

Abstract
Recent studies have helped identify multiple factors affecting increased risk for substance use disorders (SUDs) following traumatic brain injury (TBI). These factors include age at the time of injury, repetitive injury and TBI severity, neurocircuits, neurotransmitter systems, neuroinflammation, and sex differences. This review will address each of these factors by discussing 1) the clinical and preclinical data identifying patient populations at greatest risk for SUDs post-TBI, 2) TBI-related neuropathology in discrete brain regions heavily implicated in SUDs, and 3) the effects of TBI on molecular mechanisms that may drive substance abuse behavior, like dopaminergic and glutamatergic transmission or neuroimmune signaling in mesolimbic regions of the brain. Although these studies have laid the groundwork for identifying factors that affect risk of SUDs post-TBI, additional studies are required. Notably, preclinical models have been shown to recapitulate many of the behavioral, cellular, and neurochemical features of SUDs and TBI. Therefore, these models are well suited for answering important questions that remain in future investigations.
Keywords: Traumatic brain injury, substance use disorders, preclinical modeling
Graphical abstract
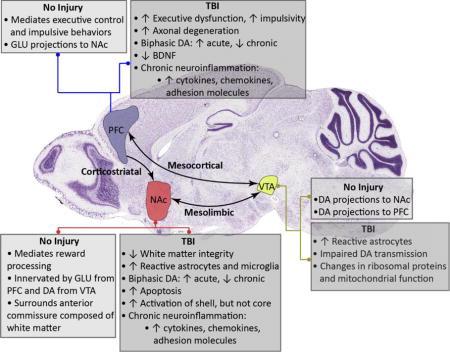
Effects of traumatic brain injury (TBI) on mechanisms implicated in substance use disorders (SUDs)
Recent studies find that TBI may affect many key brain regions implicated in the neurobiology of addiction including the prefrontal cortex (PFC), nucleus accumbens (NAc), and ventral tegmental area (VTA). Furthermore, evidence suggests that TBI-induced pathology may resemble neurochemical responses that mediate SUDs. As shown, experimental TBI disrupts many of the neural substrates that regulate substance abuse behavior. However, few studies have explicitly tested the effect of TBI on the functional status of neurocircuits that mediate drug and alcohol reward, like the corticolimbic, mesolimbic, and mesocortical pathways. This review highlights the results of recent reports that identify important factors affecting the risk of SUDs post-TBI. Moreover, this review proposes the use of preclinical models to answer questions that still remain, as animal models have been shown to recapitulate the neurochemical and behavioral effects of both SUDs and TBI. Notably, expanding upon the current literature with future studies will be essential to fully validate and understand the mechanisms underlying increased risk of SUDs post-TBI. Arrows indicate increase or decrease in neurotransmitter concentration, gene expression level, incidence of pathology, or behavioral function as listed.
DA-dopamine, GLU-glutamate, BDNF-brain-derived neurotropic factor; histology image obtained from www.alleninstitute.org.
INTRODUCTION
Traumatic brain injury (TBI) is a prominent public health concern affecting millions of Americans and their families each year. These injuries may produce lifelong deficits in physical, cognitive, social, emotional, and behavioral function1. In fact, current estimates suggest that as many as 5.3 million people living in the United States may struggle with a TBI-related disability1. Notably, this figure is likely an underestimate, as some disorders that are highly prevalent among TBI patients, like substance use disorders (SUDs), are typically considered to be pre-existing conditions rather than a consequence of TBI2. However, dedicated research in the field of head trauma rehabilitation and the emergence of the first preclinical studies to investigate the effects of experimental TBI on drug abuse behavior have led to the identification of multiple factors affecting risk of SUDs post-TBI.
Results of the 2014 National Survey on Drug Use and Health show that approximately 21.5 million Americans are currently diagnosed with SUDs3. Importantly, these disorders are the most common psychiatric diagnoses among TBI patients prior to injury, and the third most common psychiatric diagnoses post-TBI4. Alcohol is the most common drug abused by individuals with a history of TBI. In fact, estimates indicate that 37–66% of TBI patients struggle with alcohol use disorders, while 10–44% of TBI patients abuse illicit drugs5. Notably, within illicit drug use, studies have shown that TBI patients are most likely to abuse cannabis, cocaine, methamphetamine, and prescription medications, including opioids, stimulants, benzodiazepines, antidepressants, and antipsychotics6–10. Furthermore, additional studies have shown that daily cigarette use is significantly elevated in TBI patients when compared to age-matched controls, suggesting that brain injuries may also affect nicotine-dependence6, 11.
In addition, many reports find that patients with co-morbid TBI and SUD have poorer long-term outcomes5, 12. These patients experience higher mortality rates, show deficits in physical and neurological recovery, display greater brain atrophy and diminished white matter integrity, are more likely to behave aggressively, show signs of impulsivity and reduced executive function, and have higher arrest rates5, 8, 13–15. Furthermore, these patients have poorer neuropsychological outcomes, higher rates of psychiatric disease, increased risk of attempted suicide/suicidal ideation, and greater likelihood of sustaining additional TBIs8, 12, 14. Despite these findings, research in the field has met much adversity due to two commonly held beliefs: 1) that data generated to assess the risk of SUD post-TBI are very difficult to interpret because of shared risk factors in co-morbid TBI and SUD patients, and 2) that TBI is more often a consequence of substance abuse rather than a cause of SUDs2, 9. However, by refining clinical studies and utilizing animal models to assess the risk of SUDs post-TBI, biomedical researchers have identified novel factors rarely accounted for in previous studies.
This review will address each of the following factors affecting increased risk for SUDs following TBI: age at the time of injury, repetitive injury and TBI severity, neurocircuits, neurotransmitter systems, neuroinflammation, and sex differences. First, the recent clinical and preclinical studies that have helped identify patient populations at greatest risk for SUDs post-TBI will be discussed. Next, a review of TBI-related neuropathology occurring in the neurocircuits, neurotransmitter systems, and neuroimmune signals that are heavily implicated in substance abuse behavior will be presented. Finally, gaps in knowledge and critical next steps will be discussed to better understand the causal relationship that exists between TBI and SUDs. Notably, employing the use of preclinical models will be essential to expediting this process as in vivo microdialysis, electrophysiology, and drug self-administration assays are well suited to answer questions that remain illusive in the link between SUDs and TBI.
AGE AT THE TIME OF INJURY
Most studies assessing the risk of SUDs post-TBI have monitored the incidence of SUDs in adult TBI patients (typically patients age 18+). These studies have produced two key findings: 1) that SUD rates decline following adult TBI and 2) that very few SUDs are newly diagnosed in adult TBI patients4, 5. These findings are not at all surprising considering that the prevalence of SUDs in the general population peaks between the ages of 18–25 then steadily declines among individuals 26 and older3. Therefore, recognition of a downward trend in SUD rates among adult TBI patients is consistent with the trend established in the general population, and not necessarily an indication that TBI fails to be a risk factor for SUDs. Unfortunately, these findings have lead some researchers to conclude that the high prevalence of SUDs among TBI patients is primarily due to pre-existing conditions rather than a consequence of brain injury2.
Although adult TBI studies have helped identify substance abuse as a risk factor for brain injury2, 5, 12, the inability of these studies to accurately assess the risk of SUDs post-TBI becomes profoundly apparent when observing the rates of both SUDs and TBIs across a range of ages (Figure 1). First, these studies neglect the largest population of TBI patients (children and adolescence, age 0–19), and instead assess the risk of SUDs in a population that is least likely to sustain a TBI (with the exception of individuals over the age of 65)16. Furthermore, as mentioned, these studies assess the risk of substance abuse in TBI patients when the prevalence of SUDs is in decline17. However, by accounting for the incidence of early-life TBI, Corrigan et al. determined the following: the younger the age of a patient at the time of TBI, the greater the effect on problematic alcohol consumption and illicit drug use later in life18.
Figure 1. Observing the distribution of illicit drug use and TBI-related emergency department (ED) visits by age identifies understudied populations in reports that evaluate the effect of TBI on SUDs.
