

Abstract
Pump probe microscopy is a time-resolved multiphoton imaging technique capable of generating contrast between non-fluorescent pigments based on differences in excited-state lifetimes. Here we describe a fiber-based ultrafast system designed for imaging heme proteins with an independently-tunable pulse pair in the visible-wavelength regime. Starting with a 1060 nm fiber amplifier (1.3 W at 63 MHz, 140 fs pulses), visible pulses were produced in the vicinity of 488 nm and 532 nm by doubling the output of a short photonic crystal fiber with a pair of periodically-poled lithium niobate crystals, providing 5–20 mW power in each beam. This was sufficient for acquiring transient absorption images from unstained cryosectioned tissue.
OCIS codes: (170.6920) Time-resolved imaging, (190.7110) Ultrafast nonlinear optics
1. Introduction
Recent years have seen the development of numerous laser-scanning microscopy techniques for visualizing the endogenous molecular components of cells and tissues, including multiphoton-excited fluorescence and harmonic generation [1], coherent Raman scattering [2], photothermal [3], photoacoustic [4], and pump-probe (i.e. transient absorption) [5–7]. Among these techniques, pump-probe microscopy is unique in its ability to distinguish molecules with similar absorption spectra by differences in their excited-state photodynamics, typically on the timescale of femtoseconds to picoseconds. Pump-probe microscopy is an adaptation of transient absorption spectroscopy [8,9] that uses low pulse-energy, high repetition-rate ultrafast laser sources and a few selected pump/probe wavelength combinations at a time, trading broad spectral coverage for fast, high resolution, 3-dimensional imaging capabilities. Pump-probe is somewhat akin to fluorescence lifetime microscopy [10, 11], but without the need for appreciable fluorescence quantum yield. Rather, pump-probe resolves excited-state lifetimes by measuring subtle changes in optical absorption of a probe pulse following absorption of a pump pulse. To date, pump-probe microscopy has many applications in the materials sciences, but relatively few in the life sciences [7]. Of these, the only endogenous biomedical imaging targets probed so far have been melanin [12] and hemoglobin [13], even though a large number of non-fluorescent chromophores could in principle be observed with pump-probe microscopy.
An interesting potential class of imaging targets is the other heme proteins with visible-wavelength absorption bands, namely myoglobin and cytochromes a/a3, b, c. Myoglobin serves as an oxygen storage molecule in muscle tissue, and these cytochromes have redox-dependent absorption spectra that serve as an indicator of mitochondrial respiratory chain function and activity [14, 15]. Therefore, a microscopy technique that could distinguish different cytochromes and their redox states could potentially be a valuable addition to current fluorescence techniques that measure the redox state of the respiratory chain inputs NAD(P)H and FAD+ [16–18]. Cytochromes have been the target of several molecular imaging techniques, including photothermal [3], photacoustic [19], and Raman [20] microscopy. The success of pump-probe microscopy with another heme protein, hemoglobin [13, 21, 22], suggests myoglobin and cytochrome pump-probe microscopy might also be possible.
Here we present the development of an ultrafast laser source with pump and probe wavelengths suitable for transient absorption microscopy in the vicinity of the Q-band of myoglobin and the respiratory chain cytochromes. With this system, we have recorded preliminary images of unstained, mitochondria-rich tissue, with sufficient signal-to-noise ratio to provide some distinction between tissue types.
Whereas transient absorption spectroscopy (non-imaging) systems typically use pulse energies with plenty of overhead for nonlinear wavelength conversion (~1 mJ @ 1 kHz), through bulk supercontinuum generation [23] and/or optical parametric amplifiers (OPA) [24], pump-probe microscopic imaging uses significantly lower pulse energies to avoid sample damage. To maintain reasonably fast imaging speed requires the use of high repetition-rate, low pulse-energy sources (∼1 nJ @ 80 MHz) to make up for low signal levels. This regime is well below the threshold for bulk white light generation, restricting options for converting the near-infrared output of an ultrafast laser to the 450–600 nm region.
One option for producing wavelength-tunable pulses to access pump-probe contrast from the Q band of heme proteins is fiber-based supercontinuum generation [25], followed by a narrowband filter to select either the pump or the probe pulse [26]. But in order to reach < 500 nm while maintaining coherence with this approach, the supercontinuum must be seeded with short (30 fs), low-energy (1 nJ) pulses, which leaves the power spectral density exceedingly small in any given spectral band. For femtosecond transient absorption spectroscopy, this approach has been demonstrated only down to 550 nm [26]. In addition, the shot-to-shot amplitude of such broad supercontinuum is far less stable than bulk supercontinuum [27], reducing the signal-to-noise ratio (SNR) and making the limit of detection (LOD) too high for physiological concentrations.
Another option would be to employ two independently-tunable OPOs, pumped by the same source [28–30]. But this introduces the complexity and footprint of two additional optical cavities that are nontrivial to align and keep locked to the master oscillator’s repetition rate, in addition to an exceedingly high price point for a commercial system.
A third option for generating tunable visible-wavelength pulses is to perform modest spectral broadening (self-phase modulation dominated) in an all normal dispersion, polarization maintaining fiber, followed by efficient degenerate and non-degenerate sum-frequency generation in a periodically-poled lithium niobate (PPLN) crystal [31, 32]. This approach simultaneously leverages (1) the broad, stable, and coherent supercontinuum bandwidth generated at high average power (∼ 1 W) [33, 34] to provide a large tuning range, and (2) the narrow phase-matching bandwidth of PPLN to generate wavelength-tunable pulses with high spectral brightness.
Here we expand this approach to provide independently-tunable pump and probe pulses, seeded by an inexpensive homebuilt fiber oscillator and amplifier [35]. Our system currently provides 5–20 mW power in each beam (75–300 pJ / pulse), with tuning ranges of 485–495 nm, and 525–540 nm determined by phase matching conditions. The current tuning range is limited by the PPLN crystals poling period and is in principle expandable to cover > 50 nm tuning bandwidth with different PPLN crystals containing a larger array of poling periods. We also demonstrate with transient absorption spectroscopy and imaging experiments that this system meets the unique requirements of a heme pump-probe imaging system in providing two pulses in the blue/green region with independently-tunable wavelengths, at a high repetition rate.
2. Source characterization
Figure 1 illustrates the layout of the nonlinear broadening stage followed by the PPLN doubling heads and the transient absorption microscope. The high stability, coherence, and power spectral density generated in a nominally normal dispersion, polarization maintaining optical fiber [33] is the enabling component of our two-color system. We employ a large mode area photonic crystal fiber (PCF), NKT Photonics LMA-PM-5, 89 mm in length as the nonlinear fiber (γ0 = 9 W km−1 and zero-dispersion wavelength of 1175 nm). End caps of HI-1060 fiber seal the ends of the PCF (3 and 5 mm in length on the entrance and exit sides, respectively) as well as allowing the fiber end faces to be potted into angled-physical contact connectors. Commercial aspheric and triplet collimators enclose and protect the optical fiber faces, allowing robust and efficient coupling into and out of the fiber cocktail (or segmented fiber). The connector/collimator units virtually eliminate day-to-day coupling drift.
