

Abstract
Novel approaches using OMICS techniques enable a collective assessment of multiple related biological units, including genes, gene expression, proteins, and metabolites. In the past decade, next-generation sequencing (NGS) technologies were improved by longer sequence reads and the development of genome databases and user-friendly pipelines for data analysis, all accessible at lower cost. This has generated an outburst of high-throughput data. The application of OMICS has provided more depth to existing hypotheses as well as new insights in the etiology of dental caries. For example, the determination of complete bacterial microbiomes of oral samples rather than selected species, together with oral metatranscriptome and metabolome analyses, supports the viewpoint of dysbiosis of the supragingival biofilms. In addition, metabolome studies have been instrumental in disclosing the contributions of major pathways for central carbon and amino acid metabolisms to biofilm pH homeostasis. New, often noncultured, oral streptococci have been identified, and their phenotypic characterization has revealed candidates for probiotic therapy. Although findings from OMICS research have been greatly informative, problems related to study design, data quality, integration, and reproducibility still need to be addressed. Also, the emergence and continuous updates of these computationally demanding technologies require expertise in advanced bioinformatics for reliable interpretation of data. Despite the obstacles cited above, OMICS research is expected to encourage the discovery of novel caries biomarkers and the development of next-generation diagnostics and therapies for caries control. These observations apply equally to the study of other oral diseases.
Keywords: dental caries, biofilm, next-generation sequencing, microbiome, genome, metabolome
Introduction
Advances in DNA sequencing and bioinformatics have facilitated the disclosure of numerous oral microbial taxa and their associations with dental health or caries activity (Aas et al. 2008; Tanner et al. 2016). The use of next-generation sequencing (NGS) technologies has revealed the high complexity of the oral microbiome (Belda-Ferre et al. 2012; Simón-Soro et al. 2013), metatranscriptome (Benítez-Páez et al. 2014; Duran-Pinedo and Frias-Lopez 2015), metaproteome (Belda-Ferre et al. 2015; Belstrom et al. 2016), and metabolome (Washio et al. 2016) at remarkable levels. Altogether, the data collected from these OMICS approaches are providing the foundation to better understand how hundreds of microbial species coinhabit and functionally interact in oral biofilms to cause disease or to maintain homeostasis (Duran-Pinedo and Frias-Lopez 2015; Simón-Soro and Mira 2015).
A symposium entitled “How the OMICS Are Contributing to the Understanding of Caries” was held in June 2016 during the 94th general session of the International Association for Dental Research (IADR) in Seoul, South Korea. The objectives of this symposium were to 1) recognize the applications of OMICS technologies, including the advantages and disadvantages of each method; 2) gain knowledge on how the OMICS are contributing to the understanding of dental caries; and 3) outline the scientific evidence from current OMICS studies, which support the development of novel approaches for caries diagnostics and management. This review aims to summarize the information presented at the symposium and to expand the discussion of how OMICS is leading to paradigm shifts in oral microbiology and cariology.
First Era of the OMICS: Entering the Technology Hype Cycle
When a technological breakthrough leads to an advent of a new technology, it generally follows a cycle of maturation and acceptance, also known as “Gartner hype cycle” (Fig. 1), named after the company that proposed this theory (Gartner, Inc.). The hype cycle starts with a technology trigger and proof-of-principle studies, which are noticed by the media and receive significant publicity. This early publicity produces a number of successful stories while heading toward the peak of inflated expectations. However, when the initial errors and failures of the new technology become apparent, the cycle enters the phase of trough of disillusionment. The technology can survive this phase to enter the slope of enlightenment only when significant investment occurs to overcome the problems. At the slope of enlightenment, the technology will continue to develop and become more widely accepted and used by consumers. Once it becomes mainstream, the cycle enters the plateau of productivity.
Figure 1.
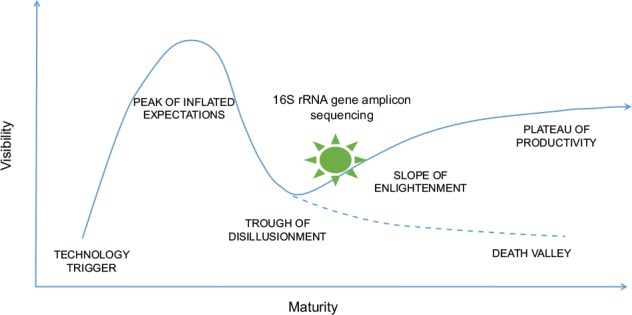
The Gartner hype cycle of technologies. rRNA, ribosomal RNA.
In general terms, the 454 Life Sciences (Roche), Illumina (Illumina), and SOLID (Life Technologies) platforms are the second generation or NGS. In 2005, the 454 pyrosequencing technology introduced parallel high-throughput sequencing that allowed simultaneous determination of hundreds of thousands of 80- to 120-bases-long sequence reads in a single machine run. This was later expanded to 250 bp with the FLX system (Roche). The initial 35-bp-long reads in the Illumina system were also increased to 70 to 200 bp. These improvements of sequencing technologies opened a new dimension for biomolecular research, which until then had relied on the traditional Sanger sequencing. The 454 technology was picked up by frontrunners in microbial ecology, and its applicability was first demonstrated on the ecosystem of deep sea water, disclosing a previously unseen microbial diversity (Sogin et al. 2006). This was followed by new findings on other ecosystems such as soil, microbial mats, and murine and human gut. The first report revealing the vast diversity of oral microbial communities was published in 2008 (Keijser et al. 2008) and succeeded by the first core oral microbiome report (Zaura et al. 2009).
