

Abstract
The genomes of the type B3 bacteriophages that infect Caulobacter crescentus are among the largest phage genomes thus far deposited into GenBank with sizes over 200 kb. In this study, we introduce six new bacteriophage genomes which were obtained from phage collected from various water systems in the southeastern United States and from tropical locations across the globe. A comparative analysis of the 12 available genomes revealed a “core genome” which accounts for roughly 1/3 of these bacteriophage genomes and is predominately localized to the head, tail, and lysis gene regions. Despite being isolated from geographically distinct locations, the genomes of these bacteriophages are highly conserved in both genome sequence and gene order. We also identified the insertions, deletions, translocations, and horizontal gene transfer events which are responsible for the genomic diversity of this group of bacteriophages and demonstrated that these changes are not consistent with the idea that modular reassortment of genomes occurs in this group of bacteriophages.
Introduction
Although they are still underrepresented in the GenBank database, bacteriophages are the most abundant organisms on the planet. The majority of the sequences deposited so far are classified as Siphoviridae. This family is noted for having a non-enveloped head and a non-contractile tail. The majority of the known bacteriophages that infect Caulobacter crescentus are type B3 Siphoviridae similar to φCbK with an elongated head and a flexible tail [16]. The prototype phage φCbK has a large genome of about 205 kb and a 65% GC content [1, 13, 21]. In addition, Gill et al. [13] published an analysis of five additional φCbK-like C. crescentus bacteriophage genome sequences showing that these phage genomes vary in size and contain long terminal repeats. They also showed that these closely related genomes were organized into three primary modules, the Structural module, the Lysis module, and the DNA replication module which would be consistent with the theory of modular evolution of phage genomes. Botstein [5] proposed that bacteriophage genomes are a collection of interchangeable genetic elements (modules). Each module is responsible for a specific function and has the ability to evolve independently of the other modules in the genome. Thus, a collection of related bacteriophages would be predicted to contain favorable combinations of the available modules. Since the φCbK-like phages contain three conserved modules, we decided to sequence six additional phage genomes to determine if modular evolution could be observed among these phages which were collected from surface water samples obtained from ponds and slow-moving streams across the southeastern United States, as well as, samples from tropical fish tanks which represent diverse geographical locations since aquarium fish are captured and raised all over the world and then shipped in their own water to commercial dealers [16]. Although our comparison of 12 φCbK-like phage genomes showed no support for the modular evolution model, we did find evidence of large numbers of insertions, deletions, and gene translocations. Thus, these phage genomes appear to evolve primarily via small changes rather than by generating recombinant genomes.
Materials and Methods
Phage Isolation and Culture
C. crescentus strain CB15 was used as the bacterial host strain for the isolation and proliferation of the Cr2, Cr5, Cr10, Cr29, Cr32, and Cr34 bacteriophages in this study [16]. Modified PYE growth medium [15] was used for all liquid cultures and soft-agar overlays. All incubations were at 30 °C with aeration. Agar plates were placed in plastic bags and refrigerated immediately after hardening, to prevent drying of the agar.
DNA Sequencing and Genome Assembly
DNA extraction was performed using a Qiagen QIAamp DNA Mini Kit. DNA sequencing was performed with both the Roche 454 and Illumina MiSeq sequencing platforms. The resulting reads were aligned into contigs using the DNASTAR Seqman software program (SeqMan NGen, DNASTAR version 11; Madison, WI). The genome sequences are available in the NCBI database under accession numbers: KY555142 to KY555147. Mauve Whole Genome Alignment software [7] was used to align the contigs against the reference sequence (ϕCbK) to discern the orientation of the contigs and to assist in assembly of the phage genomes. The extent of the terminal repeats of these bacteriophage genomes was determined using the Tablet sequence viewer [18] software to identify regions with twice as many reads as described by Gill et al. [13]. The newly sequenced phage genomes were annotated using the RAST automated annotation system [4, 19] and edited in the Artemis Genome browser) [23]. For gene comparisons, we used the BlastStation 2 software (http://www.blaststation.com) and performed a BlastP search of the amino acid sequences of all coding regions for each genome. To be considered a match, the genes had to share an e-value less than e−05.
