

Summary
Sepsis is an often lethal syndrome resulting from maladaptive immune and metabolic responses to infection, compromising host homeostasis. Disease tolerance is a defense strategy against infection that preserves host homeostasis without exerting a direct negative impact on pathogens. Here, we demonstrate that induction of the iron-sequestering ferritin H chain (FTH) in response to polymicrobial infections is critical to establish disease tolerance to sepsis. The protective effect of FTH is exerted via a mechanism that counters iron-driven oxidative inhibition of the liver glucose-6-phosphatase (G6Pase), and in doing so, sustains endogenous glucose production via liver gluconeogenesis. This is required to prevent the development of hypoglycemia that otherwise compromises disease tolerance to sepsis. FTH overexpression or ferritin administration establish disease tolerance therapeutically. In conclusion, disease tolerance to sepsis relies on a crosstalk between adaptive responses controlling iron and glucose metabolism, required to maintain blood glucose within a physiologic range compatible with host survival.
Keywords: infection, sepsis, disease tolerance, ferritin, iron, heme, gluconeogenesis, glucose-6-phosphatase, inflammation, metabolism
Graphical Abstract
Highlights
-
•
Ferritin is required to establish disease tolerance to sepsis
-
•
Iron heme represses liver G6Pase during sepsis
-
•
Ferritin counters G6Pase repression and sustains blood glucose levels after sepsis
-
•
Liver gluconeogenesis is required to establish disease tolerance to sepsis
Disease tolerance to sepsis depends on a crosstalk between iron and glucose- metabolic responses that maintain blood glucose levels within a dynamic range compatible with host survival.
Introduction
The pathologic outcome of infections is a direct consequence of the extent of metabolic dysfunction and damage imposed to tissues that sustain host homeostasis (Kotas and Medzhitov, 2015, Soares et al., 2014, Soares et al., 2017). Disease tolerance is a defense strategy that limits the pathologic outcome of infections without interfering directly with the host's pathogen load (Medzhitov et al., 2012). This defense strategy relies on tissue damage control mechanisms that preserve the functional output of parenchymal tissues, maintaining homeostatic parameters within a dynamic range compatible with host survival (Kotas and Medzhitov, 2015, Soares et al., 2014, Soares et al., 2017).
Sepsis is a clinical syndrome, affecting ∼19 million individuals per year worldwide, characterized by a maladaptive host response with ensuing organ dysfunction. Despite tremendous efforts during the last decades, no specific therapy for sepsis exists. Increasing rates of antimicrobial resistance and lack of novel antimicrobials adds to the problem and substantiates the urgent need of innovative therapeutic options (Angus and van der Poll, 2013).
The pathogenesis of sepsis is only partially explained by unfettered inflammation while metabolic deregulation, leading to organ dysfunction and eventually to organ failure, is increasingly recognized as an important component of this process (Angus and van der Poll, 2013). While the mechanisms underlying the inflammatory response that characterizes the pathogenesis of sepsis are fairly well understood, those driving metabolic deregulation and multi-organ dysfunction or failure remain elusive (Kotas and Medzhitov, 2015, Soares et al., 2014).
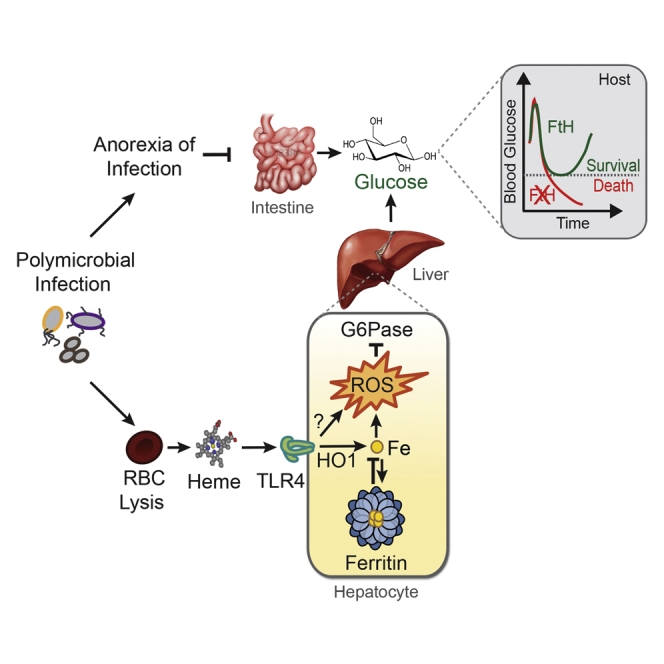
Systemic infections, including those leading to sepsis, are coupled to a host metabolic response restraining invading pathogens from accessing iron (Soares and Weiss, 2015). The large majority of iron available to pathogens is contained within the prosthetic heme groups of hemoproteins, among which hemoglobin holds the largest reservoir (Gozzelino and Soares, 2014). Upon hemolysis, extracellular hemoglobin is oxidized and releases heme (Larsen et al., 2010, Pamplona et al., 2007), eventually leading to the accumulation of labile heme in plasma (i.e., metabolic active heme that is loosely bound to a variety of plasma molecules). Accumulation of labile heme in plasma plays a central role in the pathogenesis of sepsis (Larsen et al., 2010). This is countered by the induction heme-catabolism by heme oxygenase-1 (HO-1), which contributes critically to the establishment of disease tolerance to sepsis (Larsen et al., 2010). As a trade-off, however, heme catabolism by HO-1 generates labile iron that can catalyze the production of reactive oxygen species via Fenton chemistry, eventually leading to oxidative stress. This is counteracted by ferritin, a heteropolymeric protein complex encoded by the ferritin heavy/heart chain (FTH) and light/liver (FTL) genes (Harrison and Arosio, 1996). Ferritin is composed of 24 FTH/FTL subunits, which can store and convert ∼4,500 atoms of Fe2+ into inert Fe3+ through the ferroxidase activity of FTH (Harrison and Arosio, 1996). The ferroxidase activity of ferritin is critical to the establishment of disease tolerance to infection in animals (Gozzelino and Soares, 2014) and plants (Deák et al., 1999). Here, we demonstrate that FTH establishes disease tolerance to sepsis via a mechanism that sustains the expression/activity of the liver G6Pase, a rate-limiting enzyme in the gluconeogenesis and glycogenolysis pathways (Mithieux, 1997, van Schaftingen and Gerin, 2002). This is required to support liver glucose production in response to systemic infections so that blood glucose levels are maintained within a dynamic physiologic range compatible with host survival, hence conferring disease tolerance to sepsis.
Results
FTH Is Essential to the Establishment of Disease Tolerance to Sepsis
Expression of FTH protein (Figures 1A and 1B) by liver F4/80+ macrophages and hepatocytes (Figures 1C, S1A, and S1B), was readily induced in C57BL/6 mice subjected to cecal ligation and puncture (CLP), a well-established experimental model of polymicrobial infection. Liver Fth mRNA was only slightly induced (Figure S1C), consistent with post-transcriptional regulation of FTH expression (Hentze et al., 1987).
Figure 1.

FTH Establishes Disease Tolerance to Sepsis
(A) FTH western blot from C57BL/6 mice liver extracts before (Control; Ctr) or 12 hr after a severe CLP. Representative western blot from one out of three mice per genotype.
(B) Densitometry of FTH protein expression normalized to β-actin. Data pooled from three mice per genotype.
(C) Representative immunostaining of liver FTH (green), DNA (blue), and F4/80+ Kupffer cells (red) before (Control; Ctr) or 12 hr after a severe CLP.
(D) FTH western blot in liver extracts from Fthlox/lox and Mx1CreFthΔ/Δ mice before (Control; Ctr) and 48 hr after CLP. Representative of four mice per genotype.
(E) Survival of Fthlox/lox (n = 20) and Mx1CreFthΔ/Δ (n = 17) mice subjected to CLP, pooled from four independent experiments.
(F) Aerobic (Ae) and anaerobic (An) bacterial colony forming units (CFU), 48 hr after CLP. Data pooled from three independent experiments.
(G) Survival after CLP in bone marrow chimeric mice expressing Fth in all tissues (Fth+/+ → Fth+/+, n = 14), in parenchyma only (FthΔ/Δ → Fth+/+, n = 14), or in bone marrow-derived cells only (Fth+/+ → FthΔ/Δ, n = 8). Data from three independent experiments with similar trend.
(H) Survival of control Fthlox/lox mice (n = 35), AlbCreFthΔ/Δ (n = 12), LysMCreFthΔ/Δ (n = 24), and AlbCreLysMCreFthΔ/Δ (n = 12) mice lacking FTH expression in hepatocytes, myeloid cells, or hepatocytes and myeloid cells, respectively. Data pooled from ten independent experiments.
(I and J) Clinical severity score (CSS) (Gonnert et al., 2011) (I) and survival of Fthlox/lox (n = 6) and Mx1CreFthΔ/Δ (n = 4) mice subjected to PCI (J). Data for CSS is from one independent experiment and survival is from two independent experiments.
(K) CFU for aerobic (Ae) and anaerobic (An) bacteria 24 hr after PCI in two independent experiments.
Red bars in (F) and (K) are mean values and dotted circles are individual mice. PC, peritoneal cavity. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
See also Figures S1, S2, and S3.
Figure S1.

FTH Establishes Disease Tolerance to Sepsis, Related to Figure 1
(a) Representative FTH immunostaining in the liver of C57BL/6 mice before (Control) and 12 hr after severe CLP. Red: F4/80+ Kupffer cells; Green: FTH, Blue: DNA. (b) Control staining with secondary antibodies only. (c) Quantification of Fth mRNA by qRT-PCR before (Control; Ctr.) and 12 hr after CLP. Data are shown as mean ± SD of 3 mice per group. (d) Percentage of circulating CD45.1+ versus CD45.2+ leukocytes in control (Fth+/+→Fth+/+), (Fth+/+→FthΔ/Δ) and (FthΔ/Δ→Fth+/+) bone marrow chimeras. (e) Percentage of CD19+ B cells, TCRβ+ T cells, Gr1+CD11bhigh PMN cells and CD11b+ (monocyte/macrophages and PMN cells) in the same mice as in (d). Data in (d) and (e) is shown as mean ± SD of 4-6 mice per group, pooled from 2-3 representative experiments. (f) Fth mRNA expression quantified by qRT-PCR in circulating leukocytes and expressed as % relative to (B6→B6) controls. Data shown as mean ± SD of 3-4 mice per group, pooled from 2-3 representative experiments. ∗p < 0.05.
Mx1CreFthΔ/Δ mice, in which the Fthlox/lox allele was deleted in an inducible manner in the liver and hematopoietic cells (Figure 1D), succumbed to CLP (Figure 1E), whereas control Fthlox/lox mice, expressing physiologic levels of FTH (Figures 1D and 1E), had much reduced mortality. Pathogen load of Mx1CreFthΔ/Δ mice was indistinguishable from that of Fthlox/lox mice, as assessed for aerobic and anaerobic bacteria in different organs 48 hr after CLP (Figure 1F). This demonstrates that FTH is required to establish disease tolerance to sepsis in mice.
To address the relative contribution of FTH expression in hematopoietic and parenchyma cells to disease tolerance, we generated chimeric mice in which the Fthlox/lox allele was deleted specifically in hematopoietic or parenchyma cells, respectively. Inducible Fthlox/lox deletion, specifically in hematopoietic cells, was achieved in lethally irradiated Fthlox/lox or C57BL/6 mice reconstituted with hematopoietic cells from ROSA26CreERT2Fthlox/lox mice (FthΔ/Δ → Fthlox/lox) (Figures S1D–S1F). Inducible Fthlox/lox deletion, specifically in parenchyma cells, was achieved in lethally irradiated ROSA26CreERT2Fthlox/lox mice reconstituted with hematopoietic cells from Fthlox/lox or C57BL/6 mice (Fthlox/lox → FthΔ/Δ) (Figures S1D–S1F). Fthlox/lox → FthΔ/Δ but not FthΔ/Δ → Fthlox/lox mice, succumbed more frequently to CLP, as compared to control Fthlox/lox → Fthlox/lox mice (Figure 1G). This suggests that FTH expression in parenchyma cells, but not in hematopoietic cells, is necessary to the establishment of disease tolerance to sepsis.
AlbCreLysMCreFthΔ/Δ mice, in which the Fthlox/lox allele was deleted in hepatocytes and myeloid cells, succumbed more frequently to CLP, as compared to control Fthlox/lox mice (Figure 1H). Lethality of CLP in AlbCreFthΔ/Δ and LysMCreFthΔ/Δ mice, in which the Fthlox/lox allele was deleted in hepatocytes or myeloid cells, respectively, was similar to control Fthlox/lox mice (Figure 1H). This suggests that FTH expression in hepatocytes, and presumably in macrophages, is required to establish disease tolerance to sepsis.
When subjected to a defined polymicrobial inoculum isolated from human microbiota (peritoneal contamination and infection [PCI]), Mx1CreFthΔ/Δ mice developed clinical signs of severe disease (Figure 1I) and succumbed (Figure 1J), which was not the case for control Fthlox/lox mice (Figures 1I and 1J). Pathogen load of Mx1CreFthΔ/Δ and Fthlox/lox mice was indistinguishable (Figure 1K). This suggests that the susceptibility of Mx1CreFthΔ/Δ mice to infection is not attributed to a putative modulatory effect exerted by FTH on the polymicrobial inoculum.
The systemic inflammatory response to CLP was indistinguishable in Mx1CreFthΔ/Δ versus Fthlox/lox mice, as assessed for a range of cytokines in plasma (Figure S2A). This suggests that FTH confers disease tolerance to sepsis via a mechanism that does not involve a putative immunoregulatory effect.
Figure S2.