Fig. 1.
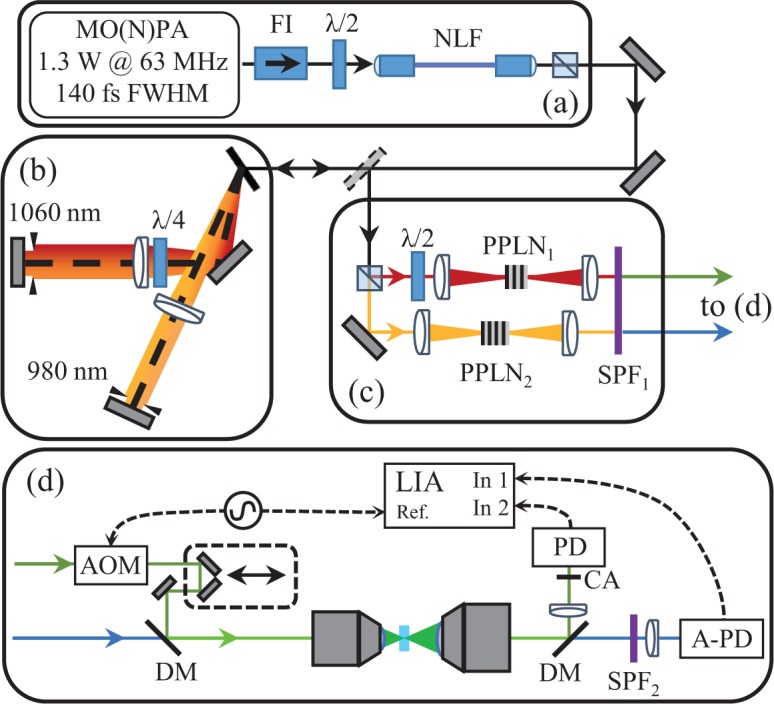
Schematic of the experiment showing (a) the near-infrared ultrafast source and supercontinuum stage, (b) the two-color Martinez, (c) the two SHG stages, and (d) the transient absorption microscope and detection system. Master Oscillator / Nonlinear Power Amplifier (MO(N)PA) (a) supplies optical pulses through a Faraday isolator (FI) and half wave plate (λ/2) to our nonlinear fiber (NLF). The broadened near-infrared spectrum is then directed to a split Martinez compressor, where one arm is rotating polarization by double-passing a quarter wave plate (λ/4). (c) Individually compressed spectral halves are then focused into periodically poled lithium niobate (PPLN) crystals for conversion to visible wavelength. A short pass filter (SPF1) rejects the remaining near-infrared fundamental before the pump-probe setup (d). An acousto-optic modulator (AOM) imparts a 200 kHz amplitude modulation to the pump pulse train, which is fed to delay line, and combined with the probe by a dichroic mirror (DM) and focused on to the sample with a microscope objective. After collection with a second objective lens, a second dichroic splits pump and probe. The pump is collected by a photodiode (PD) through a confocal aperture (CA), and the probe is collected by an amplified photodiode (A-PD) through an additional short pass filter (SPF2). A lock-in amplifier (LIA) is used to detect the transfer of modulation from the pump onto the probe via transient absorption interactions in the sample.
An all-normal dispersion fiber oscillator [36] seeds a nonlinear fiber amplifier [35], outputting 1.3 W (21 nJ / pulses at a repetition rate of 63 MHz) and pulse durations of 140 fs after compression. The net transmission efficiency through the fiber cocktail, or segmented fiber, is 60%. Since pulses de-chirped in a Martinez compressor [37] to the transform-limit before the fiber broadening stage were found to unfavorably deplete the power spectral density around 980 nm; residual, positive group delay dispersion on the amplifier pulses, imparted by tuning the compressor away from the optimal compensation position, is used to tune the structure of the nonlinear fiber broadened power spectrum. An example of the fiber broadened supercontinuum power spectrum is shown in Fig. 2(a).
Fig. 2.

(a) The near-infrared power spectrum of the supercontinuum, where the two highlighted regions are what the two-color Martinez passes and compresses. (b) Independent SHG spectral tuning via temperature tuning.
We estimate an optical wave-breaking length [38] of 50 mm for the compressed amplifier pulses in the PCF, indicating the necessity of a pulse shaper to fully compress the power spectrum generated in the nonlinear fiber due to the presence of higher-order spectral phase [33, 39, 40]. To pump the SFG stages with a dichroic pulse pair, where each pulse is approaching its respective transform-limited intensity, we use two pseudo-linear Martinez compressors each de-chirping a portion of the supercontinuum. Moving to a nonlinear fiber whose length is less than the optical wave-breaking length would enable a linear-compressor (e.g. a prism compressor) to de-chirp the entirety of the power spectrum generated in the nonlinear fiber due to an absence of higher-order spectral phase [33, 39, 40]. Additionally such a fiber would allow us to seed the nonlinear fiber with amplifier pulses de-chirped to near their transform-limit, increasing the spectral tuning range of SFG stages as well as increasing the conversion efficiency.
Fig. 1(b) shows the two-color Martinez compressor we use to both filter and compress the supercontinuum power spectrum into two pulses. The imaging nature of the Martinez compressor allows for a natural Fourier spectral filter by placing irises in the central Fourier plane (located at the fold mirror). The two-color Martinez generates two spatially separated central Fourier planes, allowing us to carve two pulses out of the supercontinuum bandwidth centered at 980 and 1060 nm, the bandwidths of which are the shaded regions in Fig. 2(a). We are able to isolate the two pulses through polarization by adding a quarter wave-plate between the grating and lens in the 1060 nm arm of the two-color Martinez. This keeps the two pulses nearly synchronous as well as in the same spatial vicinity to keep the difference in turbulent phase distortions from the laboratory environment to a minimum. The 980 and 1060 nm pulses contain 80 and 200 mW, respectively.
Two PPLN crystals compose the SFG stages [Fig. 1(c)]: Covesion’s MSHG976-0.5-1 and MSHG1064-0.5-0.3 with poling periods around 5.2 and 6.9 μm. Temperature tuning each PPLN crystal individually along with laterally scanning to other poling periods allows for independent spectral tuning of the phase-matched visible output pulses. The extent of the spectral tuning from these standard commercial crystals is shown in Fig. 2(b), although operation of the crystal ovens at high temperature is preferred to avoid damage due to the photorefractive effect when operated near the PPLN damage threshold (> 1 GW/cm2). The focus into the PPLN crystals was set close to the optimal ratio of crystal length to confocal parameter of 2.84 [41] for the 1 mm thick crystal (488 nm SFG). The same focus was used in the 0.3 mm thick crystal to keep the 1060 nm pulse fluence incident on the 532 nm SFG crystal below 10 GW/cm2.