Results
The CbK-like bacteriophages Cr2, Cr5, Cr10, Cr29, Cr32, and Cr34 are highly similar to four of the bacteriophage genomes, ϕCbK, Karma, Swift, and Magneto, described by Gill et al. [13] in terms of genetic make-up and gene location (Fig. 1). However, each genome has unique insertions and deletions. The genome size of the newly sequenced phage including the repeats ranged from 216 to 229 Kb (Table 1). The BLASTn two sequence alignment program [26] was used to determine the pairwise similarity of these genomes along with the six genomes described by Gill et al. [13]. Most phage genomes were 97–99% identical to each other over 94% to 100% of their genomes (Table 2 bottom left). In contrast, the Rogue genome was 80–84% identical to most of the other phage across 81–85% of its genome, and the Colossus genome was 30% larger than the other genomes and had only 66–69% nucleotide identity across 31–33% of its genome. We also used a genome-to-genome distance calculator (http://ggdc.dsmz.de/) to compare the 12 genomes and obtained similar results (Table 2 top right). This calculator is based upon the Genome Blast Distance Phylogeny approach (GBDP) which begins with a blast+ alignment between a query and subject sequence to establish the segments of sequence which are considered HSPs (High-scoring pairs; intergenomic matches). The distances between these pairs were calculated, and then converted to percent-wise similarities analogous to DNA-DNA Hybridization [3]. The data in Table 2 are the sum of all identities found in HSPs divided by total genome length. In this comparison, Colossus had only 13% identity across its entire genome. Together these data indicate that the larger Colossus genome not only consists of 70% unique genetic material, but also the genes that it does share with the other phage have only 67% identity at the nucleotide level. Since Colossus was so different from the other phage, we excluded it from many of the subsequent analyses, but it provided important information about the core genome of these phages.
Fig. 1.

Mauve whole genome comparison of the CbK, Cr2, Cr5, CcrKarma, and CCrswift bacteriophage genomes. Red space corresponds to similar sequences. Regions that are entirely white correspond to inserted gene regions.
Table 1.
Summary of the genomic characteristics of 12 Caulobacter crescentus bacteriophage genomes.
| CbK | Ccr2 | Ccr5 | Ccr10 | Ccr29 | Ccr32 | Ccr34 | Karma | Magneto | Swift | Rogue | Colossus | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Genome Size (bp) |
215710 | 220299 | 218729 | 219348 | 229319 | 215779 | 216240 | 221828 | 218929 | 219216 | 223720 | 296667 |
| tRNA Genes | 26 | 26 | 27 | 26 | 26 | 26 | 26 | 26 | 27 | 27 | 23 | 28 |
| # of Genes | 339 | 345 | 341 | 347 | 363 | 332 | 342 | 354 | 347 | 343 | 350 | 448 |
Table 2.
Phage genome sequence comparisons. Top right: Genome-to-Genome distances expressed as percent-wise similarities. Bottom left: Percent identity in pairwise comparisons 12 bacteriophage genomes (percent coverage).
| CbK | Ccr2 | Ccr5 | Ccr10 | Ccr29 | Ccr32 | Ccr34 | Karma | Magneto | Swift | Rogue | Colossus | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CbK | 97 | 96 | 97 | 97 | 99 | 99 | 96 | 96 | 89 | 37 | 13 | |
| Ccr2 | 97 (98) | 96 | 99 | 100 | 97 | 97 | 96 | 97 | 90 | 38 | 13 | |
