

Abstract
Direct visualization of protein-protein interactions (PPIs) at high spatial and temporal resolution in live cells is crucial for understanding the intricate and dynamic behaviors of signaling protein complexes. Recently, bimolecular fluorescence complementation (BiFC) assays have been combined with super-resolution imaging techniques including PALM and SOFI to visualize PPIs at the nanometer spatial resolution. RESOLFT nanoscopy has been proven as a powerful live-cell super-resolution imaging technique. With regard to the detection and visualization of PPIs in live cells with high temporal and spatial resolution, here we developed a BiFC assay using split rsEGFP2, a highly photostable and reversibly photoswitchable fluorescent protein previously developed for RESOLFT nanoscopy. Combined with parallelized RESOLFT microscopy, we demonstrated the high spatiotemporal resolving capability of a rsEGFP2-based BiFC assay by detecting and visualizing specifically the heterodimerization interactions between Bcl-xL and Bak as well as the dynamics of the complex on mitochondria membrane in live cells.
OCIS codes: (170.3880) Medical and biological imaging, (170.2520) Fluorescence microscopy, (170.1530) Cell analysis, (170.1420) Biology
1. Introduction
Visualizing the dynamic process of protein-protein interactions (PPIs) at high spatiotemporal resolution is of great importance for understanding molecular mechanisms [1, 2] in live cells. To date, a number of approaches have been developed for PPIs detection, including yeast two-hybrid system [3], co-immunoprecipitation [4], GST-pull down [5], FRET [6] and bimolecular fluorescence complementation (BiFC) [7, 8]. Based on the complementary reconstitution of a functional fluorescent protein from its split non-fluorescent fragments, BiFC is known for its superb sensitivity in visualizing PPIs in live cells [9]. However, the spatial resolution of conventional fluorescence microscopy (~250 nm) is limited by the diffraction of light, which precludes the detection of individual PPIs which are often spatially organized in sub-micron domains [10]. Recently, BiFC assay has been combined with super-resolution techniques to overcome the diffraction limit. For instance, by splitting photoconvertible or photoactivable fluorescent proteins, BiFC-PALM is able to probe PPIs with nanometer spatial resolution [11, 12]. However, BiFC-PALM is mainly used for detection of PPIs in fixed cells or tracking of single molecules in live cells [13]. BiFC assay has also been combined with stochastic optical fluctuation imaging (SOFI) to super-resolve the dynamics of STIM1 and ORAI1 in endoplasmic reticulum (ER)-plasma membrane [13]. As the BiFC-SOFI (refSOFI) approach [13] needs post-imaging processing and analysis to obtain the super-resolution images, it thus can’t offer real-time observation of PPIs in live cells. RESOLFT nanoscopy has been proven as a powerful live-cell super-resolution imaging technique [14–18]. In this report, we developed BiFC assay to achieve high resolution, real time detection of PPIs in live cells based on split rsEGFP2, a highly photostable and reversibly photoswitchable fluorescent protein with fast and efficient maturation previously applied in RESOLFT nanoscopy [15]. As a proof of concept, through combing with parallelized RESOLFT microscopy, we demonstrated the high spatiotemporal resolving capability of rsEGFP2-based BiFC assay by visualizing specifically the heterodimerization interactions between members of the Bcl-2 family proteins Bcl-xL and Bak [19], as well as the dynamics of the complex on mitochondria membrane in live HeLa cells. Thus, our newly developed reversibly photoswitchable fluorescent protein rsEGFP2-based BiFC assay not only expands the fluorescent protein toolbox available for BiFC but also facilitates the detection and visualization of PPIs at super-resolution level in live cells.
2. Materials and methods
2.1 Plasmids construction
To construct pBiFC-rsEGFP2-N plasmid, the cDNA sequence of rsEGFP2 (residues 1-158aa) was amplified by PCR and insert into the pBiFC-VN173 plasmid (provided by Dr. Chang-Deng Hu, Purdue University) with XbaI and BamHI sites to replace VN173 sequence. To construct pBiFC-rsEGFP2-C plasmid, the cDNA sequence of rsEGFP2 (residues 159-238aa) was amplified by PCR and insert into the pBiFC-VC155 plasmid (provided by Dr. Chang-Deng Hu, Purdue University) with XhoI and NotI sites to replace VC155 sequence. To construct pBiFC-β-Jun-rsEGFP2-N plasmid, the β-Jun (c-Jun (residues 257-334aa)) cDNA sequence was amplified by PCR and inserted into pBiFC-rsEGFP2-N plasmid by EcoRI and XbaI sites. To construct pBiFC-β-Fos-rsEGFP2-C plasmid, the β-Fos (c-Fos (residues 118-211aa)) cDNA sequence was amplified by PCR and inserted into pBiFC-rsEGFP2-C plasmid by EcoRI and KpnI sites. Accordingly, the pBiFC-β-Fos (Δ Zip)-rsEGFP2-C plasmid was constructed by inserting the interaction domain (179–193aa) truncated β-Fos into the pBiFC-rsEGFP2-C plasmid by EcoRI and KpnI sites. To construct pBiFC-FKBP-rsEGFP2-N plasmid, the cDNA sequence of FKBP was amplified by PCR and insert into pBiFC-rsEGFP2-N plasmid by EcoRI and XbaI sites. To construct pBiFC-FRB-rsEGFP2-C plasmid, the cDNA sequence of FRB was amplified by PCR and insert into pBiFC-rsEGFP2-C plasmid by EcoRI and KpnI sites. To construct pBiFC-Bak-rsEGFP2-N plasmid, the cDNA sequence of Bak was amplified by PCR and insert into the pBiFC-rsEGFP2-N plasmid by EcoRI and XbaI sites. Accordingly, the interaction domain deleted Bak cDNA sequence Bak (delete) was also inserted into the pBiFC-rsEGFP2-N plasmid with the same restriction enzyme sites. To construct pBiFC-Bcl-xL-rsEGFP2-C plasmid, the cDNA sequence of Bcl-xL was also amplified by PCR and insert into the pBiFC-rsEGFP2-C plasmid with EcoRI and KpnI sites. The flexible linker between β-Jun or Bak or Bak (delete) and rsEGFP2-N is RSIAT while the flexible linker between β-Fos or β-Fos (Δ Zip) or Bcl-xL and rsEGFP2-C is RPACKIPNDLKQKVMNH respectively. To construct CFP-Bak and CFP-Bak (delete) expression plasmid, the BH3 domain of Bak and interaction domain deleted Bak cDNA was amplified by PCR and insert into the HindIII and BamHI sites of pECFP-C1plasmid (Clontech). To construct YFP-Bcl-xL expression plasmid, the full length Bcl-xL was amplified by PCR and insert into the pEYFP-C1plasmid (Clontech). The vector pECFP-YFP coding for the CFP-YFP fusion protein was generated by inserting YFP cDNA into pECFP-C1vector using a spacer peptide GASTVPRARDPPVAT between CFP and YFP protein [20]. All the constructs were sequenced to ensure correct reading frame, orientation and sequences.
