

1. INTRODUCTION
Transition-metal-catalyzed asymmetric allylic alkylation of enolates is a powerful method for the formation of carbon–carbon bonds.1 Within this field, palladium-catalyzed allylic alkylation reactions have undoubtedly been the most studied.2 Aside from limited cases,3 palladium catalysts preferentially form the linear substitution product through alkylation at the less-substituted terminus of the allylic electrophile (Scheme 1). However, in contrast to palladium, most other transition metals (e.g., Mo, W, Fe, Ru, Co, Rh, Ni, Pt, and Ir) have been shown to favor the construction of the branched product, with iridium catalysts being some of the most efficient and selective.1 The potential application of these chiral, branched products to the synthesis of natural products and biologically active compounds has motivated the development of practical and reliable transition-metal-catalyzed methods for their construction.
Scheme 1.
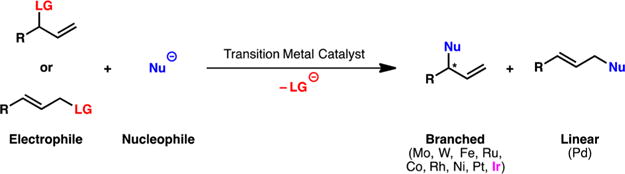
Transition-Metal-Catalyzed Asymmetric Allylic Alkylation
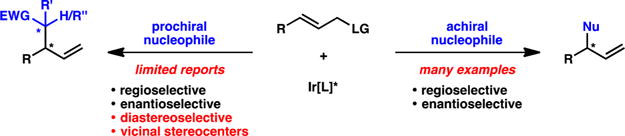
In 1997, Takeuchi and co-workers disclosed the first iridium-catalyzed allylic alkylation, which Helmchen rendered enantioselective shortly thereafter.4,5 Though the scope of the nucleophiles in these early examples was limited to stabilized enolates, iridium–phosphoramidite catalysts have emerged as privileged scaffolds which allow for the enantioselective allylic alkylation of a wide range of nonstabilized enolates as well as heteroatom nucleophiles.6 Work from the Hartwig, Helmchen, and You groups has elucidated the mechanism of the enantioselective iridium-catalyzed allylic alkylation reaction and identified the structure of the metallacyclic catalysts (Figure 1).7 These reports constitute the majority of the contributions in this field to date, wherein the allylic alkylation reactions are solely enantioselective, forming products with a single stereocenter resulting from the use of a prochiral allyl electrophile (Scheme 2, right).
Figure 1.
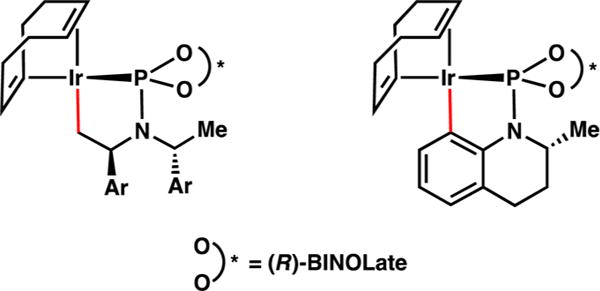
Metallacyclic iridium catalysts.
Scheme 2.
Iridium-Catalyzed Asymmetric Allylic Alkylation
Despite significant advances, methods to construct vicinal stereocenters via the union of both a prochiral nucleophile and a prochiral electrophile remain elusive (Scheme 2, left). The synthesis of these congested stereodiads is challenging not only due to the sterics of the bond-forming process but also because of the need to set two stereocenters concomitantly. The stereoselectivity in these transformations requires control of both the facial selectivity of the electrophile on the iridium catalyst as well as the facial approach of the nucleophile to the iridium π-allyl complex. This Viewpoint will give an account of the state-of-the-art of diastereo-, enantio-, and regioselective iridium-catalyzed allylic alkylation.
2. IRIDIUM CATALYST-CONTROLLED PROCESSES
2.1. Acyclic Nucleophiles
In 2003, Takemoto and co-workers reported the first example of a diastereo-, enantio-, and regioselective iridium-catalyzed allylic alkylation (Scheme 3).8 The authors showed that an iridium complex of bidentate chiral phosphite 4 and [Ir(cod)Cl]2 catalyzes the reaction of glycinate nucleophile 1 and aryl-substituted allylic phosphates 3. The corresponding branched products 5 and 7 bearing vicinal tertiary and trisubstituted stereocenters are afforded in moderate to excellent yields with good to excellent enantioselectivities, albeit with moderate diastereoselectivities. Takemoto also found that simply by employing either potassium hydroxide (KOH) or lithium hexamethyldisilazide (LiHMDS), the diastereomeric ratio could be inverted in order to preferentially access either diastereomer (5 versus 7); this synthetically useful phenomenon has yet to be reported again.
