

Abstract
Changes in climbing fiber-Purkinje cell (CF-PC) synaptic connections have been found in the essential tremor (ET) cerebellum, and these changes are correlated with tremor severity. Whether these postmortem changes are specific to ET remains to be investigated. We assessed CF-PC synaptic pathology in the postmortem cerebellum across a range of degenerative movement disorders [10 Parkinson’s disease (PD) cases, 10 multiple system atrophy (MSA) cases, 10 spinocerebellar ataxia type 1 (SCA1) cases, and 20 ET cases] and 25 controls. We observed differences in terms of CF pathological features across these disorders. Specifically, PD cases and ET cases both had more CFs extending into the parallel fiber (PF) territory, but ET cases had more complex branching and increased length of CFs in the PF territory along with decreased CF synaptic density compared to PD cases. MSA cases and SCA1 cases had the most severely reduced CF synaptic density and a marked paucity of CFs extending into the PF territory. Furthermore, CFs in a subset of MSA cases formed collateral branches parallel to the PC layer, a feature not seen in other diagnostic groups. Using unsupervised cluster analysis, the cases and controls could all be categorized into four clusters based on the CF pathology and features of PC pathology, including counts of PCs and their axonal torpedoes. ET cases and PD cases co-segregated into two clusters, whereas SCA1 cases and MSA cases formed another cluster, separate from the control cluster. Interestingly, the presence of resting tremor seemed to be the clinical feature that separated the cases into the two ET-PD clusters. In conclusion, our study demonstrates that these degenerative movement disorders seem to differ with respect to the pattern of CF synaptic pathology they exhibit. It remains to be determined how these differences contribute to the clinical presentations of these diseases.
Keywords: Essential tremor, Parkinson’s disease, Spinocerebellar ataxia, Multiple system atrophy, Climbing fiber, Purkinje cell
Introduction
Clinical and neuroimaging studies have implicated cerebellar dysfunction in essential tremor (ET) [52]. Controlled postmortem studies in recent years have documented a growing number of structural changes in the ET cerebellum, involving the Purkinje cell (PC) and neighboring neuronal populations [33]. These include an increase in torpedoes and changes in PC axonal morphology [2, 40], regressive changes in the PC dendritic arbor with a reduction in dendritic spines [37], an increase in the number of heterotopic PC bodies [30], and abnormally dense and elongated basket cell axonal processes surrounding PC soma and axon initial segments [15, 29]. PC loss has been documented in several [12, 35, 40] but not all studies [49, 54].
Harmaline-induced tremor in rodents shares a number of clinical and pharmacological response features with ET [11]. Harmaline can induce hyper-synchronous activity of neurons in the inferior olivary nucleus, which subsequently leads to synchronized PC firing via climbing fiber (CF) synapses [11, 22]. Whether there is hyper-excitability of the inferior olivary nucleus in ET remains to be empirically demonstrated; however, an extensive search for pathology in the inferior olivary nucleus in ET was unrevealing [34]. Instead, we have noted the presence of abnormalities in CF-PC synapses in ET cases: (1) decreased CF-PC synaptic density on proximal PC dendrites, the normal synaptic domain of climbing fibers and (2) abnormal CF-PC synapses in extending into the PF-PC synaptic territory [31]. The number of abnormal distal CF-PC synapses inversely correlated with tremor severity [31, 41] and could be modulated by the deep brain stimulations [28], suggesting a link between this pathological feature and tremor.
CFs and parallel fibers (PFs) are important excitatory inputs to PCs and are critical in regulating PC physiology. CF synapses express vesicular glutamate transporter type 2 (VGlut2, now known as SLC17A6), whereas PF synapses express VGlut1 (now known as SLC17A7) [18]. The proper distribution of climbing and PFs along PC dendritic arbors is strictly regulated to ensure the proper timing of excitatory synaptic activation of PCs [57]. CFs form synapses on the thick, proximal PC dendrites, whereas PFs form synapses on the thin, spiny, terminal PC branchlets [46]. The activation of PF synapses leads to short duration PC action potentials, whereas the stimulation of CFs causes long duration action potentials with complex spikes and calcium influx in PCs [14, 42, 55]. The simultaneous activation of both PF and CF synapses could trigger the long-term depression, a form of long-standing cerebellar learning [24]. Defective CF-PC synaptic connections can also occur in the setting of PC degeneration in various forms of spinocerebellar ataxias (SCAs) [25]. Specifically, CFs regressed and only formed excitatory synaptic connections in the very proximal PC dendritic region in mouse models of SCAs [3, 13, 19], which is quite different from the pattern observed in the human disease ET, in which there is an extension of CFs to the distal PC dendritic branchlets [31, 41]. To our knowledge, there are no systematic comparisons of CF and PC pathologies in the postmortem human cerebellum across a range of degenerative movement disorders.
Our goal in these analyses was to understand CF-PC synaptic pathology more broadly, by extending our studies to other cerebellar degenerative disorders as well as disorders in which cerebellum, as part of a larger tremor loop, has been postulated to be involved. In addition to ET, we analyzed postmortem cerebellar tissue from: (1) SCA type 1 (SCA1), a common genetic form of cerebellar degeneration; (2) multiple system atrophy (MSA), the most common sporadic cerebellar degenerative disorder of older age [16]; and (3) Parkinson’s disease (PD), which is the second most common tremor disorder, and in which the cerebellum has been proposed to be central in modulating tremor severity and other motor symptoms [23, 58].