2.2 Cell culture and transfection
HeLa cell line was grown in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and antibiotics in a 5% CO2 incubator. Exponentially growing cells were dispersed with trypsin, seeded at 2 × 105 cells/35mm glass bottom dish in 1.5 mL of culture medium. The transfection of fusion protein constructs were carried out using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s protocol. After transfection, the cells were grown in DMEM complete medium for 24-72 h. For RESOLFT imaging, cells were grown in IMEM (Gibco, Eggenstein, Germany) or DMEM complete medium without phenol red.
2.3 Analysis of the switching kinetics of reconstituted β-Fos-β-Jun-rsEGFP2 and full length rsEGFP2
To analyze the switching kinetics of reconstituted β-Fos-β-Jun-rsEGFP2 and full length rsEGFP2 fluorescent protein, we used an inverted fluorescent microscopy (Olympus, IX81) to acquire images. HeLa cells expressing rsEGFP2 or β-Fos-β-Jun-rsEGFP2 separately were imaged. To activate and off-switch rsEGFP2, we used 405 nm light (25 W/cm2) and 488 nm light (60 W/cm2) alternatively. The time constant (τ) for off-switching was determined by fitting the curves with a single exponential function. The residue fluorescence during each switching cycle in live cells was calculated and presented as a percentage value of remaining fluorescence intensity of on-state. The photobleaching of reconstituted β-Fos-β-Jun-rsEGFP2 and full length rsEGFP2 were measured in live HeLa cells by alternating irradiation with 405nm light for on-switching (0.1 kW/cm2, 2ms) and 488nm light for off-switching (1 kW/cm2, 24ms) for each cycle.
2.4 Characterization in mammalian cells
To compare the ensemble brightness of live HeLa cells co-expressing β-Jun-rsEGFP2-N and β-Fos-rsEGFP2-C or co-expressing β-Jun-rsEGFP2-N and β-Fos (Δzip)-rsEGFP2-C, 0.5μg plasmid of pBiFC-β-Jun-rsEGFP2-N was co-transfected with 0.5μg plasmid of pBiFC-β-Fos-rsEGFP2-C or 0.5μg plasmid of β-Fos (Δzip)-rsEGFP2-C. 24 hours after transfection, the ensemble brightness of live HeLa cells was measured by fluorescence spectrophotometer SpectraMax M5 (Molecular device, USA) instrument. The expression level of indicated protein was quantified by western blot analysis. To test the rapamycin-inducible interaction of rsEGFP2-based BiFC, exponentially growing cells were dispersed with trypsin, seeded parallelly at two 35mm glass bottom dish in 2 mL of culture medium. The next day, cells in each dish were parallelly co-transfected with 0.5μg plasmid of pBiFC-FKBP-rsEGFP2-N and 0.5μg plasmid of pBiFC-FRB-rsEGFP2-C and 0.1μg plasmid of pcDNA3.1 ( + )-mCherry (internal control). 24 hours later, cells were imaged with fluorescence microscope under the same imaging conditions. Then 100 nM rapamycin was added to one dish and cells were incubated in 37°C for the next 20 hours. The green fluorescence and red fluorescence were measured by fluorescence spectrophotometer. The expression level of indicated protein was quantified by western blot analysis.
2.5 RESOLFT nanoscopic imaging
RESOLFT experiments were carried out on a commercial parallelized RESOLFT microscope (2-color RESOLFT Parallel, Abberior Instruments, Germany). A 405 nm continuous wave diode laser was used for on-switching; a 488 nm continuous wave laser was used for off-switching and fluorescence readout. A 100 × , NA 1.45 oil-immersion objective lens (IX83, Olympus) was equipped for imaging. Two independent sCMOS cameras (Hamamatsu, Flash 4.0) were equipped for two-color RESOLFT microscope. Parallelized RESOLFT images were taken using 24nm scanning step (pixel) size and (360/24)2 steps or 36nm scanning step (pixel) size and (360/36)2 steps are required for one RESOLFT frame reconstruction.
2.6 FRET measurement with three-filter microscopy
HeLa cells were plated onto 0.17 mm thick bottom glass dishes and were transiently transfected with Lipofectamine 2000 (Invitrogen) 24 hours later. The cells were washed twice with phosphate buffer saline (pH 7.4) and covered with 1 mL fresh medium. Then, images were taken with an Olympus IX81 inverted microscopy equipped with a 100×, NA = 1.45 oil immersion objective lens and cooled-coupled device. Excitation light was delivered by an X-cite light source. For imaging, the image-pro plus software version 6.0 (Media Cybernetics) was used. In most experiments, the excitation intensity was attenuated down to 25% of the maximum power of the light source. Images were acquired using 1 × 1 binning mode and 400 ms integration times. For quantitative FRET measurements, the method of sensitized FRET was previously described in detail [20, 21]. Images were acquired sequentially through YFP, FRET and CFP filter channels. Here, the filter sets used were YFP (Excitation filter: 504/12nm; Dichroic mirror: 440/521/607/700nm; Emission filter: 529/39nm; Semrock); CFP (Excitation filter: 427/10nm; Dichroic mirror: 440/521/607/700nm; Emission filter: 472/30nm; Semrock); FRET (Excitation filter: 427/10nm; Dichroic mirror: 440/521/607/700nm; Emission filter: 529/39nm; Semrock). The background images were subtracted from the raw images before carrying out FRET calculation. Corrected FRET (FC) was calculated on a pixel-by-pixel basis for the entire image using the following equation: FC = FRET – (a × YFP) – (b × CFP), where FRET, CFP and YFP correspond to background subtracted images of cells co-expressing CFP and YFP acquired through the FRET, CFP and YFP channels respectively. The “a” and “b” are the fractions of bleed-through of YFP and CFP fluorescence through the FRET filter channel, respectively. In our system, a = 0.063 ± 0.0026(n = 28), b = 0.586 ± 0.013(n = 23). To quantify FRET efficiency, we used FR = [FRET-(b × CFP)]/ (a × YFP), a relative value that varies with changes in energy transfer to quantify the FRET signal. The FRET ratio (FR) represents the fractional increase in YFP emission due to FRET. Thus, in the absence of energy transfer, FR has a predicted value of 1 [21].