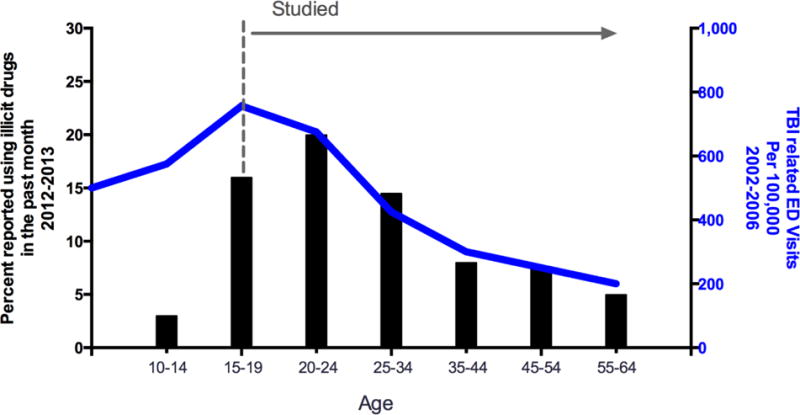
The majority of reports evaluating TBI as a risk factor for substance abuse fail to account for the incidence of early-life TBI (dashed line and arrow identify the age groups commonly included in published reports). Furthermore, these studies do not account for the initial peak in the incidence of TBI (TBI-related ED visits per 100,000, 2002–2006; blue line, right-hand y-axis), and instead have drawn conclusions based on a downward trend in drug use that exists among the general population (percent of U.S. population using illicit drugs in the past month, 2012–2013; black bars, left-hand y-axis). Therefore, future studies that account for the incidence of early-life TBI and monitor the rise in drug and alcohol use rather than its decline may help to more accurately assess the risk of SUDs post-TBI16, 17.
Similarly, a number of recent studies have identified early-life TBI as an important factor affecting the risk of SUDs. Ilie et al. reports that high school students with a history of TBI are 2–3 times more likely to participate in binge drinking, daily cigarette smoking, nonmedical use of prescription drugs, and illicit drug use6. Furthermore, Fishbein et al. found that early-life TBI was predictive of greater drug use severity and earlier onset of drug use19. Likewise, Ramesh et al. determined that 84% of cocaine-dependent research volunteers with a history of TBI sustained their first TBI prior to the onset of cocaine use20, and Olsen-Madden et al. saw that 54% of veterans with a history of TBI seeking outpatient treatment for substance abuse sustained at least one TBI prior to adulthood7. Together, these results highlight the need for future studies to account for the incidence of early-life brain injury in adult TBI patients in order to accurately assess the risk of SUDs following TBI.
Although large longitudinal, prospective clinical studies that assess the risk of SUDs throughout the life of TBI patients may be costly, time-consuming, and difficult to manage due to patient attrition, preclinical models are well suited to achieve progress in the field of TBI/SUD research. Importantly, a variety of preclinical models are available that recapitulate the behavioral, cellular, and neurochemical features of both SUDs and TBI. In fact, recent studies have produced valuable data by combining the premiere models of experimental TBI with behavioral assays of drug and alcohol dependence in order to evaluate the link between TBI and SUDs. Notably, two of these preclinical studies have assessed the effect of early-life TBI on substance abuse behavior.
Using the controlled cortical impact (CCI) model of TBI and a cocaine conditioned place preference (CPP) assay, which is a behavioral test that can be used to determine the rewarding effects of drugs of abuse, we recently observed a significant increase in the magnitude of the cocaine place preference shift of adult mice with a history of adolescent TBI (injury sustained at 6 weeks of age) when compared to the place preference shift of uninjured (naïve) controls21. Furthermore, Weil et al. found that juvenile mice sustaining a closed-head impact injury at 3 weeks of age displayed significantly greater alcohol place preference and increase alcohol consumption as adults (9 weeks of age) when tested using both an alcohol CPP assay and two-bottle choice alcohol self-administration, respectively22. Conversely, studies performed in animals with a history of adult TBI have produced conflicting results, sometimes showing an increase in the consumption of alcohol and sometimes not23–25. Importantly, these findings reflect data obtained from clinical studies and supports the use of animal models in order to answer questions that still remain, like 1) does early-life TBI interrupt developmental processes that increase the risk of SUDs, 2) are key neurocircuits that mediate drug abuse behavior more susceptible to damage in the maturing brain, or 3) does early-life TBI impair the ability to extinguish drug seeking behavior after a period of drug experimentation/binge drinking (a period which commonly occurs between the ages of 18–25 in human subjects)?
REPETITIVE INJURY AND TBI SEVERITY
As a group, adult TBI patients have a number of unique characteristics. One of these characteristics is a high prevalence of SUDs prior to injury5, 12. Another is a history of repeated TBIs12, 18, 26. Notably, these injuries seem to occur periodically throughout the life of a TBI patient, and as discussed above, a large portion of these injuries may be sustained in early-life7, 18, 26. Importantly, recent studies have identified a high percentage of repetitive TBI in patients struggling with substance abuse, suggesting that multiple TBIs may be an important risk factor for SUDs.
Using the Ohio State University Traumatic Brain Injury Identification (OSU-TBI-ID) Method, a questionnaire designed to elicit a lifetime history of TBI from current TBI patients, Corrigan et al. found that 20% of patients indexed in the TBI Model Systems National Database (a cohort of 4,464 patients with moderate or severe TBI) sustained at least one previous TBI before being added to the database (notably, 40% of these patients incurred an additional TBI before the age of 16)18. Again, using the OSU-TBI-ID, Olsen-Madden et al. determined that the number of TBIs sustained by veterans with a positive history of TBI, who entered a substance abuse treatment program, ranged from 1–12 TBIs (an average of 3.4 lifetime TBIs per person)7. Furthermore, Darke et al. reported that 37% of the heroin users in their cohort suffered multiple TBIs27, while Ramesh et al. saw that 35.7% of cocaine-dependent research volunteers also sustained two or more brain injuries20. Although there are no longitudinal, prospective clinical studies or preclinical reports that directly assess the effect of repetitive TBI on substance abuse, these results certainly suggest that some patients may enter into a cycle whereby early-life TBI begets substance abuse and substance abuse begets additional TBIs. Notably, preclinical studies are well suited to evaluate the effects of repetitive TBI on substance abuse behavior, as recent efforts have produced a variety of repetitive injury models, and would be less time consuming and costly than the studies necessary in human subjects.
In addition to repetitive injury, studies show that TBI severity may also be an important factor affecting the risk of SUDs post-TBI. In the clinic, TBIs are broadly categorized as mild, moderate, or severe by evaluating loss of consciousness, neurological impairment, post-traumatic amnesia, or the presence of structural abnormalities in the brain1. Mild TBI is the most common type of injury accounting for approximately 80% of reported TBIs, while moderate and severe injuries make up the remaining 20%18. Although the rate of moderate/severe TBI is similar between the general population and patients with comorbid SUDs and TBI7, 18, 20, 27, Corrigan et al. found that these injuries have the greatest effect on problematic alcohol consumption and illicit drug use when compared mild TBI, indicating that TBIs of greater severity may increase the risk of SUDs post-TBI18.
Similar results have been obtained in preclinical reports. Using the CCI model of experimental TBI, we tested a range of TBI severities on cocaine reward and found that only animals with a history of moderate CCI exhibited significant increases in cocaine place preference when compared to controls21. Interestingly, for each increase in the degree of TBI severity (sham to mild TBI to moderate TBI), animals displayed a stepwise enhancement in cocaine place preference, again indicating that the extent of brain injury may be an important factor affecting the risk of SUDs21.
Even though these studies suggest that moderate and severe TBIs have the greatest effect on substance abuse behavior, mild TBIs have also been shown to increase the risk of SUDs post-TBI. In a large cohort of United States airmen, Miller et al. reported increased risk for certain addiction-related disorders following mild TBI9. In a similar study, Heltemes et al. found that military service members with a combat-acquired mild TBI had a slightly higher proportion of alcohol use disorders compared to controls; however, after multivariate analysis, mild TBI was not associated with increased risk for alcohol abuse28. Although results of the multivariate analysis confirm the commonly held opinion that epidemiological data are very difficult to interpret because of shared risk factors in co-morbid TBI/SUD patients, these studies only consider the effects of a single TBI and do not assess the potential effects of a lifetime history of TBI in military service members, which was shown in the study by Olsen-Madden et al. to be very high (approximately 3 TBIs person)7. Therefore, these studies fail to account for the effects of repetitive brain injury and early-life TBI, potentially obscuring the effects of mild TBI on problematic use of drugs and alcohol when covariates are included in analyses.
Interestingly, conflicting results have also been observed in preclinical reports assessing the effect of mild TBI on substance abuse behavior. For instance, Mayeux et al. noted a significant increase in alcohol self-administration in rats following mild lateral fluid percussion injury (LFPI)25, and Weil et al. observed significant increases in both alcohol self-administration and alcohol place preference in mice with a history of mild, closed-head impact injury22. However, Lim et al. found that mice exposed to mild blast overpressure exhibited no change in voluntary ethanol intake when compared to controls23. Importantly, although each of these studies characterizes the severity of experimental TBI as mild, the conflicting results obtained in each study may actually be a function of the different experimental models of TBI employed (i.e. LFPI, closed-head impact, blast), as different models recapitulate different types of brain injury (Table 1). Perhaps then, the effects of mild TBI on substance abuse behavior are less associated with the number of TBIs or broad, arbitrary categorization of TBI severity, and more an effect of how different types of injury affect specific brain structures.
Table 1. Synopsis of studies that test the effect of experimental TBI on substance abuse behavior using preclinical models.