FTH Does Not Modulate Cytokine Levels or Tissue Damage, Related to Figure 1
(a) Cytokine levels in plasma 24 hr after CLP. Mean ± SD (n = 4 per genotype). (b) Serological markers before (Control; Ctrl) and 48 hr after CLP. Mean ± SD, n = 3-5 mice per genotype, pooled from 3 independent experiments. (c) H&E stained paraffin sections representative of at least three Fthlox/lox and Mx1CreFthΔ/Δ mice untreated (Control) and 48 hr after CLP. B: bronchus; CV: central vein; G: glomerulus. Magnification 400x.
The extent of organ dysfunction in Mx1CreFthΔ/Δ and Fthlox/lox mice subjected to CLP was similar, as monitored for liver, muscle, and kidney by the accumulation of aspartate aminotransferase (AST), alanine aminotransferase (ALT), creatine phosphokinase (CPK), lactate dehydrogenase (LDH), and blood urea nitrogen (BUN) in plasma, respectively (Figure S2B), confirmed by histological analyzes (Figure S2C). This suggests that disease tolerance to sepsis does not rely on the well-established cytoprotective effect of FTH (Berberat et al., 2003).
Cardiovascular damage (Figure S3A), cardiac function (Figure S3B), and vascular integrity (Figure S3C) were similar in Mx1CreFthΔ/Δ and Fthlox/lox mice subjected to CLP. This suggests that FTH is not required to prevent the development of cardiovascular dysfunction, a hallmark of sepsis (Angus and van der Poll, 2013).
Figure S3.

FTH Does Not Modulate Cardiovascular Function, Related to Figure 1
(a) H&E stained paraffin section of hearts from Fthlox/lox and Mx1CreFthΔ/Δ mice before and 48 hr after CLP. Images are representative of at least 3 mice per genotype per experimental conditions. Magnifications are 100 (top) and 400x. (b) Cardiac pressure-volume (PV) loop analysis of Fthlox/lox and Mx1CreFthΔ/Δ mice, 48 hr after CLP. Load-dependent, i.e. Heart rate (HR) end-systolic pressure (ESP), cardiac output (CO), relaxation (Tau-Glantz), pressure volume area (PVA) and mean arterial pressure (MAP) and load-independent, i.e. slope of end-systolic pressure volume Relationship (ESPVR) and maximum of generated energy (Emax) parameters are shown as mean ± SD from n = 3-6 mice per group pooled from 4 independent experiments. NS: non-significant. (c) Vascular leakage monitored by quantification of Evans Blue accumulation in parenchyma organs, before and 48 hr after CLP. Data are shown as mean ± SD from n = 5 mice per group, pooled from 3 independent experiments. NS: non-significant.
FTH Regulates Glucose Metabolism in Response to Infection
When subjected to CLP, Mx1CreFthΔ/Δ mice decreased body weight (Figure 2A) and temperature (Figure 2B) more so than Fthlox/lox mice. Food intake (i.e., anorexia of infection) was similar in Mx1CreFthΔ/Δ and Fthlox/lox mice (Figure S4A), suggesting that FTH regulates metabolic adaptation to infection rather than sickness behavior.
Figure 2.

FTH Regulates Glucose Metabolism in Response to Polymicrobial Infection
(A–C) Relative weight (A), temperature (B), and blood glucose levels (C) in Fthlox/lox (n = 13) and Mx1CreFthΔ/Δ (n = 15) mice subjected to CLP. Mean ± SEM from five independent experiments.
(D) Blood glucose levels in Mx1CreFthΔ/Δ mice subjected to CLP and receiving glucose (n = 15) (gavage; 2 mg/kg body weight [BW]; two times daily for 4 days and one time daily for 3 days) or vehicle (n = 12). Mean ± SEM from three independent experiments.
(E) Survival of the same mice as in (D).
(F) CFU for aerobic (Ae) and anaerobic (An) bacteria, 48 hr after CLP. Red bars represent mean values and dotted circles individual mice.
(G) Blood glucose levels in C57BL/6 mice receiving heme (n = 15) (intraperitoneally [i.p.] 30 mg/kg BW) or control protoporphyrin IX (PPIX) (n = 10). Mean ± SEM from three to five independent experiments.
(H) Blood glucose levels in Fthlox/lox (n = 8) and Mx1CreFthΔ/Δ (n = 9) mice receiving heme (i.p. 25–30 mg/kg BW). Mean ± SD from two independent experiments.
(I) Survival of Fthlox/lox (n = 8) and Mx1CreFthΔ/Δ (n = 9) mice receiving heme (i.p. 25–30 mg/kg BW).
(J) Blood glucose levels in C57BL/6 Tlr4+/+ (n = 13) and Tlr4−/− (n = 9) mice receiving heme (i.p. 30 mg/kg BW). Mean ± SEM from three to four independent experiments.
(K) Blood glucose levels in C57BL/6 Ifnr1+/+ (n = 7) and Ifnr1−/− (n = 7) mice receiving heme (i.p. 30 mg/kg BW).
Mean ± SD from two independent experiments. PC, peritoneal cavity. Numbers in gray in (A)–(D) and (H) are live/total mice at each time point. ∗p < 0.05; ∗∗p < 0.01.
See also Figure S4.
Figure S4.

FTH Expression and Glucose Metabolism, Related to Figure 2
(a) Food intake in Fthlox/lox and Mx1CreFthΔ/Δ mice before and after CLP. Data are shown as mean and SD from 3 mice per genotype. (b) Blood glucose levels in Fthlox/lox (n = 3) and Mx1CreFthΔ/Δ (n = 3) mice in the first 12 hr after CLP. Mean ± SD, data from 1 experiment. (c) Blood glucose levels in C57BL/6 mice in the first 12 hr after CLP (n = 7) or control sham operation (n = 12). Mean ± SD, data from 3-4 independent experiments. (d) Total heme levels in the peritoneal cavity in C57BL/6 mice before (n = 4) and 3 hr after CLP (n = 3). (e) Blood glucose levels in C57BL/6 mice in the first 8 hr after heme (n = 15) or protoporphyrin IX (PPIX) (n = 10) administration (i.p.; 30 mg/kg BW). Mean ± SEM, data from 3-5 independent experiments. (f) Blood glucose levels in Fthlox/lox (n = 8) and Mx1CreFthΔ/Δ (n = 9) in the first 8 hr after heme administration (i.p.; 25 to 30 mg/kg BW). Mean ± SD from 2 independent experiments. (g) Blood glucose levels in C57BL/6 Tlr4+/+ (n = 13) and Tlr4−/− (n = 9) mice in the first 8 hr after heme administration (i.p.; 30 mg/kg BW). Mean ± SEM from 3-4 independent experiments. (h) Blood glucose levels in C57BL/6 Ifnr1+/+ (n = 7) and Ifnr1−/− (n = 7) mice in the first 8 hr after heme administration (i.p.; 30 mg/kg BW). Mean ± SD from 2 independent experiments. (i) Oral glucose tolerance test (oGTT), (j) pyruvate tolerance test (PTT) and (k) glucagon challenge test (GCT) in Fthlox/lox and Mx1CreFthΔ/Δ mice. Data are shown as mean ± SEM, pooled from 3 independent experiments with n = 3 mice per genotype per experiment. (l) insulin tolerance test (ITT) in Fthlox/lox and Mx1CreFthΔ/Δ mice. Data are shown as mean ± SD, from 1 experiment with n = 5 mice per genotype. (m) Plasma insulin levels in Fthlox/lox and Mx1CreFthΔ/Δ mice after overnight fasting. Data are shown as mean ± SD from 4-5 mice per genotype. (n) Plasma insulin levels in Fthlox/lox and Mx1CreFthΔ/Δ mice before (control; Ctr.) and 48 hr after CLP. Data are shown as mean ± SD from 4-6 mice per genotype, pooled from 2-3 experiments.
Blood glucose levels 24 hr after CLP were reduced to similar levels in Mx1CreFthΔ/Δ and Fthlox/lox mice (Figure 2C). However, while within the following days blood glucose levels were partially restored in Fthlox/lox mice, this was not the case for Mx1CreFthΔ/Δ mice that went on to succumb to CLP (Figure 2C). Glucose administration to Mx1CreFthΔ/Δ mice, starting from 12 hr after CLP onward, restored blood glucose levels (Figure 2D) and survival (Figure 2E), as compared to vehicle-treated controls (Figures 2D and 2E). This was not associated with modulation of pathogen load (Figure 2F), suggesting that FTH establishes disease tolerance to sepsis via a mechanism that regulates glucose metabolism.
Of note, both Mx1CreFthΔ/Δ and Fthlox/lox mice increased blood glucose levels in the first 6 hr following CLP (Figure S4B), which was also the case in sham-operated mice (Figure S4C). This suggests that this early metabolic response is (1) likely elicited by tissue injury, and (2) not controlled by FTH.
We then asked whether extracellular accumulation of labile heme following CLP (Figure S4D) contributes to deregulate glucose metabolism. Heme administration to C57BL/6 mice decreased blood glucose levels (Figure 2G), an effect not observed in control mice receiving a synthetic protoporphyrin IX lacking iron (Figure 2G). In contrast to Fthlox/lox mice, heme administration to Mx1CreFthΔ/Δ mice led to severe hypoglycemia (Figure 2H) and death (Figure 2I). This suggests that FTH counters the development of iron heme-driven hypoglycemia in response to polymicrobial infections.
Of note, reduction of blood glucose levels in response to heme administration was preceded by a transient raise in blood glucose levels not observed in response to protoporphyrin IX (Figure S4E). This effect was similar in Mx1CreFthΔ/Δ and Fthlox/lox mice (Figure S4F), suggesting that FTH does not regulate this early iron heme-driven hyperglycemia.
Labile heme is an alarmin (Soares and Bozza, 2016) sensed by the toll-like receptor 4 (TLR4) (Figueiredo et al., 2007). TLR4-deficient (Tlr4−/−) mice failed to reduce blood glucose levels in response to heme administration, as compared to wild-type Tlr4+/+ controls (Figure 2J). This suggests that heme reduces blood glucose levels via a TLR4-dependent mechanism. In contrast, heme reduced blood glucose levels in mice lacking type 1 interferon (IFN) receptor (Ifnr1−/−), to the same extent to wild-type (Ifnr1+/+) controls (Figure 2K). This suggests that heme reduces blood glucose levels via a type 1 IFN response-independent mechanism.
The early increase in blood glucose levels observed in response to heme also occurred in Tlr4−/− (Figure S4G) and Ifnr1−/− (Figure S4H) mice, suggesting that neither TLR4 nor type 1 IFN response are involved in this metabolic response.
FTH Promotes Liver Gluconeogenesis in Response to Polymicrobial Infection
Administration of glucose (Figure S4I), glucose precursors (e.g., pyruvate [Figure S4J] or glucagon [Figure S4K]), as well as insulin (Figure S4L), resulted in similar transient elevation and normalization of blood glucose levels in Mx1CreFthΔ/Δ and Fthlox/lox mice. Levels of insulin in plasma were similar in overnight-fasted Mx1CreFthΔ/Δ and Fthlox/lox mice (Figure S4M) or Mx1CreFthΔ/Δ and Fthlox/lox mice subjected to CLP (Figure S4N). This suggests that FTH does not regulate steady-state glucose metabolism and/or regulates insulin resistance. This does not exclude insulin resistance from regulating glucose metabolism in response to acute polymicrobial infection (Wang et al., 2016).
Luminex-based analysis of major glucose regulatory hormones, including amylin, glucagon, gastric inhibitory polypeptide, and pancreatic polypeptide revealed no changes in their concentration in the serum of Fthlox/lox versus MxCreFthΔ/Δ mice at basal state or after polymicrobial infection (data not shown). While we cannot exclude that FTH may regulate the expression of these or other glucose regulatory hormones, our data are not consistent with this notion.
Pyruvate administration to Mx1CreFthΔ/Δ mice subject to CLP failed to normalize blood glucose levels (Figure 3A) or to prevent mortality (Figure 3B), as compared to control vehicle-treated Mx1CreFthΔ/Δ mice (Figures 3A and 3B). Of note, pyruvate is a glucose precursor used preferentially by the liver gluconeogenic pathway, suggesting that FTH regulates liver gluconeogenesis in response to polymicrobial infections. To test this hypothesis, we performed targeted metabolomics in the liver of Mx1CreFthΔ/Δ and Fthlox/lox mice subjected to CLP, analyzing metabolites (Figure 3C) and amino acids (Figure S5) involved in the gluconeogenesis and the Krebs cycle. The data obtained was integrated into the genome scale metabolic reconstruction of mouse metabolism iMM1415 (Sigurdsson et al., 2010) and analyzed using a flux balance analysis approach assessing the overall capability to produce ATP (Orth et al., 2010). Metabolomics, flux balance, and flux variability analysis (Mahadevan and Schilling, 2003), including simulations of single gene and reaction deletion of iMM1415 (Tables S1 and S2) (Mahadevan and Schilling, 2003), suggest that when subjected to CLP, Mx1CreFthΔ/Δ mice shifted liver glucose metabolism toward a metabolic flux characteristic of glycolysis, in detriment of gluconeogenesis, as compared to Fthlox/lox mice (Figure 3C). This indicates that induction of FTH in response to polymicrobial infection sustains liver gluconeogenesis in detriment of glycolysis. This is not associated, however, with changes in the relative levels of liver glucogenic amino acids, apart from cysteine (Figure S5), arguing that FTH regulates the expression and/or the activity of one or several enzymes partaking in these metabolic pathways.
Figure 3.