The maximum conversion efficiency of 25% to a visible 487 nm pulse demonstrates the 980 nm pump pulse is well compressed. The lower conversion efficiency at 530 nm, 5%, is due to the reduced crystal thicknesses and poorer temporal compression of the 1060 nm pulse. Even with the modest conversion efficiency at 530 nm, we are able to generate 5–20 mW of average power in either visible pulse, which is sufficient for transient absorption microscopy.
Since our system cascades several nonlinear systems (~13× spectral broadening in the amplifier fiber, supercontinuum generation, SFG), we measure the relative intensity noise (RIN) of the near-infrared amplifier and SFG pulse trains up to 500 kHz with an amplified silicon photodetector (Thorlabs PDA36A) [Fig. 3]. The RIN of the 488 nm SFG pulse train only increases by 5 dB compared to the amplifier seed pulse at frequencies above 100 kHz. The 532 nm SFG pulse train is noisier, increasing by > 12 dB over the amplifier seed above 100 kHz. The near-infrared spectral band used to seed the 532 nm SFG crystal is composed of both depleted power spectrum seeding the nonlinear fiber plus back-conversion from optical wave-breaking mediated four-wave mixing. The cascade of nonlinearities within the broadening fiber leads to an increase in noise. We estimate the shot noise for the visible SFG pulse trains to be < −105 dBc/Hz for the average power used in the imaging experiments discussed in section 4.
Fig. 3.
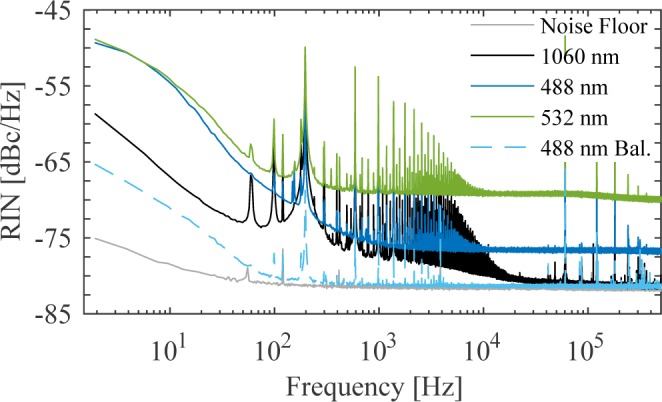
RIN of the 1060 nm amplifier SFG seed, 488 nm probe, and 532 nm pump pulses incident on an amplified photodiode as well as the noise floor of the detection system (all solid curves), normalized to the 488 nm probe. We also include the RIN of the 488 nm probe incident on a balanced photodetector (dashed curve).
Based on the RIN data, Table 1 contains estimates of the 488 nm LOD (SNR of 3× over background noise) for several lock-in amplifier (LIA) time constants tc and detector configurations, given by , where ν0 = 200 kHz is the modulation frequency imparted on the pump pulse train and we assume a weakly absorbed probe pulse (i.e. small reduction in the average power reaching the photodetector compared to the RIN measurements). For rapid stage-scanning microscope speeds, tc = 1.5 ms yields a high enough sensitivity for microscopy at typical transient absorption magnitudes (~1 part in 104−5).
Table 1.
Transient absorption sensitivity estimates
| Detector | LIA time constant [ms] | LOD [%] / [μOD] |
|---|---|---|
| amplified | 1.5 | 4.3 · 10−5 / 19 |
| amplified | 0.03 | 2 · 10−3 / 860 |
| balanced | 0.03 | 6.7 10−4 / 290 |
| balanced* | 0.03 | 6.7 · 10−6 / 3 |
The increased intensities associated with high resolution, laser scanning microscopy though, requires a reduction in pixel dwell time relative to stage-scanning speeds to prevent optical damage to the biological sample (section 3). At the faster scan times, the LOD of the current system falls to 2 parts in a 1000 (assuming tc = 30 μs), resulting in unfavorable SNR for typical transient absorption signal levels (SNR = 0.5 based on signal levels attained section 3).
We identify two routes towards increasing the sensitivity of the system to make it amendable to high resolution microscopy. One, is to move a future version of the system to a shorter nonlinear fiber, below the optical wave-breaking length, with minimized feedback into the amplifier system. In addition to the large benefits in supercontinuum pulse compressibility, the resulting SFG pulse trains should also have RIN closer to the fundamental near-infrared seed pulse trains.
The second route is to capture the probe pulse on a balanced-photodetector along with a reference copy of the probe pulse train. We demonstrate that with a balanced-photodetector with 103 trans-impedance gain (Thorlabs BPD100A), the noise on the current probe pulse train is reduced to nearly the electronic noise floor for frequencies above ~ 400 Hz, as seen in the dashed-curve in Fig. 3. At 7 parts in 104 though, we could feasibly implement a two-color laser scanning microscope.
Due to the elevated noise floor used to take the RIN measurements (-82 dBc/Hz), we suspect the LOD attainable through a low-noise LIA is lower than what is measured here. Additional analysis of the RIN of the balanced-photodetector using a radio-frequency spectrum analyzer (Hewlett Packard 8594E) indicates the noise floor of the balanced detection system above 100 kHz is close to the shot noise floor of −101 dBc/Hz. At the shot noise floor, the probe attenuation sensitivity would be reduced by an additional 20 dB [Table 1 (bottom row)].
The issue with balanced detection in an imaging system is the requirement of auto-balancing to maintain shot noise limited performance in the presence of large changes in scattering and absorption across the interrogated field-of-view. For our particular two-color system, there is a lack of commercially available auto-balanced silicon-based photodetectors with trans-impedance gain bandwidths > 200 kHz. Other researchers however, have published home-built auto-balanced detectors which could in the future be implemented to realize the increased sensitivity when operating at the shot-noise limit [42].
As the last step in characterizing the visible pulses suitability to transient absorption spectroscopy, we measure their cross-correlation through non-degenerate two-photon absorption in bismuth germanate (BGO) through modulation transfer from the pump pulse train to the probe pulse train, see Fig. 4(a) for curve, which reveals an instrument response function of 780 fs full-width at half-maximum (FWHM). This result indicates that our visible pulses are sub-ps in duration, in spite of the lack of dispersion compensation of the visible pulses. If dispersion compensation were to be added, we estimate the Fourier-limited durations to be 250 and 100 fs FWHM for the blue and green pulses, respectively. Fig. 1(d) shows the modulation transfer system used for both the cross-correlation measurement and the transient absorption imaging experiments discussed later. The 532 nm green pulse is used as the pump pulse since it overlaps with the Q-band of cytochrome c [43]. We modulate the green pump pulse train at 200 kHz using an acousto-optic modulator, achieving > 90% amplitude modulation depth. The 488 nm probe light is filtered through a 500 nm short pass filter and captured on the same amplified silicon photodetector used in the RIN measurements. A LIA is then used to detect out-of-phase modulations at 200 kHz in the signal generated by the 488 nm probe pulse, where this phase configuration is the typical transient absorption spectroscopy convention since it makes probe attenuation signals positive. 2 mW of average power in both the pump and probe pulse trains are used for these cross-correlation measurements and subsequent transient absorption measurements.