| Ccr5 | 98 (98) | 96 (97) | 96 | 96 | 96 | 96 | 95 | 95 | 90 | 37 | 13 | |
| Ccr10 | 97 (98) | 99 (98) | 96 (98) | 100 | 97 | 97 | 95 | 96 | 92 | 40 | 13 | |
| Ccr29 | 97 (99) | 99 (99) | 96 (98) | 99 (100) | 97 | 97 | 96 | 96 | 92 | 39 | 13 | |
| Ccr32 | 99 (100) | 97 (97) | 98 (97) | 97 (97) | 97 (97) | 100 | 96 | 96 | 91 | 38 | 13 | |
| Ccr34 | 99 (100) | 97 (97) | 98 (97) | 97 (97) | 97 (97) | 99 (100) | 96 | 96 | 91 | 37 | 13 | |
| Karma | 97 (98) | 97 (98) | 97 (98) | 97 (98) | 97 (98) | 97 (98) | 97 (98) | 98 | 94 | 38 | 13 | |
| Magneto | 97 (98) | 97 (98) | 96 (98) | 97 (97) | 97 (97) | 97 (98) | 97 (98) | 99 (97) | 93 | 38 | 13 | |
| Swift | 97 (94) | 97 (94) | 97 (94) | 97 (95) | 97 (96) | 97 (98) | 97 (96) | 98 (94) | 98 (95) | 42 | 13 | |
| Rogue | 83 (81) | 83 (81) | 80 (81) | 83 (81) | 83 (82) | 83 (82) | 83 (82) | 84 (81) | 83 (82) | 84 (85) | 13 | |
| Colossus | 67 (31) | 68 (30) | 66 (30) | 68 (30) | 68 (30) | 67 (31) | 67 (31) | 67 (30) | 67 (31) | 67 (31) | 69 (31) |
Taking advantage of the evolutionary distance between CbK and Colossus, we determined that only 110 genes were present in all 12 phage genomes. The location of these common genes is well conserved within the bacteriophage genomes of this group (Fig. 2). Even in a comparison between CbK and Colossus, the location of the common genes was found to be in highly similar segments and arrangements [13]. With regard to the pan genome of this phage family, the 10 similar genomes contain a total of only 9 genes that are unique to a single genome. In contrast, the more distantly-related Rogue and Colossus genomes make large contributions with 47 and 315 unique genes, respectively (Table 3). Other categories of the pan genome include CbK-like genes that were shared in all genomes except for Colossus, and genes which were present in all genomes except for Colossus and Rogue, were designated CbK-like (-Rogue). Other genes which were present in at least 2 genomes, and did not fall into the categories mentioned above, were classified as INDELS. The final category designated Unique included genes which were present in a single genome. The locations of the genes in these five categories are summarized in a DNA plot image of the bacteriophage CbK genome with each gene category color-coded (Fig. 3). The genomic layout for each of the other CbK-like genomes is similar.
Fig. 2.
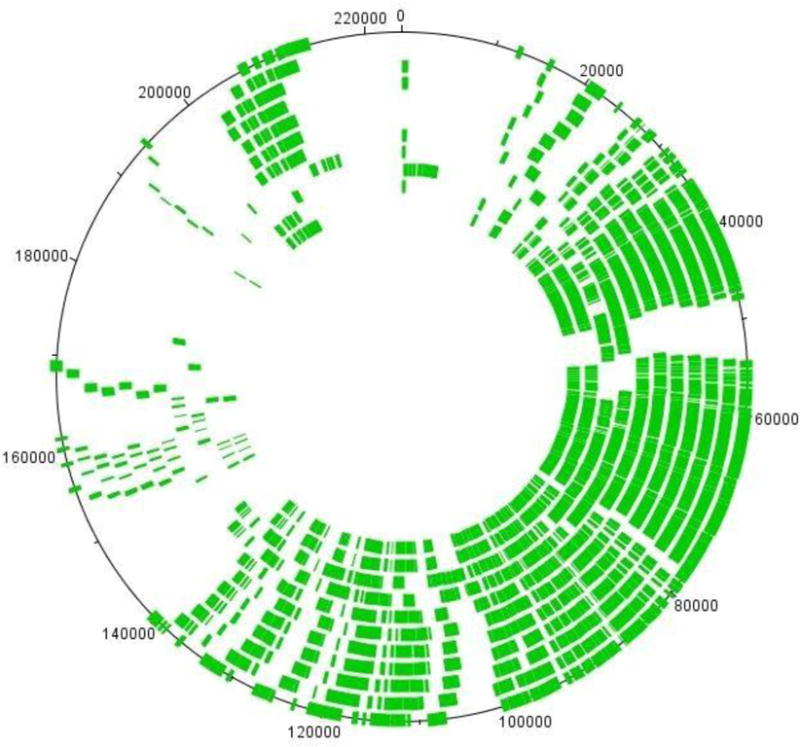
The conserved locations of genes present in all 12 CbK-like genomes. The genomes are arranged in size from largest to smallest (Table 1), with Rogue on the outside track and Cr32 on the inner most track. The Colossus genome is omitted from this comparison.
Table 3.
Classification of the bacteriophage genes based upon the prevalence of these genes among the entire group of 12 bacteriophages.
| Gene Classifications | CbK | Ccr2 | Ccr5 | Ccr10 | Ccr29 | Ccr32 | Ccr34 | Karma | Magneto | Swift | Rogue | Colossus |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Conserved Genes | 110 | 111 | 111 | 111 | 111 | 110 | 110 | 110 | 110 | 110 | 110 | 112 |
| CbK-like | 148 | 148 | 148 | 148 | 148 | 148 | 148 | 148 | 148 | 148 | 147 | N/A |
| CbK-like (-Rogue) | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | 36 | N/A | N/A |
| Indels | 45 | 50 | 44 | 52 | 68 | 38 | 47 | 58 | 49 | 48 | 46 | 21 |
| Unique | 0 | 0 | 2 | 0 | 0 | 0 | 1 | 1 | 4 | 1 | 47 | 315 |
Fig. 3.