This table provides a summary of publications that test the effect of experimental TBI on substance abuse behavior using preclinical animal models. The table identifies TBI-related mechanisms of injury, experimental models of TBI used to study the different mechanisms of injury, brain regions affected by experimental models of TBI, assays used to test substance abuse behavior after experimental TBI, and drugs of abuse evaluated in current publications. Citations for currently published reports have also been provided. As shown, many more studies will be required to fully evaluate the effect of experimental TBI on substance abuse behavior using preclinical models.
| Mechanism of injury | Experimental model of TBI available | Brain region affected by injury | Behavioral assay for drug abuse behavior | Drug of abuse studied | Study investigating SUD post TBI |
|---|---|---|---|---|---|
| Coup Contrecoup | Acceleration/Deceleration | Cortex, Striatum Midbrain, Hypothalamus | n/a | n/a | |
| Controlled cortical impact (CCI) | Cortex/PFC, Striatum/NAc, Substantia nigra/VTA | CPP | Cocaine | Merkel et al. (2016) | |
| Weight Drop | Cortex, Striatum | n/a | |||
| Closed head injury | Cortex/PFC, Striatum/NAc | CPP, DID, LRR, 2BC, QD, SD | Alcohol | Weil et al. (2016) Lowing et al. (2014) | |
| Cortex | Alcohol | ||||
| Diffuse Axonal Injury (DAI) | Lateral fluid percussion injury (LFPI) | Self-administration | Mayeux et al. (2015) | ||
| Blast Exposure | Blast-induced neurotrauma (BINT) | PFC, NAc | 2BC, DE, QD | Alcohol | Lim et al. (2015) |
| Penetrating | Stab wound | Cortex, Striatum | n/a | n/a |
n/a- indicates studies are not available
PFC = Prefrontal Cortex, NAc= Nucleus Accumbens, VTA= Ventral tegmental area, CPP = Conditioned place preference, DID = Drinking in the dark, LRR =Loss of righting reflex, 2BC = 2 bottle choice, DE = Deprivation effect, QD =Quinine discrimination, SD = Saccharine discriminiation
NEUROCIRCUITS
Studies indicate that TBI affects many brain regions implicated in the development and maintenance of SUDs. These regions include the prefrontal cortex (PFC), the nucleus accumbens (NAc), and the ventral tegmental area (VTA). Together, these brain structures comprise a series of neurocircuits that mediate substance abuse behavior29, 30. Importantly, studies suggest that each of these brain regions may be particularly vulnerable to different types of injury.
For instance, one of the mechanisms underlying TBI pathology is coup contrecoup injury (Table 1), which occurs due to movement of the brain inside of the skull. This type of injury primarily affects the frontal, temporal, and occipital cortices, as these brain regions may contact the cranial vault31. In fact, a study by Levin et al. revealed that the majority of radiological abnormalities (i.e. contusions, hematomas, etc.) observed using routine neuroimaging modalities are detected in the frontal and temporal lobes, suggesting that anterior regions of the brain like the PFC are more sensitive to injury in cases of moderate and severe TBI32. Furthermore, using functional magnetic resonance imaging (fMRI), a recent study by Eierud et al. demonstrated that the PFC is vulnerable even in cases of mild TBI31. Notably, focal (i.e. penetrating) injuries to the PFC are associated with important behavioral deficits, as studies by Cristofori et al. and McDonald et al. have shown that damage to the PFC produces executive dysfunction and heightened impulsivity, two traits commonly implicated in drug and alcohol dependence33, 34. Therefore, regardless of TBI severity, damage to the PFC may be an important factor affecting the risk of SUDs in patients with a history of TBI.
Although the most ubiquitous preclinical models of experimental TBI can be adapted to produce frontal lobe injury35, recent reports have employed the conventional open-head CCI and closed-head impact injury models to study the effects of TBI on substance abuse behavior. Importantly, these models recapitulate the cortical damage associated with coup contrecoup injury, and thus may be useful in understanding the cellular and molecular changes that occur in the PFC after TBI.
In our recent studies, we injured the right parietal somatosensory cortex using the open-head CCI model of experimental TBI. Our results show that this model produces chronic glial activation at the site of injury and upregulation of numerous immune response genes in the cortex21. Furthermore, after extracting the ipsilateral PFC (distal to the site of injury), we probed for the expression of select immune response genes finding that some targets (i.e. chemokines and interleukin-1β) were significantly overexpressed even in this distal cortical region (unpublished data). Moreover, by inducing experimental TBI at approximately the same cortical level using a closed-head impact injury model, Weil et al. show significant axonal degeneration in the forebrain and significant down-regulation of brain derived neurotrophic factor (BDNF), specifically in the PFC22. Similarly, Lowing et al. used a closed-head injury model to induce experimental TBI at the midline above the parietal cortex finding that this type of injury also causes blood brain barrier dysfunction, astroglial activation, and cell death at the site of injury (however, the report did not assess the effects of parietal midline injury directly in the PFC)24. Together, these studies show that TBI can lead to a number of cellular and molecular changes both at the site of impact and in distal cortical regions like the PFC.
Another mechanism underlying TBI pathology is the phenomenon of diffuse axonal injury (DAI) (Table 1), which occurs due to linear and rotational forces that stretch and shear axon bundles in the brain36. DAI primarily affects the white matter tracts and can lead to atrophy/reduced volume in a number of brain regions including the NAc, a subcortical region that surrounds the anterior commissure (a large white matter tract that connects the temporal lobes)37–40. Importantly, using high-resolution neuroimaging modalities, Shah et al. observed significant volumetric reductions and decreased fractional anisotropy (FA; an indicator of white matter integrity) in the NAc following severe TBI39. Furthermore, a recent study by Alhilali et al. also saw reduced FA in the NAc of mild TBI patients, indicating that white matter injury may occur in the NAc, again regardless of TBI severity40.
Although LFPI is the premiere preclinical model of DAI, the only report to use this model while assessing the effects of experimental TBI on substance abuse behavior (Mayeux et al.) focused on the characterization of cortical injuries (i.e. astrocyte and microglial activation, neurodegeneration)25 rather than regions of the brain with large white matter tracts like the NAc. This is unfortunate considering the importance of the NAc in the neurobiology of SUDs29, 30. However, additional studies have evaluated the effects of experimental TBI on the NAc using many other preclinical models including open-head CCI, closed-head impact injury, and blast-induced neurotrauma (BINT).
In our recent studies, we extensively characterized the immune response and change in glial activation status that occurs in the NAc following open-head CCI. Using a passive clarity technique and multi-photon microscopy, we observed a significant increase in GFAP intensity and the appearance of hypertrophic astrocytic processes (reactive astrocytes) in the NAc of animals sustaining moderate CCI41. Likewise, we saw significant increases in IBA-1 immunostaining and microglial volume and the appearance of amoeboid-shaped microglia in the NAc following moderate CCI, again consistent with glial reactivity41. Furthermore, using a large immune response panel, we found that 66 of 92 target genes were significantly upregulated in the NAc 2 weeks after moderate CCI, suggesting that a number of cytokines, chemokines, adhesion molecules, stress response genes, etc. may be overexpressed chronically in the NAc following TBI21.
Similarly, using a model of closed-head impact injury, Lowing et al. observed reactive astrocytes in the NAc at 3 and 7 days post-TBI24. However, unlike the chronic response observed in our studies, astrocyte reactivity in the closed-head injury model had resolved by day1524. This discrepancy is most likely an effect of the two different models of experimental TBI (open-head CCI vs. closed-head impact injury) employed in the two different studies. Interestingly, regardless of the time point post-TBI, the report by Lowing et al. also noted reactive astrocytes in the anterior commissure, suggesting that a protracted cellular response may occur in this region of the brain even in a closed-head injury24. Moreover, using cresyl violet staining, Lowing et al. saw no change in cellularity in the NAc at any time point post-TBI, suggesting that at least in a closed-head injury model of TBI, the medium spiny neurons (MSNs) of the NAc remain intact24.
Although Lowing et al. observed no cell loss in the NAc, a study by Sajja et al. revealed significant increases in Bax and caspase-3, markers of cellular apoptosis, in the NAc following experimental blast exposure (Table 1), a.k.a. BINT42. This type of TBI occurs when shockwave forces from an explosion pass through the brain. In addition to affecting the NAc, Budde et al. found that experimental BINT significantly reduced FA, increased the expression of apoptotic markers, and lead to the appearance of reactive astrocytes in the PFC43. Therefore, BINT may substantially damage two critical brain regions that mediate drug-seeking behavior. Notably, in the study by Weil et al., a crucial finding regarding the function of these two brain regions was uncovered that may explain how TBI affects the risk of SUDs post-TBI.