Fig. 4.

(a) Temporal cross-correlation of blue and green pulses from non-degenerate two-photon in BGO. The transient absorption signal of (b) reduced and (c) oxidized 0.5 mM cytochrome c (black markers) fitted with exponential decays (red curves).
3. Transient absorption spectroscopy results
To determine characteristic transient absorption delay curves at our pump and probe wavelengths, we interrogate 0.5 mM cytochrome c solutions in a cuvette with a 1 mm pathlength, using a 0.15 numerical aperture (NA) focusing objective and a 0.3 NA collection objective. Bovine heart cytochrome c (Sigma-Aldrich) was prepared in the oxidized state by exposure to atmosphere, and in the reduced state by the addition of dithiothreitol. The transient absorption delay curves for either redox state are shown in Fig. 4(b) and (c). Fitting the transient absorption delay curves to an exponential decay convolved with a Gaussian approximation of our cross-correlation response function, we calculate decay coefficients within a 95% confidence interval of 5.5–6.1 ps and 4.6–5.3 ps for the reduced and oxidized states of cytochrome c, respectively.
Unfortunately, this result differs from transient absorption delay curves found previously in Ref [43]. However, other groups have shown that there can be dramatic differences in transient absorption delay curves between kHz and MHz laser systems [44]. A goal of future work is to increase the tunability of the probe pulse down to 450 nm, to search for pump-probe wavelength combinations of increased signal contrast between redox states. Seeding the nonlinear fiber with a near-infrared pulse closer to its bandwidth-limited duration already generates the spectral bandwidth necessary to up-convert light to the 450 nm spectral band. A PPLN chip with broader poling period coverage is the remaining component needed for a future upgrade to the two-color system.
Given the success of acquiring pump-probe signals in 0.5 mM cytochrome c, which is close to the physiological concentration of cytochrome c in mitochondria [45], we moved on to a thin section of biological tissue, unstained cryogenically-sectioned brown adipose tissue (from a hibernating ground squirrel). We repeatedly exposed the tissue to determine an optical damage threshold for imaging. Line scans are repeatedly taken at constant velocity over the same tissue using a 0.25 NA focusing objective (creating a point-spread function approximately 1 μm in width). By changing the line scan velocity, we are able to vary the net pump-probe fluence (average intensity × exposure time), where for a total illumination of 4 mW we estimate the average intensity to be 500 kW/cm2. The imaging system is configured to scan across a 5 μm pixel every time constant of the lock-in amplifier with zero time delay between the pump and probe pulses. The lock-in amplifier time constant and pixel size therefore set the scan velocity and equivalently the exposure time. The large line scans allow us to incorporate the tissue heterogeneity into the measurement.
As the sample changes due to optical damage, the line scans begin to de-correlate with the initial scan. We can therefore use a spatial cross-correlation to create a parameter that changes as the sample damages due to illumination by the pump and probe pulses. Integrating the cross-correlation between the initial line scan and subsequent line scans generates a curve showing the amount of sample damage as a function of line scan number, examples of which are shown in Fig. 5(a). This integrated cross-correlation method intrinsically incorporates pixels which “burn” and dramatically increase in signal level. A simpler method is to take the sum of the signals from a line scan and analyze the rate at which it decays with line scan number, but this method requires the exclusion of burned pixels.
Fig. 5.

(a) Examples of the decay in transient absorption signal with line scan number for (red curve) 600 and (black curve) 2400 . (b) Decay coefficient fits as a function of fluence, with error bars indicating 95% confidence interval of fits.
Fitting the integrated cross-correlation decay curves as a function of line scan number with decaying exponentials, we are able to extract damage rate as a function of pump-probe fluence. Fig. 5(b) plots the decay coefficients (scan number associated with a decay of e−1) and the 95% confidence intervals of the exponential fits. The damage curve for the fastest scan velocity (1.5 ms lock-in amplifier time constant and 5 ms pixel dwell time) however, is processed using the simpler method using the summed signal decay, excluding “burned” spatial points that exhibit large increases.
Unsurprisingly, very little fluence (∼ 2000 J/cm2 or equivalently 4 mW of average power focused down to a spot size of 1 μm and a 20 ms exposure time) is necessary to drive a rapid degradation in the biological tissue. Not only do we see a reduction in the transient absorption signal but we see changes in the confocal transmittance channel, capturing changes in absorption and scattering of the pump pulse. Therefore, we maintain a fluence below 1500 J/cm2 for our pump-probe delay-scanned image acquisition. For the images in the next section, we use 2 mW each beam (4 mW total) through a 0.25 NA objective, with 12 ms/pixel dwell (1200 J/cm2).
Future applications to live tissue will require consideration of heat generated by the strongly absorbed pump beam. In the experiments here, the high repetition rate and slow scan rates lead to an accumulation of heat at the focal spot. The initial temperature rise at the center of the focal volume induced by a single pump pulse is
| (1) |
where N = 3 × 105 μm−3 is the number density of absorbing molecules (assuming 0.5 mM cytochrome c [45]), σ = 1.13 × 10−16 cm2 is the absorption cross-section [46], F0 = 20 pJ/μm2 is the pulse fluence at the center of the focal volume under our focal conditions with 2 mW average power, ρ = 911 kg/m3 is the mass density of brown adipose tissue, and C = 2503 J kg−1K1 the specific heat capacity [47]. Though such a small ΔT is negligible, the cumulative heating of the 63 MHz pulse train is much higher. A rough estimate of the steady-state temperature at the focus can be obtained by approximating the problem as a spherical volume of heat sources [48], which leads to:
| (2) |
where Pavg = 2 mW and k = 0.26 W m−1 K−1 is the thermal conductivity of brown adipose tissue [47]. Simulations in MATLAB of heat transport in the geometry of the experiments reported here (a Gaussian beam incident on 20 μm tissue slice bounded by glass on one side and air on the other) result in a ΔT = 66 K that reaches steady-state in 1 ms. The code is available at Ref. [49]. This is likely an overestimate, considering there was no evidence of vaporization/cavitation in our experiments at these power levels. In the future, mitigating this temperature rise will require either lowering the repetition rate or increasing scanning speed, to reduce the accumulation of heat at each pixel. Lowering the repetition rate is undesirable, because it would reduce SNR and require longer averaging times to compensate. Given ΔT = 0.03 K per pulse, at a 63 MHz repetition rate, a scan rate of 2 μm/μs would be required to prevent the cumulative ΔT from exceeding 1 K. This scan rate translates to a 8 kHz line rate, or 16 frames per second, given a typical microscopy field of view of 250 μm and 512 × 512 pixel frame size, which is well within the capabilities of a resonant galvo scanner.