A new and promising technology will inevitably generate a race for “a first-time application.” For the oral microbiome, this race resulted in highly cited publications with long lists of bacterial names and conclusions obtained from the analysis of a few oral samples, rather than from well-designed and hypothesis-driven studies. These limitations were not exclusive for oral microbiology research. Of all sequencing studies published in major ecological journals in 2009, only 18% analyzed replicate samples (Prosser 2010). In most cases, the lack of replicates was attributed to or justified by the high cost of these cutting-edge technologies. Later, when expenses per sequencing run decreased significantly, the determination of the microbiomes associated with oral diseases—for example, periodontal disease (Griffen et al. 2012), endodontic infections (Ozok et al. 2012), and caries (Gross et al. 2012)—became a trending topic in dental research.
The increased use of NGS technology also led to the first critical evaluations of the methodology, which described issues from bias due to DNA extraction, polymerase chain reaction (PCR), and sequencing (Lee et al. 2012; Abusleme et al. 2014) to the choice of sequencing primers (Nossa et al. 2010), sample contamination (van der Horst et al. 2013), and methods used for preprocessing and clustering data (Bonder et al. 2012). Data analysis during the first era of OMICS consisted of various custom-made pipelines, which resulted in diverging outputs (Bonder et al. 2012). Furthermore, at that time, databases for functional classification were designed for eukaryotic organisms with many categories not suitable for prokaryotic organisms, for example, cytoskeleton proteins. These databases also did not account for phage genes, many of which were assigned to categories like DNA replication. Research groups had no choice but to design their own analysis pipelines to assign short sequence reads to bacterial taxa and functions. However, the lack of computing expertise within the oral microbiology groups hampered the usability of OMICS techniques. In addition, frequent updates in the NGS technologies contributed to the difficulties in performing the analyses. Staying on top of the technological progress required regular updates in advanced bioinformatics, an essential new competence for research in oral microbiology.
The first generation of oral metabolomics also had its own challenges with problems associating specific metabolites with oral bacterial pathways. The metabolome consists of a wide range of metabolites and is the last step in the biological hierarchy from the genome through transcriptome and proteome to metabolome. While there are several databases to identify metabolites of Escherichia coli or mammalian cells, research of oral microbial metabolism has been difficult mostly because the metabolic pathways of oral bacteria are generally different from those of other organisms. Basic knowledge about oral bacterial metabolism, particularly carbohydrate metabolism by caries-associated bacteria (i.e., streptococci), was compiled by Carlsson in 1986 (Carlsson 1986). Later, Takahashi and others provided information about carbohydrate metabolism by Actinomyces (Takahashi and Yamada 1999), as well as amino acid metabolism by periodontal pathogens such as Porphyromonas gingivalis and Prevotella intermedia (Takahashi and Yamada 2000). A more comprehensive catalogue of metabolic pathways of oral bacteria was published subsequently (Takahashi 2015).
Second Era of the OMICS: On the Slope of Enlightenment
The advances in sequencing technology continued to improve the resolution of sequence analysis. For example, the Illumina platform increased once again the length of the reads to 300 bp while the 454 technology increased to 800 bp, making the taxonomic and functional assignments more reliable. Moreover, sequencing costs were reduced considerably. Currently, Illumina sequencing of the 16S ribosomal RNA (rRNA) gene by the paired-ends technology can be performed at US$20–30 per sample compared with the US$80–120 cost of 7 y ago. Following the advancement of gut microbiome research, genome databases specific to oral microorganisms were developed, including the Human Oral Microbiome database (HOMD) (Chen et al. 2010) and the CORE database (Griffen et al. 2011), which are curated to remove contaminants and improve reliability of the analysis. Equally important was the sequencing of genomes of oral bacteria by the Human Microbiome Consortium (Gevers et al. 2012). This was particularly relevant to reduce the number of unassigned reads in metagenomic analysis. Nevertheless, 40% to 50% of the metagenomic reads still remain unassigned or assigned as hypothetical proteins with unknown function; therefore, more work is still needed in this area (Belda-Ferre et al. 2012; Simón-Soro et al. 2013; Duran-Pinedo and Frias-Lopez 2015). A “wiki solution” (Salzberg 2007) and a gene knockout approach (Baba et al. 2006) were proposed to improve annotation of bacterial genomes and elucidate gene function in model organisms.
In parallel to the improvement in sequence length and development of oral databases, several user-friendly pipelines have become available that allow research groups to analyze high-throughput sequence data without requiring programming skills. For instance, the Ribosomal Database Project developed a 16S rNA gene analysis pipeline with complete quality filtering, barcode separation, taxonomic analysis, and estimation of diversity indexes in an online system (Barriuso et al. 2011). Other applications developed for metagenomic analysis include the METAREP, a microbiome-specific version of the Integrated Microbial Genomes system (Markowitz et al. 2012), and the MG-RAST, which permits users to upload sequences remotely and provides full taxonomic and functional analyses based on a well-designed pipeline (Keegan et al. 2016). In the context of interpreting bioinformatic data, it is important to consider that the availability of tools and affordability of generating genomic information have led to a false sense of simplicity, when the truth is that a vast knowledge of microbial ecology, microbial metabolism, and OMICS-related biostatistics capacity is still required.
What Have We Learned?