FTH Prevents Liver Glucose Metabolism from Shifting Toward Glycolysis in Detriment of Gluconeogenesis
(A) Blood glucose in Mx1CreFthΔ/Δ mice subjected to CLP and receiving pyruvate (n = 6) or vehicle (n = 7). Mean ± SD from two independent experiments. Numbers in gray indicate live/total mice at different time points.
(B) Survival of the same mice as in (A).
(C) Targeted metabolomics of the liver and flux balance analysis for ATP generation using data integration into a reconstructed metabolic network iMM1415 (Sigurdsson et al., 2010).
Data are shown as mean ± SD (n = 5 mice per group). DHAP, dihydroxyacetone phosphate; F6P, fructose 6-phosphate; F16P, fructose-1,6-bisphosphate; GAP, glyceraldehyde 3-phosphate; G6P, glucose 6-phosphate; 13bPG, 1,3-bisphosphoglycerate; 2PG, 2-phosphoglycerate; 3PG, 3-phosphoglycerate; PEP, phosphopyruvate. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Figure S5.

Targeted Amino Acids Metabolomics of the Liver, Related to Figure 3
Data are shown as mean ± SD of 5 mice per group. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. NS: non-significant.
FTH Counters Iron Heme-Driven Inhibition of Liver G6Pase
To address whether FTH regulates the expression of genes controlling liver metabolic pathways that might contribute to disease tolerance to polymicrobial sepsis we performed a microarray analysis. Eight genes were differentially expressed in the liver of Mx1CreFthΔ/Δ versus Fthlox/lox mice subjected to CLP (Figure 4A; GEO: GSE92703). These included Fth1, epoxide hydrolase 1 (Ephx1), glutathione S-transferase Mu 1 (Gstm1) and 2 (Gstm2), lysine-specific demethylase hairless (Hr), natriuretic peptide receptor B (Npr2), and transferrin receptor protein 1 (Tfrc), as well as the G6Pase catalytic subunit (G6pc1) (Figure 4A), encoding the final and rate-limiting enzyme of the gluconeogenic and glycogenolytic pathways (Mutel et al., 2011, van Schaftingen and Gerin, 2002).
Figure 4.

FTH Sustains Hepatic G6Pase in Response to Systemic Polymicrobial Infection
(A) Volcano plot of mean RNA expression from RNA microarray screen in the liver of Mx1CreFthΔ/Δ versus Fthlox/lox mice, 48 hr after CLP (n = 4 mice per group).
(B) Validation of G6pc1 mRNA expression by qRT-PCR in the liver of Mx1CreFthΔ/Δ mice not subjected to CLP (n = 13), 48 hr after CLP (n = 12) versus Fthlox/lox mice not subjected to CLP (n = 11), or 48 hr after CLP (n = 11). Data pooled from three independent experiments.
(C and D) Representative western blot of G6pc1 in Mx1CreFthΔ/Δ versus Fthlox/lox mice (C) and relative quantification by densitometry (D) before (Control; Ctr) or 48 hr after CLP (n = 6 per group).
(E) Quantification of liver Gpt1 mRNA by qRT-PCR, same mice as (B).
(F) Liver G6Pase enzymatic activity, same mice as (B).
(G) Quantification of G6PC1 mRNA levels by qRT-PCR in HepG2 cells untreated (−) or treated (+) with heme and/or TNF. Mean ± SEM from eight independent experiments with similar trend.
(H) G6PC1 protein levels in HepG2 cells treated as in (G).
(I) Relative quantification G6PC1 protein levels by densitometry of western blot from HepG2 cells treated as in (G) and (H).
(J) Relative luciferase units (RLU) in HepG2 cells transiently co-transfected with a rat G6pc1 firefly luciferase and CMV Renilla luciferase reporters. Control cells were transfected with a promoterless firefly luciferase reporter. Cells were treated, 48 hr after transfection, with heme and TNF. Data are shown as mean RLU ± SD from four independent experiments with similar trend.
(K) Liver mRNA quantification by qRT-PCR in C57BL/6 mice receiving heme (+; n = 8) (i.p. 30 mg/kg BW) or not (−; n = 9). Data pooled from three independent experiments.
(L) Liver mRNA quantification by qRT-PCR in Mx1CreFthΔ/Δ versus Fthlox/lox mice receiving heme (+; i.p. 15 mg/kg BW; n = 3 per genotype) or not (−; n = 6–8 per genotype). Data pooled from one to three independent experiments.
(M) Relative luciferase units (RLU) in HepG2 cells transiently co-transfected with a rat G6pc1 firefly luciferase and a CMV Renilla luciferase reporters plus a human FTH expression vector. Cells transfected with a promoterless firefly luciferase reporter were used as baseline RLU. Transfected cells were treated 48 hr thereafter with heme and TNF and analyzed 12 hr thereafter. Data shown as mean RLU ± SD from five independent experiments with similar trend.
Red bars in (B), (D)–(F), (J), and (K) represent mean values and doted circles indicate individual mice. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.01.
See also Figure S6.
Consistent with previous observations (Deutschman et al., 1997), liver G6pc1 mRNA (Figure 4B) and G6pc1 protein (Figures 4C and 4D) expression were decreased in response to CLP. This effect was exacerbated in Mx1CreFthΔ/Δ versus Fthlox/lox mice (Figures 4B–4D), confirming the microarray data (Figure 4A). Expression of the solute carrier family 37 member 4, i.e., glucose-6-phosphate transporter (G6PT), another component of the G6Pase complex (van Schaftingen and Gerin, 2002) not included in the initial RNA microarray screen (Figure 4E) and hepatic G6Pase enzymatic activity (Figure 4), were also decreased in a more pronounced manner in Mx1CreFthΔ/Δ versus Fthlox/lox mice subjected to CLP. Of note, if not subjected to CLP, Mx1CreFthΔ/Δ and Fthlox/lox mice expressed similar levels of G6pc1 and G6pt mRNA (Figures 4B and 4E). This suggests that FTH regulates the expression and activity of the hepatic G6Pase complex in response to polymicrobial infection.
Expression of G6PC1 mRNA (Figure 4G) was decreased in human hepatocytic HepG2 cells exposed to heme, as compared to vehicle-treated controls, which is consistent with previous findings (Yin et al., 2007). TNF also inhibited G6PC1 mRNA expression in HepG2 cells, an effect boosted when heme and TNF were used together (Figure 4G). This was confirmed for G6PC1 protein expression (Figures 4H and 4I).
We then asked what the mechanism is by which heme and TNF suppress G6PC1. Heme and TNF repressed G6pc1 transcription as assessed in HepG2 cells transiently transfected with G6pc1-luciferase reporter (Figure 4J). This in keeping with a previous demonstration that heme can repress G6PC1 transcription (Yin et al., 2007).
Heme administration to C57BL/6 mice inhibited liver G6pc1 and G6pt1 mRNA expression (Figure 4K), an effect exacerbated in Mx1CreFthΔ/Δ versus Fthlox/lox mice receiving a lower heme dosage (Figure 4L). This suggests that FTH counters heme-driven inhibition of liver G6Pase in vivo.
We then asked what the mechanism is by which FTH regulates G6PC1. FTH induced G6PC1 transcription as assessed in HepG2 cells transiently transfected with G6pc1-luciferase reporter (Figure 4L). Moreover, FTH countered the repression of G6pc1-luciferase reporter in response to heme and TNF (Figure 4L). This suggests that FTH promotes G6PC1 expression via a mechanism regulating its transcription.
Repression of G6PC1 protein in HepG2 cells exposed to heme and TNF was not reverted by proteasome inhibition (Figure S6A), as observed for the cyclin-dependent kinase inhibitor 1 p21 (Figure S6A). This suggests that G6PC1 protein degradation is not the mechanism by which its protein levels are reduced on heme and TNF.
Figure S6.

Liver Gluconeogenesis Is Required to Establish Disease Tolerance to Sepsis, Related to Figures 4 and 5
(a) Western Blot of G6PC1 and p21 in HepG2 cells untreated (-) or treated (+) with heme and TNF in the presence (+) or absence (-) of Lactacystin (Lact.) or MG132. Actin was used as loading control. Data are representative of three independent experiments with the same trend. (b) G6Pase enzymatic activity in HepG2 cells transduced (+) or not (-) with a G6PC1 Rec.Ad. and when indicated (+) co-transduced with a FTH Rec.Ad. Transduction with a LacZ Rec. Ad. was used as a control. NT: Not transduced. Cells were treated (+) or not (-) with heme and TNF 72 hr after Rec.Ad. transduction. Data are shown as mean ± SD, pooled from 2 independent experiments with four replicates each. (c) Temperature and (d) weight of G6pc1lox/lox (n = 12) and AlbCreERT2G6pc1Δ/Δ (n = 12) mice subjected to CLP. Data are shown as mean ± SEM, pooled from 3 independent experiments. (e) Blood glucose and (f) clinical severity score in Control (C57BL/6; n = 7) and AlbCreERT2G6pc1Δ/Δ (n = 5) mice subjected to CLP. Mean ± SD pooled from 2 independent experiments. (g) Serological markers of organ injury in C57BL/6 (n = 7) and AlbCreERT2G6pc1Δ/Δ (n = 5) mice 6 hr mice after CLP. (h) H&E stained paraffin sections representative of at least four G6pc1lox/lox and AlbCreERT2G6pc1Δ/Δ mice 9 hr after PCI. B: bronchus; CV: central vein; G: glomerulus. Magnification 400x. (i) Temperature and (h) weight and (j) blood glucose levels in G6pc1lox/lox (n = 6) and AlbCreERT2G6pc1Δ/Δ (n = 7) mice receiving heme (i.p; 30 mg/kg BW). Mean ± SD from 2 independent experiments. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Transduction of HepG2 cells with a G6PC1 recombinant adenovirus (Rec.Ad.) resulted in a robust induction of G6Pase activity, as compared to control cells not transduced or transduced with a LacZ Rec.Ad. (Figure S6B). Heme and TNF did not inhibit G6Pase activity in HepG2 cells transduced with the G6PC1 and co-transduced or not with a LacZ or FTH Rec.Ad. (Figure S6B) (Berberat et al., 2003, Gozzelino et al., 2012).
Liver Gluconeogenesis Is Required to Establish Disease Tolerance to Polymicrobial Infections
To address whether liver gluconeogenesis per se is required to establish disease tolerance to polymicrobial infections, we used AlbCreERT2G6pc1Δ/Δ mice lacking liver gluconeogenesis due to inducible and specific deletion of the G6pc1lox/lox allele in hepatocytes (Figure 5A) (Mutel et al., 2011). AlbCreERT2G6pc1Δ/Δ mice subjected to CLP developed severe hypoglycemia (Figure 5B) and succumbed (Figure 5C), as compared to control G6pc1lox/lox mice that reduced transiently blood glucose levels and survived (Figures 5B and 5C). Loss of body temperature (Figure S6C), but not body weight (Figure S6E), was also more pronounced in AlbCreERT2G6pc1Δ/Δ versus control G6pc1lox/lox mice subjected to CLP. Of note, AlbCreERT2G6pc1Δ/Δ mice failed to develop transient hyperglycemia in the 6–12 hr that followed CLP, as compared to control G6pc1lox/lox mice (Figure S6E).
Figure 5.