Scheme 3.
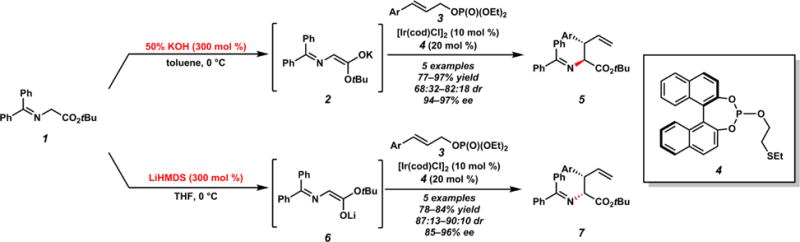
First Report of Diastereo- and Enantioselective Iridium-Catalyzed Allylic Alkylation
The different behavior of the bases is believed to be the result of contrasting enolate geometry; the hypothesis being that KOH would predominately give the E enolate 2 and LiHMDS would instead form the Z enolate 6. This observation illuminates the additional challenge that acyclic, prochiral nucleophiles present in contrast to cyclic nucleophiles in diastereo- and enantioselective allylic alkylation chemistry. In acyclic cases, the enolate geometry must be selectively controlled in addition to the facial approach of the nucleophile. This combination of challenges has resulted in significantly fewer reports of acyclic, prochiral enolate nucleophiles as compared to cyclic, prochiral nucleophiles.
A decade after Takemoto’s report, our group disclosed the second report of an iridium-catalyzed allylic alkylation of acyclic enolates (Scheme 4).9 Like Takemoto, we found chelation of the enolate with a metal cation, specifically lithium, to be crucial to the diastereoselectivity as bases lacking a chelating metal cation (i.e., 1,4-diazabicyclo[2.2.2]octane) provided significantly lower selectivity. By deprotonating ketoesters 8 with lithium tert-butoxide prior to introduction of the electrophile, products 11 bearing vicinal tertiary and all-carbon quaternary stereocenters can be obtained in moderate to excellent selectivities with the use of a catalyst derived from [Ir(cod)Cl]2 and THQphos (10). Although a majority of the cinnamyl carbonates 9 proceed with a high degree of regioselectivity (>90:10), we found that the use of electron-deficient aryl-substituted electrophiles 9 (e.g., R = C6H5NO2 or C6H5CF3) lead to a decrease in selectivity (50:50–86:14).
Scheme 4.

Allylic Alkylation of Acyclic β-Ketoesters by Stoltz
Recently, Hartwig and co-workers found that acyclic α-alkoxy ketones 12 undergo selective allylic alkylation reactions with allyl carbonates 13 in the presence of preformed metallacyclic iridium complex 14, LiHMDS, and copper(I) bromide (Scheme 5).10 In these reactions, the geometry of the acyclic enolate, formed by deprotonation with the lithium base, is controlled by chelation to a copper(I) cation. Interestingly, the identity of the cation associated with the base was also found to play an integral role in achieving high diastereoselectivities, with KHMDS providing much lower selectivities than LiHMDS. The exact nature of this cation dependence is unknown. It is also worth noting that the scope of this reaction permits the use of crotyl carbonates, albeit in lower diastereoselectivities (ca. 7:1 dr). This is the only report of alkyl-substituted electrophiles in a diastereoselective iridium-catalyzed allylic alkylation.
Scheme 5.