One of the most significant findings from OMICS studies was the identification of previously unknown, thus “new” bacterial genera/species, which appeared to be associated with dental health or caries. Such was the case for Streptococcus A12 (Huang et al. 2016) and Streptococcus dentisani (Camelo-Castillo et al. 2014), which were both isolated from supragingival plaque of caries-free individuals. A12 is able to inhibit the growth and intercellular signaling of the caries pathogen Streptococcus mutans and to buffer pH through the arginolytic pathway. Phylogenomic analyses comparing the A12 genome to reference genomes revealed that A12 is most closely related to Streptococcus australis but sufficiently different to represent a new species. Similar to A12, S. dentisani presents antimicrobial activity on S. mutans and is capable of metabolizing arginine (Fig. 2). Compelling evidence supports the role of arginine metabolism in caries prevention (Marquis et al. 1987; Casiano-Colón and Marquis 1988; Nascimento et al. 2013). Given their beneficial and health-associated properties, A12 and S. dentisani are currently being tested as probiotic strains for caries prevention. Other newly identified species are yet to be characterized, such as Schlegelella species, which were detected by high-throughput 16S rRNA gene sequence analysis of DNA and RNA of carious dentin samples. In some subjects, Schlegelella and Pseudoramibacter represented a high proportion of the total microbiota detected in the carious samples (Simón-Soro and Mira 2015). Studies have also revealed new associations of relatively unknown taxa and caries pathogenesis. For example, Scardovia wiggsiae was significantly associated with severe early childhood caries (Tanner et al. 2011). In theory, microbiome analysis and other OMICS approaches coupled with phenotypic characterization of newly identified species may support the identification of new microbial biomarkers for caries.
Figure 2.

Phenotypic characterization of Streptococcus dentisani. (A) Graphic shows the buffering of extracellular pH by Streptococcus dentisani strain 7746, which is a heath-associated oral bacteria recently identified by OMICS approaches. S. dentisani was grown in Brain Heart Infusion medium with (blue diamonds) and without (red squares) addition of 5 g/L arginine. (B) Streptococcus mutans UA159 (lab strain) cells after 30 min of exposure to the supernatant of S. dentisani strain 7746. The arrows shows pores in the cells wall formed as a consequence of the action of bacteriocins and thus potential probiotic activity of S. dentisani. This figure is available in color online.
Another accomplishment from high-throughput sequence analyses was the validation of previously suggested ecological hypotheses with solid data sets. For example, Lactobacillus species have long been associated with late stages of caries progression (Becker et al. 2002). In a metagenome analysis of the bacterial communities found at the different stages of caries development, Lactobacillus species were detected only in deep carious dentin (Simón-Soro et al. 2013). Moreover, OMICS studies (Simón-Soro and Mira 2015) confirmed the previously proposed concept that both dental caries and periodontal disease are closely related to a dysbiosis of microbial consortia rather than by individual bacterial species (Marsh 2006). The dysbiosis is driven by environmental changes, such as a sugar-frequent/acidic-pH environment in caries and a protein-rich/neutral-to-weakly alkaline-pH environment in periodontal disease (Takahashi 2015).
OMICS studies also underpinned a pivotal aspect of the caries process—the presence of bacteria does not necessarily indicate metabolic activity. Specifically, the prevalence of bacteria revealed by metagenome analysis may not correlate well with the patterns of the active microbial community revealed by metatranscriptome analysis of the same oral sample. Actinomyces, Corynebacterium, and Neisseria were the 3 most abundant taxa in the RNA-based community of a 24-h plaque sample, whereas Veillonella, Streptococcus, and Leptotrichia were the most prevalent in the total DNA-based metagenome of the same sample (Fig. 3; Benítez-Páez et al. 2014). This underlines the dynamic nature of microbial activity in biofilms by indicating that some genera were especially active at 24 h of biofilm development, whereas others were less active, albeit being present at higher proportions at that point of sampling. Interestingly, the recent use of the CLASI-FISH (Combinatorial Labeling and Spectral Imaging—Fluorescence in situ Hybridization) technique indicated that Corynebacterium could be the cornerstone of supragingival plaque architecture with long filaments that serve as anchor sites for many other microbes (Mark Welch et al. 2016). If these findings are confirmed, it could imply that Corynebacterium rather than Fusobacterium species are the bridging bacteria in biofilms. From there, novel antiplaque strategies have been proposed directed toward Corynebacterium and other key biofilm bacteria (Ferrer and Mira 2016).
Figure 3.

Total and active microbial composition in a 24-h supragingival dental plaque sample. DNA and RNA were extracted from the same sample and subject to direct sequencing to obtain the metagenome and metatranscriptome of the microbial community, respectively. Genera are ranked according to their proportion in the RNA-based, active fraction. Some organisms that are present at low levels in the DNA appear to be extremely active, such as Actinomyces and Corynebacterium. Others, like Neisseria and Streptococcus, appear to be less active at the time of sample collection, as indicated by their higher DNA to RNA ratios (Benítez-Páez et al. 2014).
Metagenome analyses have begun to elucidate some aspects related to bacterial activity in dentin caries. High expression levels of collagenases and other proteases, such as serine-proteases, glycoproteases, carboxy-terminal proteases, and metalloproteases, were detected in samples of carious dentin (Simón-Soro et al. 2013). These findings suggest that the microbial-encoded proteolytic arsenal, coupled with the activity of human metalloproteases, may play a significant role in the degradation of dentinal protein. Twenty percent of the collagenase DNA sequences from carious dentin samples corresponded to Prevotella species, but significant matches were also obtained to annotated collagenases from Bacteroides, Campylobacter, Capnocytophaga, Treponema, and others. Future RNA-based metatranscriptomic work combined with experimental assays should be performed to confirm if these bacterial-encoded collagenases are in fact able to degrade dentin proteins.