We can also estimate (order of magnitude) the sensitivity necessary for transient absorption imaging of hemes using the optical damage survey. Calculating the mean probe attenuation in the presence of the pump pulse over the first line scans gives us a loose threshold of sensitivity. The mean attenuation at zero time delay is ∼ 1 · 10−4, and is roughly invariant of illumination fluence.
The stage-scanning speed and LIA time constants for a low NA microscope allow sufficient sensitivity from the current system [Table 1 (top row)] to resolve a transient absorption image delay stack above the loose threshold of sensitivity. In a high resolution configuration (> 0.5 NA), the necessary scan speeds and LIA time constants to maintain a fluence at each pixel of 150 J/cm2 reduces the sensitivity of the current unbalanced detection system [Table 1 (middle row)] to below the threshold needed to resolve heme features using transient absorption. Adapting a balanced detector scheme to the two-color transient absorption microscope or pushing the noise floor of the 488 nm probe pulse closer to the amplifier near-infrared seed pulse are the critical improvements necessary to extend our system to high resolution microscopy.
4. Transient absorption microscopy results
We next proceeded to acquire images of cryosectioned brown adipose (from hibernating ground squirrel) and heart tissue (mouse), both known to be rich in mitochondria and thus cytochrome c in addition to cytchromes b and a/a3, and in the case of heart, myoglobin. Brown adipose and heart tissues were collected from animals in accordance with a Colorado State University Institutional Animal Care and Use Committee approved protocol #14-5137A. Tissue was flash frozen in LN2 prior to storage at −80 °C, and cyrosectioned at −15 °C at 20 μm in thickness. For each field of view, a series of images at different pump-probe delays were acquired (a delay stack), as shown in Fig. 6. As expected, a transient absorption response is observed in the tissue (which has a faint brown hue in the conventional brightfield image), and is absent outside the tissue. The transient absorption response has a peak around 0.5 ps, and rapidly decays thereafter. There is a small signal at -1.0 ps, which may reflect a process with nanosecond (or longer) relaxation times (possibly a long-lived electronic state or thermal lensing) [22, 50].
Fig. 6.

Transient absorption image delay stack of brown adipose tissue. Negative signal shown in black, positive signal white. Conventional brightfield microscope image shown for comparison, exhibits characteristic brown color from heme content. Scale bar 500 μm. Colorbar units arbitrary.
The pump-probe signal in Fig. 6 necessitates an optical interaction with pigments that absorb at the pump wavelength (532 nm); in brown adipose tissue, the only candidates are the various mitochondrial cytochromes and hemoglobin from the blood supply, both of which contribute to the characteristic brown color [51], which is evident in the brightfield image in Fig. 6. We estimate, based on recent measurements of blood vessel density in brown adipose tissue [52], that the relative contribution of hemoglobin in this pump-probe image is only ∼ 2%. Therefore, the overwhelming majority of the signal is attributable to respiratory chain cytochromes, particularly cytochromes b and c, whose absorption peaks overlap with our pump wavelength [15].
The averaged, peak-normalized responses for two brown adipose tissue delay stacks and two heart tissue delay stacks are shown in Fig. 7. Differences between the two types of tissue can be observed in the 2–5 ps window. The averaged response for heart tissue lies about 1σ below the distribution for brown adipose tissue, and vice-versa. One possible explanation for these differences is the presence of myoglobin in heart tissue (which is absent in adipose tissue).
Fig. 7.
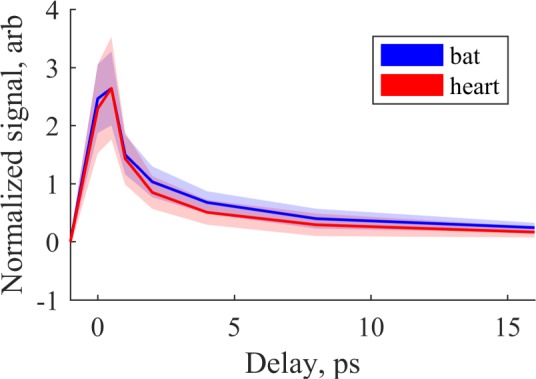
Averaged pump-probe response from image stacks of brown adipose and heart tissue, after 9 × 9 binning and automatic foreground/background separation with the MATLAB imbinarize() function. Signals are offset-corrected and normalized by root-mean-square. Shaded area indicates region within ± 1 standard deviation of the mean for each probe delay.
In order to determine whether the differences in signal were sufficient to differentiate tissue type, we performed nonnegative least-squares fitting (using the MATLAB lsqnonneg() function) of each pixel to the representative curves for heart and brown adipose tissue shown in Fig 7, plus positive and negative offset terms. Before fitting, 9×9 binning was performed to increase SNR by a factor of 9. In essence, this produces an estimate of concentrations [BAT] and [heart] for each pixel. The results are shown in Fig 8, where each pixel is colored blue in proportion to its estimated similarity to brown adipose tissue, and red in proportion to estimated similarity to heart muscle. Though imperfect, the brown adipose tissue shows up generally more blue, while the muscle tissue shows up more red, indicating that the pump-probe response at these wavelengths has the potential to classify unstained tissue. Further evidence of this is given by the box plots in Fig. 8(b), which shows the distribution of y = [BAT]/([heart] + [BAT]) for each pixel above threshold. A Wilcoxon rank-sum test confirms these are different distributions with p-value of 5 × 10−61. Furthermore, a simple classifier was constructed that categorized y ≥ 0.5 as brown adipose tissue, and y < 0.5 as heart tissue. Individual pixels from the brown adipose tissue images were classified correctly at a 79% rate, with a 21% misclassification rate; individual pixels from the heart tissue images were classified correctly at a 66% rate with a 34% misclassification rate.
Fig. 8.

(left) Pixel-by-pixel non-negative least-squares fitting to representative response for brown adipose tissue (BAT) and heart tissue, after 9 × 9 binning. Magnitude (arbitrary scale) of fit to heart response in red, fit to BAT in blue. (right) Boxplot of normalized fit coefficients by tissue type. p = 5 × 10−61.
