM, Meehan M, Foster TJ. The Staphylococcus aureus surface protein SasG and its homologues promote bacterial adherence to human desquamated nasal epithelial cells. Microbiology. 2003;149:2759–2767. doi: 10.1099/mic.0.26412-0. [DOI] [PubMed] [Google Scholar]
- Rohde H, Burandt EC, Siemssen N, Frommelt L, Burdelski C, Wurster S, Scherpe S, Davies AP, Harris LG, Horstkotte MA, Knobloch JK, Ragunath C, Kaplan JB, Mack D. Polysaccharide intercellular adhesin or protein factors in biofilm accumulation of Staphylococcus epidermidis and Staphylococcus aureus isolated from prosthetic hip and knee joint infections. Biomaterials. 2007;28:1711–1720. doi: 10.1016/j.biomaterials.2006.11.046. [DOI] [PubMed] [Google Scholar]
- Rohde H, Burdelski C, Bartscht K, Hussain M, Buck F, Horstkotte MA, Knobloch JK, Heilmann C, Herrmann M, Mack D. Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Molecular microbiology. 2005;55:1883–1895. doi: 10.1111/j.1365-2958.2005.04515.x. [DOI] [PubMed] [Google Scholar]
- Rupp ME. Clinical characteristics of infections in humans due to Staphylococcus epidermidis. Methods Mol Biol. 2014;1106:1–16. doi: 10.1007/978-1-62703-736-5_1. [DOI] [PubMed] [Google Scholar]
- Schaeffer CR, Hoang TN, Sudbeck CM, Alawi M, Tolo IE, Robinson DA, Horswill AR, Rohde H, Fey PD. Versatility of Biofilm Matrix Molecules in Staphylococcus epidermidis Clinical Isolates and Importance of Polysaccharide Intercellular Adhesin Expression during High Shear Stress. mSphere. 2016;1 doi: 10.1128/mSphere.00165-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer CR, Woods KM, Longo GM, Kiedrowski MR, Paharik AE, Buttner H, Christner M, Boissy RJ, Horswill AR, Rohde H, Fey PD. Accumulation-associated protein enhances Staphylococcus epidermidis biofilm formation under dynamic conditions and is required for infection in a rat catheter model. Infection and immunity. 2015;83:214–226. doi: 10.1128/IAI.02177-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nature methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schommer NN, Christner M, Hentschke M, Ruckdeschel K, Aepfelbacher M, Rohde H. Staphylococcus epidermidis uses distinct mechanisms of biofilm formation to interfere with phagocytosis and activation of mouse macrophage-like cells 774A.1. Infection and immunity. 2011;79:2267–2276. doi: 10.1128/IAI.01142-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw L, Golonka E, Potempa J, Foster SJ. The role and regulation of the extracellular proteases of Staphylococcus aureus. Microbiology. 2004;150:217–228. doi: 10.1099/mic.0.26634-0. [DOI] [PubMed] [Google Scholar]
- Sievert DM, Ricks P, Edwards JR, Schneider A, Patel J, Srinivasan A, Kallen A, Limbago B, Fridkin S. Antimicrobial-resistant pathogens associated with healthcare-associated infections: summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2009-2010. Infection control and hospital epidemiology : the official journal of the Society of Hospital Epidemiologists of America. 2013;34:1–14. doi: 10.1086/668770. [DOI] [PubMed] [Google Scholar]
- Sihto HM, Tasara T, Stephan R, Johler S. Validation of reference genes for normalization of qPCR mRNA expression levels in Staphylococcus aureus exposed to osmotic and lactic acid stress conditions encountered during food production and preservation. FEMS microbiology letters. 2014;356:134–140. doi: 10.1111/1574-6968.12491. [DOI] [PubMed] [Google Scholar]
- Sturenburg E, Sobottka I, Mack D, Laufs R. Cloning and sequencing of Enterobacter aerogenes OmpC-type osmoporin linked to carbapenem resistance. International journal of medical microbiology : IJMM. 2002;291:649–654. doi: 10.1078/1438-4221-00175. [DOI] [PubMed] [Google Scholar]