Our knowledge of the bacterial metabolic pathways involved in caries was also expanded. The metabolism of dietary carbohydrates to acids by oral bacteria has long been known as the driving force of the caries process. Metabolomics is beginning to disclose the central carbon metabolism pathways in operation in supragingival plaque, including the Embden-Meyerhof-Parnas pathway (EMP pathway; glycolysis), the pentose-phosphate pathway, and the tricarboxylic acid cycle (TCA cycle). Contrary to dietary carbohydrates, proteins, peptides, and amino acids are continuously supplied by saliva and gingival crevicular fluid in the oral cavity, and they serve as vital metabolic substrates for the growth of plaque bacteria. Nowadays, it is possible to measure amino acid metabolism along with carbohydrate metabolism (Washio et al. 2016). Certain amino acids that are relatively strong acids can be converted into relatively weak organic acids, carbon dioxide, and ammonia, which can neutralize acids in the biofilm. However, metabolic pathways responsible for alkalization that can counteract cariogenic acidification as well as salivary clearance and buffering are yet to be fully clarified.
A recent study showed the contribution of major pathways for amino acid metabolism to the pH homeostasis processes in oral biofilm (Washio et al. 2016). In resting supragingival plaque, glutamate exhibited the highest concentration levels, but various metabolites related to amino acid metabolism, such as citrulline, ornithine, putrescine, γ-aminobutyric acid (GABA), and β-alanine, were also detected (Fig. 4). The relatively high concentrations of citrulline and ornithine along with the relatively low concentration of arginine detected in plaque support that the arginine deiminase system (ADS) functions to convert arginine to citrulline and ornithine and may contribute to acid neutralization in vivo. Likewise, the high concentration of glutamate and relatively high level of ammonia production from glutamate indicate that glutamate metabolism might also function as an acid neutralizer in vivo. Intriguingly, these end products of amino acid metabolism are known to be cytotoxic, induce tissue inflammation by modulating immune responses, and promote apoptosis (Kurita-Ochiai et al. 2008), and these processes contribute to periodontal disease. The role of amino acid metabolism by supragingival versus subgingival plaque bacteria in the development of oral diseases deserves further investigation. Hence, metabolomic analysis might help to identify new biomarkers of oral diseases.
Figure 4.
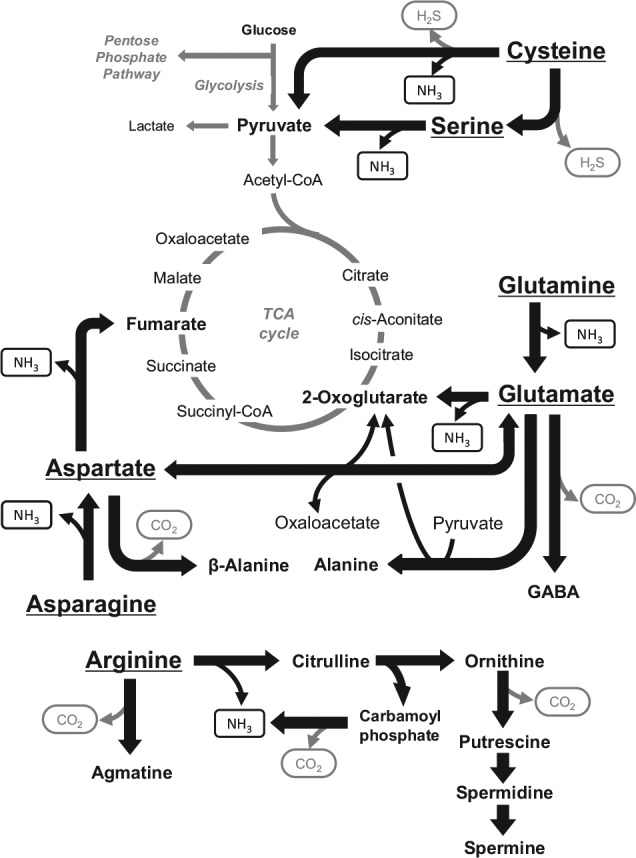
Proposed metabolic network of the representative amino acids: glutamine, glutamate, asparagine, aspartate, cysteine, serine, and arginine in dental plaque (Washio et al. 2016).
Metabolomic analyses can also be used to evaluate the effects of existing oral care products and future caries therapies. For example, the metabolome of supragingival plaque collected after oral rinsing with a mixture of sodium fluoride and glucose confirmed that fluoride reduces bacterial acid production from glucose by inhibiting the glycolytic enzyme enolase (Takahashi and Washio 2011) as previously proposed (Guha-Chowdhury et al. 1997). Xylitol was also shown to have no effect on glucose metabolism or acid production in vivo (Takahashi and Washio 2011).