Materials and methods
Subject selection
Subject selection was determined by available control tissue. We identified 15 older controls above age of 75 from the New York Brain Bank, and selected 20 ET cases to age-match with these controls. None of these ET cases had brainstem Lewy body pathology [40] or pathology consistent with progressive supranuclear palsy [36]. We obtained ten SCA1 cases from Dr. Arnulf Koeppen (Veterans Affairs Medical Center, Albany, New York) and ten MSA cases from the New York Brain Bank for which we could obtain paraffin-embedded sections. Since ET cases had much older ages of death than SCA1 and MSA cases, we additionally selected from the New York Brain Bank ten younger controls to match with ten SCA1 cases and ten MSA cases. The average age of death of PD cases was in between that of ET cases and SCA1 cases or MSA cases; therefore, we did not select separate controls to match with PD cases. Ten PD cases were selected, because these cases were initially recorded to have tremor of any types based on the preliminary clinical data. All ten PD cases had been evaluated by movement disorder neurologists during life with detailed examination of rest, postural, and action tremors. All subjects signed informed consent of the Institutional Review Board of respective institutions.
Clinical diagnosis
The clinical diagnosis of ET was first made by treating physicians, and then confirmed by an ET Centralized Brain Repository movement disorders neurologist (EDL) using data from clinical questionnaires, medical records, videotaped neurological examination, and assessment of standardized Archimedes spirals [2]. ET cases had a standardized, videotaped neurological examination to assess tremor characteristics and severity in detail, including whether resting tremor was present or not [2]. The severity of action tremor in the upper extremities was quantified using the total tremor score [TTS] (range 0–36), based on clinical ratings of tremor on videotaped examination [41], which generally occurred within 6 months of death.
The control brains were from individuals who participated in the Alzheimer’s Disease Research Center and the Washington Heights Inwood Columbia Aging Project at Columbia University. They were followed prospectively with serial neurological examinations, and were clinically free of Alzheimer’s disease, ET, PD, Lewy body dementia, or progressive supranuclear palsy.
MSA brains and PD brains were from patients seen in the Movement Disorders Clinic at Columbia University, and movement disorder neurologists made the diagnoses of respective diseases during life. Although we chose PD cases based on the presence of tremor in preliminary clinical data, additional scrutiny of the complete medical records revealed only seven out of ten PD cases had tremor (six had resting tremor and four had action and postural tremor); therefore, we studied seven PD cases with tremor and three PD cases without tremor.
Tissue processing and confirmation of pathological diagnosis
All brains, except for the SCA1 cases, were processed the same way in the New York Brain Bank [56] and received ratings of neurofibrillary tangles using Braak and Braak staging [5, 6], and Consortium to Establish a Registry for Alzheimer’s disease ratings for neuritic plaques [44]. Postmortem interval was the number of hours between death and placement of the brain in a cold room or on ice.
All ET cases were examined to determine whether there was any Lewy body pathology in the brain stem (substantia nigra, locus ceruleus, and dorsal vagal nucleus) or cerebrum by α-synuclein staining [40] or any pathological changes consistent with progressive supranuclear palsy (i.e., tau-positive tufted astrocytes, globose neuronal tangles, or tau-positive glial cytoplasmic inclusions in the cerebellar cortex, inferior olivary nucleus, dentate nucleus, or basal ganglia) [36]. We excluded rare ET cases with ubiquitin-positive intranuclear inclusions [38, 39]. The pathological diagnosis of PD was made by assessing the substantic nigra: moderate or severe neuronal loss in the substantia nigra with Luxol fast blue/haematoxylin and eosin stained sections and the presence of Lewy bodies [40]. All PD cases were further staged by α-synuclein staining and the application of Braak PD stages as previously described [7, 43]. The pathological confirmation of MSA was made by the presence of typical glial cytoplasmic inclusions and neuronal loss in the striatum, substantia nigra, cerebellum, and pons assessed by α-synuclein and Luxol fast blue/haematoxylin staining, respectively [50]. Eight MSA cases had predominant olivopontocerebellar atrophy, whereas two MSA cases had mixed pathology of striatonigral degeneration and olivopontocerebellar atrophy. The diagnosis of SCA1 cases was confirmed by the determination of pathological CAG repeat numbers in the ATXN1 gene and the presence of typical intranuclear inclusions, which are p62 and ubiquitin positive, in neurons [51].
A standard 3 × 20 × 25 mm parasagittal neocerebellar block was obtained from a 0.3-cm-thick parasagittal slice located 1 cm from the cerebellar midline, corresponding to the anterior and posterior quadrangulate lobules in the anterior lobe of the cerebellar cortex [1, 56]. This region of the cerebellar cortex is involved in motor control [21, 45]. Paraffin sections (7 μm thick) were stained with Luxol fast blue hematoxylin and eosin, and the PC counts and torpedo counts were quantified as previously described [40].
Cerebellar immunohistochemistry
Seven micrometer paraffin-embedded cerebellar sections were rehydrated and incubated with 3 % hydrogen peroxide, followed by antigen retrieval in 0.1 M Tris-base Urea solution (pH 9.5) for 20 min at 95 °C, and suppression block with 10 % normal donkey serum and 0.5 % bovine serum albumin. The sections were incubated with polyclonal rabbit anti-VGlut2 antibody (1:250) [26, 27] at 4 °C for 48 h followed by incubation with biotinylated anti-rabbit IgG (Vector labs, Burlingame, CA, 15 μg/ml), and the signals were amplified by avidin/biotinylated complex (Vector, Burlingame, CA). The sections were developed with 3,3′-diaminobenzidine precipitation. Images were acquired with bright field microscopy (Zeiss AxioPlan 2 wth AxioCam HR digital camera). We also performed dual immunofluorescence staining of monoclonal mouse anti-CalbindinD28K (Sigma-Aldrich, St. Louis, MO, 1:1000) or anti-neurofilament (SMI31, Convance, 1:1000) and polyclonal rabbit anti-VGlut2 antibody (1:250) with secondary Alexa 594 goat anti-mouse IgG and Alexa 488 donkey anti-rabbit IgG antibody (Life technologies, Eugene, OR, both 20 μg/ml) to visualize the CF-PC synapses. All immunofluorescence images were taken using confocal laser scanning microscopy (Leica TSC SP2 two photon microscope).