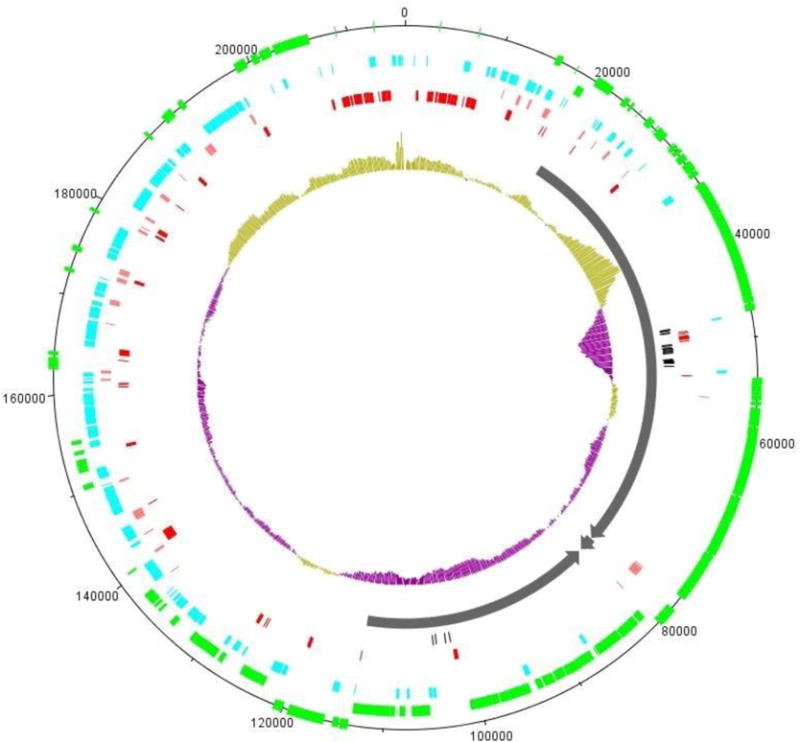
DNA plot image of the CbK genome gene categories. Gene positions correspond to the CbK genome. Starting at the outer perimeter of the circle, the green blocks represent the genes found in all 12 genomes, the light blue are conserved throughout the 11 CbK-like genomes. The pink blocks are conserved in the 10 CbK-like genomes excluding Rogue. Dark red blocks represent the indels in the CbK genome. Black blocks are the tRNA locations. The dark grey arrows represent the location of the Genomic modules, the structural module, lysis module, and DNA replication module respectively, clockwise from the top of the circle. The center plot is the GC content of the CbK genome with gold bars marking regions of above average %GC and purple bars marking the regions with below average %GC.
Discussion
Approximately 60% of the core genes are contained within the three genomic modules defined by Gill et al. [13], the structural genes, the replication genes, and the genes involved in lysis. The location of these modules is well conserved across all 12 genomes and the phylogenetic trees of the individual modules are identical to the phylogenetic tree of the wholes genomes (Fig. 4). Thus, we see no evidence of alternate combinations of modules as predicted by the theory of modular evolution [5]. This conservation of genomic modules has been observed with other phage as well. For example, the T4 bacteriophage superfamily has a highly conserved gene order with limited genetic exchange [6].
Fig. 4.
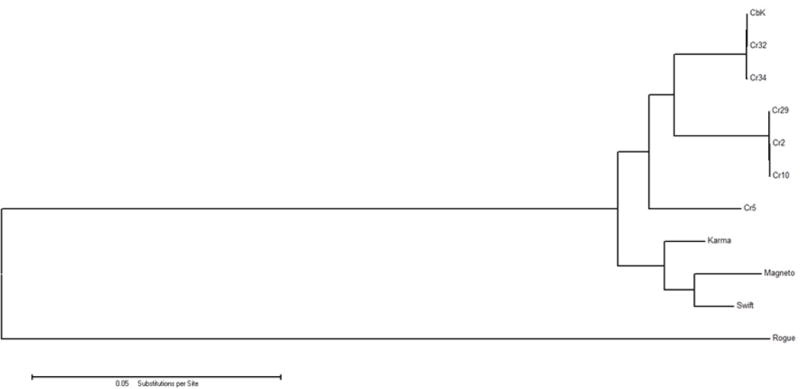
Molecular Phylogenetic analysis by Maximum Likelihood method. The evolutionary relationships of the 11 CbK-like genomes were inferred by using the Maximum Likelihood method based on the Tamura-Nei model [25]. The tree with the highest log likelihood (−498908.1958) is shown. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. The tree is drawn to scale, with branch lengths proportional to the number of substitutions per site. The analysis involved 11 genome nucleotide sequences. Codon positions included were 1st+2nd+3rd+Noncoding. All positions containing gaps and missing data were eliminated. There were a total of 179597 positions in the final dataset. Evolutionary analyses were conducted in MEGA6 [26].