The Next Era of OMICS: Reaching the Plateau of Productivity
Having passed the various stages of the hype cycle of OMICS technologies, it is time to further reflect on future applications. To unravel causal relationships between the microbiome and changes in oral health status, there is an absolute need to perform well-designed longitudinal clinical studies. As with any clinical study, particular efforts should be made when planning the workflow of OMICS studies (Fig. 5). Study planning should start with defining the hypothesis and objectives followed by the development of an appropriate study design that should include power analysis, before any resources are spent on sequencing and data analysis (Vincent et al. 2016). Several tools are available for power and sample size calculations based on preexisting OMICS data sets or users’ pilot data (Kelly et al. 2015). However, reliability of these tools still needs to be demonstrated, and the development of a simple user-interface is required to facilitate the applicability of these calculations. In addition, both positive and negative controls should be included in the assessments to allow identification of bias introduced in the different sample processing steps (Brooks 2016). Finally, OMICS data sets require multivariate statistical analysis followed by correction for multiple testing. The selection of the most appropriate statistical tools will depend on the study design, for example, cross-sectional or longitudinal. Available tools that are aimed to compare independent groups include profile similarity analysis using ANOSIM or PERMANOVA or the biomarker selection tool LefSe (Segata et al. 2011). Some methods have been recently proposed to assess changes in OMICS data sets over time (Faust et al. 2015), but adaptation of these methods into simple user-friendly tools is still necessary. In summary, OMICS data will be only as good or as poor as the quality of the samples used for creating the data. Appropriate experimental design and standardization at every level will prevent from the “garbage in, garbage out” scenario.
Figure 5.
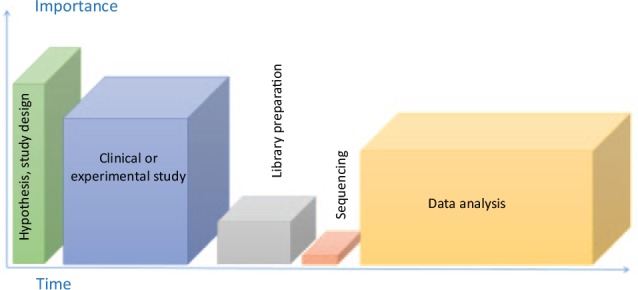
The workflow of an OMICS study, adapted from Vincent et al. (2016) by the importance of and time needed to accomplish each step of this process. The first and most important step is formulating a hypothesis and choosing the study design. The second, relatively time-consuming and highly important step is the study itself, which includes proper test and control sample collection and storage. The third step is sample processing, including DNA extraction and purification and amplicon library preparation. This step is highly sensitive to methodology bias. Next is sequencing itself—the lowest in the rank of importance and also the least time-consuming step of the workflow, followed by the “bottleneck” of OMICS studies regarding the time: the data analysis step. The time needed for the analyses will depend on the OMICS approach used, the study design, and the complexity of the metadata available.
The field of sequencing is one of the fastest-evolving technological advances in biotechnology. While the 454 pyrosequencing technology will be discontinued in 2017, new technologies of single-molecule sequencing devices are emerging for the new OMICS era. The Nanopore technology (Oxford Nanopore Technologies) has already been applied to sequence full-length 16S rRNA genes, but the high sequencing error rate may yet be a problem (Benítez-Páez et al. 2016). The new PACBIO sequencer (Pacific Biosciences) is also able to achieve reads of several kb in length with a much reduced error rate. These new technologies have the potential to revolutionize the field by improving genetic annotations through the assembly of more accurate assignments and gap-free full-genome sequences.
If we take the renowned Keyes’s diagram of dental caries as a starting point, 3 main pillars are collectively responsible for the disease: host features (e.g., immune system, genetic nature that predisposes the enamel structure, salivary composition, and buffering effect), environmental components (e.g., dietary sugars, fluoride, oral hygiene habits, and personal factors influenced by socioeconomic status and lifestyle), and microbiological features (e.g., acidogenicity of dental plaque, presence of pH-buffering bacteria, levels of pathogenic microorganisms). A “perfect storm” will be formed when the confluence of these 3 pillars leads to an increased risk for caries development (Fig. 6). Currently, high-throughput analysis of epidemiological data can be used to better understand the environmental effects on oral diseases, and whole-genome association (WGA) studies can be applied to determine genetic predisposition to caries, such as abnormalities in enamel formation (Wright 2010). However, multiple genes may be involved in caries predisposition, which complicates the diagnostic value of gene-based studies (Shaffer et al. 2013). Previous work regarding human oral microbiome has been merely descriptive, and the taxonomic characterization of healthy and diseased oral samples has many limitations. Future efforts should be directed toward associating the composition with the function of the oral microbiota. The relevant questions may be answered by elucidating what the microbes are doing rather than focusing primarily on who is performing those actions (Takahashi 2015). Also deserving more attention are the microbial interactions with the host (e.g., adhesion mechanisms between microbes and salivary proteins) (Nobbs et al. 2011) and the recognition patterns with the oral immune system (Simón-Soro and Mira 2015).
Figure 6.

The “perfect storm” of dental caries and how the postgenomics era can help to understand caries. ITS, internal transcribed spacer; WGA, whole-genome association.
Recently, the ecobiological heterogeneity of the salivary ecosystem and the relationships between microbiome, metabolome, and host-related salivary parameters were demonstrated based on the analysis of saliva samples (Zaura et al. 2017). Given the dichotomy observed when correlating salivary bacteria and metabolites, it was suggested that individual salivary ecosystems are adapted to either saccharolytic or proteolytic metabolisms. The association between salivary ecotypes and risk for development of oral diseases remains to be assessed in clinical interventional studies. In summary, the use of OMICS approaches can decipher microbial composition and function, and relating those to host or environmental features will unravel the interactions between the 3 pillars of caries disease and how to modify them to reduce caries risk in the clinical practice from a holistic approach.