Again, using a closed-head injury model, Weil et al. observed constitutive expression of c-Fos (an immediate early response gene product) in a subdivision of the NAc called the accumbens shell, but only in animals that exhibited increased alcohol self-administration and conditioned place preference following TBI22. Furthermore, Weil et al. failed to see increased c-Fos expression in another subdivision of the NAc called the accumbens core22. These findings are important because one of the main theories in addiction biology proposes that activation of the NAc shell facilitates drug-seeking behavior, while activation of the NAc core inhibits drug-seeking behavior30. Importantly, activation of either subdivision in the NAc is controlled by presynaptic glutamatergic neurons that originate in the PFC and synapse onto MSNs in NAc forming the corticolimbic circuits30. Therefore, TBI may increase the risk of SUDs by altering the function of the corticolimbic circuits in a manner that either promotes activation of the NAc shell or impairs activation of the NAc core (or potentially both as presented in Weil et al.).
Importantly, the PFC and NAc are both innervated by the VTA, a third key brain region heavily implicated in substance abuse behavior. The VTA is comprised of dopaminergic fibers that project to either the PFC or the NAc forming two neurocircuits globally affected by drugs of abuse: the mesocortical and mesolimbic neurocircuits, respectively29. Although dopaminergic deficits have been widely studied following TBI, few studies have expressly assessed the effects of TBI in the VTA. In one study, Shin et al. performed a gene microarray on the VTA/substantia nigra finding that only 25 transcripts were differentially expressed when compared to controls one month after open-head CCI44. These transcripts were from the following 3 categories: ribosomal proteins, mitochondrial function, and non-inflammatory immune factors. Furthermore, using the same experimental model of TBI, we investigated glial activation status in the VTA also at one month post-injury. Results of these studies show significant astrocyte reactivity in the VTA following moderate CCI, but no change in microglial activation status21.
As astrocytes are known to play a major role in regulating synapses, these changes may have profound effects on the tonic inhibition of VTA neurons and in turn affect dopamine concentrations in both the PFC and NAc following TBI. Notably, dopaminergic fluctuations in the PFC also affect glutamate homeostasis in the NAc via the corticolimbic circuits30. Therefore, damage to any one of these brain regions (i.e. PFC, NAc, or VTA) as a result of TBI may disrupt the function of the principle neuronal networks that mediate drug-seeking behavior. This is especially true in the NAc where glutamate projections from the PFC and dopaminergic projections from the VTA affect the activation of MSNs, which as discussed above, may facilitate or inhibit drug-seeking behavior in a region-specific manner. Interestingly, although not explicitly tested in the NAc, previous reports have shown that TBI affects both dopaminergic and glutamatergic transmission similar to chronic substance abuse, suggesting that another factor affecting the increased risk of SUDs post-TBI may be the “priming” of neurotransmitter systems that pathologically activate/inhibit MSNs in the NAc.
NEUROTRANSMITTER SYSTEMS
Acutely, drugs of abuse increase extracellular dopamine levels in the PFC and NAc45, 46. However, studies focusing on drug withdrawal and relapse have shown that dopaminergic transmission can actually be decreased following abstinence from repeated drug administration29, 47, 48. This deficit produces a dysfunctional reward circuit that facilitates drug relapse49. Notably, dopaminergic deficits also occur after brain injury.
Using single-photon emission tomography, Donnemiller et al. observed impaired dopaminergic transmission in the striatum of TBI patients several months after injury50. Importantly, the striatum is a large subcortical region in the forebrain that consists of many brain nuclei including the ventral striatum, or NAc. Furthermore, preclinical studies have expanded upon these data and shown that there may actually be two phases of dopaminergic adaptation that occur following TBI. Using an acceleration-deceleration model of experimental TBI (Table 1), Huger and Patrick report an increase in tissue dopamine levels in the cortex, striatum, midbrain, and hypothalamus within 1 hour of injury51. However, after this initial increase, data suggests that dopamine levels fall in multiple brain regions. Using fast scan cyclic voltammetry, Wagner et al. observed significant reductions in evoked dopamine release and Vmax, a measure of synaptic dopamine clearance through the dopamine reuptake transporter (DAT), in the striatum two weeks after open-head CCI52. Furthermore, consistent with the falloff in Vmax, Wagner et al. also saw significant downregulation of striatal DAT expression52. In addition, studies by Yan et al. show that tyrosine hydroxylase (TH), the rate limiting enzyme in the process of dopamine synthesis, is significantly increased in the frontal cortex, striatum, and substantia nigra nearly one month after open-head CCI53, 54. Together, the downregulation of DAT and increase in TH expression may represent a compensatory adaptation to boost extracellular dopamine concentrations in these brain regions following TBI. Importantly, although these studies have observed dopaminergic fluctuations in many brain regions, no preclinical studies have expressly evaluated dopaminergic transmission in the NAc following TBI where dopaminergic deficiency is known to play a role in regulating substance abuse behavior49.
Interestingly, despite the falloff in dopaminergic transmission, Lowing et al. found that phosphorylated dopamine- and cAMP-regulated neuronal phosphoprotein, 32kDa (pDARPP-32), a downstream mediator of the dopamine D1 receptor, was significantly elevated in the striatum two weeks after closed-head impact injury24. Although increases in pDARPP-32 may seem paradoxical given the evidence of dopaminergic depression following TBI, DARPP-32 phosphorylation depends on both dopaminergic and glutamatergic tone24. Therefore, increases in pDARPP-32 expression may actually be an effect of glutamatergic signaling in the striatum following TBI. Importantly, glutamate signaling and DARPP-32 phosphorylation are associated with neuroplastic adaptions in the NAc that mediate drug-seeking behavior30, 55.
Using enzyme-based microelectrode arrays, Hinzman et al. observed significant increases in extracellular glutamate in the striatum 2 days after midline fluid percussion injury56. Furthermore, Hinzman et al. found that the increase in extracellular glutamate levels was a function of decreased cellular glutamate reuptake through excitatory amino acid transporters, including glutamate transporter subtype 1 (GLT-1), the principle means by which glutamate is cleared from the extracellular compartment, and the glutamate aspartate transporter (GLAST), rather than an increase in neuronal release56. Although qPCR analysis by Hinzman et al. revealed a non-significant reduction in GLT-1 and GLAST expression at 2 days post-TBI56, Goodrich et al. observed significant downregulation of GLT-1 expression in the cortex 7 days after LFPI57. Notably, these findings are similar to changes that occur during repeated drug use.
Glutamate signaling in the reward circuitry is disrupted by both acute and chronic exposure to drugs of abuse and contributes to drug craving/reinforcement and triggers relapse to drug-seeking behavior58, 59. One glutamate pathway that is critical to drug craving and relapse is the corticolimbic circuit that connects the prelimbic cortex to the NAc core60. Prior studies have shown that acute drug administration activates glutamatergic neurons in the PFC leading to excessive glutamate release in the NAc61. This spike in glutamate release disrupts glutamate homeostasis in the NAc and is counterbalanced in part by a reduction in GLT-1 expression after repeated drug administration30. Therefore, the increase in extracellular glutamate and downregulation of GLT-1 observed after TBI may evoke neuroplastic adaptions that essentially prime regions of the brain for drug-seeking behavior. Furthermore, the effects of DARPP-32 phosphorylation on glutamatergic signaling may preclude the possibility of counter-adaptation, as pDARPP-32 is associated with neuroplastic deficiencies in the NAc24, 55.
Furthermore, in rat models, abstinence from chronic exposure to psychostimulants, such as cocaine and methamphetamine, causes a decrease in corticolimbic activity that leads to decreased extracellular glutamate levels in the NAc core30. As a result, the loss of extrasynaptic glutamatergic tone in the NAc core is a key neurochemical feature of chronic drug exposure and thought to facilitate drug craving and mood changes that trigger relapse. Interestingly, in rodent self-administration assays that model drug relapse, reinstatement of drug-seeking behavior, which is produced by presentation of a drug-associated cue (i.e., a cue previously associated with cocaine, nicotine, alcohol, or heroin), is facilitated by an increase in glutamate release from corticolimbic terminals that leads to increased extracellular glutamate in the NAc core30. Therefore, increased glutamate release from prelimbic cortex terminals, and subsequent elevation in extracellular glutamate in the NAc core, may be a shared consequence of chronic drug exposure and TBI, making this pathway an attractive target for developing therapies that treat not only drug addiction and TBI alone, but also the comorbid occurrence of both TBI and SUD.
Collectively, these results show that TBI affects two of the primary neurotransmitter systems implicated in substance abuse behavior, and moreover, that the effects of TBI are similar to changes that occur during repeated drug use. Importantly, although these effects have been observed in the cortex, striatum, and substantia nigra, explicitly testing the effect of experimental TBI on these neurotransmitter systems in the PFC, NAc, and VTA using preclinical models is necessary to further understand how these changes may affect the risk of SUDs post-TBI.