2.7 Statistics
All results are expressed as means ± SD values. Data were analyzed using two-tailed Student’s t test for comparison of independent samples. Differences were considered significant at p < 0.05.
3. Results
3.1 Split rsEGFP2 for BiFC imaging in live cells
To determine the optimal cleavage site for generating non-fluorescent fragments of rsEGFP2 for BiFC imaging, we referred to EGFP which was successfully used in BiFC with a cleavage site at 158Q [22, 23] as the amino acids sequence of rsEGFP2 shares 98% identity with that of EGFP (Fig. 1(a)). We then used a previously established mVenus-based BiFC system [24] to test the reconstitution performance of split rsEGFP2. Specifically, we genetically fused the two fragments, rsEGFP2-N (residues 1-158aa) to the C-terminus of c-Jun residues 257-334aa (β-Jun) and rsEGFP2-C (residues 159-238aa) to the C-terminus of c-Fos residues 118-211aa (β-Fos), respectively. β-Fos and β-Jun are known to form a heterodimer through leucine zipper interaction, which often serves as a model system to examine the fluorescence complementation of BiFC [24]. To test the specificity of rsEGFP2-based BiFC assay, we also fused rsEGFP2-C to the C-terminus of β-Fos (Δ Zip), a β-Fos mutant with the interaction domain (residues 179–193aa) deleted (Fig. 1(b)). The flexible linker sequence was RSIAT between β-Jun and rsEGFP2-N and RPACKIPNDLKQKVMNH between β-Fos/β-Fos (Δ Zip) and rsEGFP2-C, respectively.
Fig. 1.

Sequence alignment of rsEGFP2 with EGFP and constructs design for BiFC assay. (a) Amino acids sequence alignment of rsEGFP2 (black) with EGFP(green), amino acids different from EGFP sequence are highlighted with dark red color. The 158Q splitting site of rsEGFP2 or EGFP for BiFC assay is indicated by red arrows. (b) β-Fos/β-Jun heterodimer model was used in establishing rsEGFP2-based BiFC assay. β-Jun was fused to the rsEGFP2-N with RSIAT linker while β-Fos or β-Fos(Δ Zip) was fused to the rsEGFP2-C with RPACKIPNDLKQKVMNH linker.
As shown in Fig. 2, no fluorescence signal could be detected in HeLa cells individually expressing rsEGFP2-N or rsEGFP2-C fragments (Fig. 2(a) and Fig. 2(b)). In contrast, without lower temperature pretreatment (e.g. 30°C), HeLa cells co-transfected with β-Jun-rsEGFP2-N and β-Fos-rsEGFP2-C showed strong nucleus localized fluorescence signal (Fig. 2(c)) under 37°C physiological temperature, while HeLa cells co-transfected with β-Jun-rsEGFP2-N and β-Fos (Δzip)-rsEGFP2-C showed much lower spontaneously reconstituted fluorescence signal (Fig. 2(d)), which indicated the specific BiFC signal for detecting PPIs under physiological temperature in mammalian cells using rsEGFP2-based BiFC system. Quantitatively measured by fluorescence spectrophotometer, live HeLa cells co-expressing β-Jun-rsEGFP2-N and β-Fos-rsEGFP2-C were about 6 to 8 times brighter than cells co-expressing β-Jun-rsEGFP2-N and β-Fos (Δzip)-rsEGFP2-C (Fig. 2(e) and Fig. 2(f)).
Fig. 2.

Split rsEGFP2 for BiFC analysis in live HeLa cells. HeLa cells individually expressing rsEGFP2-N fragment (a), rsEGFP2-C fragment (b), or co-expressing β-Jun-rsEGFP2-N and β-Fos-rsEGFP2-C (c) or co-expressing β-Jun-rsEGFP2-N and β-Fos(Δzip)-rsEGFP2-C (d) were imaged under a fluorescence microscope with a 100 × objective lens. The same intensity scale was applied to (c) and (d). Scale bar: 20μm. The relative brightness of cells expressing indicated plasmids were measured with fluorescence spectrophotometer in (e), *p<0.01 compared with cells expressing co-expressing β-Jun-rsEGFP2-N and β-Fos(Δzip)-rsEGFP2-C and the data were from three independent measurements. (f) Comparable expression level of the fusion proteins in (c) and (d) determined by western blotting with anti-Flag and anti-HA antibodies. (g) 5 consecutive off-switching curves of reconstituted rsEGFP2 (blue) in live HeLa cells co-expressing β-Jun-rsEGFP2-N and β-Fos-rsEGFP2-C by alternating irradiation with 405 nm light (25 W/cm2, 100 ms) and 488 nm (60 W/cm2, 5000 ms). Fluorescence was recorded only during irradiation of 488 nm light. (h) Single off-switching curve of reconstituted rsEGFP2 mediated by β-Jun/β-Fos interaction (blue) and full length rsEGFP2 (red) in live HeLa cells by alternating irradiation with 405 nm light (25 W/cm2, 100 ms) and 488 nm (60 W/cm2, 5000 ms). Fluorescence was recorded only during irradiation of 488 nm light. Each curve is an average of 10 switching cycles. (i) The photobleaching curves of reconstituted β-Fos-β-Jun-rsEGFP2 and full length rsEGFP2 were measured in live HeLa cells by alternating irradiation with 405nm light for on-switching (0.1 kW/cm2, 2ms) and 488nm light for off-switching (1 kW/cm2, 24ms) for each cycle.