In addition to focusing on bacteria (e.g., which is the most abundant microbial component of the oral cavity), studies involving other kingdoms, such as viruses, fungi, Archaea, and protozoa, should provide a more realistic picture of the complex interactions contributing to the compositional and functional stability of the oral ecosystem. It was recently demonstrated that bacterial viruses (bacteriophages) might assist in maintaining a stable, healthy ecosystem compared with the dysbiosis of periodontal diseases (Wang et al. 2016). OMICS approaches have been successfully used to assess composition and functionality of complex communities grown in an in vitro biofilm model (Edlund et al. 2015). OMICS could also be used in search for optimal growth conditions of the so-called unculturable organisms, which may grow in the presence of certain helper strains and/or compounds with siderophore activity (Vartoukian et al. 2016).
As OMICS approaches become more affordable and readily accessible, there is a clear need to integrate the massive amount of data being generated so far as to fully understand the interplay of the different oral microorganisms with the function of their metabolic mechanisms in dental health and caries disease. Of great priority, future efforts should aim to explore the roles of the thousands of bacterial genes, proteins, and metabolites revealed by OMICS to ascertain their value as risk factors, biomarkers, or therapeutic targets. Data generated from the OMICS studies remain to be carefully tested by the construction of new and well-designed study models. The plea for integrating genomic methodologies is not new and is also not exclusive to dental research (Rotroff and Motsinger-Reif 2016). The recent application of multi-OMICS methods in clinically relevant time frames has opened new opportunities for clinical interventions (Quinn et al. 2016). Noticeably, the tools needed to interpret and translate the OMICS data into clinical practice demand expertise from multiple disciplines, such as biology, dentistry, mathematics, statistics, and bioinformatics.
Conclusion
The application of OMICS techniques has provided more depth to existing hypotheses as well as new insights in the etiology of dental caries, which emphasizes that some long-held caries paradigms should be revised. These would include the infectious nature of caries, the validity of the classical Koch’s postulates to the disease, and the antimicrobial effect of fluoride in addition to its enamel protection features. Large-scale clinical studies involving OMICS and other oral physiological parameters should reveal new aspects of the heterogeneity of caries. Future efforts should also aim to investigate the association between the composition and the function of the oral microbiota, including bacteria, viruses, fungi, Archaea, and protozoa. Despite current problems with study design and data analysis, OMICS research is expected to encourage the discovery of novel caries biomarkers and the development of next-generation diagnostics and therapies for caries control. These observations apply equally to the study of other oral diseases.
Author Contributions
M.M. Nascimento, contributed to conception, design, data analysis, and interpretation, drafted and critically revised the manuscript; E. Zaura, A. Mira, N. Takahashi, contributed to conception, design, data analysis, and interpretation, critically revised the manuscript; J.M. ten Cate, contributed to data analysis and interpretation, critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of the work.
Footnotes
Research support has been provided to M.M.N. by the National Institute of Dental and Craniofacial Research K23 DE023579, to A.M. by the Spanish MINECO BIO2015-68711-R, and to N.T. by the Japan Society for the Promotion of Science, KAKENHI 60183852.
M.M.N. holds a provisional patent through the University of Florida on methodologies to identify arginolytic oral isolates that might eventually be incorporated into probiotic formulations to promote oral health. A.M. holds a patent through the FISABIO Foundation protecting the use of S. dentisani to prevent dental caries. The authors declare no other potential conflicts of interest with respect to the authorship and/or publication of this article.
References
- Aas JA, Griffen AL, Dardis SR, Lee AM, Olsen I, Dewhirst FE, Leys EJ, Paster BJ. 2008. Bacteria of dental caries in primary and permanent teeth in children and young adults. J Clin Microbiol. 46(4):1407–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abusleme L, Hong BY, Dupuy AK, Strausbaugh LD, Diaz PI. 2014. Influence of DNA extraction on oral microbial profiles obtained via 16s rRNA gene sequencing. J Oral Microbiol. 6:23990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli k-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2:2006.0008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriuso J, Valverde JR, Mellado RP. 2011. Estimation of bacterial diversity using next generation sequencing of 16s rDNA: a comparison of different workflows. BMC Bioinformatics. 12:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker MR, Paster BJ, Leys EJ, Moeschberger ML, Kenyon SG, Galvin JL, Boches SK, Dewhirst FE, Griffen AL. 2002. Molecular analysis of bacterial species associated with childhood caries. J Clin Microbiol. 40(3):1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belda-Ferre P, Alcaraz LD, Cabrera-Rubio R, Romero H, Simón-Soro A, Pignatelli M, Mira A. 2012. The oral metagenome in health and disease. ISME J. 6(1):46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belda-Ferre P, Williamson J, Simón-Soro A, Artacho A, Jensen ON, Mira A. 2015. The human oral metaproteome reveals potential biomarkers for caries disease. Proteomics. 15(20):3497–3507. [DOI] [PubMed] [Google Scholar]