Rationale for studying CF pathology
CF terminals that form synapses with PCs express VGlut2 [18]; therefore, VGlut2 puncta in the molecular layer represent CF-PC synapses. We chose to study CF synaptic density, which could reflect the degree of synaptic pruning often encountered in cerebellar degeneration [3, 13, 19]. In addition, the CF synaptic territory along proximal and distal domains of PC dendrites is tightly controlled and can be altered in response to abnormal synaptic activities [8] and/or cerebellar degeneration [3, 13, 19]. We, therefore, studied CF synapses abnormally present in the PF territory by quantifying the percentage of CFs extending to the outer 20 % of the molecular layer [31]. We further studied additional features of these CFs in the outer 20 % of the molecular layer by quantifying the CF length, the number of CF synapses, and the number of CF branches in this region. Another way to study CF synapses in the PF territory is to investigate the percentage of CF synapses on the thin, spiny PC dendritic branchlets [31, 41], which normally form synapses with PFs. The intra- and inter-rater reliabilities of a number of pathological readouts have been published [12, 31].
Assessment of VGlut2 (i.e., CF) synaptic density
Trained raters (SHK and DL) performed image acquisition and readout quantification without the knowledge of the diagnosis of each subject. We used random digits to select 25 fields in the cerebellar cortex in each subject and quantified the total number of visualized VGlut2 puncta on proximal PC dendrites in a 200× field directly above the PC layer. The total visualized CF length in the same 200X field was measured with NeuriteTracer (Neuron J) [48], a plugin of Image J (NIH, Besthesda, MD). CF synaptic density was defined as the total number of visualized VGlut2 puncta divided by the total CF length in a 200× field. Twenty-five fields were quantified to obtain an average CF synaptic density in each subject.
Assessment of CFs in the outer portion of the molecular layer of the cerebellar cortex
To assess the distribution of CFs across the height of the molecular layer, we investigated the percentage of CFs abnormally extending into the outer 20 % of the molecular layer. We randomly imaged 15 200× fields, which included the whole thickness of the molecular layer, in each subject, and imported images into Image J. Trained raters (SHK and CYL) first measured the total thickness of the molecular layer and then drew a line at the border between the outer 20 % and inner 80 % of the molecular layer in the cerebellar cortex. We quantified the number of CF extending above this line into the outer 20 % of the molecular layer. We calculated the percentage of CFs extending into the outer 20 % of the molecular layer in a given field (CFs in outer 20 % of the molecular layer/total CF in the field × 100). A rater (SHK) additionally quantified several features of CFs in the outer 20 % of the molecular layer, including: (1) total CF length; (2) the number of CF synapses; and (3) the number of CF branches in different diagnostic groups.
Assessment of the distribution of CF synapses on distal PC dendrites
We studied the CF synapses on the thin PC dendritic branchlets, previously defined as VGlut2 puncta on the PC thin, spiny branchlets <1 μm in diameter [17, 46]. For each subject, trained raters (CYL and JYL) used random digits to choose PC dendritic trees for image acquisition using confocal microscopy (63× objective and 5× digital zoom, Leica). Once the field was chosen, we acquired serial images of both green and red channels from the base of the molecular layer to the pial surface to reconstruct complete dendritic arborizations of five PCs per subject. We calculated the percentage of these abnormal CF-PC synapses (VGlut2 puncta on the PC thin spiny branchlets divided by the total VGlut2 puncta). MSA cases and SCA cases had markedly damaged PC dendrites; therefore, it was not possible to identify the complete PC dendritic structures or to differentiate between thick dendritic shafts and dendritic spines. Therefore, we only quantified this feature in ET cases, PD cases, and a subset of controls (n = 12, age 81.3 ± 7.1).
Statistical analyses
Analyses were performed in SPSS (version 22). Demographic and clinical characteristics were compared using the analysis of variance and Chi-square tests. Our primary outcome measures [(1) CF synaptic density, (2) the percentage of the CFs extending to the outer 20 % of the molecular layer, and (3) the percentage of CF synapses on the thin PC dendritic branchlets] and age and brain weight followed a normal statistical distribution (Kolmogorov– Smirnov test p values >0.05); therefore, we used parametric tests (analysis of variance followed by LSD post hoc analyses and Student t test). For the non-normally distributed primary measures: (1) the number of CF synapses in the outer 20 % of the molecular layer; (2) the CF length in the outer 20 % of the molecular layer; and (3) the number of CF branches in the outer 20 % of the molecular layer, and in postmortem intervals, we used Kruskal–Wallis test. We constructed linear regression models using CF synaptic density or the percentage of CFs extending to the outer 20 % of the molecular layer as the outcome variables, and age, sex, postmortem intervals, and the dummy variables of each diagnosis as the predictive variables to assess the effects of age, sex, and postmortem intervals on these pathological features. All tests of statistical significance were conducted at the two-tailed α level of 0.05.
Unsupervised cluster analyses
We performed unsupervised hierarchical clustering analyses and stratified all 75 cases based on four pathological features: (1) CF synaptic density; (2) the percentage of the CFs extending to the outer 20 % of the molecular layer; (3) PC counts; and (4) torpedo counts. We did not include the following pathological features in the analysis, because these did not follow normal distributions and thus would not be suitable for the unsupervised cluster analysis: (1) CF length in the outer 20 % of the molecular layer; (2) the number of CF synapses in the outer 20 % of the molecular layer; and (3) the number of CF branches in the outer 20 % of the molecular layer. We could not adequately assess the percentage of CF synapses on the thin PC branchlets in SCA1 cases and MSA cases because of the marked fragmentation of PC dendrites; therefore, this feature was not included in the unsupervised cluster analyses. PC pathology has been a key feature for ET [40]; therefore, we included in this analysis the two most extensively studied PC pathological features: PC counts and torpedo counts. We performed statistical analyses to assess whether the diagnoses and pathological features segregate using the MATLAB software.