5. Conclusions and outlook
Here we have demonstrated pump-probe microscopy is capable of generating label-free images of mitochondria-rich tissue. The pump-probe response we measured from tissue was likely a combination of responses from cytochromes a/a3, b, c and myoglobin, though it should be noted the ∼ 5 ps excited-state lifetime is similar to that which we measured for cytochrome c. This was enabled by a high repetition-rate ultrafast source with suitable wavelengths for exciting and probing in the vicinity of the Q-band of heme proteins. We have demonstrated that the approach of modest supercontinuum generation followed by independent frequency doubling stages is a viable alternative to a pair of frequency-doubled OPOs, for these experiments. By sealing the ends of the PCF and enclosing the fiber cocktail between collimators, we achieved robust, stable day-to-day operation. Currently, the limiting factor in SNR is the relative intensity noise of the visible-wavelength output. This problem can be mitigated with a balanced detector, which we demonstrated to effectively lower the noise floor to near the shot noise limit. To carry over this solution for imaging will require an auto balanced circuit with sufficient bandwidth and dynamic range to keep up with variations in absorption and scattering as the beam scans across the sample; such high-performance autobalanced detectors are not commercially available, but working designs have been published [42]. Furthermore, we expect that significant improvements in noise characteristics and pulse compressibility can be achieved by by manufacturing a short, sealed collimator-PCF-collimator, to restrict fiber propagation to below the wave-breaking length. With these improvements, this fiber-based system may become a viable alternative to visible-wavelength OPOs for a wide range of applications.
The results in Fig. 8 show that the differences in transient absorption response between brown adipose and heart muscle tissue are great enough to permit some degree of separation. Further improvements in SNR are expected to produce greater separation between tissue types. However, it still remains to be seen whether pump-probe microscopy can sufficiently differentiate redox states of respiratory chain hemes in a tissue imaging scenario. The results in Fig. 4 show that, given the current limit on SNR, reduced and oxidized cytochrome c differ too little in lifetime to be distinguished by a measurement at λpu = 532 nm, λpr = 488 nm. However, previous experiments at a 1 kHz repetition rate [43] indicate it is likely that other wavelength combinations will yield better redox contrast. This will require extending the tuning range of our system with a different set of PPLN crystals (the output wavelength is set by the PPLN poling period and temperature). In addition, the presence of lifetime components as short as 0.2 ps (Table 2) indicates that dispersion compensation following the PPLN stage may be worthwhile. It should also be noted that though some differences were noted in lifetime between redox states of cytochrome c with our system (Fig. 4), they were much more subtle than expected from Ref [43]. This is not the first instance of disagreement in measuring transient absorption at kHz versus MHz repetition rates [44]. Possible explanations include buildup of long-lived states that do not completely relax on the timescale of our pulse repetition rate, and also the accumulation of heat. While heat accumulation can be mitigated by a fast galvo-scanning microscope, contributions from long-lived electronic states requires further study. Long-lived electronic states, however are unlikely, given a lack of evidence from prior studies [43]. Also note that residual heating and photodamage will be dependent on thermal conductivity, and will be different between bulk solution and different types of tissue. Once these limitations are overcome, transient absorption imaging of cytochromes should in principle be extendable to thick tissue, through detection of backscattered probe light [7,53].
In conclusion, we have developed a laser source that could lead to a new approach for label-free metabolic microscopy. Here we have demonstrated the use of a fiber-based laser system for obtaining pump-probe images from mitochondrial respiratory chain hemes, and laid out a number of options to further refine the technique and obtain better contrast between heme redox states and different tissue types. This could serve as a replacement for dyes such as TMRM, which require high concentrations for imaging in thick tissue in vivo [54], which introduces problems with heterogeneous dye loading [54] and phototoxicity [55], by providing a redox measurement with which to calculate membrane potential [56]. Even without redox sensitivity, pump-probe imaging could be applied for, e.g., tracking spatial dynamics of cytochrome c release during apoptosis, as an alternative to genetically expressed fluorescent labels, which are bulky (∼ 27 kDa) compared to cytochrome c (∼ 12 kDa) and may interfere with the motion under study [20]. Therefore, pump-probe imaging would be a valuable extension to current label-free metabolic imaging methods.
Acknowledgments
The authors would like to thank Jennifer Ogilvie for discussions on pump-probe microscopy of heme proteins and Robert Balaban for discussions on mitochondria and cytochrome imaging. Tissue cryosections were prepared by Caleb Worker.
Funding
We gratefully acknowledge funding support from Colorado State University.
References and links
- 1.Zipfel W. R., Williams R. M., Webb W. W., “Nonlinear magic: multiphoton microscopy in the biosciences,” Nature Biotech. 21, 1369–1377 (2003). 10.1038/nbt899 [DOI] [PubMed] [Google Scholar]
- 2.Cheng J.-X., Xie X. S., “Vibrational spectroscopic imaging of living systems: An emerging platform for biology and medicine,” Science 350, aaa8870 (2015). 10.1126/science.aaa8870 [DOI] [PubMed] [Google Scholar]
- 3.Nedosekin D. A., Galanzha E. I., Ayyadevara S., Shmookler Reis R. J., Zharov V. P., “Photothermal confocal spectromicroscopy of multiple cellular chromophores and fluorophores,” Biophys. J. 102, 672–681 (2012). 10.1016/j.bpj.2011.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu S., “Emerging concepts in functional and molecular photoacoustic imaging,” Curr. Opin. Chem. Biol. 33, 25–31 (2016). 10.1016/j.cbpa.2016.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grumstrup E. M., Gabriel M. M., Cating E. E., Van Goethem E. M., Papanikolas J. M., “Pump-Probe Microscopy: Visualization and Spectroscopy of Ultrafast Dynamics at the Nanoscale,” Chem. Phys. 458, 30–40 (2015). 10.1016/j.chemphys.2015.07.006 [DOI] [Google Scholar]