- Belstrom D, Jersie-Christensen RR, Lyon D, Damgaard C, Jensen LJ, Holmstrup P, Olsen JV. 2016. Metaproteomics of saliva identifies human protein markers specific for individuals with periodontitis and dental caries compared to orally healthy controls. PeerJ. 4:e2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benítez-Páez A, Belda-Ferre P, Simón-Soro A, Mira A. 2014. Microbiota diversity and gene expression dynamics in human oral biofilms. BMC Genomics. 15:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benítez-Páez A, Portune KJ, Sanz Y. 2016. Species-level resolution of 16s rRNA gene amplicons sequenced through the minion portable nanopore sequencer. Gigascience. 5:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonder MJ, Abeln S, Zaura E, Brandt BW. 2012. Comparing clustering and pre-processing in taxonomy analysis. Bioinformatics. 28(22):2891–2897. [DOI] [PubMed] [Google Scholar]
- Brooks JP. 2016. Challenges for case-control studies with microbiome data. Ann Epidemiol. 26(5):336–341. [DOI] [PubMed] [Google Scholar]
- Camelo-Castillo A, Benitez-Paez A, Belda-Ferre P, Cabrera-Rubio R, Mira A. 2014. Streptococcus dentisani sp. nov., a novel member of the mitis group. Int J Syst Evol Microbiol. 64(Pt 1):60–65. [DOI] [PubMed] [Google Scholar]
- Carlsson J. 1986. Metabolic activities of oral bacteria. In: Thylstrup A. CFO, editor. Textbook of cariology. Copenhagen (Denmark): Munksgaard; p. 74–106. [Google Scholar]
- Casiano-Colón A, Marquis RE. 1988. Role of the arginine deiminase system in protecting oral bacteria and an enzymatic basis for acid tolerance. Appl Environ Microbiol. 54(6):1318–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Yu WH, Izard J, Baranova OV, Lakshmanan A, Dewhirst FE. 2010. The human oral microbiome database: a web accessible resource for investigating oral microbe taxonomic and genomic information. Database (Oxford: ). 2010:baq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran-Pinedo AE, Frias-Lopez J. 2015. Beyond microbial community composition: functional activities of the oral microbiome in health and disease. Microbes Infect. 17(7):505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund A, Yang Y, Yooseph S, Hall AP, Nguyen DD, Dorrestein PC, Nelson KE, He X, Lux R, Shi W, et al. 2015. Meta-omics uncover temporal regulation of pathways across oral microbiome genera during in vitro sugar metabolism. ISME J. 9(12):2605–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust K, Lahti L, Gonze D, de Vos WM, Raes J. 2015. Metagenomics meets time series analysis: unraveling microbial community dynamics. Curr Opin Microbiol. 25:56–66. [DOI] [PubMed] [Google Scholar]
- Ferrer MD, Mira A. 2016. Oral biofilm architecture at the microbial scale. Trends Microbiol. 24(4):246–248. [DOI] [PubMed] [Google Scholar]
- Gevers D, Knight R, Petrosino JF, Huang K, McGuire AL, Birren BW, Nelson KE, White O, Methe BA, Huttenhower C. 2012. The human microbiome project: a community resource for the healthy human microbiome. PLoS Biol. 10(8):e1001377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, Podar M, Leys EJ. 2012. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16s pyrosequencing. ISME J. 6(6):1176–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffen AL, Beall CJ, Firestone ND, Gross EL, Difranco JM, Hardman JH, Vriesendorp B, Faust RA, Janies DA, Leys EJ. 2011. Core: a phylogenetically-curated 16s rDNA database of the core oral microbiome. PLoS One. 6(4):e19051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross EL, Beall CJ, Kutsch SR, Firestone ND, Leys EJ, Griffen AL. 2012. Beyond Streptococcus mutans: dental caries onset linked to multiple species by 16s rRNA community analysis. PLoS One. 7(10):e47722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha-Chowdhury N, Clark AG, Sissons CH. 1997. Inhibition of purified enolases from oral bacteria by fluoride. Oral Microbiol Immunol. 12(2):91–97. [DOI] [PubMed] [Google Scholar]
- Huang X, Palmer SR, Ahn SJ, Richards VP, Williams ML, Nascimento MM, Burne RA. 2016. A highly arginolytic streptococcus species that potently antagonizes Streptococcus mutans. Appl Environ Microbiol. 82(7):2187–2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keegan KP, Glass EM, Meyer F. 2016. Mg-rast, a metagenomics service for analysis of microbial community structure and function. Methods Mol Biol. 1399:207–233. [DOI] [PubMed] [Google Scholar]
- Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, ten Cate JM, Crielaard W. 2008. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res. 87(11):1016–1020. [DOI] [PubMed] [Google Scholar]
- Kelly BJ, Gross R, Bittinger K, Sherrill-Mix S, Lewis JD, Collman RG, Bushman FD, Li H. 2015. Power and sample-size estimation for microbiome studies using pairwise distances and permanova. Bioinformatics. 31(15):2461–2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita-Ochiai T, Seto S, Suzuki N, Yamamoto M, Otsuka K, Abe K, Ochiai K. 2008. Butyric acid induces apoptosis in inflamed fibroblasts. J Dent Res. 87(1):51–55. [DOI] [PubMed] [Google Scholar]