NEUROINFLAMMATION
In addition to affecting key neurocircuits and neurotransmitter systems implicated in the development and maintenance of SUDs, reports show that TBI also produces a chronic neuroinflammatory response in the cortex, NAc, and white matter tracts. Notably, although neurocircuits and neurotransmitter systems may comprise the machinery that enables substance abuse behaviors, recent reports suggest that the expression of innate immune factors (i.e. toll-like receptors62–64, adapter proteins65, chemokines41, 66, 67, interleukins68, etc.) may actually be the driving force that fuels the neurobiology of addiction69, 70.
Together with the primary mechanisms of injury (Table 1) that can directly introduce damage to brain structures like the PFC, NAc, and VTA, neuroinflammation (one of the secondary mechanisms of injury) may also chronically affect neural function post-TBI in areas of the reward pathway. Using post-mortem brain tissue and immunohistochemistry, Johnson et al. observed reactive microglia accumulation throughout the corpus callosum (CC, a large white matter tract that connects the cerebral hemispheres) in samples obtained from patients with a history of moderate/severe TBI71. Surprisingly, reactive microglia could still be seen in the CC up to 18 years after a single TBI. Notably, microglial accumulation coincided with white matter degeneration in the CC71. As reports by Shah et al. and Alhilali et al. also observed white matter injury in the NAc of TBI patients39, 40, similar immune cell activation and accumulation may occur in all white matter tracts including the anterior commissure, which runs directly through the NAc. In fact, as mentioned, our recent studies show that reactive microglia and chronic neuroinflammation occur not only at the cortical site of injury, but also in distal brain regions like the PFC and NAc (Figure 2)21, 41.
Figure 2. Glial reactivity and immune cell responses in the nucleus accumbens (NAc) post-injury using the controlled cortical impact (CCI) model of experimental TBI.
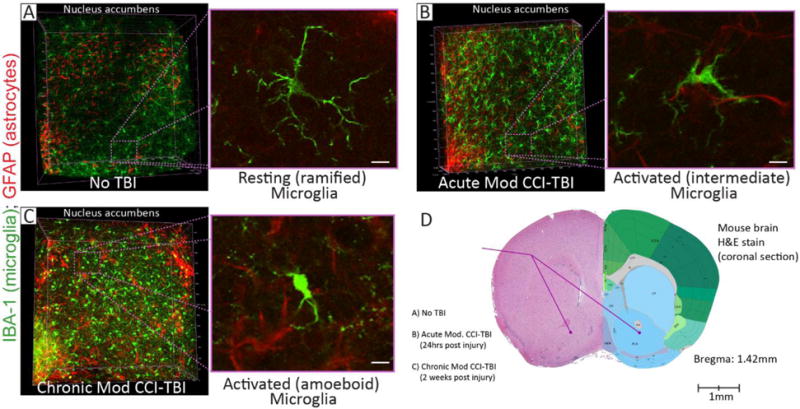
Adolescent (six-week old) male C57BL/6 mice incurred moderate (Mod) CCI-TBI with the following impact parameters: velocity = 4.5m/s, depth = 2mm, dwell time = 500ms. Brains were then harvested at 24 hours (Acute) and 2 weeks (Chronic) post-injury. Brains from naïve (No TBI) mice were used for control. Next, the ipsilateral NAc was excised from 2mm thick coronal brain segments using a 1.25mm microdissection tool and processed using a passive CLARITY technique. Cleared NAc tissue was then immunolabeled for IBA-1 (microglia) and GFAP (astrocytes). Glial activation status in the NAc was then assessed using multi-photon microscopy. (A-C) Left-hand panels: Representative three-dimensional volumetric renderings (approximately 300μm thick) show IBA-1 (green) and GFAP (red) immunostaining in the NAc for No TBI (A), Acute, (B), and Chronic (C) time points post-injury. Compared to the No TBI control, Mod CCI-TBI shows an increase in the intensity of both GFAP and IBA-1 staining, along with an increase in the number of glial cells, at both acute and chronic time points. Furthermore, cellular morphology in the NAc is drastically changed following Mod CCI-TBI. Note, region selected for analysis of microglial morphology outlined in purple, dotted lines. Right-hand panels: High-resolution images of microglial activation status in the NAc. No TBI controls reveal resting (ramified) microglia (A). However, Mod CCI-TBI shows activated (intermediate) microglia at 24 hours (B) and activated (amoeboid) microglia at 2 weeks (C) post-injury. Importantly, the effect of CCI-TBI on glial activation status and the increased number of microglia observed in these studies post-injury might affect the function of neurocircuits that mediate substance abuse behavior (i.e. the corticolimbic and mesolimbic pathways) post-TBI. (D) Image provided to show the subcortical region where the NAc was extracted from each sample used in multi-photon microscopy (A–C). Left hemisphere: Coronal section (Bregma: 1.42mm) of a mouse brain (No TBI) stained with hematoxylin & eosin (H&E). Right hemisphere: A drawing of neuroanatomical regions observed in the mouse brain at Bregma: 1.42mm (image obtained from www.alleninstitute.org). For a more comprehensive presentation of these studies, please refer to the following publication41.
Additionally, we recently reported on the effect of neuroimmune activation as a mediator of substance abuse behavior by administering a potent anti-inflammatory therapy to animals beginning 24 hours after open-head CCI41. As previous studies have shown that immunosuppressive agents can modulate the severity of substance abuse behavior72, we wanted to determine whether neuroinflammation plays an important role in enhancing the rewarding effects of cocaine following adolescent brain injury. Our results show that administration of a synthetic, anti-inflammatory glucocorticoid receptor agonist (dexamethasone) significantly attenuates the enhanced rewarding effects of cocaine and returns drug-seeking behavior to control levels41. Furthermore, we show that dexamethasone therapy significantly reduced the expression of select immune response genes (i.e. CD163, CCL2, and I-CAM) in the NAc to control levels at two weeks post-TBI, suggesting that mononuclear phagocytes of the innate immune system (i.e. monocytes, perivascular macrophages, microglia) may play an important role in enhancing the rewarding effect of cocaine following TBI41. Therefore, these results show that TBI-induced neuroinflammation may increase the risk of SUDs, and moreover, that a brief regimen of anti-inflammatory therapeutics may help to attenuate this increased risk.
SEX DIFFERENCES
Another factor potentially affecting increased risk of SUDs post-TBI includes sex differences. Surveillance reports suggest that males are nearly 2 times more likely to sustain a brain injury than females16. Therefore, many clinical studies are bias towards the effect of TBI on males, and preclinical studies typically only utilize male rodents to study brain injury. However, as females become more active in contact sports and military combat, sex differences are emerging as an important risk factor to consider when studying outcomes following TBI.
Epidemiological studies indicate that the characteristics of female TBI patients are similar to males with regards to cause/mechanism of injury (i.e. falls, motor vehicle accidents, etc.), age at time of injury, distribution of TBIs by severity, and rate of repetitive TBI1, 16. Although these TBI-associated characteristics follow similar trends in males and females, substance abuse research shows that sex differences may play an important role in drug and alcohol addiction, as females exhibit different pharmacokinetics and sensitivity to drugs of abuse, are more likely to abuse different classes of drugs, and have greater rates of seeking out and succeeding in treatment for SUDs than males3, 73, 74. Consequently, understanding how sex differences affect the response to drugs of abuse following TBI may help to accurately assess risk of SUDs in males versus females.
Notably, Weil et al. is the only study to evaluate sex differences using both a preclinical model of experimental TBI and behavioral assays of substance abuse. Weil et al. found that specifically female mice with a history of juvenile closed-head impact injury displayed increased alcohol consumption and alcohol-induced CPP as adults when compared to males, suggesting that females with a history of early-life TBI may be at greater risk for alcohol use disorders in adulthood22. Furthermore, these behavioral changes coincided with changes in cellular activation in the NAc following TBI. As described above, only female mice that exhibited increased alcohol consumption and CPP showed positive c-fos immunolabeling in the NAc shell and c-fos negativity in the NAc core22. Although no other studies directly test the effect of sex differences on the development of SUD following brain injury, sex-specific changes have been observed in many preclinical studies investigating the neural substrates implicated in substance abuse behavior following TBI including dopaminergic transmission, DAT expression, glial activation, and neuroinflammation75, 76. While it is clear that more studies are required, these results suggest that sex differences may also play an important role in assessing the risk of SUDs post-TBI.
CONCLUSIONS AND FUTURE DIRECTIONS
In summary, recent reports have identified multiple factors that affect the risk of SUDs post-TBI. These factors include a history of early-life TBI, repetitive injury and TBIs of increasing severity (i.e. moderate/severe TBI), and sex differences. Furthermore, TBI has been shown to affect many of the neural substrates that mediate substance abuse behavior. These include: 1) neurocircuits that connect specific brain regions like the PFC, NAc, and VTA, 2) neurotransmitter systems, like dopamine and glutamate, and 3) immune cell activation and the expression of innate immune response genes in the brain. Importantly, not only does TBI affect these substrates, but as discussed, it also seems to affect them in a manner similar to chronic substance abuse. Therefore, in order to validate these hypotheses, future investigations explicitly testing how TBI affects neurotransmitter systems and influences neuroplastic adaptations specifically in key brain regions like the PFC, NAc, and VTA (both with and without exposure to drugs of abuse) may be useful for identifying the molecular mechanisms that increase the risk of SUDs following TBI.