Results
Demographics and clinical comparisons
As listed in Table 1, the age of controls, ET cases, PD cases, MSA cases, and SCA1 cases was significantly different (p < 0.001), because MSA cases and SCA1 cases had a shorter lifespan. However, the age of older controls was not significantly different from ET cases (p = 0.23) and the age of younger controls was not significantly different from either MSA cases (p = 0.37) or SCA1 cases (p = 0.89). The five diagnostic groups did not differ in sex and Consortium to Establish a Registry for Alzheimer’s Disease ratings for neuritic plaques. The five diagnostic groups differed with respect to postmortem intervals (p < 0.001), with the highest in SCA1 cases and controls. ET cases had a higher Braak Alzheimer’s disease stage than other diagnostic groups (ET vs. controls, PD cases, and MSA cases combined, Chi square, p = 0.04, Table 1), consistent with a previous report [47].
Table 1.
Clinical and pathological features of controls, essential tremor, Parkinson’s disease, multiple system atrophy, and spinocerebellar ataxia type 1 cases
| Controls | PD cases | ET cases | MSA cases | SCA1 cases | p value | |
|---|---|---|---|---|---|---|
| n | 25 | 10 | 20 | 10 | 10 | |
| Age at death (years) | 74.7 ± 14.0 | 72.8 ± 7.5 | 85.2 ± 6.2 | 66.8 ± 5.4 | 61.6 ± 7.1 | <0.001a |
| 82.9 ± 4.5 | p = 0.23b | |||||
| Old (n = 15) | ||||||
| 62.3 ± 14.5 | p = 0.37b | p = 0.89b | ||||
| Young (n = 10) | ||||||
| Female sex | 11 (44.0 %) | 2 (20.0 %) | 13 (65.0 %) | 6 (60.0 %) | 3 (30.0 %) | 0.12c |
| Postmortem interval (hours) | 11.5 ± 13.0 (Median = 5.7) | 4.0 ± 4.1 (Median = 2.3) | 2.7 ± 1.9 (Median = 2.4) | 3.5 ± 3.0 (Median = 2.9) | 15.5 ± 8.8 (Median = 16.5) | <0.001d |
| Braak AD stage | N/A | 0.11c | ||||
| 0 | 9 (36.0 %) | 2 (20.0 %) | 1 (5.0 %) | 5 (50.0 %) | ||
| I | 7 (28.0 %) | 1 (10.0 %) | 3 (15.0 %) | 4 (40.0 %) | ||
| II | 6 (24.0 %) | 3 (30.0 %) | 7 (35.0 %) | 0 (0.0 %) | ||
| III | 2 (8.0 %) | 2 (20.0 %) | 7 (35.0 %) | 1 (10.0 %) | ||
| IV | 1 (4.0 %) | 1 (10.0 %) | 1 (5.0 %) | 0 (0.0 %) | ||
| V | 0 (0.0 %) | 1 (10.0 %) | 1 (5.0 %) | 0 (0.0 %) | ||
| VI | 0 (0.0 %) | 0 (0.0 %) | 0 (0.0 %) | 0 (0.0 %) | ||
| CERAD plaque score | N/A | 0.13c | ||||
| 0 | 17 (68.0 %) | 6 (60.0 %) | 8 (40.0 %) | 9 (100.0 %)g | ||
| A | 6 (24.0 %) | 2 (20.0 %) | 7 (35.0 %) | 0 (0.0 %) | ||
| B | 2 (8.0 %) | 2 (20.0 %) | 3 (15.0 %) | 0 (0.0 %) | ||
| C | 0 (0.0 %) | 0 (0.0 %) | 2 (10.0 %) | 0 (0.0 %) | ||
| Purkinje cell counts | 11.3 ± 2.0 | 11.0 ± 3.0e | 8.7 ± 1.4e,**** | 6.0 ± 3.0e,**** | 5.8 ± 2.0e,**** | <0.001a |
| Torpedo counts | 5.8 ± 8.1 (Median = 3.0) | 8.8 ± 6.3f,** (Median = 8.5) | 12.7 ± 8.8f,*** (Median = 15.0) | 106.0 ± 80.0f,**** (Median = 105.0) | 47.9 ± 39.2f,**** (Median = 39.0) | <0.001d |
| CF synaptic density (puncta/100 μm) | 23.8 ± 3.6 | 23.9 ± 5.0e | 20.1 ± 2.7e,*** | 17.2 ± 4.1e,**** | 16.6 ± 2.5e,**** | <0.001a |
| Percentage of CF extending into outer 20 % ML | 16.4 ± 5.4 | 25.1 ± 6.4e,**** | 27.4 ± 7.6e,**** | 8.6 ± 5.4e,*** | 5.1 ± 1.8e,**** | <0.001a |
| CF length in outer 20 % ML (μm) | 1.8 ± 1.2 (Median = 1.4) | 3.6 ± 1.4f,**** (Median = 3.2) | 9.2 ± 5.6f,**** (Median = 7.0) | 0.5 ± 0.6f,**** (Median = 0.2) | 0.8 ± 0.7f,** (Median = 0.6) | <0.001d |
| CF synaptic number in outer 20 % ML | 2.5 ± 1.6 (Median = 2.2) | 5.5 ± 3.0f,**** (Median = 4.6) | 9.7 ± 4.8f,**** (Median = 8.9) | 1.0 ± 1.5f,*** (Median = 0.2) | 1.5 ± 1.9f,* (Median = 0.8) | <0.001d |
| CF branches in outer 20 % ML | 0.1 ± 0.1 (Median = 0.1) | 0.2 ± 0.1f,**** (Median = 0.2) | 0.8 ± 0.6f,**** (Median = 0.7) | 0.0 ± 0.0f,* (Median = 0.0) | 0.1 ± 0.2 (Median = 0.1) | <0.001d |
| Percentage of CF synapses on the thin PC dendritic branchlets | 13.8 ± 6.6 | 22.3 ± 4.8e,* | 34.4 ± 11.8e,**** | N/A | N/A | <0.001a |