Additionally, we also use rapamycin-inducible FRB/FKBP interaction system [25] to test the specificity of rsEGFP2-based BiFC in detecting PPIs under physiological temperature in live mammalian cells. We expressed complementary fragments of rsEGFP2 (rsEGFP2-N and rsEGFP2-C) fused to the C-terminal ends of FKBP and FRB in HeLa cells. As shown in Fig. 3(b) and Fig. 3(f), prior to rapamycin treatment, HeLa cells showed low fluorescence signal that was not significantly different from the scatter produced by non-transfected cells. 20 hours after addition of saturating concentration of rapamycin, the fluorescence signal at 37°C physiological temperature was significantly higher than the control sample without rapamycin addition (Fig. 3(d) and Fig. 3(h)). Quantitatively, the normalized intensity ratio increased 7 times after treatment of rapamycin (Fig. 3(i)) despite the expression levels of protein were similar (Fig. 3(j)), indicating that the fluorescence is highly interaction-inducible and the newly developed rsEGFP2-based BiFC assay can be reliably applied to detect PPIs in live cells.
Fig. 3.

Characterization of rsEGFP2-based BiFC assay by rapamycin-inducible FRB/FKBP interaction system. Two dish of HeLa cells co-transfected parallelly with the same amount of pBiFC-FKBP-rsEGFP2-N, pBiFC-FRB-rsEGFP2 and pcDNA3.1( + )-mCherry plasmids (internal control) were incubated at 37°C for 24 hours, and then the cells were imaged with the same imaging conditions (a, b, e, f).(a, e) DIC channel, (b, f) GFP channel, the intensity scale was set to the same from 150 to 500 for (b) and (f). After taking images of (a, b, e, f), 100 nM rapamycin was added to only one dish (a, b) and then the two dish of HeLa cells were maintained at 37°C for the next 20 hours and imaged with the same imaging conditions (c, d, g, h). (c, g) DIC channel, (d, h) GFP channel, the intensity scale was set to the same from 150 to 5000 for (d) and (h). Scale bar: 300μm. (i) After 20 hours rapamycin induction, the ensemble green and red fluorescence were measured by fluorescence spectrophotometer and the normalized green-to-red ratios were calculated. *p<0.01 compared with cells without rapamycin induction. The data were from three independent measurements. (j) Comparable expression level of the fusion proteins in (d) and (h) determined by western blotting with anti-Flag and anti-HA antibodies.
3.2 Photoswitching kinetics reserved for complemented rsEGFP2
To test whether complemented rsEGFP2 fluorescent protein mediated by PPIs still preserve the photoswitching kinetics that are crucial for RESOLFT imaging in live cells [15, 26], we co-transfected HeLa cells with β-Jun-rsEGFP2-N and β-Fos-rsEGFP2-C and compared the switching kinetics of complemented rsEGFP2 with that of full length rsEGFP2. As shown in Fig. 2(g), multiple photoswitching cycles were generated by alternating irradiation with 488 nm and 405 nm light and recorded in living HeLa cells expressing complemented rsEGFP2 fluorescent protein. Under the same illumination conditions, both the ensemble off-switching time constant τ(~200ms) and residual fluorescence (~8-10%) were same between complemented rsEGFP2 and full length rsEGFP2 in live cells (Fig. 2(h)). We also measured the photobleaching characteristic of rsEGFP2 and complemented rsEGFP2 fluorescent protein during 2000 times on-off switching cycles, and the curves (Fig. 2i) showed that complemented rsEGFP2 is as photostable as original full length rsEGFP2.
3.3 Detection and super-resolution visualization of Bcl-xL and Bak heterodimerization by combining parallelized RESOLFT microscopy with a rsEGFP2-based BiFC assay in live cells
In order to test whether the newly developed rsEGFP2-based BiFC assay can faithfully detect and visualize PPIs with high spatial resolution in live cells, we used the previously reported homodimerization of Lifeact targeting to polymerized G-actin [13] as a model. As shown in Fig. 4(a)-4(e), HeLa cells co-expressing Lifeact-rsEGFP2-N and Lifeact-rsEGFP2-C showed expected green fluorescence labelling F-actin filaments. The super-resolved raw RESOLFT image (Fig. 4(b) and Fig. 4(d)) of fast moving thick F-actin bundles showed an effective spatial resolution about ∼100 nanometer in live cell (Fig. 4(e)). We also detect and visualize the heterodimerization of Bcl-xL and Bak [19] in live HeLa cells. Bak was tagged to the rsEGFP2-N fragment and Bcl-xL was tagged to rsEGFP2-C fragment respectively. Truncated Bak (Bak (delete)) which is lack of BH3 interaction domain was used as a negative control. Similarly, as shown in in Fig. 4(f)-4(j), HeLa cells co-expressing Bak-rsEGFP2-N and Bcl-xL-rsEGFP2-C showed expected reconstituted green fluorescence at the mitochondria while no reconstituted fluorescence signal could be detected on cells co-expressing Bak (delete)-rsEGFP2-N and Bcl-xL-rsEGFP2-C. Compared with the conventional fluorescence images (Fig. 4(f) and Fig. 4(h)), the super-resolved RESOLFT images clearly showed that Bak and Bcl-xL heterodimerized at the membrane of mitochondria in live HeLa cells (Fig. 4(g) and Fig. 4(i)). This result is also consistent with the previous findings that proapoptotic Bak is sequestered by Bcl-xL via its BH3 domain at the mitochondrial membrane [19]. Additionally, continuous time-lapse imaging and tracking of Bak and Bcl-xL heterodimerization could also be performed in live cells (Fig. 4(k)-4(p)).
Fig. 4.