Liver Gluconeogenesis Is Critical to Establish Disease Tolerance to Sepsis
(A) G6pc1 protein expression in the liver of control (G6pc1lox/lox; Ctr) and AlbCreERT2G6pc1Δ/Δ (G6pc1Δ/Δ) mice.
(B) Blood glucose levels of G6pc1lox/lox (n = 12) and AlbCreERT2G6pc1Δ/Δ (n = 12) mice subjected to CLP. Data are shown as mean ± SEM from three independent experiments.
(C) Survival of control (G6pc1lox/lox; n = 10) and AlbCreERT2G6pc1Δ/Δ (n = 10) mice subjected to CLP. Data were pooled from two independent experiments with similar trend.
(D) Blood glucose of control (C57BL/6; n = 7) and AlbCreERT2G6pc1Δ/Δ (n = 5) mice subjected to PCI. Data are shown as mean ± SD from two independent experiments with the same trend.
(E) Survival of control (C57BL/6; n = 10) and AlbCreERT2G6pc1Δ/Δ (n = 8) subjected to PCI. Data pooled from three independent experiments with similar trend.
(F) CFU for aerobic (Ae) and anaerobic (An) bacteria, 6 hr after PCI. Red bars represent mean values and dotted circles represent individual mice. PC, peritoneal cavity.
(G) Blood glucose levels in G6pc1lox/lox (n = 6) and AlbCreERT2G6pc1Δ/Δ (n = 7) mice receiving heme (i.p. 30 mg/kg BW). Mean ± SD from two independent experiments.
(H) Survival of same mice as (G).
(I) Blood glucose levels in G6pc1lox/lox (n = 6) and AlbCreERT2G6pc1Δ/Δ (n = 9) mice receiving LPS (i.p. 5 mg/kg BW). Mean ± SD from two independent experiments.
(J) Survival of same mice as (I).
(K) Blood glucose levels in G6pc1lox/lox (n = 9) and AlbCreERT2Gpc1Δ/Δ (n = 10) mice receiving poly(I:C) (intra-retro orbital [i.r.o.] 30 mg/kg BW). Mean ± SD from two independent experiments.
(L) Survival of same mice as (K).
Numbers in gray (B, D, G, I, and K) are live/total mice at each time point. ∗p < 0.05; ∗∗p < 0.01.
See also Figure S6.
AlbCreERT2G6pc1Δ/Δ mice also developed severe hypoglycemia (Figure 5D) and succumbed (Figure 5E) from severe disease (Figure S6F) when subjected to PCI, as compared to control G6pc1lox/lox mice (Figures 5D, 5E, and S6F). Pathogen load was similar in AlbCreERT2G6pc1Δ/Δ versus G6pc1lox/lox mice subjected to PCI (Figure 5F). With the exception of the liver, the extent of organ damage was also similar in AlbCreERT2G6pc1Δ/Δ versus G6pc1lox/lox mice, as assessed serologically (Figure S6G) and by histology (Figure S6H). This shows that liver gluconeogenesis is required to establish disease tolerance to sepsis in mice.
Heme administration to AlbCreERT2G6pc1Δ/Δ mice led to more severe hypoglycemia (Figure 5G), loss of body temperature (Figure S6I) and body weight (Figure S6J), as well as higher incidence of mortality (Figure 5H), as compared to control G6pc1lox/lox mice. Transient elevation of blood glucose levels in heme-treated G6pc1lox/lox mice was not observed in AlbCreERT2G6pc1Δ/Δ mice (Figure S6K). This suggests that liver G6Pase is required to counter the development of lethal hypoglycemia in response to heme.
Taking into consideration that heme reduces blood glucose levels via a TLR4-dependent mechanism (Figure 2J), we examined whether liver G6Pase is also required to counter the pathogenic effects of a bona fide TLR4 agonist such as bacterial lipopolysaccharide (LPS). AlbCreERT2G6pc1Δ/Δ mice developed severe hypoglycemia (Figure 5I) and succumbed (Figure 5J) when subjected to a sub-lethal dosage of LPS inducing mild hypoglycemia in G6pc1lox/lox mice (Figures 5I and 5J). This further supports the notion that liver glucose production is strictly required to prevent the development of lethal hypoglycemia in response to systemic TLR4 signaling.
We also tested whether liver glucose production is required to counter the pathogenic effect exerted by other TLR agonists, such as polyinosinic-polycytidylic acid (Poly(I:C)) a TLR3 agonist that triggers a type 1 interferon response. In contrast to heme or LPS, Poly(I:C) administration was associated with the development of mild hypoglycemia in G6pc1lox/lox and AlbCreERT2G6pc1Δ/Δ mice, resulting in similar levels of mortality (Figures 5K and 5L). This argues that host adaptive responses regulating glucose metabolism are tailored to specific inflammatory insults (Soares et al., 2017, Wang et al., 2016).
The Ferroxidase Activity of FTH Establishes Disease Tolerance to Sepsis
We hypothesized that FTH overexpression in the liver should be sufficient to re-establish disease tolerance to sepsis. Overexpression of FTH in the liver of C57BL/6 mice, via transduction of a FTH Rec.Ad. (Berberat et al., 2003, Gozzelino et al., 2012), was protective against a severe form of CLP, as compared to control C57BL/6 mice not transduced or transduced with a LacZ Rec.Ad. (Figure 6A). Transduction of a mutated FTH (FTHm) lacking ferroxidase activity was not protective against CLP (Figure 6A). The protective effect of FTH was not associated with modulation of pathogen load (Figure 6B), showing that FTH overexpression establishes disease tolerance to lethal form of polymicrobial infection via a mechanism that relies on its ferroxidase activity. Of note, FTH overexpression did not prevent the development of organ dysfunction/damage in response to CLP (Figure S7A), arguing that the protective mechanism of FTH does not rely on its cytoprotective effect (Berberat et al., 2003, Gozzelino et al., 2012).
Figure 6.

FTH Overexpression or Ferritin Administration Induce Disease Tolerance to Sepsis
(A) Survival of C57BL/6 mice transduced 48 hr before severe CLP with FTH Rec.Ad. (n = 11), ferroxidase-deficient FTH Rec.Ad. (FTHm; n = 11), LacZ Rec.Ad. (n = 12), or receiving vehicle (PBS; n = 19). Data pooled from six independent experiments with similar trend.
(B) CFU for aerobic (Ae) and anaerobic (An) bacteria, 12 hr after CLP in mice treated as in (A).
(C) Representative western blot in HepG2 cells transduced with LacZ, FTH, or mutated FTH (FTHm) Rec.Ad.
(D and E) Densitometry of G6PC1 (D) and FTH protein (E) expression normalized to α-tubulin (n = 4 per group).
(F) Expression of G6pc1 mRNA quantified by qRT-PCR in HepG2 cells exposed (+) or not (−) to heme, TNF, and deferoxamine (DFO). Mean ± SEM from three independent experiments done in duplicates.
(G) Survival of C57BL/6 mice receiving apoferritin (n = 16), ferritin (n = 10), BSA (n = 12), or saline (n = 12), 24 hr before severe CLP. Data pooled from five independent experiments.
(H) CFU for aerobic (Ae) and anaerobic (An) bacteria 12 hr after severe CLP in mice treated as in (G). Data from three independent experiments.
(I) Survival of C57BL/6 mice receiving apoferritin (n = 22), BSA (n = 20), or saline (n = 16) 6 hr after severe CLP. Data pooled from seven independent experiments.
Red bars represent mean values and dotted circles represent individual mice (B and G). PC, peritoneal cavity; NS, non-significant. ∗p < 0.05, ∗∗p < 0.01.
See also Figure S7.
Figure S7.

FTH Overexpression and Apo-Ferritin Administration Induce Disease Tolerance to Sepsis, Related to Figure 6
(a) Serological markers of organ injury, before (Control; Ctr) and 12 hr after severe CLP in C57BL/6 mice transduced with Rec.Ad. encoding FTH, ferroxidase-deficient FTH (FTHm), LacZ or receiving vehicle (PBS). Data are shown as mean ± SD from n = 4-5 mice per group, pooled from 3 independent experiments. (b) Serological markers of organ injury, before (Control; Ctr) and 12 hr after severe CLP C57BL/6 mice receiving apoferritin, ferritin, bovine serum albumin (BSA) or saline. Data are shown as mean ± SD from n = 4-5 mice per group, samples were pooled from 3 independent experiments. ∗p < 0.05, ∗∗p < 0.01; ∗∗∗∗p < 0.001.
We then addressed whether the ferroxidase activity of FTH regulates the expression of G6PC1 in hepatocytes. Expression of G6PC1 protein was higher in HepG2 cells transduced with FTH Rec.Ad., as compared to control HepG2 cells transduced with LacZ Rec.Ad (Figures 6C and 6D). This was not the case for HepG2 cells transduced with the FTHm Rec.Ad. (Figures 6C and 6D) despite similar levels of FTHm and FTH expression (Figures 6C and 6E). This suggests that the ferroxidase activity of FTH promotes G6PC1 protein expression in hepatocytes.
We then investigated whether iron chelation by DFO promotes G6PC1 expression. Inhibition of G6PC1 mRNA expression in HepG2 cells exposed to heme plus TNF was countered by DFO (Figure 6F), as compared to vehicle-treated controls. This reinforces the notion that the ferroxidase activity of FTH, mimicked by DFO, promotes G6PC1 expression in hepatocytes.
Soluble Ferritin Promotes the Establishment of Disease Tolerance to Sepsis
Ferritin complexes can be secreted and presumably exert biologic effects in bystander cells (Fisher et al., 2007). We questioned whether soluble ferritin complexes can be used pharmacologically to enforce the establishment of disease tolerance to sepsis. Administration of non-iron loaded apoferritin protected C57BL/6 mice from succumbing to severe CLP, as compared to controls receiving vehicle or bovine serum abumin (BSA) (Figure 6G). In contrast, administration of iron-loaded ferritin was not protective (Figure 6G). Apoferritin and iron-loaded ferritin-treated mice had similar pathogen loads, as compared to controls receiving vehicle or BSA (Figure 6H). Apoferritin was still protective against mortality when administered up to 6 hr after severe CLP, as compared to control mice receiving vehicle or BSA (Figure 6I). This suggests that apoferritin administration after the onset of infection can act therapeutically to establish disease tolerance to sepsis.
Of note, apoferritin administration did not prevent the development of liver, muscle, or kidney dysfunction/damage, as compared to controls receiving vehicle or BSA (Figure S7B). This argues further that the mechanism by which ferritin confers disease tolerance to sepsis does not rely on its cytoprotective effects (Berberat et al., 2003, Gozzelino et al., 2012).
Antioxidants Restore Normoglycemia and Disease Tolerance to Sepsis
Given that the ferroxidase activity of FTH sustains G6Pase expression in hepatocytes (Figures 6C–6E), we asked whether a pharmacologic anti-oxidant such as the glutathione precursor N-acetylcysteine (NAC) can re-establish disease tolerance in Mx1CreFthΔ/Δ mice. NAC administration to Mx1CreFthΔ/Δ mice restored body weight (Figure 7A), blood glucose levels (Figure 7B), and survival (Figure 7C), as compared to control vehicle-treated Mx1CreFthΔ/Δ mice subjected to CLP. NAC- and vehicle-treated Mx1CreFthΔ/Δ mice had similar pathogen loads (Figure 7D).
Figure 7.