Values represent mean ± standard deviation or number (percentage), and for variables with non-normal distribution, the Median is reported as well
Since essential tremor (ET) cases had much older age of death than spinocerebellar ataxia type 1 (SCA1) cases, we selected 15 older controls to match with 20 ET cases and 10 younger controls to match with 10 SCA1 cases to study climbing fiber synaptic pathology during aging
AD Alzheimer’s disease, CERAD the Consortium to establish a Registry for Alzheimer’s disease, CF climbing fiber, ET essential tremor, ML molecular layer, MSA multiple system atrophy, N/A not available, PC Purkinje cells, PD Parkinson’s disease, SCA1 spinocerebellar ataxia type 1, VGlut2 vesicular glutamate transporter type 2
p < 0.05,
p < 0.01,
p < 0.005,
p < 0.001, when compared to controls
One-way analysis of variance (ANOVA)
2 independent samples t test
Chi-square test
Kruskal-Wallis test
Analysis of variance followed by LSD post hoc analyses
Mann-Whitney U test
One data point missing
CF synaptic density
We first quantified the CF synaptic density in different diagnostic groups. These five diagnostic groups showed heterogeneous CF synaptic density (analysis of variance, p < 0.001) (Fig. 1). PD cases had a similar CF synaptic density to controls (Fig. 1a, b, f; Table 1), whereas ET cases had a 16 % reduction in CF synaptic density compared to controls (Fig. 1a, c, f; Table 1). Both MSA cases and SCA1 cases had very small amounts of CFs in the molecular layer, consistent with a previous report [27] (Fig. 1d, e). In residual CFs present, MSA cases and SCA1 cases also had 28 and 30 % reduced CF synaptic density, respectively, as compared to controls, and this was also further reduced compared to ET cases (Fig. 1; Table 1) but not different from each other (p = 0.71, LSD post hoc analysis).
Fig. 1.
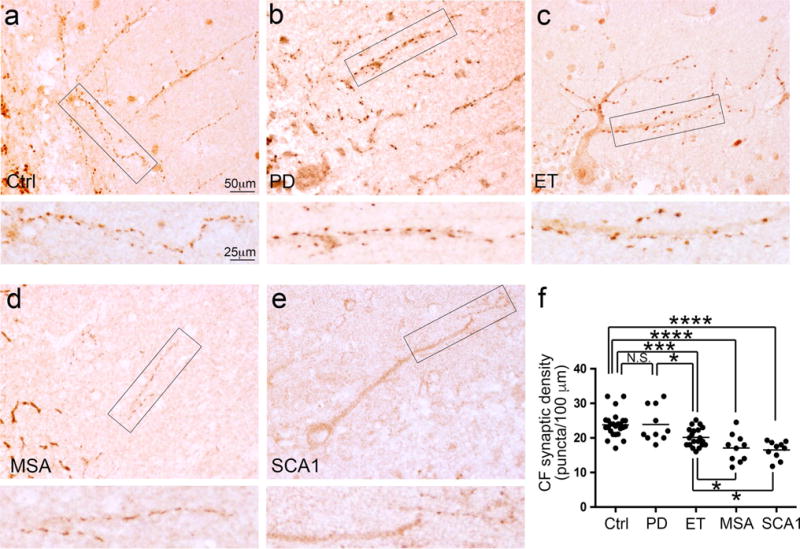
Decreased CF synaptic density on PC dendritic shafts in the molecular layer of tremor and cerebellar degenerative disorders. Representative cerebellar cortical sections labeled with anti-VGlut2 antibody of a control (a), a PD case (b), an ET case (c), an MSA case (d), and an SCA1 case (e) are shown. The rectangular areas (a–e) are shown below at higher magnification and demonstrate the decreased CF linear synaptic density in the ET case, the MSA case, and the SCA1 case as compared to the control and the PD case. The quantifications of the CF synaptic density across different diagnostic groups are shown (f). Ctrl control, PD Parkinson’s disease, ET essential tremor, MSA multiple system atrophy, SCA1 spinocerebellar ataxia type 1, CF climbing fiber. NS non-significant, *p < 0.05, ***p < 0.005, ****p < 0.001
CFs extending into the outer molecular layer
We next investigated the distribution of CFs across the height of the molecular layer. The percentage of CFs extending into the outer 20 % of molecular layer also differed across diagnostic groups (analysis of variance, p < 0.001) (Fig. 2). PD cases had 1.53-fold higher number of CFs in the outer molecular layer when compared to controls (Fig. 2a, b, f; Table 1). ET cases had 1.67-fold higher number of CFs extending into the outer molecular layer as compared to controls but similar to PD cases (Fig. 2a–c, f; Table 1). Both MSA cases and SCA1 cases had few CFs extending into the outer portion of the molecular layer, being 0.48- and 0.69-fold lower compared to controls, respectively (Fig. 2d–f; Table 1). When compared with ET cases, both MSA cases and SCA1 cases also had 0.69-fold and 0.81-fold less CFs extending into the outer molecular layers, respectively (LSD post hoc analyses, both p < 0.001) (Fig. 2c–f; Table 1), but MSA cases and SCA1 cases were similar to each other in this measure (p = 0.19, LSD post hoc analysis).