Super-resolution imaging of protein-protein interactions by combing BiFC with parallelized RESOLFT in live HeLa cells. HeLa cells co-transfected with Lifeact-rsEGFP2-N and Lifeact-rsEGFP2-C or Bak-rsEGFP2-N and Bcl-xL-rsEGFP2-C were imaged with a parallelized RESOLFT microscope. (a) Conventional wide-field fluorescence image and (b) RESOLFT image of cells expressing reconstituted rsEGFP2 signal mediated by Lifeact-rsEGFP2-N and Lifeact-rsEGFP2-C homodimerization. (c) The magnified image of boxed area of (a) and (d) the magnified image of boxed area of (b) are presented and intensity profiles measured of arrowed regions in (c) and (d) are presented in (e) with black and red curves, respectively. (f) Conventional wide-field image and (g) RESOLFT image of cells expressing reconstituted rsEGFP2 signal mediated by Bcl-xL and Bak heterodimerization. (h) The magnified image of boxed area of (f) and (i) the magnified image of boxed area of (g) are presented and intensity profiles measured of arrowed regions in (h) and (i) are presented in (j) with black and red curves, respectively. (k) Conventional wide-field image of a HeLa cell expressing reconstituted rsEGFP2 signal mediated by Bcl-xL and Bak heterodimerization. (l-p) Continuous time-lapse imaging of the HeLa cell in (k) with parallelized RESOLFT. A 405 nm continuous wave diode laser was used for on-switching (2 ms; 12 mW measured at the back focal plane of the objective, corresponding to 0.1 kW/cm2); a 488 nm continuous wave laser was used for off-switching (20 ms; 50 mW measured at the back focal plane of the objective, corresponding to 1 kW/cm2) and fluorescence readout (4 ms; 50 mW measured at the back focal plane of the objective). RESOLFT images were taken using a 24 nm scanning step (pixel) size and (360/24)2 steps were required for one RESOLFT frame in (b) and 36 nm scanning step (pixel) size and (360/36)2 steps were required for one RESOLFT frame in (g) and (l-p). Each RESOLFT frame was taken within ∼6s in (b) and ∼3s in (g) and (l-p). Scale bar: 5μm in (a) and (b),10μm in (f) and (g) and 1μm in (c),(d),(h),(i) and 5μm in (k-p). The images are displayed using linear intensity scale without adjusting γ factor. All the RESOLFT images are raw images without deconvolution.
3.4 Verification of Bcl-xL and Bak heterodimerization by three-filter FRET microscopy
To verify the heterodimerization between Bak and Bcl-xL directly in live cells, we quantitatively detect their interactions using the three-filter FRET microscopy [27–29] (Fig. 5). CFP (FRET donor: cyan fluorescent protein) was tagged to the N-terminus of Bak or Bak (delete) and YFP (FRET acceptor: yellow fluorescent protein) was tagged to the N-terminus of Bcl-xL and FRET signal were quantitatively gauged by the FR algorithm [21]. To ensure that our recording system could reliably detect FRET, we first carried out several control experiments. As shown in Fig. 5(b), cells co-expressing CFP and YFP, serving as a negative control, showed no FRET with FR = 1.30 ± 0.06 (n = 14). On the other hand, cell co-expressing the CFP-YFP concatemer, a positive control for FRET, showed significantly increased FRET signal with FR = 10.78 ± 0.35(n = 27). The corrected FRET (FC) images in Fig. 5(a) showed strong FRET signal in both cytoplasm and nucleus of cells expressing CFP–YFP fusion protein, while cells co-expressing CFP and YFP showed no FRET signal. We next detected the interaction between Bcl-xL and Bak proteins by FRET. Cells co-expressing CFP-Bak and YFP-Bcl-xL showed significantly higher FR = 4.03 ± 0.54 (n = 15) compared to cells co-expressing CFP and YFP or cells co-expressing CFP-Bak (delete) and YFP-Bcl-xL with FR = 1.30 ± 0.05 (n = 10). The FC images also showed strong FRET signal in mitochondria of live HeLa cells. These results clearly indicated that Bak interacts with Bcl-xL at the mitochondria of live cells.
Fig. 5.
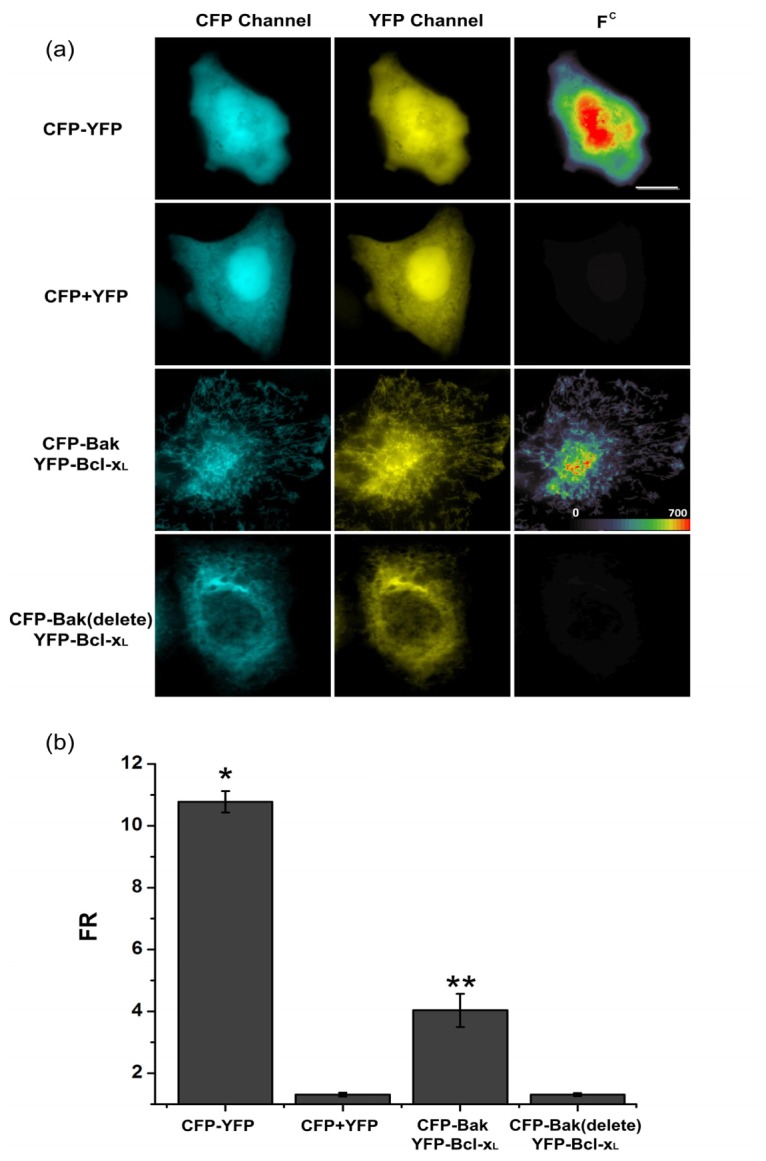
Detection of Bcl-xL and Bak heterodimerization by FRET microscopy in live cells. Detection of Bcl-xL and Bak interactions in live HeLa cells (a) cells expressing CFP-YFP or co-expressing CFP and YFP or co-expressing CFP-Bak and YFP-Bcl-xL or co-expressing CFP-Bak (delete) and YFP-Bcl-xL were imaged by three-filter FRET microscopy. Corrected FRET (FC) images were calculated as described in “materials and methods” and presented as pseudo-color images. All colors are arbitrarily assigned to indicate signal strength. HeLa cells expressing CFP–YFP served as a positive control, while cells co-expressing CFP and YFP as a negative control. Scale bar: 10μm. (b) FR values calculated from individual experimental groups. *p<0.01 compared with cells co-expressing CFP and YFP, **p<0.01 compared with cells co-expressing CFP-Bak (delete) and YFP-Bcl-xL.