- 6.Davydova D., de la Cadena A., Akimov D., Dietzek B., “Transient absorption microscopy: advances in chemical imaging of photoinduced dynamics,” Las. Photon. Rev. 10, 62–81 (2015). 10.1002/lpor.201500181 [DOI] [Google Scholar]
- 7.Fischer M. C., Wilson J. W., Robles F. E., Warren W. S., “Invited Review Article: Pump-probe microscopy,” Rev. Sci. Instrum. 87, 031101 (2016). 10.1063/1.4943211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berera R., van Grondelle R., Kennis J. T. M., “Ultrafast transient absorption spectroscopy: Principles and application to photosynthetic systems,” Photosyn. Res. 101, 105–118 (2009). 10.1007/s11120-009-9454-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruckebusch C., Sliwa M., Pernot P., de Juan a., Tauler R., “Comprehensive data analysis of femtosecond transient absorption spectra: A review,” J. Photochem. Photobiol., C Photochem. Rev. 13, 1–27 (2012). 10.1016/j.jphotochemrev.2011.10.002 [DOI] [Google Scholar]
- 10.Becker W., “Fluorescence lifetime imaging - techniques and applications,” J. of Microsc. 247, 119–136 (2012). 10.1111/j.1365-2818.2012.03618.x [DOI] [PubMed] [Google Scholar]
- 11.Suhling K., Hirvonen L. M., Levitt J. A., Chung P.-H., Tregido C., le Marois A., Rusakov D. A., Zheng K., Ameer-Beg S., Poland S., Coelho S., Dimble R., “Fluorescence Lifetime Imaging (FLIM): Basic Concepts and Recent Applications BT - Advanced Time-Correlated Single Photon Counting Applications,” in “Advanced Time-Correlated Single Photon Counting Applications,” Becker W., ed. (Springer International Publishing, 2015), chap. 3, pp. 119–188. 10.1007/978-3-319-14929-5_3 [DOI] [Google Scholar]
- 12.Matthews T. E., Wilson J. W., Degan S., Simpson M. J., Jin J. Y., Zhang J. Y., Warren W. S., “In vivo and ex vivo epimode pump-probe imaging of melanin and microvasculature,” Biomed. Opt. Express 2, 1576–1583 (2011). 10.1364/BOE.2.001576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fu D., Ye T., Matthews T. E., Chen B. J., Yurtserver G., Warren W. S., “High-resolution in vivo imaging of blood vessels without labeling,” Opt. Lett. 32, 2641–2643 (2007). 10.1364/OL.32.002641 [DOI] [PubMed] [Google Scholar]
- 14.Spinazzi M., Casarin A., Pertegato V., Salviati L., Angelini C., “Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells,” Nature Protocols 7, 1235–1246 (2012). 10.1038/nprot.2012.058 [DOI] [PubMed] [Google Scholar]
- 15.Chess D. J., Billings E., Covian R., Glancy B., French S., Taylor J., De Bari H., Murphy E., Balaban R. S., “Optical spectroscopy in turbid media using an integrating sphere: Mitochondrial chromophore analysis during metabolic transitions,” Anal. Biochem. 439, 161–172 (2013). 10.1016/j.ab.2013.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang S., Heikal A. A., Webb W. W., “Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein,” Biophys. J. 82, 2811–2825 (2002). 10.1016/S0006-3495(02)75621-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinn K. P., Sridharan G. V., Hayden R. S., Kaplan D. L., Lee K., Georgakoudi I., “Quantitative metabolic imaging using endogenous fluorescence to detect stem cell differentiation,” Sci. Rep. 3, 3432 (2013). 10.1038/srep03432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Walsh A. J., Cook R. S., Manning H. C., Hicks D. J., Lafontant A., Arteaga C. L., Skala M. C., “Optical metabolic imaging identifies glycolytic levels, subtypes, and early-treatment response in breast cancer,” Cancer Res. 73, 6164–6174 (2013). 10.1158/0008-5472.CAN-13-0527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang C., Zhang Y. S., Yao D.-K., Xia Y., Wang L. V., “Label-free photoacoustic microscopy of cytochromes,” J. Biomed. Opt. 18, 20504 (2013). 10.1117/1.JBO.18.2.020504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okada M., Smith N. I., Palonpon A. F., Endo H., Kawata S., Sodeoka M., Fujita K., “Label-free Raman observation of cytochrome c dynamics during apoptosis,” PNAS 109, 28–32 (2012). 10.1073/pnas.1107524108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fu D., Matthews T. E., Ye T., Piletic I. R., Warren W. S., “Label-free in vivo optical imaging of microvasculature and oxygenation level,” J. Biomed. Opt. 13, 040503 (2008). 10.1117/1.2968260 [DOI] [PubMed] [Google Scholar]
- 22.Flynn D. C., Bhagwat A. R., Ogilvie J. P., “Chemical-contrast imaging with pulse-shaping based pump-probe spectroscopy,” in “Multiphoton Microscopy in the Biomedical Sciences XIII,”, vol. 8588Periasamy A., König K., So P. T. C., eds. (SPIE, San Francisco, California, USA, 2013), vol. 8588, p. 85881Z. 10.1117/12.2003218 [DOI] [Google Scholar]
- 23.Alfano R. R., The Supercontinuum Laser Source (Springer-Verlag, 2016), 3rd ed. 10.1007/978-1-4939-3326-6 [DOI] [Google Scholar]
- 24.Cerullo G., De Silvestri S., “Ultrafast optical parametric amplifiers,” Rev. Sci. Instrum. 74, 1–18 (2003). 10.1063/1.1523642 [DOI] [Google Scholar]
- 25.Dudley J. M., Coen S., Genty G., “Supercontinuum generation in photonic crystal fiber,” Rev. Mod. Phys. 78, 1135–1184 (2006). 10.1103/RevModPhys.78.1135 [DOI] [Google Scholar]
- 26.Zheltikov A. M., “Let there be white light: supercontinuum generation by ultrashort laser pulses,” Physics-Uspekhi 49, 605 (2006). 10.1070/PU2006v049n06ABEH005975 [DOI] [Google Scholar]
- 27.Carroll E. C., Hill M. P., Madsen D., Malley K. R., Larsen D. S., “A single source femtosecond-millisecond broadband spectrometer,” Rev. Sci. Instrum. 80, 2–5 (2009). 10.1063/1.3070516 [DOI] [PubMed] [Google Scholar]
- 28.Min W., Lu S., Chong S., Roy R., Holtom G. R., Xie X. S., “Imaging chromophores with undetectable fluorescence by stimulated emission microscopy,” Nature 461, 1105–1109 (2009). 10.1038/nature08438 [DOI] [PubMed] [Google Scholar]
- 29.Brustlein S., Ferrand P., Walther N., Brasselet S., Billaudeau C., Marguet D., Rigneault H., “Optical parametric oscillator-based light source for coherent Raman scattering microscopy: practical overvie,” J. Biomed. Opt. 16, 021106 (2011). 10.1117/1.3533311 [DOI] [PubMed] [Google Scholar]
- 30.Yang S., Adhikari S., Dobbala M., Adusumilli S., Rowley J. D., Ganikhanov F., Zhang L., Marrs G., Wysolmerski R., Spirou G., “Multi-color ultrafast laser platform for nonlinear optical imaging based on independently tunable optical parametric oscillators,” Appl. Phys. B., Lasers and Optics 111, 617–625 (2013). 10.1007/s00340-013-5381-x [DOI] [Google Scholar]