Antioxidants Bypass the Requirement of FTH in the Establishment of Normoglycemia and Disease Tolerance to Sepsis
(A–C) Relative weight (A), blood glucose (B), and survival (C) of Mx1CreFthΔ/Δ mice subjected to CLP and receiving NAC (n = 7; 15 mg/kg) or vehicle (n = 6). Data pooled from two independent experiments.
(D) Colony forming units (CFU) for aerobic (Ae) and anaerobic (An) bacteria, 48 hr after CLP. PC, peritoneal cavity. Red bars represent mean values and doted circles represent individual mice. Pooled from two independent experiments.
(E) Expression of G6PC1 mRNA quantified by qRT-PCR in HepG2 cells exposed (+) or not (−) to heme, TNF, and/or NAC. Data shown as mean ± SD from four independent experiments.
(F and G) Blood glucose (F) and survival (G) of Mx1CreFthΔ/Δ mice subjected to CLP and receiving BHA (n = 11; 50 mg/kg) or vehicle (n = 9). Data shown as mean ± SD. Pooled from three independent experiments.
(H–J) Relative weight (H), blood glucose (I), and survival (J) of AlbCreERT2G6pc1Δ/Δ mice subjected to CLP and receiving NAC (n = 5) or vehicle (n = 5). Mean ± SD from one experiment.
NS, non-significant. ∗p < 0.05, ∗∗p < 0.01. Numbers in gray (A, B, F, I, and J) indicate live/total mice.
We then asked whether NAC modulates G6PC1 expression. Inhibition of G6PC1 mRNA expression in HepG2 cells exposed to heme and TNF was prevented by NAC, as compared to vehicle-treated controls (Figure 7E). This suggests that heme and TNF inhibit G6Pase via a pro-oxidant effect reverted by NAC. Of note, NAC can also inhibit the activation of nuclear factor kappa B (NF-κB), a transcription factor that represses G6PC1 expression in hepatocytes (Grempler et al., 2004). Therefore, we cannot exclude that NAC promotes G6PC1 expression via a mechanism involving NF-κB inhibition, which is probably not dissociated from the anti-oxidant effect of NAC.
In a similar manner to NAC, the lipophilic anti-oxidant butylated hydroxyanisole (BHA) also restored blood glucose levels (Figure 7F) and survival (Figure 7G) in Mx1CreFthΔ/Δ mice subjected to CLP, as compared to vehicle-treated controls. This suggests that Mx1CreFthΔ/Δ mice succumb to CLP due to the development of iron-driven oxidative stress, compromising liver glucose production and leading to lethal hypoglycemia. Presumably, iron chelation by FTH acts in an anti-oxidant manner to sustain liver G6Pase expression and glucose production, contributing to establish disease tolerance to polymicrobial infections.
Finally, we asked whether anti-oxidants restore glucose metabolism in mice lacking liver glucose production. NAC administration to AlbCreERT2G6pc1Δ/Δ mice subjected to CLP failed to restore body weight (Figure 7I), blood glucose levels (Figure 7J), or improve survival (Figure 7K), as compared to control vehicle-treated AlbCreERT2G6pc1Δ/Δ mice. This confirms that the anti-oxidant activity of FTH acts upstream of G6Pase to restore blood glucose levels and establish disease tolerance to sepsis.
Discussion
Host metabolic responses to systemic infections are associated with induction of hypoferremia (i.e., low levels of circulating iron concomitant with iron accumulation in macrophages and parenchyma cells) (Soares and Weiss, 2015). While critical to limit iron availability and regulate immune-driven resistance to extracellular pathogens, hypoferremia can promote the development of oxidative damage in host parenchyma tissues (Soares and Weiss, 2015). This pathologic effect is fueled by the accumulation of labile heme in plasma, released from extracellular hemoglobin as a byproduct of hemolysis (Larsen et al., 2010). Presumably, for this reason, the accumulation of extracellular hemoglobin in plasma is associated with poor clinical outcome of sepsis (Adamzik et al., 2012). Moreover, high levels of the hemoglobin scavenger haptoglobin (Janz et al., 2013) or the heme scavenger hemopexin (Janz et al., 2013, Larsen et al., 2010) in plasma are associated with a better clinical outcome of sepsis.
Heme catabolism by HO-1 is critical to prevent the pathogenic effects of labile heme and establish disease tolerance to sepsis (Larsen et al., 2010). However, under inflammatory conditions, the intracellular iron generated via heme catabolism cannot be readily exported, owed to systemic inhibition of the iron cellular exporter ferroportin by hepcidin (Drakesmith and Prentice, 2012, Nemeth et al., 2004). Presumably, this explains why FTH becomes essential to restrict the participation of intracellular iron in the generation of reactive oxygen species via Fenton chemistry (Gozzelino and Soares, 2014, Soares and Bozza, 2016, Soares and Weiss, 2015) and to establish of disease tolerance to sepsis (Figure 1).
While essential to establish disease tolerance to acute infections, the induction of HO-1 (Larsen et al., 2010) and FTH (Figures 1A–1F) is likely associated with tradeoffs, presumably explaining why the expression of these stress-responsive genes is not constitutively high at steady state. In the case of HO-1, this has been associated with the development of insulin resistance in response to chronic metabolic inflammation (Jais et al., 2014).
FTH does not regulate glucose metabolism in response to sustained inflammation, as assessed in Mx1CreFthΔ/Δ versus Fthlox/lox subjected to repeated low-dose LPS administration (data not shown) (Okin and Medzhitov, 2016). However, we cannot exclude that FTH might partake in the development of insulin resistance in response to chronic metabolic inflammation.
Induction of FTH expression in response to acute polymicrobial infections prevents liver metabolism from shifting toward glucose consumption, via glycolysis, in detriment of glucose production, via gluconeogenesis (Figure 3C). We propose that this is required to compensate for reduced nutritional glucose intake associated with anorexia of infection (Wang et al., 2016), maintaining blood glucose levels within a dynamic physiologic range compatible with host survival (Figures 2C–2E and 5B–5F) (Hermanides et al., 2010). The anti-oxidant effect associated with the ferroxidase activity of FTH is central to this adaptive response, preventing pro-oxidant iron-heme from inhibiting liver G6Pase (Figure 4).
While iron can catalyze oxidative modifications in G6PC1 and G6PT1 thiol (SH) groups (Clottes and Burchell, 1998), which represses G6Pase expression and/or activity (Witzleben and Chaffey, 1962), this does not appear to be involved in the mechanism in which heme and TNF repress G6Pase and/or the mechanism in which FTH promotes G6Pase expression and/or activity (Figures S6A and S6B). Instead, heme and FTH modulate G6Pase expression via a mechanism that relies on the modulation of G6PC1 transcription (Figures 4J and 4M).
The relative contribution of heme-activated TLR4 signaling and iron heme-induced oxidative stress to the inhibition of liver G6Pase remains unclear. At least three mechanisms might contribute to this effect: (1) TLR4 signaling in response to heme (Figueiredo et al., 2007), (2) unfettered free radical production via the participation of heme iron in Fenton chemistry, or (3) synergic effect of TLR4 signaling and heme iron-driven oxidative stress.
Deregulation of glucose metabolism is thought to play a central role in the development of metabolic and energetic failure associated with the pathogenesis of sepsis (Lee and Hüttemann, 2014, Pravda, 2014). Development of insulin resistance (Yki-Järvinen et al., 1989) is perceived as an adaptive response that counters unfettered glucose utilization by host tissues, presumably preventing the development of lethal hypoglycemia in response to acute bacterial infections (Wang et al., 2016). Our data suggest that regulation of liver glucose production by FTH is a critical component of this metabolic response, which is required to prevent the development of lethal hypoglycemia in response to acute infection.
Consistent with the notion that the accumulation of labile heme in plasma plays a central role in the pathogenesis of sepsis (Larsen et al., 2010), we found that labile heme is sufficient per se to reduce blood glucose levels (Figures 2C and 2G), an effect associated with the repression of liver G6pc1 and G6pt mRNA expression (Figures 4H–4K). Reduction of blood glucose levels in response to heme is TLR4-dependent (Figure 2J), which is consistent with previous findings showing that heme can signal via TLR4 (Figueiredo et al., 2007), and that signaling via TLR4 in response to LPS reduces blood glucose levels and represses liver G6Pase (Raetzsch et al., 2009). However, it remains to be established how signaling via the TLR4 represses the liver G6Pase and induces hypoglycemia.
FTH does not regulate tissue hypoperfusion or anaerobic glycolysis, as Fthlox/lox and MxCreFthΔ/Δ mice have similar low levels of lactate in serum, reflecting tissue hypoperfusion or anaerobic glycolysis, both at steady state and in response to polymicrobial infection (data not shown). There is also no apparent role of FTH in preventing cardiovascular failure or fluid overload, as assessed by stroke volume (Figure 4A), end-diastolic and end-systolic volumes (Figures 4B and 4C), or mean arterial pressure (data not shown) in Fthlox/lox versus MxCreFthΔ/Δ mice, both at baseline or when subjected to polymicrobial infection.
A significant proportion of iron-loaded ferritin is secreted, and the physiologic role of soluble ferritin was related mainly to iron distribution between cells (Meyron-Holtz et al., 2011). The observation that pharmacologic use of apoferritin, but not iron-loaded ferritin, establishes disease tolerance to sepsis (Figures 6G and 6I) argues that soluble ferritin also works as an iron chelator. This is in keeping with a previous report showing that ferritin administration in also protective against Escherichia coli infection in mice (Lipiński et al., 1991).
In conclusion, disease tolerance to sepsis relies on a crosstalk between host iron and glucose metabolic responses in which ferritin sustains liver glucose production to maintain blood glucose levels in response to infection within a dynamic range compatible with survival. These pathways can be targeted pharmacologically to promote the establishment of disease tolerance to systemic infections, possibly providing a novel therapeutic approach against sepsis.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse G6PC1 (1:1000) | N/A | Kind gift from Fabienne Rajas |
| Anti-FTH1 (1:1000), clone D1D4 | Cell Signaling Technology | Cat# 4393S; RRID:AB_11217441 |
| Anti-p21 | Cell Signaling Technology | Cat# 2947S; RRID: AB_823586 |
| Anti-β-actin | Sigma | Cat#: A5441; RRID:AB_476744 |
| Anti-β-tubulin | Sigma | Cat#: T9026; RRID:AB_AB_477593 |
| F4/80 BM8 | Biolegend | Cat#: 123102; RRID:AB_893506 |
| Bacterial and Virus Strains | ||
| pAC.CMV-pLpASR+ßGal (LacZ) | Berberat et al., 2003, Gozzelino et al., 2012 | Kind gift from Paolo Arosio, University of Brescia, Italy |
| pAC.CMV-pLpASR+FTH | Berberat et al., 2003, Gozzelino et al., 2012 | Kind gift from Paolo Arosio, University of Brescia, Italy |
| pAC.CMV-pLpASR+FTHm | Berberat et al., 2003, Gozzelino et al., 2012 | Kind gift from Paolo Arosio, University of Brescia, Italy |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Apoferritin from equine spleen | Sigma-Aldrich | Cat#: A3641; CAS: 9013-31-4 |
| Ferritin from equine spleen | Sigma-Aldrich | Cat#: F4503; CAS: 9007-73-2 |
| Lipopolysaccharide (Escherichia coli O55:B5) | Sigma-Aldrich | Cat#: L2880 |
| Poly(I:C) | InvivoGen | Cat#: tlrl-pic; CAS: 31852-29-6 |
| Tamoxifen | Sigma-Aldrich | Cat#: T5648; CAS: 10540-29-1 |
| D-glucose | Sigma-Aldrich | Cat#: G-8270; CAS: 50-99-7 |
| Pyruvate | Sigma-Aldrich | Cat#: P4562 CAS: 113-24-6 |
| Hemin | Frontier Scientific | Cat#: H651-9 |
| Protoporhpyhrin IX (PPIX) | Frontier Scientific | Cat#: P562-9 |
| recombinant human TNF-α | R&D Systems | Cat#: 210-TA-20 |
| Critical Commercial Assays | ||
| Nucleospin RNA II | Macherey-Nagel | Cat#: 740955 |
| Transcriptor First Strand cDNA Synthesis Kit | Roche | Cat#: 04897030001 |
| Luciferase assay system Dual-Glo | Promega | Cat#: E2920 |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat#: 1725125 |
| Milliplex MAP Mouse Metabolic Magnetic Bead Panel | EMD Millipore | Cat#: MMHAG-44K |
| Mouse Cytokine Chemokine Magnetic Bead Panel Immunoassay | EMD Millipore | Cat#: MCYTIMAG-70K-16 |
| TargetAmp-Nano Labeling Kits | Epicenter | Cat#: TAN091096 |
| Illumina mouseRef8 V2 Chips | Illumina | Cat#: BD-202-0602 |
| Mouse Ultrasensitive Insulin ELISA | ALPCO | Cat#: 80-INSMSU-E01 |
| EnzyChrom Alanine Transaminase Assay Kit | Bioassay Systems | Cat#: EALT-100 |
| EnzyChrom Aspartat Transaminase Assay Kit | Bioassay Systems | Cat#:EAST- EASTR-100 |
| EnzyChromTM Creatine Kinase Assay Kit | Bioassay Systems | Cat#: ECPK-100 |
| QuantiChrom Lactate Dehydrogenase Kit | Bioassay Systems | Cat#: DLDH-100 |
| QuantiChrom Urea Assay Kit | Bioassay Systems | Cat#: DIUR-500 |
| EnzyChrom L-Lactate Assay Kit | Bioassay Systems | Cat#: ECLC-100 |
| Deposited Data | ||
| Raw and analyzed data | This paper and the Gene Expression Omnibus database | GEO: GSE92703; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE92703. |
| Experimental Models: Cell Lines | ||