- Lee CK, Herbold CW, Polson SW, Wommack KE, Williamson SJ, McDonald IR, Cary SC. 2012. Groundtruthing next-gen sequencing for microbial ecology-biases and errors in community structure estimates from PCR amplicon pyrosequencing. PLoS One. 7(9):e44224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, Chen IM, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, et al. 2012. Img: The integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 40(Database issue):D115–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark Welch JL, Rossetti BJ, Rieken CW, Dewhirst FE, Borisy GG. 2016. Biogeography of a human oral microbiome at the micron scale. Proc Natl Acad Sci USA. 113(6):E791–E800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquis RE, Bender GR, Murray DR, Wong A. 1987. Arginine deiminase system and bacterial adaptation to acid environments. Appl Environ Microbiol. 53(1):198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh PD. 2006. Dental plaque as a biofilm and a microbial community—implications for health and disease. BMC Oral Health. 6(Suppl 1):S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nascimento MM, Liu Y, Kalra R, Perry S, Adewumi A, Xu X, Primosch RE, Burne RA. 2013. Oral arginine metabolism may decrease the risk for dental caries in children. J Dent Res. 92(7):604–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobbs AH, Jenkinson HF, Jakubovics NS. 2011. Stick to your gums: mechanisms of oral microbial adherence. J Dent Res. 90(11):1271–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nossa CW, Oberdorf WE, Yang L, Aas JA, Paster BJ, Desantis TZ, Brodie EL, Malamud D, Poles MA, Pei Z. 2010. Design of 16S rRNA gene primers for 454 pyrosequencing of the human foregut microbiome. World J Gastroenterol. 16(33):4135–4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozok AR, Persoon IF, Huse SM, Keijser BJ, Wesselink PR, Crielaard W, Zaura E. 2012. Ecology of the microbiome of the infected root canal system: a comparison between apical and coronal root segments. Int Endod J. 45(6):530–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prosser JI. 2010. Replicate or lie. Environ Microbiol. 12(7):1806–1810. [DOI] [PubMed] [Google Scholar]
- Quinn RA, Navas-Molina JA, Hyde ER, Song SJ, Vazquez-Baeza Y, Humphrey G, Gaffney J, Minich JJ, Melnik AV, Herschend J, et al. 2016. From sample to multi-omics conclusions in under 48 hours. mSystems. 1(2):pii:e00038-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotroff DM, Motsinger-Reif AA. 2016. Embracing integrative multiomics approaches. Int J Genomics. 2016:1715985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg SL. 2007. Genome re-annotation: a wiki solution? Genome Biol. 8(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. 2011. Metagenomic biomarker discovery and explanation. Genome Biol. 12(6):R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer JR, Feingold E, Wang X, Lee M, Tcuenco K, Weeks DE, Weyant RJ, Crout R, McNeil DW, Marazita ML. 2013. GWAS of dental caries patterns in the permanent dentition. J Dent Res. 92(1):38–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simón-Soro A, Belda-Ferre P, Cabrera-Rubio R, Alcaraz LD, Mira A. 2013. A tissue-dependent hypothesis of dental caries. Caries Res. 47(6):591–600. [DOI] [PubMed] [Google Scholar]
- Simón-Soro A, Mira A. 2015. Solving the etiology of dental caries. Trends Microbiol. 23(2):76–82. [DOI] [PubMed] [Google Scholar]
- Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, Arrieta JM, Herndl GJ. 2006. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc Natl Acad Sci USA. 103(32):12115–12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N. 2015. Oral microbiome metabolism: from “who are they?” to “what are they doing?” J Dent Res. 94(12):1628–1637. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Washio J. 2011. Metabolomic effects of xylitol and fluoride on plaque biofilm in vivo. J Dent Res. 90(12):1463–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Yamada T. 1999. Glucose and lactate metabolism by Actinomyces naeslundii. Crit Rev Oral Biol Med. 10(4):487–503. [DOI] [PubMed] [Google Scholar]
- Takahashi N, Yamada T. 2000. Pathways for amino acid metabolism by Prevotella intermedia and Prevotella nigrescens. Oral Microbiol Immunol. 15(2):96–102. [DOI] [PubMed] [Google Scholar]
- Tanner AC, Kressirer CA, Faller LL. 2016. Understanding caries from the oral microbiome perspective. J Calif Dent Assoc. 44(7):437–446. [PubMed] [Google Scholar]
- Tanner AC, Mathney JM, Kent RL, Chalmers NI, Hughes CV, Loo CY, Pradhan N, Kanasi E, Hwang J, Dahlan MA, et al. 2011. Cultivable anaerobic microbiota of severe early childhood caries. J Clin Microbiol. 49(4):1464–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst J, Buijs MJ, Laine ML, Wismeijer D, Loos BG, Crielaard W, Zaura E. 2013. Sterile paper points as a bacterial DNA-contamination source in microbiome profiles of clinical samples. J Dent. 41(12):1297–1301. [DOI] [PubMed] [Google Scholar]
- Vartoukian SR, Adamowska A, Lawlor M, Moazzez R, Dewhirst FE, Wade WG. 2016. In vitro cultivation of ‘unculturable’ oral bacteria, facilitated by community culture and media supplementation with siderophores. PLoS One. 11(1):e0146926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent AT, Derome N, Boyle B, Culley AI, Charette SJ. 2016. Next-generation sequencing (NGS) in the microbiological world: how to make the most of your money. J Microbiol Methods [epub ahead of print 16 March 2016] in press. doi: 10.1016/j.mimet.2016.02.016 [DOI] [PubMed] [Google Scholar]
- Wang J, Gao Y, Zhao F. 2016. Phage-bacteria interaction network in human oral microbiome. Environ Microbiol. 18(7):2143–2158. [DOI] [PubMed] [Google Scholar]
- Washio J, Ogawa T, Suzuki K, Tsukiboshi Y, Watanabe M, Takahashi N. 2016. Amino acid composition and amino acid-metabolic network in supragingival plaque. Biomed Res. 37(4):251–257. [DOI] [PubMed] [Google Scholar]
- Wright JT. 2010. Defining the contribution of genetics in the etiology of dental caries. J Dent Res. 89(11):1173–1174. [DOI] [PubMed] [Google Scholar]
- Zaura E, Brandt BW, Prodan A, Teixeira de, Mattos MJ, Imangaliyev S, Kool J, Buijs MJ, Jagers FL, Hennequin-Hoenderdos NL, Slot DE, et al. 2017. On the ecosystemic network of saliva in healthy young adults. ISME J. 11(5):1218-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaura E, Keijser BJ, Huse SM, Crielaard W. 2009. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 9:259. [DOI] [PMC free article] [PubMed] [Google Scholar]