The DNA ligase gene is not included in our list of core phage genes since this CbK-like gene is not present in the Colossus genome. However Colossus does have a different DNA ligase gene (gp191). A Blast comparison of gp191 produced the best matches with genes in six bacterial genomes and two bacteriophage genomes. The best phage gene match was to the DNA ligase gene of Cr30, the T4-like bacteriophage used for transduction in C. crescentus genetic experiments [9, 10, 11]. The other matching phage gene was the DNA ligase gene from phiM12 which is the closest known relative of Cr30 [10]. Gp191 is located in the same location of the Colossus genome as the DNA ligase genes in the CbK-like genomes (e. g. CbK gp151) but complex gene rearrangements have occurred in this region of the Colossus genome (Fig. 5). The homologues of CbK gp150 and gp151 genes are not present in the Colossus genome, but the CbK gp134 homologue, Colossus gp190, has been translocated adjacent to the new DNA ligase gene. In addition the two other Colossus gene insertions at this location, gp188 and gp189, do not match any known phage genes. The Colossus region corresponding to the location of CbK gp134 is missing four genes that are present in the CBK genome and contains an insertion that codes for 11 proteins including gp169, a T4-like protein that has 65% amino acid identity to the corresponding phiM12 T4 30.3-like protein. Thus, it appears that the Colossus Cbk-like DNA ligase gene was replaced by a DNA ligase gene from a T4-like phage that co-infected a C. crescentus host along with a Colossus ancestor. At the same time, or in separate events, a translocation brought the Colossus gp190 gene from its position 12 kb away to its current location, and two additional genes were inserted as well. Since the distant locus also has a deletion and an insertion that includes a phiM12-like gene, there could have been a simultaneous complex rearrangement involving at least two phage genomes. This rearrangement could have created the Colossus current gene arrangement or the current arrangement could be the result of additional insertions or deletions that occurred after an initial complex rearrangement involving these two regions of the genome.
Fig. 5.
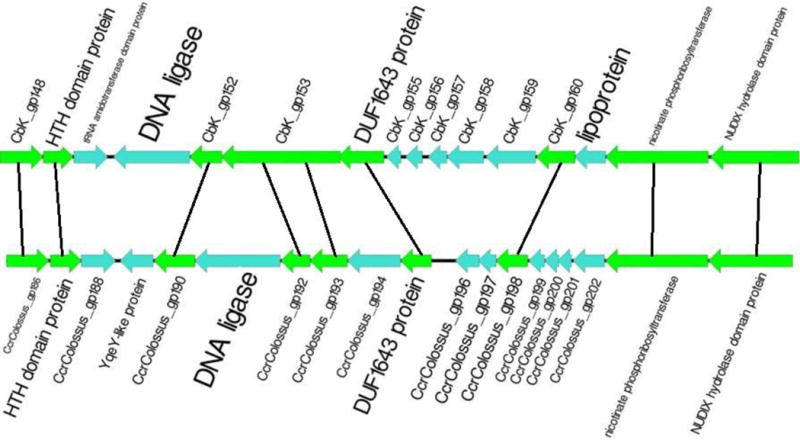
The DNA ligase regions of the CbK (top) and Colossus genomes. The green arrows represent the genes which are found in all 12 genomes. Homologous genes are connected by lines.
This scenario with the DNA ligase gene could be repeated in other gene sets which cannot be detected at this time due to the fact only about 20% of the genes in the phage genomes have a match to a gene with a predicted function and only 43% of the conserved genes have a predicted function. In addition, the replacement of the DNA ligase gene illustrates that the conserved phage genome is not equivalent to an essential phage genome. An essential phage genome would have to be determined experimentally, but it is likely that there would be substantial overlap between genes in the conserved genome and those in the essential genome.