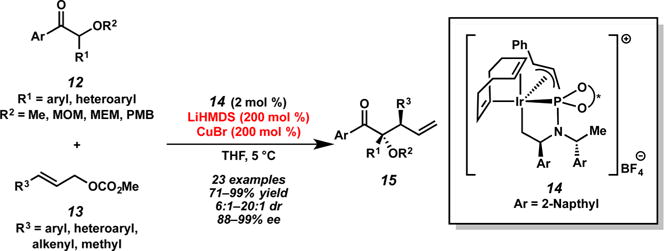
Allylic Alkylation with Acyclic α-Alkoxy Ketones by Hartwig
2.2. Cyclic Nucleophiles
In 2013, Hartwig and co-workers published the first example of a diastereoselective iridium-catalyzed allylic alkylation of cyclic, prochiral nucleophiles (Scheme 6).11 In this report, vicinal tertiary and tetra-substituted stereodiads are created via the allylic alkylation of azlactones 18 with aryl- and alkenyl-substituted allylic carbonates 19 in the presence of catalytic amounts of achiral silver phosphate 17, 3 Å molecular sieves, and an iridium catalyst generated in situ from [Ir(cod)Cl]2 and ligand 16. Through a series of control experiments, the authors were able to determine that both the phosphate and methyl carbonate anions, but not the silver cation, are key to the high reaction diastereoselectivity (up to >20:1 dr). The counteranions are presumed to deprotonate and control the facial attack of the nucleophile, whereas the silver cation is believed to sequester chloride and promote the formation of the active metallacyclic iridium catalyst.
Scheme 6.
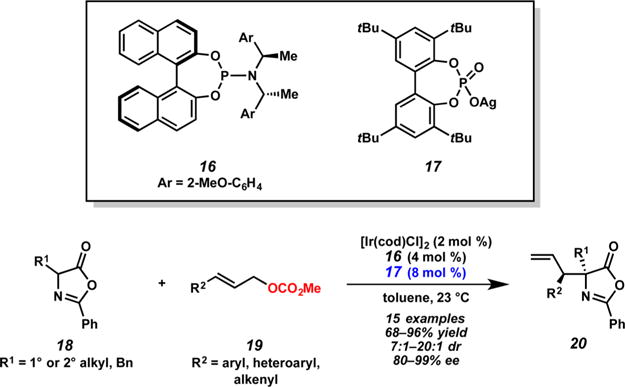
Counterion-Assisted Iridium-Catalyzed Allylic Alkylation of Azlactones by Hartwig
This counterion strategy did not prove fruitful in the iridium-catalyzed allylic alkylation of substituted 5H-oxazol-4-ones 21 or 5H-thiazol-4-ones 25 (Scheme 7).12 Application of the previously developed conditions to the reaction of 5H-oxazol-4-ones with cinnamyl carbonates failed to yield the desired allylic alkylation products 24 (Scheme 7a). The yields were improved by examining a number of organic and inorganic bases in combination with silver phosphate 17. However, it was not until a substoichiometric amount of diethyl zinc, allylic acetate 22, and preformed catalyst 23 were used in place of the silver phosphate, that good diastereoselectivities were achieved (up to 18:1 dr). The authors propose that the addition of diethyl zinc leads to the formation of zinc enolate aggregates ranging from dimers to tetramers, which in turn impart facial selectivity to the prochiral nucleophile.
Scheme 7.
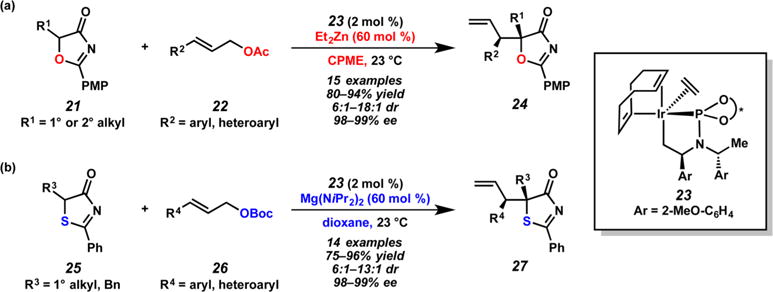
Cation Control of Diastereoselectivity in Iridium-Catalyzed Allylic Alkylation of (a) 5H-Oxazol-4-ones 21 and (b) 5H-Thiazol-4-ones 25 by Hartwig
Application of the optimal conditions developed for 5H-oxazol-4-ones 21 using diethyl zinc does not afford a diastereoselective reaction (1.3:1 dr) with substituted 5H-thiazol-4-ones 25 (Scheme 7b). Instead, the authors achieved diastereoselectivity using a magnesium enolate formed from magnesium bis(diisopropyl)amide. The aggregation states of magnesium enolates, which are generally thought to be higher-order aggregates than that of the corresponding zinc enolates, are believed to be responsible for the difference in diastereoselectivity. The authors also found that tert-butyl carbonate electrophiles 26 further improve the diastereoselectivities (up to 13:1 dr) for this nucleophile class.