4. Discussion and conclusion
In this work, we developed BiFC assay to visualize protein-protein interactions with high spatiotemporal resolution based on split rsEGFP2, a reversibly photoswitchable fluorescent protein which previously demonstrated good performance in RESOLFT nanoscopy [15]. We chose the cleave site as well as the flexible linkers of split rsEGFP2 according to the previous knowledge of EGFP as rsEGFP2 and EGFP share high sequence similarity (98%). The high specificity of the rsEGFP2-based BiFC system was then confirmed using both the classic β-Fos/β-Jun heterodimer interaction model [24] and rapamycin-inducible FRB/FKBP interaction system [25]. It is also worth noting that although we adopted the same flexible linker composition and tagging directions as the previously established mVenus-based BiFC system to detect β-Fos/β-Jun heterodimerization in live cells, it is necessary to optimize the linker length and compositions or even tagging directions between the non-fluorescent rsEGFP2 fragments and proteins of interest for specific PPIs detections [30].
Besides the apparent reconstituted fluorescence which is important for conventional protein labelling and visualization of protein-protein interactions, reversibly photoswitchable characteristics (switching speed, residual fluorescence, and photostability) of RSFPs are also crucial for RESOLFT imaging [17, 26]. We quantified these characteristics of reconstituted rsEGFP2 and compared them with those of full length rsEGFP2; the data showed that these crucial switching characteristics remain the same as original. Those results also indicated that we could apply the same RESOLFT imaging parameters for complemented rsEGFP2 mediated by protein interactions as full length rsEGFP2.
As a proof of concept, by combing with parallelized RESOLFT microscopy, we applied the newly developed rsEGFP2-based BiFC assay to visualize actin filaments labeled by homodimerization of Lifeact. Measuring the filamentous structure such as fast moving thick F-bundles shows significant improvement of spatial resolution compared with wide-field imaging (Fig. 4(e)). In order to demonstrate this assay could be reliably applied to visualize PPIs at super-resolution level in live cells, we use anti-apoptotic Bcl-xL and pro-apoptotic Bak heterodimerization as a model. Previous reports have showed that anti-apoptotic protein Bcl-xL localized to the cytosolic side of outer mitochondria membrane heterodimerized with pro-apoptotic Bak to prevent membrane permeabilization and cell apoptosis [31]. We co-transfected HeLa cells with Bcl-xL-rsEGFP2-C and Bak-rsEGFP2-N plasmids and the results clearly show that the reconstituted green fluorescence signal was mainly localized to the mitochondria. With parallelized RESOLFT imaging, Bcl-xL-Bak heterodimerized complexes could be well visualized at the mitochondria membrane which appeared blurry under conventional fluorescence imaging (Fig. 4(h)). HeLa cells co-transfected with Bcl-xL-rsEGFP2-C and Bak (delete)-rsEGFP2-N showed no reconstituted signal, indicating the specificity of rsEGFP2-based BiFC assay. We also used another classical and straightforward method, three-filter FRET microscopy, to verify our results in detecting PPIs directly in live cells [20, 27, 32]. As demonstrated, FRET signal from CFP-Bak to YFP-Bcl-xL heterodimerization could also be reliably detected. However, compared with the super-resolved RESOLFT images, the corrected FRET images (Fc) was not able to reveal the membrane localization of Bak-Bcl-xL heterodimeric complexes due to limited spatial resolution. Taken together, these results demonstrate that compared with previously established conventional fluorescent protein-based BiFC assays (e.g. mCerulean, EGFP, mVenus, et al.), our newly developed rsEGFP2-based BiFC assay can be used to detect and visualize PPIs not only with high specificity (Fig. 4(k)) but also with high spatiotemporal resolution in live cells (Fig. 4(l)-4(p)).
Recently, several super-resolution BiFC assays have been reported to detect PPIs with nanometer resolution. For example, BiFC-PALM approaches have been developed using photoactivable fluorescent protein PA-mCherry [11] or photoconvertible fluorescent protein mEos3.2 [12] and mIrisFP [33]. Compared with BiFC-PALM, rsEGFP2-based BiFC assay combing with parallelized RESOLFT microscopy can provide better temporal resolution for PPIs detection and imaging in large fields of view (100μm × 100μm) in live cells. In comparison with the recently developed BiFC-SOFI (refSOFI) approach [13] which needs post image processing and analysis to get super-resolved PPIs images, combing BiFC with parallelized RESOLFT microscopy can get super-resolved PPIs images robustly in real time [14]. Thus, rsEGFP2-based BiFC assay combing with RESOLFT microscopy can serve as a complementary strategy to the existing super-resolution techniques combined with BiFC assays to achieve real-time PPIs detection and imaging with both high spatiotemporal resolution and large fields of view in live cells.