Comparisons of the CbK-like genomes also provide evidence of gene fusion events. One example is Colossus gene gp212 which is 924 base pairs in length and codes for a 308 amino acid protein. The first 77 amino acids coded by this gene correspond to the first 77 amino acids coded by CbK gene, gp174 and amino acids 99 to 269 correspond to amino acids 27 to 195 coded by CbK gene gp169. Thus, the Colossus gp212 protein may combine the functions of both genes since it includes 77% CbK gp174 protein and the entire metal-dependent phosphohydrolase domain of the CbK gp169 protein plus some flanking amino acids. The CbK genes that are found between gp174 and gp169 are not present in the Colossus genome suggesting that the Colossus gp212 fusion occurred as the result of a deletion event.
We also examined the region between CbK gp192 and gp193 where an insertion was observed in the genomes described by Gill et al. [13]. We found that one extra gene was present in this region in the Cr32 and Cr34 genomes, but nine extra genes were present in most of the other phage genomes, with 10 extra genes in Cr29. The tenth gene in Cr29, gp197, is a duplication of gene Cr29 gp144 which is classified as a core gene. Cr29 gp144 is 100% identical to its CbK counterpart (gp141), while Cr29 gp197 is only 30% identical to gp141 with 94% coverage. Therefore this duplication set could be a classic example of an evolutionary event where one gene of the duplication is well conserved and maintains the original function and the other quickly accumulates mutations [12]. Alternatively, the Cr29 gp197 gene could have been acquired from a distant relative. The much smaller one gene insert found in the Cr32 and Cr34 genomes suggests that a deletion event may have occurred in these phage genomes to remove the eight genes found in the other phage genomes. Evidence to support this idea was obtained when we examined the region between CbK gp192 and gp193 and found an open reading frame with an amino acid sequence that is identical to the first 50 amino acids of the 67 amino acids found in the corresponding gene in most of the other CbK-like phage genomes. The presence of a truncated gene indicates that, at least in this case, a deletion event is likely to have occurred. The matching gene in Rogue (Rogue gp196) is nearly twice as long as the gene in the other phages suggesting the presence of another gene fusion. However, the distal portion of Rogue gp196 does not have a significant match to any other gene in the GenBank database.
The inconsistencies seen in the numbers of genes in each category (Table 3) are due to the gene duplications found within this collection of genomes. Rogue has two gene duplications, Colossus has three, and Cr29 contains seven gene duplication pairs. These duplications correspond to a total of 11 different CbK genes. Of the Cr29 duplications, two are core gene duplications that correspond to CbK gp76 and CbK gp141 (discussed above). Genes Cr29 gp237 through gp240 also have been duplicated and the duplications are located directly downstream in genes Cr29 gp241 through gp245. However Cr29 gp243 is missing from one of the duplicated gene sets. Since Cr29 gp243 is only present in the CbK, Cr32, Cr34, and Karma genomes and Cr29 is more closely related to Cr2 and Cr10 (Fig. 4), the Cr29 ancestral genome probably would not have included Cr29 gp243. Therefore, we hypothesize that the duplication was created by an HGT event from a CbK-like genome that contained the Cr29 gp243 equivalent. The inserted DNA would have begun with the second half of CbK gp224 and continued through CbK gp228, and would have been inserted into the Cr29 genome immediately after gene gp240.
With regard to the other duplications, the Rogue core gene duplicates correspond to CbK gp67 and CbK gp68 (the major capsid protein) and the core gene duplicates for Colossus correspond to CbK gp68, CbK gp73, and CbK gp99 (a 128 kd tail protein). The duplications that correspond to core genes are moderately similar with percent amino acid identity ranging from 30% to 82%. The non-core gene duplications of Cr29 share high similarity ranging from 78% to 100% identity. This high level of identity suggests that the non-core gene duplications of Cr29 are relatively new on an evolutionary time scale. It is tempting to speculate that the major capsid gene duplication facilitates the synthesis of a larger Colossus head structure [13]. However, the major capsid protein gene is duplicated in Rogue as well and the Rogue head structure is the same size as that of the other phages which do not contain the gene duplication [13].