Fig. 2.
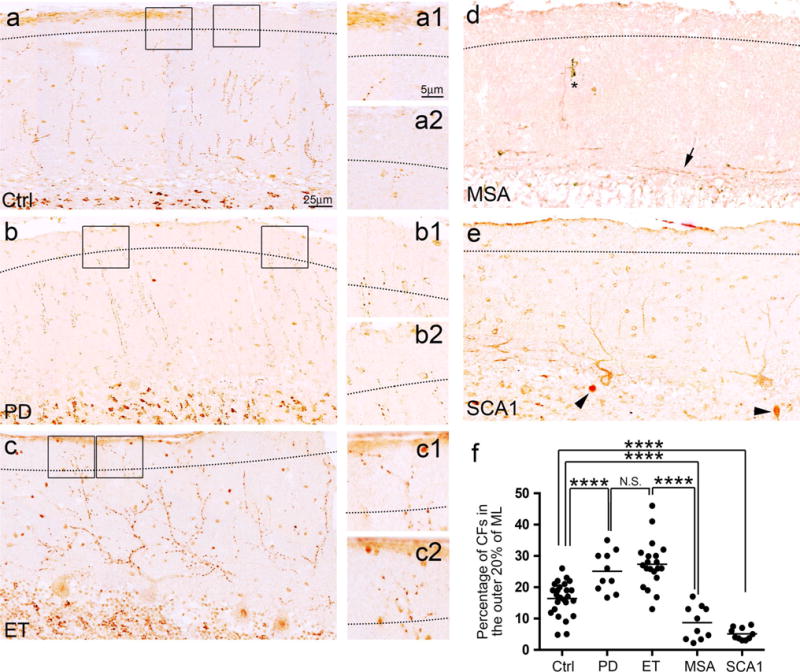
CFs in the outer portion of the molecular layer in the cerebellar cortex of tremor and cerebellar degenerative disorders. The dotted line indicates the border between the outer 20 % and inner 80 % of the molecular layer (a–e). Representative cerebellar sections of a control (a), a PD case (b), an ET case (c), an MSA case (d), and an SCA1 case (e) labeled with anti-VGlut2 antibodies are shown. The square areas are shown at higher magnification, to the right of each panel (a–c), and demonstrate that an ET case and a PD case have an increased number of CFs passing the dotted line as compared to a control. Representative cerebellar sections of an MSA case (d) and an SCA case (e) demonstrate that the CFs regressed from the outer portion of the molecular layers. Abnormal CFs parallel to the PC layer (d, an arrow) and a dystrophic CF (d, asterisk) can be observed in the MSA case. Torpedoes are seen in the SCA1 case (e, arrowheads). When compared to controls, ET cases and PD cases have a significantly increased percentage of total CF profiles extending into the outer 20 % of the molecular layer, whereas this pathology is markedly decreased in MSA cases and SCA1 cases. Ctrl control, PD Parkinson’s disease, ET essential tremor, MSA multiple system atrophy, SCA1 spinocerebellar ataxia type 1, CF climbing fiber. NS non-significant, ****p < 0.001
CF synaptic pathology in the outer molecular layer
ET cases and PD cases had a similar percentage of CFs extending into the outer molecular layer, but we often observed more complex CF branching and longer CFs in the outer molecular layer in ET cases (Figs. 2c, 3a). Thus, we further assessed CF length, the number of CF synapses, and the number of CF branches in the outer 20 % of the molecular layer. We found that ET cases had significantly longer CF length, increased number of CF synapses, and more CF branching in the outer 20 % of the molecular layer as compared to controls and all other diagnostic groups (Fig. 3a–d; Table 1). Interestingly, PD cases also had significantly longer CF length, increase in the number of CF synapses, and more CF branching in the outer 20 % of the molecular layer than controls, but to a lesser degree than in ET cases (Fig. 3a–d; Table 1). On the other hand, MSA cases and SCA1 cases had shorter CF length and a smaller number of CF synapses in the outer 20 % of the molecular layer than controls (all p < 0.05, Mann–Whitney tests), and MSA cases also had less CF branching in the outer 20 % of molecular layer than controls (p = 0.02, Mann-Whitney test) (Fig. 3a–d; Table 1). MSA cases and SCA1 cases were not different in CF length, the number of CF synapses, and the number of CF branches in the outer 20 % of molecular layer (p = 0.35, p = 0.06, p = 0.22, respectively, Mann–Whitney test).
Fig. 3.