Apart from super-resolution imaging discussed above, rsEGFP2-based BiFC assay could also rival previously established Dronpa-based BiFC assay [34, 35] in PPIs detection and tracking applications, such as tracking of organelles and intracellular molecules in living cells mainly because of excellent photostability and larger on-off contrast ratio of rsEGFP2 over Dronpa [36].
In summary, we report the first time to our knowledge the development, characterization and application of BiFC assay based on reversibly photoswitchable fluorescent protein rsEGFP2 to detect and visualize PPIs in living cells at super-resolution level by combing BiFC with RESOLFT techniques. As a proof of concept, through combing with parallelized RESOLFT microscopy we demonstrated the high spatiotemporal resolving capability rsEGFP2-based BiFC assay by detecting and visualizing specifically the heterodimerization interactions between members of the Bcl-2 family proteins, Bcl-xL and Bak as well as the dynamics of the complex on mitochondria membrane in live HeLa cells. Our newly developed rsEGFP2-based BiFC assay combing with parallelized RESOLFT microscopy not only expands the fluorescent protein toolbox available for BiFC but also facilitates the detection and visualization of PPIs at super-resolution level simultaneously with large fields of view in live cells.
Disclosures
The authors declare that there are no conflicts of interest related to this article.
Acknowledgments
The authors would like to thank Dr. Chang-Deng Hu for providing following plasmids: pBiFC-β-Jun-VN173, pBiFC-β-Fos-VC155, pBiFC-β-Fos (Δ Zip)-VC155, pBiFC-VN173, pBiFC-CrN173, pBiFC-VC155 and pBiFC-CC155.
Funding
National Natural Science Foundation of China (NSFC) (21573013, 21390412, 31271423, and 31327901); Program 863 (SS2015AA020406); Chinese Academy of Sciences (CAS) Interdisciplinary Innovation Team for Y.S.
References and links
- 1.Yang J., Wagner S. A., Beli P., “Illuminating spatial and temporal organization of protein interaction networks by mass spectrometry-based proteomics,” Front. Genet. 6, 344 (2015). 10.3389/fgene.2015.00344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Klingström T., Plewczynski D., “Protein-protein interaction and pathway databases, a graphical review,” Brief. Bioinform. 12(6), 702–713 (2011). 10.1093/bib/bbq064 [DOI] [PubMed] [Google Scholar]
- 3.Yang Q. H., Li L., “Yeast two-hybrid system and its application on proteomics,” Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao (Shanghai) 31(3), 221–225 (1999). [PubMed] [Google Scholar]
- 4.Masters S. C., “Co-immunoprecipitation from transfected cells,” Methods Mol. Biol. 261, 337–350 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Einarson M. B., Pugacheva E. N., Orlinick J. R., “GST Pull-down,” CSH protocols 2007, pdb prot4757 (2007). 10.1101/pdb.prot4757 [DOI] [PubMed] [Google Scholar]
- 6.Mahajan N. P., Linder K., Berry G., Gordon G. W., Heim R., Herman B., “Bcl-2 and Bax interactions in mitochondria probed with green fluorescent protein and fluorescence resonance energy transfer,” Nat. Biotechnol. 16(6), 547–552 (1998). 10.1038/nbt0698-547 [DOI] [PubMed] [Google Scholar]
- 7.Hu C. D., Grinberg A. V., Kerppola T. K., Visualization of Protein Interactions In Living Cells Using Bimolecular Fluorescence Complementation (BiFC) Analysis (Wiley,2005),Chap.19. [DOI] [PubMed] [Google Scholar]
- 8.Vidi P. A., Przybyla J. A., Hu C. D., Watts V. J., Visualization of G Protein-Coupled Receptor (GPCR) Interactions in Living Cells using Bimolecular Fluorescence Complementation (BiFC) (Wiley,2010),Chapter 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wong K. A., O’Bryan J. P., “Bimolecular fluorescence complementation,” J. Vis. Exp. 50, 2643 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tian T., Harding A., Inder K., Plowman S., Parton R. G., Hancock J. F., “Plasma membrane nanoswitches generate high-fidelity Ras signal transduction,” Nat. Cell Biol. 9(8), 905–914 (2007). 10.1038/ncb1615 [DOI] [PubMed] [Google Scholar]
- 11.Nickerson A., Huang T., Lin L. J., Nan X., “Photoactivated localization microscopy with bimolecular fluorescence complementation (BiFC-PALM) for nanoscale imaging of protein-protein interactions in cells,” PLoS One 9(6), e100589 (2014). 10.1371/journal.pone.0100589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Z., Xing D., Su Q. P., Zhu Y., Zhang J., Kong X., Xue B., Wang S., Sun H., Tao Y., Sun Y., “Super-resolution imaging and tracking of protein-protein interactions in sub-diffraction cellular space,” Nat. Commun. 5, 4443 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hertel F., Mo G. C., Duwé S., Dedecker P., Zhang J., “RefSOFI for Mapping Nanoscale Organization of Protein-Protein Interactions in Living Cells,” Cell Reports 14(2), 390–400 (2016). 10.1016/j.celrep.2015.12.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chmyrov A., Keller J., Grotjohann T., Ratz M., d’Este E., Jakobs S., Eggeling C., Hell S. W., “Nanoscopy with more than 100,000 ‘doughnuts’,” Nat. Methods 10(8), 737–740 (2013). 10.1038/nmeth.2556 [DOI] [PubMed] [Google Scholar]
- 15.Grotjohann T., Testa I., Reuss M., Brakemann T., Eggeling C., Hell S. W., Jakobs S., “rsEGFP2 enables fast RESOLFT nanoscopy of living cells,” eLife 1, e00248 (2012). 10.7554/eLife.00248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee S. A., Ponjavic A., Siv C., Lee S. F., Biteen J. S., “Nanoscopic cellular imaging: confinement broadens understanding,” ACS Nano 10(9), 8143–8153 (2016). 10.1021/acsnano.6b02863 [DOI] [PubMed] [Google Scholar]