The high degree of homogeneity amongst the common genes of the CbK-like bacteriophage genomes was unexpected considering their diverse geographical origins and the level of genomic rearrangements observed within the genus Caulobacter [2]. An explanation could be linked to the lack of bacteriophage immunity genes within the genome of the C. crescentus CB15 host. CB15 does have a pair of toxin-antitoxin genes that are not found in the other sequenced genomes of Caulobacter species [24]. However, these genes seem to have little impact on the 12 phage since they were all isolated with CB15 as the host. In contrast, a CRISPR-cas adaptive immunity system in Streptococcus thermophilus has been shown to be a driving force in the evolution of the bacteriophages which are specific for this host bacterium [20]. The lack of detectable phage immunity genes does not mean the host lacks bacteriophage immunity. Rather Caulobacter has an innate immunity to these bacteriophages since they have only been observed to infect swarmer cells [16]. The mechanism of infection for φCbK begins with attachment to the flagellum of the host bacterium via a head filament [14]. The bacterial phage tail attaches to the pilus and it is hypothesized that retraction of the pilus filament facilitates genome insertion into the host cell [8, 17]. Therefore, due to the asymmetrical cell division of Caulobacter, a bacteriophage particle would only be capable of infecting fewer than half of the cells in a population of Caulobacter. Further, the generation time of C. crescentus has been determined to be 1.5–2 hours with the swarmer cell stage lasting less than an hour [22]. Therefore, during the lifespan of a typical C. crescentus cell, it is only susceptible to infection by these bacteriophages for a brief window of time. Once the swarmer cell has matured into a stalked cell, it would never be affected by the presence of these bacteriophages. Thus, if a small percentage of the swarmer cells managed to mature before being infected, they could replace the stalked cell as they are lost from the population. From the perspective of the bacteriophage, evolution and adaption would not be necessary for survival against this innate immunity of Caulobacter because the mature stalked cells are like stem cells that continuously produce daughter cells susceptible to ϕCbK-like phage infection, yet the stalked cells maintain their resistance to ϕCbk-like phage predation. Therefore, we propose that there is no need for an evolutionary arms race between Caulobacter crescentus and these type B3 bacteriophages. Instead, selection acts on the bacteriophage genomes to maintain a system of genes and genome organization that works efficiently to produce progeny after infection of an immature swarmer cell.
The large number of indels and unique genes in each of the genomes indicate that the gene pool for CbK-like phages is quite large and the number of identified genes is likely to grow as we sequence additional CbK-like phage genomes. Since, we have already observed evidence for the presence of two T4-like phage genes in the Colossus genome, it will be important to sequence additional nonCbK-like phage genomes to see if it is common for the CbK-like phages to obtain genes from other types of phages that can co-infect the CB15 host. It will also be interesting to see if some of the additional CbK-like genomes are similar to the highly divergent Rogue and Colossus genomes.
Acknowledgments
This work was funded in part by National Science Foundation grant EF-0826792 and NIH grants R25GM066526 and R25GM076277 to BE.
Footnotes
Competing Interests The authors declare that there are no competing interests.
References
- 1.Agabian-Keshishian N, Shapiro L. Stalked bacteria: properties of deoxyribonucleic acid bacteriophage phiCbK. J Virol. 1970;5(6):795–800. doi: 10.1128/jvi.5.6.795-800.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ash K, Brown T, Watford T, Scott LE, Stephens C, Ely B. A comparison of the Caulobacter NA1000 and K31 genomes reveals extensive genome rearrangements and differences in metabolic potential. Open Biol. 2014;4(10) doi: 10.1098/rsob.140128. doi:rsob.140128 [pii] 10.1098/rsob.140128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auch AF, von Jan M, Klenk HP, Goker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand Genomic Sci. 2010;2(1):117–134. doi: 10.4056/sigs.531120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, Meyer F, Olsen GJ, Olson R, Osterman AL, Overbeek RA, McNeil LK, Paarmann D, Paczian T, Parrello B, Pusch GD, Reich C, Stevens R, Vassieva O, Vonstein V, Wilke A, Zagnitko O. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. doi:1471-2164-9-75 [pii] 10.1186/1471-2164- 9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Botstein D. A theory of modular evolution for bacteriophages. Ann N Y Acad Sci. 1980;354:484–490. doi: 10.1111/j.1749-6632.1980.tb27987.x. [DOI] [PubMed] [Google Scholar]