Notably, a variety of preclinical technologies are available to achieve these goals. Intracerebral microdialysis and whole-cell patch clamp/in vivo electrophysiology are equipped to determine extracellular dopamine and glutamate levels and long-term potentiation/depression in mesolimbic, mesocortical, and corticolimbic neurocircuits following experimental TBI. These assays can be enhanced with the use of small molecule reagents that can be used to pinpoint the neural substrates that mediate these effects. Furthermore, these assays can be combined with a number of behavioral assays to assess which factors play the greatest role in regulating substance abuse behavior following TBI. For instance, our recent report suggests that suppressing neuroinflammation following TBI can attenuate enhancement of the rewarding effects of cocaine in animals with a history of moderate, adolescent CCI injury41. However, these results can be refined by dissecting the systemic immune response to TBI from the parenchymal immune response by delivering anti-inflammatory reagents directly into brain structures like the NAc through the use of intracerebral injections and then testing animals for the development and expression of cocaine CPP.
In addition, many behavioral assays that assess important traits relevant to the study of SUDs, such as motivation to obtain drug reward, impulsivity and executive dysfunction, severity of drug withdrawal, etc. have yet to be tested in models of experimental TBI. In fact, most preclinical studies have utilized either CPP or voluntary ethanol consumption, yet no studies have utilized the gold standard intravenous self-administration model of substance abuse for drugs such as nicotine, psychostimulants, opiates, etc. These assays would allow for better assessment of substance abuse vulnerability (e.g., acquisition of self-administration) in animals sustaining experimental TBI. Moreover, as seen in Table 1, only two drugs of abuse (alcohol and cocaine) have been evaluated using preclinical models. Therefore, expanding upon these studies to include other drugs of abuse may be especially important to safeguard patient health, as recent studies have promoted the idea that some drugs of abuse (i.e. methamphetamine77, methylenedioxymethamphetamine78, 79, and alcohol80, 81) may possess therapeutic utility in cases of TBI, while other drugs of abuse (opiates, hypnotics/sedatives) may be prescribed to patients for pain or sleep disturbances following brain injury. Although clinical reports have helped identify patient populations at increased risk for SUDs post-TBI and popularized the importance of substance abuse/psychiatric referral and follow-up after brain injury, future preclinical studies may help to characterize the molecular mechanisms that drive substance abuse behavior post-TBI. Furthermore, these studies may be used to validate novel therapeutics to treat patients with co-morbid TBI and SUDs, or avoid medical mishaps related to the use of potentially addictive prescription drugs following TBI.
HIGHLIGHTS.
Evaluation of data identifying populations at increased risk for SUDs post-TBI
Evidence of TBI-related neuropathology in key brain regions implicated in SUDs
Discussion of mechanisms that may underlie increased risk of SUDs following TBI
Focus on the utilization of preclinical models to study links between TBI and SUDs
Acknowledgments
This work was supported by National Institutes of Health/National Institute on Drug Abuse (NIH/NIDA) R01 DA039139-02 (SMR), T32 DA007237 (LAC), and P30 DA013429-16 (SHR), NIH/National Institute of Neurological Disorders and Stroke (NINDS) R01 NS086570-01 (SHR), and The Shriners Hospitals for Children 85110-PHI-14 (SHR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prevention, C.f.D.C.a. Report to Congress on Traumatic Brain Injury in the United States: Epidemiology and Rehabilitation. National Center for Injury Prevention and Control; Division of Unintentional Injury Prevention; Atlanta, GA: 2015. [Google Scholar]
- 2.Rogers JM, Read CA. Psychiatric comorbidity following traumatic brain injury. Brain injury. 2007;21:1321–1333. doi: 10.1080/02699050701765700. [DOI] [PubMed] [Google Scholar]
- 3.Quality, C.f.B.H.S.a. (National Survey on Drug Use and Health (HSS Publication No. SMA 15-4927, NSDUH Series H-50).Behavioral health trands in the United States: Results from the 2014. 2015 [Google Scholar]
- 4.Whelan-Goodinson R, Ponsford J, Johnston L, Grant F. Psychiatric disorders following traumatic brain injury: their nature and frequency. The Journal of head trauma rehabilitation. 2009;24:324–332. doi: 10.1097/HTR.0b013e3181a712aa. [DOI] [PubMed] [Google Scholar]
- 5.Parry-Jones BL, Vaughan FL, Miles Cox W. Traumatic brain injury and substance misuse: a systematic review of prevalence and outcomes research (1994–2004) Neuropsychological rehabilitation. 2006;16:537–560. doi: 10.1080/09602010500231875. [DOI] [PubMed] [Google Scholar]
- 6.Ilie G, Mann RE, Hamilton H, Adlaf EM, Boak A, Asbridge M, Rehm J, Cusimano MD. Substance Use and Related Harms Among Adolescents With and Without Traumatic Brain Injury. The Journal of head trauma rehabilitation. 2015;30:293–301. doi: 10.1097/HTR.0000000000000101. [DOI] [PubMed] [Google Scholar]
- 7.Olson-Madden JH, Brenner L, Harwood JE, Emrick CD, Corrigan JD, Thompson C. Traumatic brain injury and psychiatric diagnoses in veterans seeking outpatient substance abuse treatment. The Journal of head trauma rehabilitation. 2010;25:470–479. doi: 10.1097/HTR.0b013e3181d717a7. [DOI] [PubMed] [Google Scholar]
- 8.West SL. Substance use among persons with traumatic brain injury: a review. NeuroRehabilitation. 2011;29:1–8. doi: 10.3233/NRE-2011-0671. [DOI] [PubMed] [Google Scholar]
- 9.Miller SC, Baktash SH, Webb TS, Whitehead CR, Maynard C, Wells TS, Otte CN, Gore RK. Risk for addiction-related disorders following mild traumatic brain injury in a large cohort of active-duty U.S. airmen. The American journal of psychiatry. 2013;170:383–390. doi: 10.1176/appi.ajp.2012.12010126. [DOI] [PubMed] [Google Scholar]
- 10.Farinde A. An examination of co-occurring conditions and management of psychotropic medication use in soldiers with traumatic brain injury. J Trauma Nurs. 2014;21:153–157. doi: 10.1097/JTN.0000000000000058. quiz 158–159. [DOI] [PubMed] [Google Scholar]
- 11.Ilie G, Adlaf EM, Mann RE, Ialomiteanu A, Hamilton H, Rehm J, Asbridge M, Cusimano MD. Associations between a History of Traumatic Brain Injuries and Current Cigarette Smoking, Substance Use, and Elevated Psychological Distress in a Population Sample of Canadian Adults. Journal of neurotrauma. 2015;32:1130–1134. doi: 10.1089/neu.2014.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corrigan JD. Substance abuse as a mediating factor in outcome from traumatic brain injury. Archives of physical medicine and rehabilitation. 1995;76:302–309. doi: 10.1016/s0003-9993(95)80654-7. [DOI] [PubMed] [Google Scholar]
- 13.Fazel S, Wolf A, Pillas D, Lichtenstein P, Langstrom N. Suicide, fatal injuries, and other causes of premature mortality in patients with traumatic brain injury: a 41-year Swedish population study. JAMA Psychiatry. 2014;71:326–333. doi: 10.1001/jamapsychiatry.2013.3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olson-Madden JH, Forster JE, Huggins J, Schneider A. Psychiatric diagnoses, mental health utilization, high-risk behaviors, and self-directed violence among veterans with comorbid history of traumatic brain injury and substance use disorders. The Journal of head trauma rehabilitation. 2012;27:370–378. doi: 10.1097/HTR.0b013e318268d496. [DOI] [PubMed] [Google Scholar]
- 15.Unsworth DJ, Mathias JL. Traumatic brain injury and alcohol/substance abuse: A Bayesian meta-analysis comparing the outcomes of people with and without a history of abuse. Journal of clinical and experimental neuropsychology. 2016:1–16. doi: 10.1080/13803395.2016.1248812. [DOI] [PubMed] [Google Scholar]
- 16.Faul M, Xu L, Wald MM, Coronado VG. Traumatic Brain Injury in the United States: Emergency Department Visits, Hospitalizations and Deaths 2002–2006. National Center for Injury Prevention and Control, Centers Disease Control; Atlanta, GA: 2010. [Google Scholar]