- 31.Moutzouris K., Sotier F., Adler F., Leitenstorfer a., “Sum frequency generation of continuously tunable blue pulses from a two-branch femtosecond fiber source,” Opt. Commun. 274, 417–421 (2007). 10.1016/j.optcom.2007.02.049 [DOI] [Google Scholar]
- 32.Molotokaite E., Kumar V., Manzoni C., Polli D., Cerullo G., Marangoni M., “Raman-induced Kerr effect microscopy with balanced detection,” J. Raman Spectrosc. 44, 1385–1392 (2013). 10.1002/jrs.4250 [DOI] [Google Scholar]
- 33.Domingue S. R., Bartels R. A., “Overcoming temporal polarization instabilities from the latent birefringence in all-normal dispersion, wave-breaking-extended nonlinear fiber supercontinuum generation,” Opt. Express 21, 13305–13321 (2013). 10.1364/OE.21.013305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Domingue S. R., Bartels R. A., “Nearly transform-limited sub-20-fs pulses at 1065 nm and 10 nJ enabled by a flat field ultrafast pulse shaper,” Opt. Lett. 40, 253 (2015). 10.1364/OL.40.000253 [DOI] [PubMed] [Google Scholar]
- 35.Domingue S. R., Bartels R. A., “Nonlinear fiber amplifier with tunable transform limited pulse duration from a few 100 to sub-100-fs at watt-level powers,” Opt. Lett. 39, 359–362 (2014). 10.1364/OL.39.000359 [DOI] [PubMed] [Google Scholar]
- 36.Chong A., Buckley J., Renninger W., Wise F., “All-normal-dispersion femtosecond fiber laser,” Opt. Express 14, 10095 (2006). 10.1364/OE.14.010095 [DOI] [PubMed] [Google Scholar]
- 37.Martinez O. E., “3000 times grating compressor with positive group velocity dispersion: Application to fiber compensation in 1.3–1.6 um region,” IEEE J. Quant. Electron. QE-23, 59–64 (1987). 10.1109/JQE.1987.1073201 [DOI] [Google Scholar]
- 38.Finot C., Kibler B., Provost L., Wabnitz S., “Beneficial impact of wave-breaking for coherent continuum formation in normally dispersive nonlinear fibers,” J. Opt. Soc. Am. B 25, 1938 (2008). 10.1364/JOSAB.25.001938 [DOI] [Google Scholar]
- 39.Liu Y., Tu H., Boppart S. A., “Wave-breaking-extended fiber supercontinuum generation for high compression ratio transform-limited pulse compression,” Opt. Lett. 37, 2172 (2012). 10.1364/OL.37.002172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heidt A. M., Rothhardt J., Hartung A., Bartelt H., Rohwer E. G., Limpert J., Tünnermann A., “High quality sub-two cycle pulses from compression of supercontinuum generated in all-normal dispersion photonic crystal fiber,” Opt. Express 19, 13873–13879 (2011). 10.1364/OE.19.013873 [DOI] [PubMed] [Google Scholar]
- 41.Boyd G. D., Kleinman D. A., “Parametric interaction of focused Gaussian light beams,” J. Appl. Phys. 39, 3597–3639 (1968). 10.1063/1.1656831 [DOI] [Google Scholar]
- 42.Freudiger C. W., Yang W., Holtom G. R., Peyghambarian N., Xie X. S., Kieu K. Q., “Stimulated Raman scattering microscopy with a robust fibre laser source,” Nat. Photonics 8, 153–159 (2014). 10.1038/nphoton.2013.360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Consani C., Bräm O., Van Mourik F., Cannizzo A., Chergui M., “Energy transfer and relaxation mechanisms in Cytochrome c,” Chem. Phys. 396, 108–115 (2012). 10.1016/j.chemphys.2011.09.002 [DOI] [Google Scholar]
- 44.Park J. K., Fischer M. C., Susumu K., Therien M. J., Warren W. S., “Femtosecond pulse train shaping improves two-photon excited fluorescence measurements,” Opt. Lett. 39, 5606–5609 (2014). 10.1364/OL.39.005606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Beek-Harmsen B. J., “Immunohistochemical Determination of Cytosolic Cytochrome c Concentration in Cardiomyocytes,” J. Histochem. Cytochem. 53, 803–807 (2005). 10.1369/jhc.4C6527.2005 [DOI] [PubMed] [Google Scholar]
- 46.Van Gelder B., Slater E. C., “The extinction coefficient of cytochrome c,” Biochimica et Biophysica Acta 58, 593–595 (1962). 10.1016/0006-3002(62)90073-2 [DOI] [PubMed] [Google Scholar]
- 47.Rodrigues D. B., Maccarini P. F., Louie S., Colebeck E., Topsakal E., Pereira P. J. S., Limão-Vieira P., Stauffer P. R., “Numerical 3D modeling of heat transfer in human tissues for microwave radiometry monitoring of brown fat metabolism,” in “Proc. SPIE 8584, Energy-based Treatment of Tissue and Assessment VII,” vol. 8584 (2013), vol. 8584, p. 85840S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Keblinski P., Cahill D. G., Bodapati A., Sullivan C. R., Taton T. A., “Limits of localized heating by electromagnetically excited nanoparticles,” J. Appl. Phys. 100, 054305 (2006). 10.1063/1.2335783 [DOI] [Google Scholar]
- 49.Wilson J., “Simulation of laser heating of brown adipose tissue (bat),” https://gitlab.com/wilsonjwcsu/batLaserHeatSim (2016).
- 50.Piletic I. R., Matthews T. E., Warren W. S., “Probing near-infrared photorelaxation pathways in eumelanins and pheomelanins,” J. Phys. Chem. A 114, 11483–11491 (2010). 10.1021/jp103608d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Flatmark T., Pedersen J. I., “Brown adipose tissue mitochondria,” Biochimica et Biophysica Acta 416, 53–103 (1975). 10.1016/0304-4173(75)90013-0 [DOI] [PubMed] [Google Scholar]
- 52.Clerte M., Baron D. M., Brouckaert P., Ernande L., Raher M. J., Flynn A. W., Picard M. H., Bloch K. D., Buys E. S., Scherrer-Crosbie M., “Brown adipose tissue blood flow and mass in obesity: a contrast ultrasound study in mice,” J. Am. Soc. Echocardiogr. 26, 1465–1473 (2013). 10.1016/j.echo.2013.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Saar B. G., Freudiger C. W., Reichman J., Stanley C. M., Holtom G. R., Xie X. S., “Video-rate molecular imaging in vivo with stimulated Raman scattering,” Science 330, 1368–1370 (2010). 10.1126/science.1197236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hall A. M., Rhodes G. J., Sandoval R. M., Corridon P. R., Molitoris B. a., “In vivo multiphoton imaging of mitochondrial structure and function during acute kidney injury,” Kidney International 83, 72–83 (2012). 10.1038/ki.2012.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perry S. W., Norman J. P., Barbieri J., Brown E. B., Gelbard H. a., “Mitochondrial membrane potential probes and the proton gradient: A practical usage guide,” BioTechniques 50, 98–115 (2011). 10.2144/000113610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim N., Ripple M. O., Springett R., “Measurement of the mitochondrial membrane potential and pH gradient from the redox poise of the hemes of the bc 1 complex,” Biophys. J. 102, 1194–1203 (2012). 10.1016/j.bpj.2012.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]