| HepG2 | N/A | Kind gift from Robert Menard, Institut Pasteur, France |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6.C57BL/6 Fthfl/fl | Darshan et al., 2009 | from Prof. Lukas Kuhn, ETH, Switzerland |
| Mouse: B6.Mx1CreFthfl/fl, | Kühn et al., 1995 | from Prof. Lukas Kuhn, ETH, Switzerland JAX stock 003556 |
| Mouse: B6.Cg-Tg(Mx1-cre)1Cgn/J (LysMCre) | Clausen et al., 1999 | JAX stock 004781 |
| Mouse: B6.Cg-Tg(Alb-cre)21Mgn/J (AlbCre) | Postic et al., 1999 | JAX stock 003574 |
| Mouse: B6.129-Gt(ROSA)26Sortm1(cre/ERT2)Tyj/J (ROSA26CreERT2) | Vooijs et al., 2001 | JAX stock 008463 |
| Mouse: B6.LyzMCreFth−/− | This paper | N/A |
| Mouse: B6.AlbCreFth−/− | This paper | N/A |
| Mouse: B6.LyzMCreAlbCreFthfl/fl | This paper | N/A |
| Mouse: B6.ROSA26CreERT2Fthfl/fl | This paper | N/A |
| Mouse: AlbCreERT2G6pc1fl/fl | Mutel et al., 2011 | N/A |
| Mouse: B6.Tlr4−/− | N/A | originally from Shizuo Akira, Osaka University, Japan |
| Mouse: B6.Ifnr1−/− | N/A | MMRRC Stock No: 32045-JAX |
| Oligonucleotides | ||
| Mouse Primer: ArbpO | This paper | N/A |
| Fwd: 5′CTTTGGGCATCACCACGAA3′ | ||
| Rev: 5′GCTGGCTCCCACCT TGTCT3′ | ||
| Mouse Primer: Fth | This paper | N/A |
| Fwd: 5′CCATCAACCGCCAGATCAAC3′ | ||
| Rev: 5′GCCACAT CATCTCGGTCAAA3′ | ||
| Mouse Primer: G6pc1 | This paper | N/A |
| Fwd: 5′TGGGGCAGAAATATGACTCCT3′ | ||
| Rev: 5′TGCAAATCAGCCGAGGCA3′ | ||
| Mouse Primer: Gpt1 | This paper | N/A |
| Fwd: 5′ CTTCTCTC GCCTACGCCTTG3′ | ||
| Rev: 5′GGAACCCAGTGTCTGCGG3′ | ||
| Human Primer: G6PC1 | This paper | N/A |
| Fwd:5-TTCAGGAAGCTGTGGGCATT3′ | ||
| Rev: 5′TCTTTCCCTGGTCCAGTCTCA3′ | ||
| Human Primer: ACTIN | This paper |
N/A |
| Fwd 5′GAGCACAGAGCCTCGCCTTT3′ | ||
| Rev 5′TCATCAT CCATGGTGAGCTGG3′ | ||
| Recombinant DNA | ||
| pGL2B-G6PC | Rajas et al., 2002 | Kind gift from Amandine Gautier-Stein; Université Claude Bernard Lyon, France |
| pSG5-FtH | Berberat et al., 2003 | Kind gift from Paolo Arosio, University of Brescia, Italy |
| pGL2B | Rajas et al., 2002 | Kind gift from Amandine Gautier-Stein; Université Claude Bernard Lyon, France |
| Software and Algorithms | ||
| ImageJ | Rasband, W.S., ImageJ, U. S. NIH, USA | https://imagej.nih.gov/ij/, 1997-2014) |
| Labscribe 2 (Version 2.342000) | iWorksSystem Incorp. | http://labscribe2.software.informer.com/2.0/ |
| GenomeStudio (v.1.9, and annotation MouseRef-8_V2_0_R3_11278551_A) | Illumina | https://support.illumina.com/array/array_software/genomestudio/downloads.html |
| Gene Expression Omnibus database, GEO: GSE92703 | This paper | https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE92703. |
| Genome-scale metabolic model iMM1415 of Mus musculus | Sigurdsson et al., 2010 | N/A |
| MATLAB toolbox COBRA | Schellenberger et al., 2011 | https://opencobra.github.io/cobratoolbox/latest/ |
| Other | ||
| TrypticaseSoy Agar II with 5% Sheep Blood plates | Becton Dickinson | Cat#: 254053 |
| GasPak anaerobe container system | Becton Dickinson | Cat#: 260678 |
| ACCU-CHEK Performa 50ct Strip | Roche | Cat#: 6454011052 |
| Rodent Thermometer | Bioseb, France | Cat#: BIO-TK8851 |
| Volume-Conductance-Katheter | Scisense | Cat#: FTS-1912B-8018 |
| Scisense Pressure-Volume Control Unit FV896B | Scisense | N/A |
Contact for Reagents and Resource Sharing
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Contact, Miguel P. Soares (mpsoares@igc.gulbenkian.pt).
Experimental Model and Subject Details
Mice
C57BL/6 Fthlox/lox, Mx1CreFthΔ/Δ, LyzMCreFthΔ/Δ, AlbCreFthΔ/Δ, LyzMCreAlbCreFthΔ/Δ, and ROSA26ERT2Fthlox/lox mice were generated by intercrossing C57BL/6 Fthlox/lox mice (Gozzelino et al., 2012) obtained from Prof. Lukas Kuhn (ETH, Switzerland) with C57BL/6 Mx1Cre (Jackson Laboratories; stock 003556), LysMCre (Jackson Laboratories; stock 004781), AlbCre (Jackson Laboratories; stock 003574), ROSA26CreERT2 (Jackson Laboratories; stock 008463) mice, respectively. Conditional deletion (Δ) of Fthlox/lox allele in Mx1CreFthΔ/Δ mice was achieved at 5 weeks of age by Poly(I:C)(InvivoGen Europe, Toulouse, France Ref: tlrl-pic) administration (i.p.; 1 mg/kg BW; 2x at 3 day intervals) (Gozzelino et al., 2012). Deletion of the Fthlox/lox allele in ROSA26CreERT2Fthlox/lox mice was achieved by Tamoxifen (Sigma-Aldrich, Sintra, Portugal. Ref: T5648) administration (i.p.; 50mg/kg in 100 μL corn oil (Sigma, Ref C8267)/5% ethanol for 5 consecutive days). Deletion of the Fthlox/lox allele was monitored in peripheral blood by qRT-PCR (Gozzelino et al., 2012, Larsen et al., 2010) and confirmed in the liver by western blot (Figure 1D). AlbCreLysMCreFthΔ/Δ mice were generated by intercrossing LysMCreFthΔ/Δ and AlbCreFthΔ/Δ mice and used at 10 weeks of age. C57BL/6 wild-type, Tlr4−/− (obtained originally from Shizuo Akira, Osaka University, Japan) and Ifnr1−/− (MMRRC Stock No: 32045-JAX) were breed at the Instituto Gulbenkian de Ciência. AlbCreERT2G6pc1Δ/Δ mice (were previously described (Mutel et al., 2011). Deletion of the G6pc1lox/lox allele in AlbCreERT2G6pc1Δ/Δ mice was achieved by Tamoxifen administration (i.p.; 50mg/kg in 100 μL corn oil/5% ethanol for 5 consecutive days). Deletion was confirmed by western blot and genomic PCR of liver extracts (Figure 5A). All animal protocols used were approved by the Instituto Gulbenkian de Ciência ethical committee and the “Órgão Responsável pelo Bem-estar dos Animais (ORBEA)” and consequently licenced by the Direccão Geral de Alimentação e Veterinária (DGAV). All animal experiments follow the Portuguese (Portaria n° 1005/92, Decreto-Lei n° 113/2013) and European (Directive 2010/63/EU) legislations, concerning housing, husbandry and animal welfare.
Cecal Ligation and Puncture
CLP was performed essentially as described (Larsen et al., 2010). Procedures were performed routinely at the same time frame, starting at 9 AM. CLP consisted of 20%–30% cecum ligation and double puncture with a 23 Gauge (G) needle. Severe CLP consisted of 60%–70% cecum ligation and double puncture with a 23 Gauge (G) needle. A small amount of faeces was extruded and the cecum was carefully placed back into the abdominal cavity. All animals received 0.9% saline (40 mL/kg, i.p.) and Imipenem/Cilastatin (25 mg/kg, i.p.), starting 2 hr after CLP and every 12 hr for 3 days). Survival was assessed twice daily for the first 3 days and once daily for a total of 14 days. Weight and temperature (Rodent Thermometer BIO-TK8851, Bioseb, France) were monitored daily for up to 7 days. Blood glucose levels (ACCU-CHECK Aviva, Roche, Amadora, Portugal) using tail vein blood was monitored in fed mice at several time points on the first day of analysis, twice daily on the second and then daily for up to 7 days. AlbCreERT2G6pc1Δ/Δ mice were subjected to CLP at least 3 weeks after the last Tamoxifen injection. Mx1CreFthΔ/Δ mice were subjected to CLP 6-8 weeks after the first poly(I:C) administration.
Peritoneal Contamination and Infection
PCI was induced as previously described (Gonnert et al., 2011). Briefly, mice were injected i.p. with 2,2 μl/g body-weight of a human stool suspension. Body weight, blood glucose and disease severity were assessed twice daily. Disease severity was graded in five severity grads using a scoring system as described (Gonnert et al., 2011): grade 1: no signs of disease; grade 2: mild disease; grade 3: medium severe disease; grade 4: severe disease, grade 5: death. Animals received 25 μg/g body weight Meropenem in 25 μL 0,9% NaCl/g body weight s.c., starting 12 hr after PCI, twice daily for a total of 6 injections.
Bone Marrow Chimeras
Fthlox/lox or ROSA26CreERT2FthΔ/Δ were lethally irradiated (900 rad) and immediately reconstituted with bone marrow cells (500,000 cells in 200 μL i.v. into the tail vein) freshly isolated from 8 week old donor Fthlox/lox or ROSA26CreERT2FthΔ/Δ mice. Hematopoietic cell reconstitution was confirmed 4-6 weeks after bone marrow transfer by flow cytometry, assessing the relative proportion of peripheral blood leukocytes expressing the CD45.1/CD5.2 haplotypes and corresponding to cells derived from donor and recipient hematopoietic progenitors, respectively. Deletion of the Fth allele was achieved as described above. Mice were allowed to recover for up to 30 days after deletion before undergoing CLP. Deletion of the Fthlox/lox allele in bone marrow chimeras was achieved by Tamoxifen administration (gavage; 50mg/kg in 100 μL corn oil/5% ethanol for 5 consecutive days).
Cell Lines
HepG2 cells were kindly provided from Robert Menard, Institut Pasteur, France.
Method Details
In Vivo Recombinant Adenovirus (Rec.Ad.)
The LacZ, FTH and FTHm Rec.Ad. were previously described (Berberat et al., 2003, Gozzelino et al., 2012). Mice were transduced (i.p.; 200 μL in PBS) with 5x108 plaque forming units, 48 hr before CLP. Non-transduced control mice received vehicle (PBS). Expression of the genes encoded by the different Rec.Ad. was confirmed in the liver by western blot.
Pharmacologic Approaches
Apoferritin: Purified horse spleen ferritin (Sigma Ref: F4503) or apoferritin (Sigma Ref: A3641) in sterile 0.9% saline was administered (i.p.; 200 μL in saline) 24 hr before or 6 hr after CLP. Vehicle (0.9% saline) or bovine serum albumin (BSA, Sigma Ref: A3059) at the same dosage were administered to control mice. D-glucose (Sigma Ref: G-8270) (200 μL 2 mg/g BW in water) or Pyruvate (Sigma Ref: P4562) (200 μL 4 mg/g BW in water) were administered via gavage twice daily for 4 days starting 12 hr after CLP and then once daily for 3 days. N-acetylcystein (NAC) was administered (i.p.; 200 μL, 15mg/kg in sterile PBS) twice daily for 12 consecutive days starting 48 hr before CLP. Butylated Hydroxianisole (BHA; Sigma, ref: B1253) was administered (gavage.; 100 μL, 50mg/kg in corn oil) twice daily for 12 consecutive days starting 48 hr before CLP. Hemin (Frontier Scientific Ref: H651-9) or empty protoporphyrin (Frontier Scientific Ref: P562) were administered to C57BL/6, Tlr4−/− and Ifnr1−/− (i.p.; 30 mg/kg) or to Fthlox/lox and Mx1CreFthΔ/Δ (i.p.; 25-30 mg/kg). Poly(I:C) was administered to G6pclox/lox and to AlbCreERT2G6pc1Δ/Δ (intra-retro orbitally; 30 mg/kg BW).
Pathogen Load
Peritoneal fluid was obtained by peritoneal lavage (5 mL sterile PBS). Whole organs (i.e., spleen and liver) were harvested and homogenized under sterile conditions using a dounce tissue grinder (Sigma Ref: D8939-1SET). Heparanized blood was obtained by intracardial puncture. Serial dilutions were plated onto TrypticaseSoy Agar II with 5% Sheep Blood plates (Becton Dickinson Ref: 254053) and incubated (24 hr at 37°C) in air 5% CO2 (aerobes) or in an air tight container equipped with the GasPak anaerobe container system (Becton Dickinson Ref 260678). Anaerobic conditions were confirmed in all experiments using BBL Dry Anaerobic Indicator Strips (Becton Dickinson Ref: 271051).
Histology
Mice were perfused in toto with ice cold PBS (10mL). Liver samples were harvested, fixed in 10% buffered formaldehyde for 48 hr at room temperature, then embedded in paraffin and stained with Hematoxylin & Eosin, essentially as described (Gozzelino et al., 2012).
Cardiovascular Function
Cardiovascular function was measured using pressure–volume conductance catheter technique (Pacher et al., 2008). Briefly, 48 hr after CLP mice were anesthetized with isoflurane, tracheotomized and artificially ventilated. Temperature was recorded continuously and kept stable. The apex of the left ventricle was punctured with a 27 G needle using the open chest approach and a FTS-1912B-8018; pressure-volume conductance catheter (Scisense, London, Canada) was inserted in the left ventricle. Mice were allowed to stabilize for 3-10 min. Baseline values, values with varying preload caused by inferior vena cava clamps using a blunt forceps and aortic pressures, were recorded with the Scisense pressure-volume control unit FV896B and analyzed using the Labscribe2 (Labscribe, iWorx Systems, USA) software. The machine was calibrated with internal and cuvette calibration, as described (Pacher et al., 2008).
Food Intake
Mice were housed individually and remaining food weight was determined daily.
Serology/ELISA
Alanine amino transferase (ALT), aspartate amino transferase (AST), blood urea nitrogen (BUN), creatinephosphokinase (CPK) Lactate Dehydrogenase (LHD)(BioAssay Systems, CA, USA) and Insulin (Mouse Ultrasensitive Insulin ELISA, ALPCO, USA) serum concentrations were measured by ELISA according to manufacturer instructions. Alternatively, serological parameters were determined by DNATech, Lisbon, Portugal.
Glucose, Pyruvate, Insulin, and Glucagon Tolerance Tests
Oral glucose tolerance test (oGTT), pyruvate tolerance test (PTT) and insulin tolerance test (ITT) were performed in fasted mice (14 hr) while the glucagon challenge test was performed without fasting. Glucose (Sigma) was administered orally (2 mg/g BW in 200 μL 0.9% saline). Pyruvate (Sigma; 2mg/g BW in 200 μL 0.9% saline), Insulin (Humulin R100 UI/mL; Eli Lilly and Company) and Glucagon (Glucagen; Novo Nordisk A/S, Denmark; 16 μg/g of BW) were administered intraperitoneally (i.p.). Tests were performed in before and 48 hr after CLP.
Cell Culture