- 17.Administration, S.A.M.H.S. Results from the 2013 National Survey on Drug Use and Health: Summary of National Findings. Administration, S.A.a.M.H.S.; Rockville, MD: 2014. (NSDUH Series H-48, HHS Publication No. (SMA) 14-4863). [Google Scholar]
- 18.Corrigan JD, Bogner J, Mellick D, Bushnik T, Dams-O’Connor K, Hammond FM, Hart T, Kolakowsky-Hayner S. Prior history of traumatic brain injury among persons in the Traumatic Brain Injury Model Systems National Database. Archives of physical medicine and rehabilitation. 2013;94:1940–1950. doi: 10.1016/j.apmr.2013.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fishbein D, Dariotis JK, Ferguson PL, Pickelsimer EE. Relationships Between Traumatic Brain Injury and Illicit Drug Use and Their Association With Aggression in Inmates. Int J Offender Ther Comp Criminol. 2016;60:575–597. doi: 10.1177/0306624X14554778. [DOI] [PubMed] [Google Scholar]
- 20.Ramesh D, Keyser-Marcus LA, Ma L, Schmitz JM, Lane SD, Marwitz JH, Kreutzer JS, Moeller FG. Prevalence of traumatic brain injury in cocaine-dependent research volunteers. The American journal on addictions / American Academy of Psychiatrists in Alcoholism and Addictions. 2015;24:341–347. doi: 10.1111/ajad.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Merkel SF, Razmpour R, Lutton EM, Tallarida CS, Heldt NA, Cannella LA, Persidsky Y, Rawls SM, Ramirez SH. Adolescent Traumatic Brain Injury Induces Chronic Mesolimbic Neuroinflammation with Concurrent Enhancement in the Rewarding Effects of Cocaine in Mice during Adulthood. Journal of neurotrauma. 2016 doi: 10.1089/neu.2015.4275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weil ZM, Karelina K, Gaier KR, Corrigan TE, Corrigan JD. Juvenile Traumatic Brain Injury Increases Alcohol Consumption and Reward in Female Mice. Journal of neurotrauma. 2016;33:895–903. doi: 10.1089/neu.2015.3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim YW, Meyer NP, Shah AS, Budde MD, Stemper BD, Olsen CM. Voluntary Alcohol Intake following Blast Exposure in a Rat Model of Mild Traumatic Brain Injury. PloS one. 2015;10:e0125130. doi: 10.1371/journal.pone.0125130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowing JL, Susick LL, Caruso JP, Provenzano AM, Raghupathi R, Conti AC. Experimental traumatic brain injury alters ethanol consumption and sensitivity. Journal of neurotrauma. 2014;31:1700–1710. doi: 10.1089/neu.2013.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayeux JP, Teng SX, Katz PS, Gilpin NW, Molina PE. Traumatic brain injury induces neuroinflammation and neuronal degeneration that is associated with escalated alcohol self-administration in rats. Behavioural brain research. 2015;279:22–30. doi: 10.1016/j.bbr.2014.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Corrigan JD, Bogner J, Holloman C. Lifetime history of traumatic brain injury among persons with substance use disorders. Brain injury. 2012;26:139–150. doi: 10.3109/02699052.2011.648705. [DOI] [PubMed] [Google Scholar]
- 27.Darke S, McDonald S, Kaye S, Torok M. Prevalence and correlates of traumatic brain injury amongst heroin users. Addiction research and theory. 2012;20:522–528. [Google Scholar]
- 28.Heltemes KJ, Dougherty AL, MacGregor AJ, Galarneau MR. Alcohol abuse disorders among U.S. service members with mild traumatic brain injury. Mil Med. 2011;176:147–150. doi: 10.7205/milmed-d-10-00191. [DOI] [PubMed] [Google Scholar]
- 29.Nestler EJ. Is there a common molecular pathway for addiction? Nature neuroscience. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- 30.Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nature reviews. Neuroscience. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- 31.Eierud C, Craddock RC, Fletcher S, Aulakh M, King-Casas B, Kuehl D, LaConte SM. Neuroimaging after mild traumatic brain injury: Review and meta-analysis. NeuroImage Clinical. 2014;4:283–294. doi: 10.1016/j.nicl.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levin HS, Williams DH, Eisenberg HM, High WM, Jr, Guinto FC., Jr Serial MRI and neurobehavioural findings after mild to moderate closed head injury. Journal of neurology, neurosurgery, and psychiatry. 1992;55:255–262. doi: 10.1136/jnnp.55.4.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonald V, Hauner KK, Chau A, Krueger F, Grafman J. Networks underlying trait impulsivity: Evidence from voxel-based lesion-symptom mapping. Human brain mapping. 2016 doi: 10.1002/hbm.23406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cristofori I, Zhong W, Chau A, Solomon J, Krueger F, Grafman J. White and gray matter contributions to executive function recovery after traumatic brain injury. Neurology. 2015;84:1394–1401. doi: 10.1212/WNL.0000000000001446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chou A, Morganti JM, Rosi S. Frontal Lobe Contusion in Mice Chronically Impairs Prefrontal-Dependent Behavior. PloS one. 2016;11:e0151418. doi: 10.1371/journal.pone.0151418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Povlishock JT, Erb DE, Astruc J. Axonal response to traumatic brain injury: reactive axonal change, deafferentation, and neuroplasticity. Journal of neurotrauma. 1992;9(Suppl 1):S189–200. [PubMed] [Google Scholar]
- 37.Maller JJ, Thomson RH, Pannek K, Rose SE, Bailey N, Lewis PM, Fitzgerald PB. The (Eigen)value of diffusion tensor imaging to investigate depression after traumatic brain injury. Human brain mapping. 2014;35:227–237. doi: 10.1002/hbm.22171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Warner MA, Youn TS, Davis T, Chandra A, Marquez de la Plata C, Moore C, Harper C, Madden CJ, Spence J, McColl R, Devous M, King RD, Diaz-Arrastia R. Regionally selective atrophy after traumatic axonal injury. Archives of neurology. 2010;67:1336–1344. doi: 10.1001/archneurol.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah S, Yallampalli R, Merkley TL, McCauley SR, Bigler ED, Macleod M, Chu Z, Li X, Troyanskaya M, Hunter JV, Levin HS, Wilde EA. Diffusion tensor imaging and volumetric analysis of the ventral striatum in adults with traumatic brain injury. Brain injury. 2012;26:201–210. doi: 10.3109/02699052.2012.654591. [DOI] [PubMed] [Google Scholar]
- 40.Alhilali LM, Delic JA, Gumus S, Fakhran S. Evaluation of White Matter Injury Patterns Underlying Neuropsychiatric Symptoms after Mild Traumatic Brain Injury. Radiology. 2015:142974. doi: 10.1148/radiol.2015142974. [DOI] [PubMed] [Google Scholar]
- 41.Merkel SF, Andrews AM, Lutton EM, Razmpour R, Cannella LA, Ramirez SH. Dexamethasone Attenuates the Enhanced Rewarding Effects of Cocaine Following Experimental Traumatic Brain Injury. Cell Transplantation. 2016 doi: 10.1177/0963689717714341. epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sajja VS, Galloway M, Ghoddoussi F, Kepsel A, VandeVord P. Effects of blast-induced neurotrauma on the nucleus accumbens. Journal of neuroscience research. 2013;91:593–601. doi: 10.1002/jnr.23179. [DOI] [PubMed] [Google Scholar]
- 43.Budde MD, Shah A, McCrea M, Cullinan WE, Pintar FA, Stemper BD. Primary blast traumatic brain injury in the rat: relating diffusion tensor imaging and behavior. Front Neurol. 2013;4:154. doi: 10.3389/fneur.2013.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shin SS, Bales JW, Yan HQ, Kline AE, Wagner AK, Lyons-Weiler J, Dixon CE. The effect of environmental enrichment on substantia nigra gene expression after traumatic brain injury in rats. Journal of neurotrauma. 2013;30:259–270. doi: 10.1089/neu.2012.2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:5274–5278. doi: 10.1073/pnas.85.14.5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2004;24:1551–1560. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Koob GF. Negative reinforcement in drug addiction: the darkness within. Current opinion in neurobiology. 2013;23:559–563. doi: 10.1016/j.conb.2013.03.011. [DOI] [PubMed] [Google Scholar]
- 48.Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology. 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koob GF, Le Moal M. Drug abuse: hedonic homeostatic dysregulation. Science. 1997;278:52–58. doi: 10.1126/science.278.5335.52. [DOI] [PubMed] [Google Scholar]
- 50.Donnemiller E, Brenneis C, Wissel J, Scherfler C, Poewe W, Riccabona G, Wenning GK. Impaired dopaminergic neurotransmission in patients with traumatic brain injury: a SPECT study using 123I-beta-CIT and 123I-IBZM. European journal of nuclear medicine. 2000;27:1410–1414. doi: 10.1007/s002590000308. [DOI] [PubMed] [Google Scholar]