Our group pioneered the discovery of diastereo- and enantioselective iridium-catalyzed allylic alkylation chemistry of cyclic enolates for the formation of vicinal tertiary and all-carbon quaternary stereocenters (Scheme 8).13 Initial investigations involving tetralone 28 revealed that a wide variety of aryl- and heteroaryl-substituted allylic carbonates 29 react to provide products 30 with high yields and stereoselectivities (Scheme 8a). The use of THQphos (10) as the chiral ligand and lithium bromide as a stoichiometric additive were found to be critical in attaining high selectivity. We postulate that lithium bromide results in lithium enolate aggregates, similar to those proposed by Hartwig.12 In further investigations, we found that reactions involving various monocyclic substrates 31 afford the corresponding products 33 with similarly high yields and selectivities to those of the tetralone-based nucleophiles (Scheme 8b). Of note, the use of electron-deficient aryl-substituted electrophiles leads to an erosion in regioselectivity (50:50 to 71:29 branched/linear) in reactions of bicyclic nucleophiles.
Scheme 8.
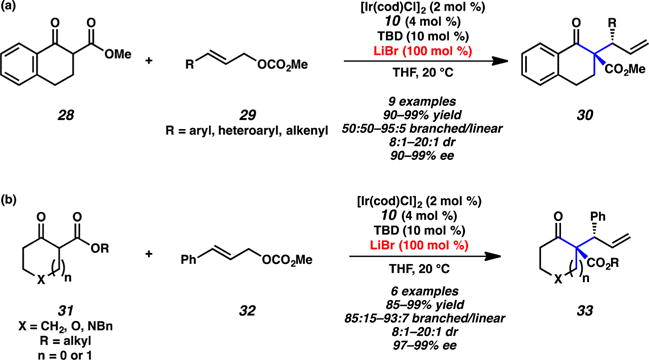
Allylic Alkylation of (a) Bicyclic β-Ketoesters 28 and (b) Monocyclic β-Ketoesters 31 by Stoltz
Studies toward substrate scope expansion of this reaction led to the successful allylic alkylation of extended enolates derived from unsaturated β-ketoester 34 (Scheme 9).14 Though the yields and selectivities were generally found to be lower than the corresponding saturated analogues (vide supra), allylic alkylation at the α-carbon atom of the extended enolate was achieved to provide products 36 bearing an additional olefin for further functionalization of the molecule.
Scheme 9.
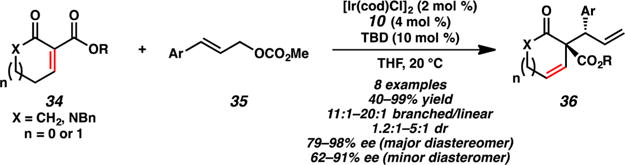
Allylic Alkylation of Extended Enolates by Stoltz
Hartwig and co-workers have found that nonstabilized enolates 37 undergo selective iridium-catalyzed allylic alkylation reactions to form vicinal tertiary and all-carbon quaternary stereocenters using preformed iridium complex 23 and barium tert-butoxide (Scheme 10).15 In all cases, products 39 are afforded with excellent enantioenrichment (>98% ee), and even simple cyclohexanone derivatives provide good selectivity for allylic alkylation of the corresponding thermodynamic enolate. This protocol is currently the only reported set of conditions for cyclic nucleophiles that is not limited to softer enolate equivalents (e.g., malonates and β-ketoesters).
Scheme 10.
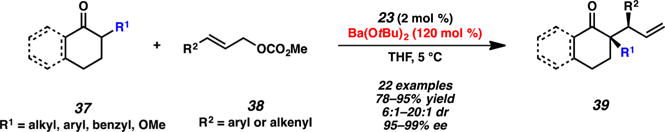
Allylic Alkylation of Non-Stabilized Enolates by Hartwig
3. DUAL CATALYST-CONTROLLED PROCESSES
Methodology that allows for the selective synthesis of all four branched stereoisomers from one set of starting materials is highly sought after in the field of allylic alkylation chemistry. However, controlling the facial selectivity of both the nucleophile and the electrophile is a daunting task for a single catalyst.16 Thus, the use of two catalysts has been required to achieve stereodivergence.