HepG2 cells were grown in low glucose (1g/L) DMEM (GIBCO® Ref: 21885-108), 10% heat inactivated FBS, 1% Penicilin/Streptomycin (GIBCO® Ref: 15140-122). Before seeding, plates were coated with collagen I rat protein (GIBCO, ThermoFisher Scientific; 5μg/ml in 20mM acetic) acid. At 100% confluence cells were treated with heme (Frontier Scientifc; 40 μM, 1 hr) in HBSS, human recombinant TNF (R&D Systems Europe Ref: 210-TA-20) was added in media (50 ng/mL; 4 hr) and cells were collected. When indicated cells were pre-treated with N-acetyl-L-cysteine (NAC; Sigma, Ref: A9165; 7.5mM in media; 3 hr) before and during heme treatment in HBSS and subsequently throughout the experiment. When indicated cells were pre-treated Deferoxamine (DFO, Sigma Ref: D9533; 60 μM; 16 hr) before and during heme treatment in HBSS and subsequently throughout the experiment. At 80%–90% of confluence, cells were transduced at 50 or 100 pfu with LacZ, G6pc1 (Vector Biolabs; Ref: SKU#: ADV-259766), FTH and FTHm Rec.Ad. (Berberat et al., 2003, Gozzelino et al., 2012). Medium was replaced 24 hr after Rec.Ad. transduction. G6Pase mRNA, protein and activity were analyzed 48 or 72 hr after Rec.Ad. transduction, as described below.
Luciferase Assays
HepG2 cells were seeded onto 6-well plates and transfected 24h thereafter (60%–80% confluency) with pGL2B-G6PC (containing −1320/+60 from the rat G6pc1 promoter, kind gift from Amandine Gautier-Stein; Université Claude Bernard Lyon, France) (Rajas et al., 2002), or the empty vector pGL2B alone or co-transfected with pSG5-FtH (Berberat et al., 2003) (kind gift from Dr. P. Arosio, University of Brescia, Brescia, Italy) using Lipofectamine LTX Reagent with PLUS Reagent (Invitrogen, Thermo Scientific), according to manufacturer instructions. A Renilla Luciferase expressing vector (kind gift from Amandine Gautier-Stein; Université Claude Bernard Lyon, France) was co-transfected as transfection control. Forty-eight hours thereafter cells were treated with heme (40 μM, 1h) in HBSS, and subsequently exposed to human recombinant TNF (50 ng/mL; 4 hr) in culture medium and collected for luciferase activity. Alternatively cells were treated with heme (40 μM, 1h) in HBSS, exposed to human recombinant TNF (50 ng/mL; 3 hr) in culture medium and collected for luciferase activity, 12 hr after exposure to TNF. Luciferase activity was determined using the Luciferase assay system Dual-Glo (Promega) was used according to manufacturer’s intructions. Luminescence was measured using a microplate reader (Victor3 Multilabel Readers, Perkin Elmer). Firefly Luciferase was normalized to Renilla Luciferase activity and expressed as relative light units (RLU) using pGL2B activity as background, essentially as described (Rajas et al., 2002).
Immunohistochemistry
Livers were harvested 12 hr after severe CLP. Livers of control (untreated) mice were also collected. All livers were fixed with 10% buffered formalin and embedded in paraffin. Paraffin sections were used for immunohistochemistry. Briefly, for antigen retrieval slides were placed in citrate buffer and heated to sub-boiling conditions for 6 min. After cooling down, slides were briefly washed in TBST and then permeabilized with 0.3% Triton (Sigma) in PBS. Slides were blocked for 1h at room temperature with blocking solution (goat serum in TBST, 1:50). Slides were incubated with primary antibodies (FTH D1D4 Cell Signaling and F4/80 BM8 Biolegend) prepared in blocking solution, overnight at 4°C. Slides were quickly washed with TBST and incubated with secondary antibodies (Alexa Fluor® 568 Goat Anti-Rabbit and Alexa Fluor® 647 Goat Anti-Rat, Life Technologies), prepared in blocking solution, for 2 hr at room temperature. Slides were washed first with TBST and then with PBS and mounted with Mowiol-Dabco containing 10μg/mL of DAPI. Images were acquired on a Leica DMRA2 upright microscope, equipped with a CoolSNAP HQ CCD camera, using a 40x HCX PL FLUOTAR objective, DAPI + TRITC + CY5 fluorescence filtersets, controlled with the MetaMorph V7.5.1/ software. The analysis was done with ImageJ (Rasband, W.S., ImageJ, U. S. NIH, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997-2014).
Heme Measurement
Total heme levels in the peritoneal cavity in C57BL/6 mice before or 3 hr after CLP were measured. Briefly, samples were diluted in H2O in 96 well plates and heme concentration was determined by comparison to a hemin standard curve (0.25-16 μM in H2O). Formic acid (150 μL/well; 98%–100%, Merck) was added and absorbance measured at 405nm using a microplate reader (Victor3 Multilabel Readers, Perkin Elmer).
qRT-PCR
RNA was isolated from cells or livers using the NucleoSpin RNA kit (Macherey-Nagel). cDNA was transcribed from total RNA with transcriptor first strand cDNA synthesis kit (Roche). Quantitative real-time PCR (qRT-PCR) was performed using 1μg cDNA and Syber Green Master Mix (Applied Biosystems, Foster City, CA, USA) in triplicate on a 7500 Fast Real-Time PCR System (Applied Biosystems) under the following conditions: 95°C/10 min, 40 cycles/95°C/15 s, annealing at 60°C/30 s and elongation 72°C/30 s. Real-time primer sequences for mouse samples were: ArbpO Fwd: 5′CTTTGGGCATCACCACGAA3′, Rev: 5′GCTGGCTCCCACCT TGTCT3′; Fth Fwd: 5′CCATCAACCGCCAGATCAAC3′, Rev: 5′GCCACAT CATCTCGGTCAAA3′. G6pc1 Fwd: 5′TGGGGCAGAAATATGACTCCT3′; Rev: 5′TGCAAATCAGCCGAGGCA3′; Gpt1 Fwd: 5′ CTTCTCTC GCCTACGCCTTG3′, Rev: 5′GGAACCCAGTGTCTGCGG3′. Real-time primer sequences for in vitro experiments with human HepG2 cells were: G6PC1 Fwd:5-TTCAGGAAGCTGTGGGCATT3′, Rev: 5′TCTTTCCCTGGTCCAGTCTCA3′ and actin Fwd 5′GAGCACAGAGCCTCGCCTTT3′ and Rev 5′TCATCAT CCATGGTGAGCTGG3′. Primers were design using Primer Blast (Ye et al., 2012).
Western Blot
Proteins were extracted, electrophoresed, and electrotransferred essentially as described (Gozzelino et al., 2012). Alternatively, for G6PC detection, samples were collected in HEPES-sucrose buffer/CHAPS and directly snap frozen. Total protein was quantified using Bradford assay. Anti-mouse G6PC1 (1:5000), anti-FTH1 (clone D1D4) (1:1000) (Cell Signaling), anti-p21 (1:1000) (2947, Cell Signaling), anti- β-actin (1:5000) (A5441, Sigma) and anti-α-tubulin-HRP (clone DM1A) (Sigma) (1:10000) were detected using peroxidase conjugated secondary antibodies (1 hr; room temperature) and developed with ECL western blotting substrate (ThermoFisher Scientific) for anti-β-tubulin and SuperSignal West Pico Chemiluminiscent substrate (ThermoFisher Scientific) for anti-G6PC and anti-FTH1 antibodies.
G6Pase Activity
Livers from Fthlox/lox and Mx1CreFthΔ/Δ mice without or 48 hr after being subjected to CLP were homogenized in liquid nitrogen and sonicated in HEPES-Sucrose (10 mM HEPES / 0.25 M Sucrose buffer). G6Pase enzymatic activity was determined essentially as described elsewhere (Mutel et al., 2011). Absorption was measured using a Bio-Rad 3550-UV Reader at 700 nm. For G6Pase activity, HepG2 cells were collected 72h after Rec.Ad. transduction and when indicated 5 hr after heme and TNF treatment. Cells were collected in 10mM HEPES buffer, subjected to three cycles of freeze-thaw and G6Pase enzymatic activity was determined essentially as described elsewhere (Mutel et al., 2011).
Cytokine Measurement
Cytokines were measured in serum 24 hr after CLP using Magpix® Luminex® (http://www.luminexcorp.com) according to manufacturer instructions.
Targeted Metabolomics
Targeted metabolomics for amino acid and glucose metabolism metabolites were determined by MetabolomicDiscoveries (Potsdam, Germany) using high-throughput Gas chromatography–mass spectrometry and accurate mass Quadrupole Time-of-Flight coupled to an Ultra Performance Liquid Chromatography.
Microarray Screening
Samples were processed by standard protocols: Briefly, RNA was extracted (RNeasy minikit; QIAGEN, Germany) and biotinylated probes were prepared using the TargetAmp-Nano Labeling Kit for Illumina® Expression. BeadChip. Illumina mouseRef8 V2 chips were scanned using the Illumina BeadArray Reader 500 X (Illumina). Arrays were read by GenomeStudio (v.1.9, and annotation MouseRef-8_V2_0_R3_11278551_A) yielding 25,697 bead types (probes) for each sample.
Metabolic Model Analysis
The metabolome data were analyzed in the context of an in silico metabolic model representation, using ratios between the 48 hr after CLP and 0 hr data points for every quantified metabolite. This was done independently for the metabolome datasets of both genotypes Fthlox/lox and Mx1CreFthΔ/Δ. Ratios were integrated into the genome-scale metabolic model iMM1415 of Mus musculus (Sigurdsson et al., 2010). Metabolic network reconstructions are concise representations of all stoichiometrically accurate metabolic reactions in a given organism. Reaction specific lower and upper thermodynamically feasible flux bounds were integrated if such information is available, otherwise were set to infinite. Of note, the exchange reaction subset comprises reactions that interchange metabolites with the environment or between different compartments within the model. If no exchange reaction existed for one of the measured metabolites a respective artificial exchange reaction was introduced into iMM1415 resulting in iMM1415∗. In a first step, a flux variability analysis (FVA) (Mahadevan and Schilling, 2003) of iMM1415∗ with ATP exchange as objective function was performed. This provides the feasible flux boundaries of all model reactions, including exchange reactions, that allow for an optimal ATP yield as calculated by a flux balance analysis (FBA, 5% tolerance from optimal value accepted) (Orth et al., 2010). Next, the feasible flux boundaries of all metabolome associated exchange reaction fluxes resulting from the FVA were constrained once by the Fthlox/lox 48h/0h and once by the Mx1CreFthΔ/Δ 48h/0h ratios. Subsequently, FBA optimization was performed for ATP exchange for both Fthlox/lox 48h/0h and Mx1CreFthΔ/Δ 48h/0h constrained models and differences in the flux distribution were assessed.
In silico single gene and reaction deletion studies are realized by setting the respective feasible reaction flux range to zero. Of note, the former leads to one or multiple reaction knock outs, if a gene is essential for these reactions. Impact of gene and reaction deletions was studied by comparing FBA derived ATP yields for Fthlox/lox 48h/0h and Mx1CreFthΔ/Δ 48h/0h constrained models. Model modification, FVA, FBA and single gene/reaction deletion studies have been performed with the MATLAB toolbox COBRA (Schellenberger et al., 2011).
Quantification and Statistical Analysis
Survival Plots were analyzed using the Log-rank test. Two-Way-ANOVA with a post hoc Bonferroni comparison and without for metabolomics was used when analyzing more than one group and time point. Mann-Whitney-U Test was used for pairwise comparisons. Analysis of microarray data were performed using R software version 3.02 (http://www.r-project.org/) and Bioconductor (Gentleman et al., 2004) packages. Raw data were subjected to robust spline normalization, quality control, and background correction with the R package Lumi (Du et al., 2008). Transcripts with detection values p < 0.05 in at least four samples were called ‘present’ and taken for further analysis. Differentially expressed genes (DEG) were filtered according to microarray quality control criteria (Shi et al., 2006) by at least average two-fold changes and p values < 0.05 from Welch-modified t tests.
Data and Software Availability
The accession number for the raw data files of the RNA Illumina screen reported in this paper are deposited in the GEO database (GEO: GSE92703), https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE92703.
Author Contributions
M.P.S. formulated the original hypothesis, drove the study design, and wrote the manuscript with S.W. and A.R.C. S.W. and A.R.C. contributed critically to study design, performed and/or contributed to all experiments, and analyzed data. M.R.M. assisted A.R.C. R.L. performed the initial experiments of the project. B.B. performed experiments involving chimeras and associated readouts. S. Singh performed experiments involving polymicrobial infection and associated readouts. S.C. crossed, bred, and when required, characterized different mouse strains. S.R. performed G6Pase activities, analyzed data, and characterized different mouse strains. L.D.B. performed experiments related to G6Pc expression. G.M. and F.R. provided mice, tools, and protocols for G6Pc experiments and contributed to experimental design. M.B. planned and supervised the systems biology approach. S. Schäuble performed constrained-based modeling and analysis. S.L. did bioinformatics analysis. All authors read and approved the manuscript.
Acknowledgments
We thank members of the Inflammation Group (IGC) for insightful discussions, Tânia Carvalho and Jasmin Tischer for histopathological examination, Amandine Gautier-Stein (Université Claude Bernard Lyon, France) for luciferase reporter constructs and advise, Luis F. Moita (IGC) and Rui Martins (Medical University, Vienna, Austria) for critical appraisal of the manuscript as well as Marta Monteiro (IGC, Flow Cytometry). Support was provided by Fundação para a Ciência e Tecnologia (PTDC/SAU-TOX/116627/2010 and HMSP-ICT/0018/2011 to M.P.S. and SFRH/BPD/101608/2014 to A.R.C.), the European Community 7th Framework (ERC-2011-AdG 294709-DAMAGECONTROL to M.P.S.), the Deutsche Forschungsgemeinschaft (DFG; WE 4971/3 to S.W.), the German Federal Ministry of Education and Research (BMBF; 01EO1002, Center for Sepsis Control and Care to M.B.), Meta-ZIK (03Z2J52 to M.B.), the German Ministry for Research and Education e:Med initiative GlioPATH (FKZ 01ZX1402C to S. Schäuble), the French National Research Agency (ANR-11-BSV1-009 and ANR-15-CE14-0026-03 to F.R.), and the Medical Research Foundation (DRM20101220448 to G.M.).