- 51.Huger F, Patrick G. Effect of concussive head injury on central catecholamine levels and synthesis rates in rat brain regions. Journal of neurochemistry. 1979;33:89–95. doi: 10.1111/j.1471-4159.1979.tb11710.x. [DOI] [PubMed] [Google Scholar]
- 52.Wagner AK, Sokoloski JE, Ren D, Chen X, Khan AS, Zafonte RD, Michael AC, Dixon CE. Controlled cortical impact injury affects dopaminergic transmission in the rat striatum. Journal of neurochemistry. 2005;95:457–465. doi: 10.1111/j.1471-4159.2005.03382.x. [DOI] [PubMed] [Google Scholar]
- 53.Yan HQ, Kline AE, Ma X, Hooghe-Peters EL, Marion DW, Dixon CE. Tyrosine hydroxylase, but not dopamine beta-hydroxylase, is increased in rat frontal cortex after traumatic brain injury. Neuroreport. 2001;12:2323–2327. doi: 10.1097/00001756-200108080-00009. [DOI] [PubMed] [Google Scholar]
- 54.Yan HQ, Ma X, Chen X, Li Y, Shao L, Dixon CE. Delayed increase of tyrosine hydroxylase expression in rat nigrostriatal system after traumatic brain injury. Brain research. 2007;1134:171–179. doi: 10.1016/j.brainres.2006.11.087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maldve RE, Zhang TA, Ferrani-Kile K, Schreiber SS, Lippmann MJ, Snyder GL, Fienberg AA, Leslie SW, Gonzales RA, Morrisett RA. DARPP-32 and regulation of the ethanol sensitivity of NMDA receptors in the nucleus accumbens. Nature neuroscience. 2002;5:641–648. doi: 10.1038/nn877. [DOI] [PubMed] [Google Scholar]
- 56.Hinzman JM, Thomas TC, Quintero JE, Gerhardt GA, Lifshitz J. Disruptions in the regulation of extracellular glutamate by neurons and glia in the rat striatum two days after diffuse brain injury. Journal of neurotrauma. 2012;29:1197–1208. doi: 10.1089/neu.2011.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goodrich GS, Kabakov AY, Hameed MQ, Dhamne SC, Rosenberg PA, Rotenberg A. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. Journal of neurotrauma. 2013;30:1434–1441. doi: 10.1089/neu.2012.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Scofield MD, Kalivas PW. Astrocytic dysfunction and addiction: consequences of impaired glutamate homeostasis. Neuroscientist. 2014;20:610–622. doi: 10.1177/1073858413520347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Scofield MD, Heinsbroek JA, Gipson CD, Kupchik YM, Spencer S, Smith AC, Roberts-Wolfe D, Kalivas PW. The Nucleus Accumbens: Mechanisms of Addiction across Drug Classes Reflect the Importance of Glutamate Homeostasis. Pharmacol Rev. 2016;68:816–871. doi: 10.1124/pr.116.012484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kalivas PW, Volkow ND. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol Psychiatry. 2011;16:974–986. doi: 10.1038/mp.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Suska A, Lee BR, Huang YH, Dong Y, Schluter OM. Selective presynaptic enhancement of the prefrontal cortex to nucleus accumbens pathway by cocaine. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:713–718. doi: 10.1073/pnas.1206287110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J, van Steeg K, Kopajtic TA, Loram LC, Sfregola C, Galer E, Miles NE, Bland ST, Amat J, Rozeske RR, Maslanik T, Chapman TR, Strand KA, Fleshner M, Bachtell RK, Somogyi AA, Yin H, Katz JL, Rice KC, Maier SF, Watkins LR. Opioid activation of toll-like receptor 4 contributes to drug reinforcement. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32:11187–11200. doi: 10.1523/JNEUROSCI.0684-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, Reddy A, Somogyi AA, Hutchinson MR, Watkins LR, Yin H. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6325–6330. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schwarz JM, Bilbo SD. Adolescent morphine exposure affects long-term microglial function and later-life relapse liability in a model of addiction. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2013;33:961–971. doi: 10.1523/JNEUROSCI.2516-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, Harris RA. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain, behavior, and immunity 25 Suppl. 2011;1:S92–S105. doi: 10.1016/j.bbi.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Adler MW, Rogers TJ. Are chemokines the third major system in the brain? Journal of leukocyte biology. 2005;78:1204–1209. doi: 10.1189/jlb.0405222. [DOI] [PubMed] [Google Scholar]
- 67.Heinisch S, Palma J, Kirby LG. Interactions between chemokine and mu-opioid receptors: anatomical findings and electrophysiological studies in the rat periaqueductal grey. Brain, behavior, and immunity. 2011;25:360–372. doi: 10.1016/j.bbi.2010.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schwarz JM, Hutchinson MR, Bilbo SD. Early-life experience decreases drug-induced reinstatement of morphine CPP in adulthood via microglial-specific epigenetic programming of anti-inflammatory IL-10 expression. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31:17835–17847. doi: 10.1523/JNEUROSCI.3297-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Crews FT, Zou J, Qin L. Induction of innate immune genes in brain create the neurobiology of addiction. Brain, behavior, and immunity. 2011;25(Suppl 1):S4–S12. doi: 10.1016/j.bbi.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kelley KW, Dantzer R. Alcoholism and inflammation: neuroimmunology of behavioral and mood disorders. Brain, behavior, and immunity. 2011;25(Suppl 1):S13–20. doi: 10.1016/j.bbi.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain : a journal of neurology. 2013;136:28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dafny N, Dougherty PM, Drath D. Immunosuppressive agent modulates the severity of opiate withdrawal. NIDA Res Monogr. 1990;105:553–555. [PubMed] [Google Scholar]
- 73.Swendsen J, Burstein M, Case B, Conway KP, Dierker L, He J, Merikangas KR. Use and abuse of alcohol and illicit drugs in US adolescents: results of the National Comorbidity Survey-Adolescent Supplement. Arch Gen Psychiatry. 2012;69:390–398. doi: 10.1001/archgenpsychiatry.2011.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gandhi M, Aweeka F, Greenblatt RM, Blaschke TF. Sex differences in pharmacokinetics and pharmacodynamics. Annu Rev Pharmacol Toxicol. 2004;44:499–523. doi: 10.1146/annurev.pharmtox.44.101802.121453. [DOI] [PubMed] [Google Scholar]
- 75.Wagner AK, Chen X, Kline AE, Li Y, Zafonte RD, Dixon CE. Gender and environmental enrichment impact dopamine transporter expression after experimental traumatic brain injury. Experimental neurology. 2005;195:475–483. doi: 10.1016/j.expneurol.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 76.Caplan HW, Cox CS, Bedi SS. Do microglia play a role in sex differences in TBI? Journal of neuroscience research. 2017;95:509–517. doi: 10.1002/jnr.23854. [DOI] [PubMed] [Google Scholar]
- 77.Rau TF, Kothiwal AS, Rova AR, Brooks DM, Poulsen DJ. Treatment with low-dose methamphetamine improves behavioral and cognitive function after severe traumatic brain injury. The journal of trauma and acute care surgery. 2012;73:S165–172. doi: 10.1097/TA.0b013e318260896a. [DOI] [PubMed] [Google Scholar]
- 78.Edut S, Rubovitch V, Rehavi M, Schreiber S, Pick CG. A study on the mechanism by which MDMA protects against dopaminergic dysfunction after minimal traumatic brain injury (mTBI) in mice. Journal of molecular neuroscience : MN. 2014;54:684–697. doi: 10.1007/s12031-014-0399-z. [DOI] [PubMed] [Google Scholar]
- 79.Edut S, Rubovitch V, Schreiber S, Pick CG. The intriguing effects of ecstasy (MDMA) on cognitive function in mice subjected to a minimal traumatic brain injury (mTBI) Psychopharmacology. 2011;214:877–889. doi: 10.1007/s00213-010-2098-y. [DOI] [PubMed] [Google Scholar]
- 80.Wang T, Chou DY, Ding JY, Fredrickson V, Peng C, Schafer S, Guthikonda M, Kreipke C, Rafols JA, Ding Y. Reduction of brain edema and expression of aquaporins with acute ethanol treatment after traumatic brain injury. Journal of neurosurgery. 2013;118:390–396. doi: 10.3171/2012.8.JNS12736. [DOI] [PubMed] [Google Scholar]
- 81.Kanbak G, Kartkaya K, Ozcelik E, Guvenal AB, Kabay SC, Arslan G, Durmaz R. The neuroprotective effect of acute moderate alcohol consumption on caspase-3 mediated neuroapoptosis in traumatic brain injury: the role of lysosomal cathepsin L and nitric oxide. Gene. 2013;512:492–495. doi: 10.1016/j.gene.2012.10.012. [DOI] [PubMed] [Google Scholar]