Carreira and co-workers disclosed the first dual catalytic allylic alkylation to construct vicinal tertiary and all-carbon quaternary stereocenters via the use of cinchona alkaloid-derived primary amine 42 and phosphoramidite ligand 43 (Scheme 11).17 With the appropriate combination of enantiomers of ligand 43 and pseudoenantiomers of 42, functionalized aldehyde 44 or any of the three other corresponding stereoisomers can be obtained with little erosion of selectivity due to mismatched catalysts from aldehyde 40 and branched alcohol 41. The authors propose that the lack of an apparent mismatched pair of catalysts likely arises from a high degree of planarity between the two chiral intermediates in the carbon–carbon bond-forming event. During the course of reaction optimization, the authors discovered that an acid cocatalyst was extremely important for high stereoselectivity, with trichloroacetic acid being optimal. This pioneering study set the foundation for future work using the powerful combination of enamine and iridium catalysis.
Scheme 11.
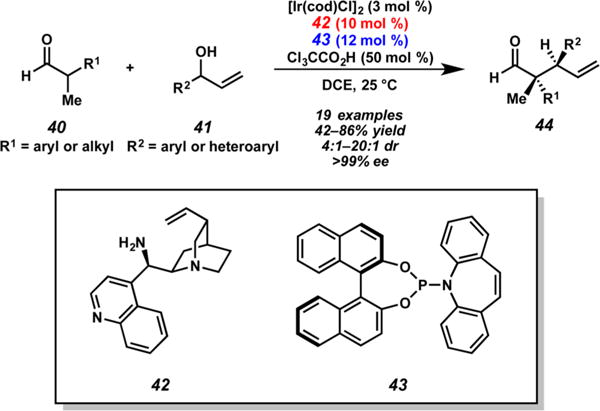
Dual-Catalyst-Promoted Allylic Alkylation of Aldehydes by Carreira
In a second-generation protocol, Carreira utilized proline-derived amine 45 for the formation of vicinal tertiary stereocenters (Scheme 12).18 While seemingly simpler from a steric perspective, this transformation in fact produces a number of additional challenges; specifically, both the starting material and allylic alkylation products are potential electrophiles for aldol processes, and the products are susceptible to epimerization. Nevertheless, the authors found that the appropriate selection of the enantiomers of 43 and 45 allows for the full complement of stereoisomers to be accessed with good to excellent selectivity. Unlike with the cinchona alkaloid-derived catalyst 42 (Scheme 11), a significant mismatched pair of catalysts is observed (7:1 dr mismatched versus 20:1 dr matched). In this work, the authors found dimethyl hydrogen phosphate to be the optimal acid cocatalyst (47, R = alkyl). In a follow-up study, the authors reported that this method tolerates α-heteroatoms (47, R = NPhth or OBn) in similarly high yields and selectivities, though trichloroacetic acid is required as the cocatalyst.19
Scheme 12.
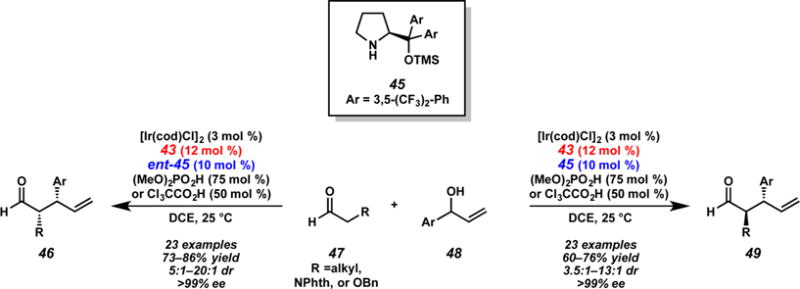
Proline-Derived Dual Catalysis by Carreira
Our group has developed a two-pot procedure for the divergent synthesis of various stereoisomers of cyclohexanone derivatives (Scheme 13).13 Trimethylsilyl ethyl ester 52 is readily accessible via the aforementioned iridium-catalyzed allylic alkylation (Scheme 8b). The use of the two pseudoenantiomeric phosphinooxazoline (PHOX) ligands 51 and 53 allows for a fluoride-triggered diastereoablative palladium-catalyzed allylic alkylation, delivering either 50 or 54 selectively. While there is a significant difference in selectivities between the two pathways (8:1 dr mismatched versus 18:1 dr matched), both palladium-catalyzed allylic alkylation products are obtained in good yields with no loss in enantioselectivity. Currently, this report represents the only available strategy for selective access to all four stereoisomers of a ketone substrate.