- 6.Comeau AM, Bertrand C, Letarov A, Tetart F, Krisch HM. Modular architecture of the T4 phage superfamily:aconserved core genome and a plastic periphery. Virology. 2007;362:384–396. doi: 10.1016/j.virol.2006.12.031. [DOI] [PubMed] [Google Scholar]
- 7.Darling AE, Mau B, Perna NT. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dillard RS, Storms RE, De Masi L, Hampton C, Panis G, Patrick H, Viollier PH, Wright ER. Analysis of Phage-Pilus Interactions in Caulobacter crescentus. Microsc Microanal. 2016;22(Suppl 3):202–203. doi: 10.1017/S1431927616001860. [DOI] [Google Scholar]
- 9.Ely B. Genetics of Caulobacter crescentus. Methods Enzymol. 1991;204:372–384. doi: 10.1016/0076-6879(91)04019-k. doi:0076-6879(91)04019-K. [DOI] [PubMed] [Google Scholar]
- 10.Ely B, Gibbs W, Diez S, Ash K. The Caulobacter crescentus transducing phage Cr30 is a unique member of the T4-Like family of myophages. Curr Microbiol. 2015;70(6):854–858. doi: 10.1007/s00284-015-0799-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ely B, Johnson RC. Generalized transduction in Caulobacter crescentus. Genetics. 1977;87:391–99. doi: 10.1093/genetics/87.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151(4):1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gill JJ, Berry JD, Russell WK, Lessor L, Escobar-Garcia DA, Hernandez D, Kane A, Keene J, Maddox M, Martin R, Mohan S, Thorn AM, Russell DH, Young R. The Caulobacter crescentus phage phiCbK: genomics of a canonical phage. BMC Genomics. 2012;13:542. doi: 10.1186/1471-2164-13-542. doi:1471-2164-13-542 [pii] 10.1186/1471-2164-13-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guerrero-Ferreira RC, Viollier PH, Ely B, Poindexter JS, Georgieva M, Jensen GJ, Wright ER. Alternative mechanism for bacteriophage adsorption to the motile bacterium Caulobacter crescentus. Proc Natl Acad Sci U S A. 2011;108(24):9963–9968. doi: 10.1073/pnas.1012388108. doi:1012388108 [pii] 10.1073/pnas.1012388108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson RC, Ely B. Isolation of spontaneously derived mutants of Caulobacter crescentus. Genetics. 1977;86(1):25–32. doi: 10.1093/genetics/86.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson RC, Wood NB, Ely B. Isolation and characterization of bacteriophages for Caulobacter crescentus. Journal of General Virology. 1977;37(2):323–335. doi: 10.1099/0022-1317-37-2-323. [DOI] [Google Scholar]
- 17.Lagenaur C, Farmer S, Agabian N. Adsorption properties of stage-specific Caulobacter phage phiCbK. Virology. 1977;77:401–407. doi: 10.1016/0042-6822(77)90436-6. [DOI] [PubMed] [Google Scholar]
- 18.Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. Tablet - next generation sequence assembly visualization. Bioinformatics. 2010;26(3):401–402. doi: 10.1093/bioinformatics/btp666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, Vonstein V, Wattam AR, Xia F, Stevens R. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST) Nucleic Acids Res. 2013;42(Database issue):D206–214. doi: 10.1093/nar/gkt1226. doi:gkt1226 [pii] 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Paez-Espino D, Sharon I, Morovic W, Stahl B, Thomas BC, Barrangou R, Banfield JF. CRISPR immunity drives rapid phage genome evolution in Streptococcus thermophilus. MBio. 2015;6(2) doi: 10.1128/mBio.00262-15. doi:mBio.00262-15[pii] 10.1128/mBio.00262-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panis G, Lambert C, Viollier PH. Complete genome sequence of Caulobacter crescentus bacteriophage phiCbK. J Virol. 2012;86(18):10234–10235. doi: 10.1128/JVI.01579-12. doi:86/18/10234 [pii] 10.1128/JVI.01579-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Poindexter JS. Biological properties and classification of the Caulobacter group. Bacteriol Rev. 1964;28:231–295. doi: 10.1128/br.28.3.231-295.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B. Artemis: sequence visualization and annotation. Bioinformatics. 2000;16(10):944–945. doi: 10.1093/bioinformatics/16.10.944. [DOI] [PubMed] [Google Scholar]
- 24.Scott D, Ely B. Conservation of the essential genome among Caulobacter and Brevundimonas species. Current Microbiology. 2016;72:503–510. doi: 10.1007/s00284-015-0964-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–526. doi: 10.1093/oxfordjournals.molbev.a040023. [DOI] [PubMed] [Google Scholar]
- 26.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. doi:mst197 [pii] 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]