- Sugimoto S, Iwamoto T, Takada K, Okuda K, Tajima A, Iwase T, Mizunoe Y. Staphylococcus epidermidis Esp degrades specific proteins associated with Staphylococcus aureus biofilm formation and host-pathogen interaction. Journal of bacteriology. 2013;195:1645–1655. doi: 10.1128/JB.01672-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teufel P, Gotz F. Characterization of an extracellular metalloprotease with elastase activity from Staphylococcus epidermidis. Journal of bacteriology. 1993;175:4218–4224. doi: 10.1128/jb.175.13.4218-4224.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis T, Skurray RA, Brown MH. Identification of suitable internal controls to study expression of a Staphylococcus aureus multidrug resistance system by quantitative real-time PCR. Journal of microbiological methods. 2007;70:355–362. doi: 10.1016/j.mimet.2007.05.011. [DOI] [PubMed] [Google Scholar]
- Tormo MA, Marti M, Valle J, Manna AC, Cheung AL, Lasa I, Penades JR. SarA is an essential positive regulator of Staphylococcus epidermidis biofilm development. Journal of bacteriology. 2005;187:2348–2356. doi: 10.1128/JB.187.7.2348-2356.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang LH, Cassat JE, Shaw LN, Beenken KE, Smeltzer MS. Factors contributing to the biofilm-deficient phenotype of Staphylococcus aureus sarA mutants. PloS one. 2008;3:e3361. doi: 10.1371/journal.pone.0003361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3--new capabilities and interfaces. Nucleic acids research. 2012;40:e115. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorregaard M. Informatics and Mathematical Modelling. Kongens Lyngby, Denmark: Technical University of Denmark; 2008. Comstat2 - a modern 3D image analysis environment for biofilms. [Google Scholar]
- Weiser J, Henke HA, Hector N, Both A, Christner M, Buttner H, Kaplan JB, Rohde H. Sub-inhibitory tigecycline concentrations induce extracellular matrix binding protein Embp dependent Staphylococcus epidermidis biofilm formation and immune evasion. International journal of medical microbiology : IJMM. 2016 doi: 10.1016/j.ijmm.2016.05.015. [DOI] [PubMed] [Google Scholar]
- Whitfield WG, Chaplin MA, Oegema K, Parry H, Glover DM. The 190 kDa centrosome-associated protein of Drosophila melanogaster contains four zinc finger motifs and binds to specific sites on polytene chromosomes. Journal of cell science. 1995;108(Pt 11):3377–3387. doi: 10.1242/jcs.108.11.3377. [DOI] [PubMed] [Google Scholar]
- Winstel V, Kuhner P, Krismer B, Peschel A, Rohde H. Transfer of plasmid DNA to clinical coagulase-negative staphylococcal pathogens by using a unique bacteriophage. Applied and environmental microbiology. 2015;81:2481–2488. doi: 10.1128/AEM.04190-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winstel V, Kuhner P, Rohde H, Peschel A. Genetic engineering of untransformable coagulase-negative staphylococcal pathogens. Nature protocols. 2016;11:949–959. doi: 10.1038/nprot.2016.058. [DOI] [PubMed] [Google Scholar]
- Yanisch-Perron C, Vieira J, Messing J. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985;33:103–119. doi: 10.1016/0378-1119(85)90120-9. [DOI] [PubMed] [Google Scholar]
- Ziebuhr W, Heilmann C, Gotz F, Meyer P, Wilms K, Straube E, Hacker J. Detection of the intercellular adhesion gene cluster (ica) and phase variation in Staphylococcus epidermidis blood culture strains and mucosal isolates. Infection and immunity. 1997;65:890–896. doi: 10.1128/iai.65.3.890-896.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]