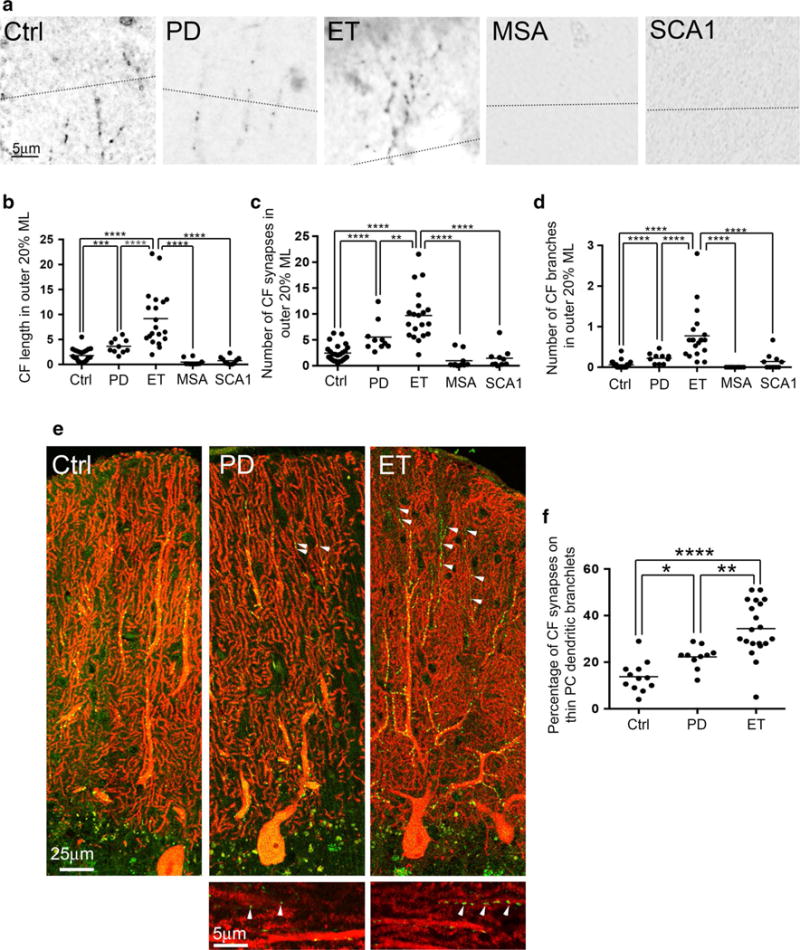
CF morphology in the outer 20 % of the molecular layer and CF synapses on the thin PC dendritic branchlets. The dotted line indicates the border between the outer 20 % and inner 80 % of the molecular layer (a). Representative cerebellar sections labeled with anti-VGlut2 antibody of a control, a PD case, an ET case, an MSA case, and an SCA1 case demonstrate more complex CF synaptic pathology in the ET case (a). In ET cases, there are increased CF length (b), increased CF synapses (c), and increased number of CF branches (d) in the outer 20 % of the molecular layer when compared to other diagnostic groups. Representative images of dual immunofluorescence with anti-VGlut2 (Alexa 488, green) and anti-calbindinD28k antibody (Alexa 594, red) of cerebellar sections of a control, an ET case and a PD case (e) and CF synapses on the thin PC dendritic branchlet <1 μm in diameter are shown (arrowheads in e). ET cases have increased percentage of CF synapses on the thin PC dendritic branchlets <1 μm in diameter when compared to PD cases and controls (f), and this feature is also increased, but to a lesser extent, in PD compared to controls. Ctrl control, PD Parkinson’s disease, ET essential tremor, MSA multiple system atrophy, SCA1 spinocerebellar ataxia type 1, CF climbing fiber. ML molecular layer, *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001
CF synapses on the thin PC dendrites
Finally, we investigated the percentage of CF synapses on the thin PC dendritic branchlets across the diagnostic groups. We found that ET cases had a 2.5-fold increase in the percentage of CF synapses on the thin PC dendritic branchlets than controls (Fig. 3e, f; Table 1), consistent with our previous finding [31]. Furthermore, ET cases had a 1.5-fold increase in the percentage of CF synapses on the thin PC dendritic branchlets than PD cases (Fig. 3e, f; Table 1). Finally, PD cases had a 1.6-fold increase in the percentage of CF synapses on the thin PC dendritic branchlets than controls (Fig. 3e, f; Table 1).
CF pathology adjusted for aging and other demographics
We assessed the association between CF synaptic density and age in the control group: Pearson’s r = −0.32, p = 0.12. In addition, we observed that age was inversely correlated with the percentage of CFs extending into the outer 20 % of the molecular layer (Pearson’s r = −0.56, p < 0.01), indicating that the regression of CF extensions is part of the normal aging process. Because sex could also affect the CF pathology [41], in a series of linear regression models, we assessed each CF pathological feature after adjusting for age, sex, as well as a potential confounder, postmortem interval. We found that the case-control differences in CF pathology in each diagnostic group still remained similar as in the unadjusted comparisons (Table 2).
Table 2.
Climbing fiber synaptic pathology measures adjusting for age, sex, and postmortem interval
| Variables | Regression coefficients of climbing fiber pathological features variablesa
|
|||||
|---|---|---|---|---|---|---|
| CF synaptic density (pucta/100 μm) | Percentage of CF extending into outer 20 % ML | CF length in outer 20 % ML | CF synaptic number in outer 20 % ML | CF branches in outer 20 % ML | Percentage of CF synapses on the thin PC dendrite branchlets | |
| Age at death (years) | −0.12** | −0.07 | 0.02 | −0.01 | −0.00 | 0.48 |
| Sexb | −0.06 | −0.88 | 0.04 | −0.07 | 0.06 | −1.71 |
| Postmortem interval (hours) | 0.01 | 0.04 | −0.03 | −0.02 | −0.00 | 0.21 |
| PD cases vs. controls | −0.81 | 10.30**** | 2.03 | 3.33* | 0.14 | 13.43** |
| ET cases vs. controls | −2.49* | 12.64*** | 7 27**** | 7.40**** | 0 73**** | 20.20**** |
| MSA cases vs. controls | −7 08**** | −7.00** | −1.23 | −1.48 | −0.08 | N/A |
| SCA1 cases vs. controls | −8 98**** | −12.19**** | −0.57 | −0.91 | 0.08 | N/A |
CF climbing fibers, ET essential tremor, ML molecular layers, MSA multiple system atrophy, N/A not available, PC Purkinje cells, PD Parkinson’s disease, SCA1 spinocerebellar ataxia type 1, VGlut2 vesicular glutamate transporter type 2
p < 0.05,
p< 0.01,
p < 0.005,
p < 0.001
All regression coefficients and p value were calculated in linear regression model, adjusting for age at death, sex, and postmortem interval
Men = 0, women = 1
CF collaterals in MSA
Interestingly, we observed unique CF morphology in MSA cases. These elongated VGlut2-positive fibers distributed parallel to the PC layer, very different from the normal CFs distributions (Figs. 2d, 4a). Where intact PC dendrites were identified, these thickened fibers extended from the granule cell layer to proximal PC dendrites before transitioning into normal CFs, supporting that these VGlut2-positive fibers are indeed abnormal CFs (Fig. 4b). With dual immunofluorescence of CalbindinD28K and VGlut2, we found that most of the PC dendrites were fragmented and the elongated CFs distributed perpendicular to the PC dendrites (Fig. 4c–e). Some of the remaining PC dendrites still had CF innervation, which was much regressed and restricted only in the very proximal part of the PC dendritic shafts (Fig. 4f–h). In addition, some of the fragmented PC dendrites still had a few CF synapses (Fig. 4i–k). The elongated CFs were partially co-localized with neurofilament, consistent with a neuronal axon identity (Fig. 4l–n). This unique parallel-aligned CF morphology was not observed in any of the controls, ET cases, PD cases, and SCA1 cases but was found in 6/10 of our MSA cases, and correlated with more severe cerebellar pathology (e.g., PC loss, decreased VGlut2-lableled cerebellar glomeruli, and cerebellar white matter destruction; data not shown) in these cases.