Scheme 13.
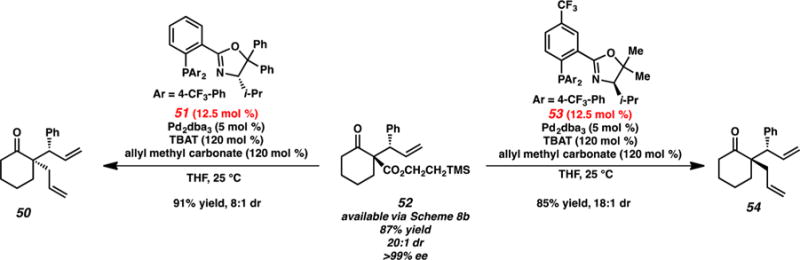
Diastereoablative Allylic Alkylation of 52 by Stoltz
4. SUMMARY AND FUTURE OUTLOOK
Over the last two decades, diastereo- and enantioselective iridium-catalyzed allylic alkylation protocols have been developed gradually, providing access to branched α-allylated carbonyl derivatives. These reactions include the synthesis of vicinal tertiary/trisubstituted, tertiary/tetrasubstituted, and tertiary/quaternary stereogenic centers using acyclic prochiral nucleophiles. For cyclic nucleophiles, only vicinal tertiary/tetrasubstituted and tertiary/quaternary stereocenter-forming methodologies have been disclosed thus far.
This emerging field is not without shortcomings. Namely, with respect to the electrophilic partner, the scope is almost exclusively limited to aryl- and alkenyl-substitution, with alkyl-substituted electrophiles remaining elusive. Moreover, reaction conditions are highly optimized for specific nucleophile classes, thus any changes to the nucleophile mandate reoptimization of the methodology.
These limitations inspire further development within this field. Moving forward, one might imagine that this technology could be extended further to alkyl-substituted electrophiles or even to the synthesis of vicinal all-carbon quaternary stereogenic centers. Perhaps new ligand classes and additives need to be explored in order to accomplish these goals. Ideally, a single catalyst or a dual catalytic system will be developed to provide good yields, diastereo-, enantio-, and regioselectivities for any combination of electrophiles and nucleophiles. However, at the present, this goal is far from being obtained. As a result, the broader synthetic field has yet to embrace diastereo- and enantioselective iridium-catalyzed allylic alkylation chemistry in the same way they have adopted the palladium-catalyzed variant; perhaps with continued time and effort, this aim will be achieved.
Acknowledgments
The authors thank the many past and present co-workers whose efforts have made our contributions in iridium-catalyzed allylic alkylation possible: Prof. Wen-Bo Liu, Dr. Corey M. Reeves, Dr. Scott C. Virgil, Dr. Noriko Okamoto, Eric J. Alexy, Dr. Allen Y. Hong, and Dr. Kristy Tran. Support for our program has been made available from the NIH-NIGMS (R01GM080269) and Caltech. Additionally, J.C.H. thanks the Camille and Henry Dreyfus postdoctoral program in Environmental Chemistry, and S.E.S. thanks the NIH-NIGMS for a predoctoral fellowship (F31GM120804).