Published: June 15, 2017
Footnotes
Supplemental Information includes seven figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2017.05.031.
Supplemental Information
References
- Adamzik M., Hamburger T., Petrat F., Peters J., de Groot H., Hartmann M. Free hemoglobin concentration in severe sepsis: methods of measurement and prediction of outcome. Crit. Care. 2012;16:R125. doi: 10.1186/cc11425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angus D.C., van der Poll T. Severe sepsis and septic shock. N. Engl. J. Med. 2013;369:2063. doi: 10.1056/NEJMc1312359. [DOI] [PubMed] [Google Scholar]
- Berberat P.O., Katori M., Kaczmarek E., Anselmo D., Lassman C., Ke B., Shen X., Busuttil R.W., Yamashita K., Csizmadia E. Heavy chain ferritin acts as an antiapoptotic gene that protects livers from ischemia reperfusion injury. FASEB J. 2003;17:1724–1726. doi: 10.1096/fj.03-0229fje. [DOI] [PubMed] [Google Scholar]
- Clausen B.E., Burkhardt C., Reith W., Renkawitz R., Förster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8:265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- Clottes E., Burchell A. Three thiol groups are important for the activity of the liver microsomal glucose-6-phosphatase system. Unusual behavior of one thiol located in the glucose-6-phosphate translocase. J. Biol. Chem. 1998;273:19391–19397. doi: 10.1074/jbc.273.31.19391. [DOI] [PubMed] [Google Scholar]
- Darshan D., Vanoaica L., Richman L., Beermann F., Kühn L.C. Conditional deletion of ferritin H in mice induces loss of iron storage and liver damage. Hepatology. 2009;50:852–860. doi: 10.1002/hep.23058. [DOI] [PubMed] [Google Scholar]
- Deák M., Horváth G.V., Davletova S., Török K., Sass L., Vass I., Barna B., Király Z., Dudits D. Plants ectopically expressing the iron-binding protein, ferritin, are tolerant to oxidative damage and pathogens. Nat. Biotechnol. 1999;17:192–196. doi: 10.1038/6198. [DOI] [PubMed] [Google Scholar]
- Deutschman C.S., Andrejko K.M., Haber B.A., Bellin L., Elenko E., Harrison R., Taub R. Sepsis-induced depression of rat glucose-6-phosphatase gene expression and activity. Am. J. Physiol. 1997;273:R1709–R1718. doi: 10.1152/ajpregu.1997.273.5.R1709. [DOI] [PubMed] [Google Scholar]
- Drakesmith H., Prentice A.M. Hepcidin and the iron-infection axis. Science. 2012;338:768–772. doi: 10.1126/science.1224577. [DOI] [PubMed] [Google Scholar]
- Du P., Kibbe W.A., Lin S.M. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- Figueiredo R.T., Fernandez P.L., Mourao-Sa D.S., Porto B.N., Dutra F.F., Alves L.S., Oliveira M.F., Oliveira P.L., Graça-Souza A.V., Bozza M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007;282:20221–20229. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- Fisher J., Devraj K., Ingram J., Slagle-Webb B., Madhankumar A.B., Liu X., Klinger M., Simpson I.A., Connor J.R. Ferritin: a novel mechanism for delivery of iron to the brain and other organs. Am. J. Physiol. Cell Physiol. 2007;293:C641–C649. doi: 10.1152/ajpcell.00599.2006. [DOI] [PubMed] [Google Scholar]
- Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonnert F.A., Recknagel P., Seidel M., Jbeily N., Dahlke K., Bockmeyer C.L., Winning J., Lösche W., Claus R.A., Bauer M. Characteristics of clinical sepsis reflected in a reliable and reproducible rodent sepsis model. J. Surg. Res. 2011;170:e123–e134. doi: 10.1016/j.jss.2011.05.019. [DOI] [PubMed] [Google Scholar]
- Gozzelino R., Soares M.P. Coupling heme and iron metabolism via ferritin H chain. Antioxid. Redox Signal. 2014;20:1754–1769. doi: 10.1089/ars.2013.5666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozzelino R., Andrade B.B., Larsen R., Luz N.F., Vanoaica L., Seixas E., Coutinho A., Cardoso S., Rebelo S., Poli M. Metabolic adaptation to tissue iron overload confers tolerance to malaria. Cell Host Microbe. 2012;12:693–704. doi: 10.1016/j.chom.2012.10.011. [DOI] [PubMed] [Google Scholar]
- Grempler R., Kienitz A., Werner T., Meyer M., Barthel A., Ailett F., Sutherland C., Walther R., Schmoll D. Tumour necrosis factor alpha decreases glucose-6-phosphatase gene expression by activation of nuclear factor kappaB. Biochem. J. 2004;382:471–479. doi: 10.1042/BJ20040160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison P.M., Arosio P. Ferritins - molecular properties, iron storage function and cellular regulation. Biochim. Biophys. Acta. 1996;1275:161–203. doi: 10.1016/0005-2728(96)00022-9. [DOI] [PubMed] [Google Scholar]
- Hentze M.W., Caughman S.W., Rouault T.A., Barriocanal J.G., Dancis A., Harford J.B., Klausner R.D. Identification of the iron-responsive element for the translational regulation of human ferritin mRNA. Science. 1987;238:1570–1573. doi: 10.1126/science.3685996. [DOI] [PubMed] [Google Scholar]
- Hermanides J., Bosman R.J., Vriesendorp T.M., Dotsch R., Rosendaal F.R., Zandstra D.F., Hoekstra J.B., DeVries J.H. Hypoglycemia is associated with intensive care unit mortality. Crit. Care Med. 2010;38:1430–1434. doi: 10.1097/CCM.0b013e3181de562c. [DOI] [PubMed] [Google Scholar]
- Jais A., Einwallner E., Sharif O., Gossens K., Lu T.T., Soyal S.M., Medgyesi D., Neureiter D., Paier-Pourani J., Dalgaard K. Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell. 2014;158:25–40. doi: 10.1016/j.cell.2014.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janz D.R., Bastarache J.A., Sills G., Wickersham N., May A.K., Bernard G.R., Ware L.B. Association between haptoglobin, hemopexin and mortality in adults with sepsis. Crit. Care. 2013;17:R272. doi: 10.1186/cc13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotas M.E., Medzhitov R. Homeostasis, inflammation, and disease susceptibility. Cell. 2015;160:816–827. doi: 10.1016/j.cell.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn R., Schwenk F., Aguet M., Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- Larsen R., Gozzelino R., Jeney V., Tokaji L., Bozza F.A., Japiassú A.M., Bonaparte D., Cavalcante M.M., Chora A., Ferreira A. A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2010;2:51ra71. doi: 10.1126/scitranslmed.3001118. [DOI] [PubMed] [Google Scholar]
- Lee I., Hüttemann M. Energy crisis: the role of oxidative phosphorylation in acute inflammation and sepsis. Biochim. Biophys. Acta. 2014;1842:1579–1586. doi: 10.1016/j.bbadis.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipiński P., Jarzabek Z., Broniek S., Zagulski T. Protective effect of tissue ferritins in experimental Escherichia coli infection of mice in vivo. Int. J. Exp. Pathol. 1991;72:623–630. [PMC free article] [PubMed] [Google Scholar]
- Mahadevan R., Schilling C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003;5:264–276. doi: 10.1016/j.ymben.2003.09.002. [DOI] [PubMed] [Google Scholar]
- Medzhitov R., Schneider D.S., Soares M.P. Disease tolerance as a defense strategy. Science. 2012;335:936–941. doi: 10.1126/science.1214935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyron-Holtz E.G., Moshe-Belizowski S., Cohen L.A. A possible role for secreted ferritin in tissue iron distribution. J. Neural Transm. (Vienna) 2011;118:337–347. doi: 10.1007/s00702-011-0582-0. [DOI] [PubMed] [Google Scholar]
- Mithieux G. New knowledge regarding glucose-6 phosphatase gene and protein and their roles in the regulation of glucose metabolism. Eur. J. Endocrinol. 1997;136:137–145. doi: 10.1530/eje.0.1360137. [DOI] [PubMed] [Google Scholar]
- Mutel E., Abdul-Wahed A., Ramamonjisoa N., Stefanutti A., Houberdon I., Cavassila S., Pilleul F., Beuf O., Gautier-Stein A., Penhoat A. Targeted deletion of liver glucose-6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J. Hepatol. 2011;54:529–537. doi: 10.1016/j.jhep.2010.08.014. [DOI] [PubMed] [Google Scholar]
- Nemeth E., Tuttle M.S., Powelson J., Vaughn M.B., Donovan A., Ward D.M., Ganz T., Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- Okin D., Medzhitov R. The effect of sustained inflammation on hepatic mevalonate pathway results in hyperglycemia. Cell. 2016;165:343–356. doi: 10.1016/j.cell.2016.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth J.D., Thiele I., Palsson B.O. What is flux balance analysis? Nat. Biotechnol. 2010;28:245–248. doi: 10.1038/nbt.1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P., Nagayama T., Mukhopadhyay P., Bátkai S., Kass D.A. Measurement of cardiac function using pressure-volume conductance catheter technique in mice and rats. Nat. Protoc. 2008;3:1422–1434. doi: 10.1038/nprot.2008.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamplona A., Ferreira A., Balla J., Jeney V., Balla G., Epiphanio S., Chora A., Rodrigues C.D., Gregoire I.P., Cunha-Rodrigues M. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat. Med. 2007;13:703–710. doi: 10.1038/nm1586. [DOI] [PubMed] [Google Scholar]
- Postic C., Shiota M., Niswender K.D., Jetton T.L., Chen Y., Moates J.M., Shelton K.D., Lindner J., Cherrington A.D., Magnuson M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274:305–315. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- Pravda J. Metabolic theory of septic shock. World J. Crit. Care Med. 2014;3:45–54. doi: 10.5492/wjccm.v3.i2.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raetzsch C.F., Brooks N.L., Alderman J.M., Moore K.S., Hosick P.A., Klebanov S., Akira S., Bear J.E., Baldwin A.S., Mackman N., Combs T.P. Lipopolysaccharide inhibition of glucose production through the Toll-like receptor-4, myeloid differentiation factor 88, and nuclear factor kappa b pathway. Hepatology. 2009;50:592–600. doi: 10.1002/hep.22999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajas F., Gautier A., Bady I., Montano S., Mithieux G. Polyunsaturated fatty acyl coenzyme A suppress the glucose-6-phosphatase promoter activity by modulating the DNA binding of hepatocyte nuclear factor 4 alpha. J. Biol. Chem. 2002;277:15736–15744. doi: 10.1074/jbc.M200971200. [DOI] [PubMed] [Google Scholar]
- Schellenberger J., Que R., Fleming R.M., Thiele I., Orth J.D., Feist A.M., Zielinski D.C., Bordbar A., Lewis N.E., Rahmanian S. Quantitative prediction of cellular metabolism with constraint-based models: the COBRA Toolbox v2.0. Nat. Protoc. 2011;6:1290–1307. doi: 10.1038/nprot.2011.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L., Reid L.H., Jones W.D., Shippy R., Warrington J.A., Baker S.C., Collins P.J., de Longueville F., Kawasaki E.S., Lee K.Y., MAQC Consortium The MicroArray Quality Control (MAQC) project shows inter- and intraplatform reproducibility of gene expression measurements. Nat. Biotechnol. 2006;24:1151–1161. doi: 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson M.I., Jamshidi N., Steingrimsson E., Thiele I., Palsson B.O. A detailed genome-wide reconstruction of mouse metabolism based on human Recon 1. BMC Syst. Biol. 2010;4:140. doi: 10.1186/1752-0509-4-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares M.P., Bozza M.T. Red alert: labile heme is an alarmin. Curr. Opin. Immunol. 2016;38:94–100. doi: 10.1016/j.coi.2015.11.006. [DOI] [PubMed] [Google Scholar]
- Soares M.P., Weiss G. The Iron age of host-microbe interactions. EMBO Rep. 2015;16:1482–1500. doi: 10.15252/embr.201540558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares M.P., Gozzelino R., Weis S. Tissue damage control in disease tolerance. Trends Immunol. 2014;35:483–494. doi: 10.1016/j.it.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Soares M.P., Teixeira L., Moita L.F. Disease tolerance and immunity in host protection against infection. Nat. Rev. Immunol. 2017;17:83–96. doi: 10.1038/nri.2016.136. [DOI] [PubMed] [Google Scholar]
- van Schaftingen E., Gerin I. The glucose-6-phosphatase system. Biochem. J. 2002;362:513–532. doi: 10.1042/0264-6021:3620513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vooijs M., Jonkers J., Berns A. A highly efficient ligand-regulated Cre recombinase mouse line shows that LoxP recombination is position dependent. EMBO Rep. 2001;2:292–297. doi: 10.1093/embo-reports/kve064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A., Huen S.C., Luan H.H., Yu S., Zhang C., Gallezot J.D., Booth C.J., Medzhitov R. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell. 2016;166:1512–1525. doi: 10.1016/j.cell.2016.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witzleben C.L., Chaffey N.J. A study of iron-induced liver damage. J. Exp. Med. 1962;115:723–729. doi: 10.1084/jem.115.4.723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin L., Wu N., Curtin J.C., Qatanani M., Szwergold N.R., Reid R.A., Waitt G.M., Parks D.J., Pearce K.H., Wisely G.B., Lazar M.A. Rev-erbalpha, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318:1786–1789. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- Yki-Järvinen H., Sammalkorpi K., Koivisto V.A., Nikkilä E.A. Severity, duration, and mechanisms of insulin resistance during acute infections. J. Clin. Endocrinol. Metab. 1989;69:317–323. doi: 10.1210/jcem-69-2-317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.