- 17.Wang S., Chen X., Chang L., Xue R., Duan H., Sun Y., “GMars-Q enables long-term live-cell parallelized reversible saturable optical fluorescence transitions nanoscopy,” ACS Nano 10(10), 9136–9144 (2016). 10.1021/acsnano.6b04254 [DOI] [PubMed] [Google Scholar]
- 18.Hofmann M., Eggeling C., Jakobs S., Hell S. W., “Breaking the diffraction barrier in fluorescence microscopy at low light intensities by using reversibly photoswitchable proteins,” Proc. Natl. Acad. Sci. U.S.A. 102(49), 17565–17569 (2005). 10.1073/pnas.0506010102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Willis S. N., Chen L., Dewson G., Wei A., Naik E., Fletcher J. I., Adams J. M., Huang D. C., “Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins,” Genes Dev. 19(11), 1294–1305 (2005). 10.1101/gad.1304105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiu S., Hua Y. L., Yang F., Chen Y. Z., Luo J. H., “Subunit assembly of N-methyl-d-aspartate receptors analyzed by fluorescence resonance energy transfer,” J. Biol. Chem. 280(26), 24923–24930 (2005). 10.1074/jbc.M413915200 [DOI] [PubMed] [Google Scholar]
- 21.Erickson M. G., Alseikhan B. A., Peterson B. Z., Yue D. T., “Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells,” Neuron 31(6), 973–985 (2001). 10.1016/S0896-6273(01)00438-X [DOI] [PubMed] [Google Scholar]
- 22.Valencia-Burton M., McCullough R. M., Cantor C. R., Broude N. E., “RNA visualization in live bacterial cells using fluorescent protein complementation,” Nat. Methods 4(5), 421–427 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Ozawa T., Natori Y., Sato M., Umezawa Y., “Imaging dynamics of endogenous mitochondrial RNA in single living cells,” Nat. Methods 4(5), 413–419 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Shyu Y. J., Liu H., Deng X., Hu C. D., “Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions,” Biotechniques 40(1), 61–66 (2006). 10.2144/000112036 [DOI] [PubMed] [Google Scholar]
- 25.Banaszynski L. A., Liu C. W., Wandless T. J., “Characterization of the FKBP.rapamycin.FRB ternary complex,” J. Am. Chem. Soc. 127(13), 4715–4721 (2005). 10.1021/ja043277y [DOI] [PubMed] [Google Scholar]
- 26.Shcherbakova D. M., Sengupta P., Lippincott-Schwartz J., Verkhusha V. V., “Photocontrollable fluorescent proteins for superresolution imaging,” Annu. Rev. Biophys. 43(1), 303–329 (2014). 10.1146/annurev-biophys-051013-022836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S., Li K. J., Lin X. W., Jiang C. Z., Chen D. H., Wu Q., Hua Z. C., “Using c-Fos/c-Jun as hetero-dimer interaction model to optimize donor to acceptor concentration ratio range for three-filter fluorescence resonance energy transfer (FRET) measurement,” J. Microsc. 248(1), 58–65 (2012). 10.1111/j.1365-2818.2012.03650.x [DOI] [PubMed] [Google Scholar]
- 28.Verveer P. J., Rocks O., Harpur A. G., Bastiaens P. I., “Measuring FRET by sensitized emission,” CSH protocols 2006(2006). 10.1101/pdb.ip14 [DOI] [PubMed] [Google Scholar]
- 29.Chen Y., Periasamy A., “Intensity range based quantitative FRET data analysis to localize protein molecules in live cell nuclei,” J. Fluoresc. 16(1), 95–104 (2006). 10.1007/s10895-005-0024-1 [DOI] [PubMed] [Google Scholar]
- 30.Hu C. D., Kerppola T. K., “Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis,” Nat. Biotechnol. 21(5), 539–545 (2003). 10.1038/nbt816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flores M. L., Castilla C., Ávila R., Ruiz-Borrego M., Sáez C., Japón M. A., “Paclitaxel sensitivity of breast cancer cells requires efficient mitotic arrest and disruption of Bcl-xL/Bak interaction,” Breast Cancer Res. Treat. 133(3), 917–928 (2012). 10.1007/s10549-011-1864-9 [DOI] [PubMed] [Google Scholar]
- 32.Wang S., Chen Y., Wu Q., Hua Z. C., “Detection of Fas-associated death domain and its variants’ self-association by fluorescence resonance energy transfer in living cells,” Mol. Imaging 12(2), 111–120 (2013). [PubMed] [Google Scholar]
- 33.Chen M., Liu S., Li W., Zhang Z., Zhang X., Zhang X. E., Cui Z., “Three-fragment fluorescence complementation coupled with photoactivated localization microscopy for nanoscale imaging of ternary complexes,” ACS Nano 10(9), 8482–8490 (2016). 10.1021/acsnano.6b03543 [DOI] [PubMed] [Google Scholar]
- 34.Lee Y. R., Park J. H., Hahm S. H., Kang L. W., Chung J. H., Nam K. H., Hwang K. Y., Kwon I. C., Han Y. S., “Development of bimolecular fluorescence complementation using Dronpa for visualization of protein-protein interactions in cells,” Mol. Imaging Biol. 12(5), 468–478 (2010). 10.1007/s11307-010-0312-2 [DOI] [PubMed] [Google Scholar]
- 35.Agustina L., Hahm S. H., Han S. H., Tran A. H., Chung J. H., Park J. H., Park J. W., Han Y. S., “Visualization of the physical and functional interaction between hMYH and hRad9 by Dronpa bimolecular fluorescence complementation,” BMC Mol. Biol. 15(1), 17 (2014). 10.1186/1471-2199-15-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X., Zhang M., Li D., He W., Peng J., Betzig E., Xu P., “Highly photostable, reversibly photoswitchable fluorescent protein with high contrast ratio for live-cell superresolution microscopy,” Proc. Natl. Acad. Sci. U.S.A. 113(37), 10364–10369 (2016). 10.1073/pnas.1611038113 [DOI] [PMC free article] [PubMed] [Google Scholar]