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.(a) Trost BM, Van Vranken DL. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Crawley ML. Chem Rev. 2003;103:2921–2944. doi: 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]; (c) Lu Z, Ma S. Angew Chem, Int Ed. 2008;47:258–297. doi: 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]
- 2.Milhau L, Guiry PJ. Top Organomet Chem. 2011;38:95–154. [Google Scholar]
- 3.(a) Pretó̂t R, Pfaltz A. Angew Chem, Int Ed. 1998;37:323–325. doi: 10.1002/(SICI)1521-3773(19980216)37:3<323::AID-ANIE323>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]; (b) Hayashi T, Kawatsura M, Uozumi Y. J Am Chem Soc. 1998;120:1681–1687. [Google Scholar]; (c) You SL, Zhu XZ, Luo YM, Hou XL, Dai LX. J Am Chem Soc. 2001;123:7471–7472. doi: 10.1021/ja016121w. [DOI] [PubMed] [Google Scholar]; (d) Zheng WH, Sun N, Hou XL. Org Lett. 2005;7:5151–5154. doi: 10.1021/ol051882f. [DOI] [PubMed] [Google Scholar]; (e) Zheng WH, Zheng BH, Zhang Y, Hou XL. J Am Chem Soc. 2007;129:7718–7719. doi: 10.1021/ja071098l. [DOI] [PubMed] [Google Scholar]; (f) Liu W, Chen D, Zhu XZ, Wan XL, Hou XL. J Am Chem Soc. 2009;131:8734–8735. doi: 10.1021/ja902410w. [DOI] [PubMed] [Google Scholar]; (g) Fang P, Ding CH, Hou XL, Dai LX. Tetrahedron: Asymmetry. 2010;21:1176–1178. [Google Scholar]; (h) Chen JP, Ding CH, Liu W, Hou XL, Dai LX. J Am Chem Soc. 2010;132:15493–15495. doi: 10.1021/ja106703y. [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi R, Kashio M. Angew Chem, Int Ed Engl. 1997;36:263–265. [Google Scholar]
- 5.Janssen JP, Helmchen G. Tetrahedron Lett. 1997;38:8025–8026. [Google Scholar]
- 6.(a) Helmchen G, Dahnz A, Dübon P, Schelwies M, Weihofen R. Chem Commun. 2007:675–691. doi: 10.1039/b614169b. [DOI] [PubMed] [Google Scholar]; (b) Hartwig JF, Pouy MJ. Top Organomet Chem. 2011;34:169–208. [Google Scholar]; (c) Liu WB, Xia JB, You SL. Top Organomet Chem. 2011;38:155–208. [Google Scholar]
- 7.(a) Madrahimov ST, Markovic D, Hartwig JF. J Am Chem Soc. 2009;131:7228–7229. doi: 10.1021/ja902609g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Spiess S, Raskatov JA, Gnamm C, Brödner K, Helmchen G. Chem - Eur J. 2009;15:11087–11090. doi: 10.1002/chem.200902065. [DOI] [PubMed] [Google Scholar]; (c) Hartwig JF, Stanley LM. Acc Chem Res. 2010;43:1461–1475. doi: 10.1021/ar100047x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Liu WB, Zheng C, Zhuo CX, Dai LX, You SL. J Am Chem Soc. 2012;134:4812–4821. doi: 10.1021/ja210923k. [DOI] [PubMed] [Google Scholar]
- 8.Kanayama T, Yoshida K, Miyabe H, Takemoto K. Angew Chem, Int Ed. 2003;42:2054–2056. doi: 10.1002/anie.200250654. [DOI] [PubMed] [Google Scholar]
- 9.Liu WB, Reeves CM, Stoltz BM. J Am Chem Soc. 2013;135:17298–17301. doi: 10.1021/ja4097829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang X, Chen W, Hartwig JF. Angew Chem, Int Ed. 2016;55:5819–5823. doi: 10.1002/anie.201600235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen W, Hartwig JF. J Am Chem Soc. 2013;135:2068–2071. doi: 10.1021/ja311363a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W, Hartwig JF. J Am Chem Soc. 2014;136:377–382. doi: 10.1021/ja410650e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu WB, Reeves CM, Virgil SC, Stoltz BM. J Am Chem Soc. 2013;135:10626–10629. doi: 10.1021/ja4052075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu WB, Okamoto N, Alexy EJ, Hong AY, Tran K, Stoltz BM. J Am Chem Soc. 2016;138:5234–5237. doi: 10.1021/jacs.6b02153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen W, Chen M, Hartwig JF. J Am Chem Soc. 2014;136:15825–15828. doi: 10.1021/ja506500u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Allen AE, MacMillan DWC. Chem Sci. 2012;3:633–658. doi: 10.1039/C2SC00907B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krautwald S, Sarlah D, Schafroth MA, Carreira EM. Science. 2013;340:1065–1068. doi: 10.1126/science.1237068. [DOI] [PubMed] [Google Scholar]
- 18.Krautwald S, Schafroth MA, Sarlah D, Carreira EM. J Am Chem Soc. 2014;136:3020–3023. doi: 10.1021/ja5003247. [DOI] [PubMed] [Google Scholar]
- 19.Sandmeier T, Krautwald S, Zipfel HF, Carreira EM. Angew Chem, Int Ed. 2015;54:14363–14367. doi: 10.1002/anie.201506933. [DOI] [PubMed] [Google Scholar]