Fig. 4.
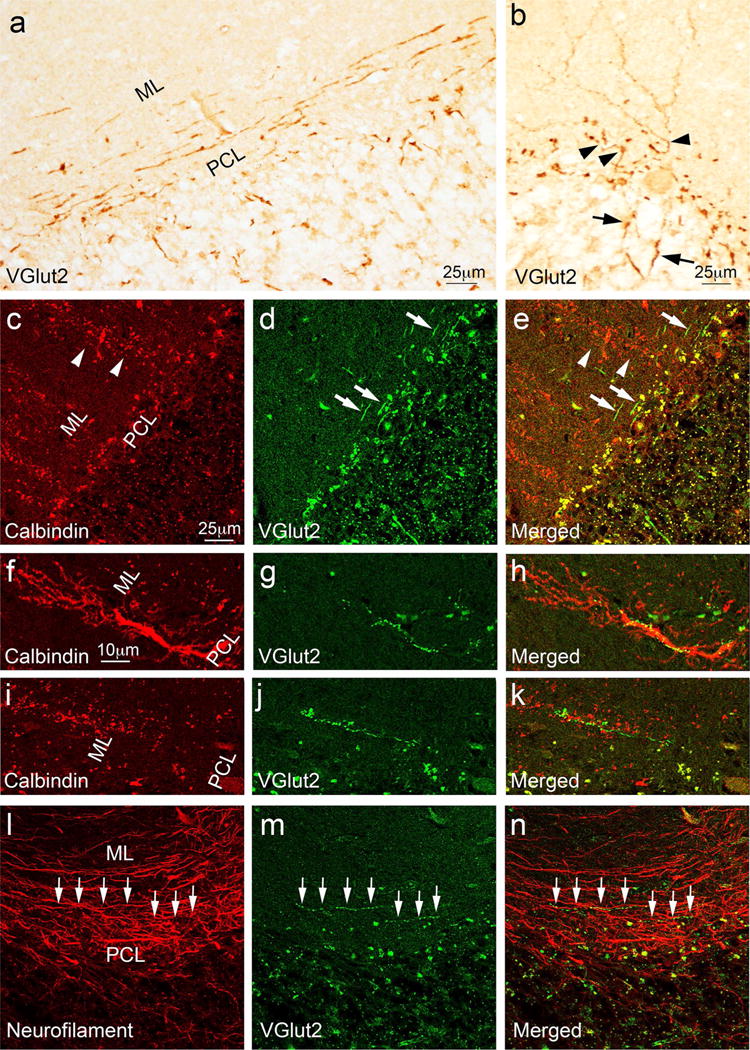
Abnormal CFs in MSA cases. The cerebellar cortical sections labeled with anti-VGlut2 antibody of MSA cases demonstrated abnormal CFs oriented parallel to the PC layer (a). Thickened CFs extended from the granule cell layer (arrows) to the proximal part of PC dendrites (arrowheads) before transitioning into normal appearing CFs (b). Dual immunofluorescence with anti-calbindinD28k antibody (Alexa 594, red) (c, f, i) and anti-VGlut2 (Alexa 488, green) (d, g, j) of cerebellar sections of MSA cases show that abnormal CFs (d, e, arrows) run perpendicular to fragmented PC dendrites (c, e, arrowheads). In addition, some abnormal CFs (g, h, j, k) form synapses with the very proximal part of the dystrophic PC dendrites (f, h) or fragmented PC dendritic dendrites (i, k) in the cerebellar cortex. The abnormal CF synapses (arrows, m, n) that are parallel to PC layers partially colocalize with neurofilament, supporting that these fibers are CF axons (arrows, l, n). ML molecular layer. PCL Purkinje cell layer
Unsupervised cluster analyses
Using unsupervised cluster analyses, we assessed whether these five diagnostic groups segregated based on four cerebellar pathological features: (1) CF synaptic density; (2) the percentage of the CFs extending into the outer 20 % of the molecular layer; (3) PC counts; and (4) torpedo counts. There were four main clusters with distinct pathological features (Fig. 5; Table 3). The blue cluster consisted mainly of MSA cases and SCA1 cases, and was characterized by the lowest PC counts, very high torpedo counts, low CF synaptic density, and a very low percentage of CFs extending into the outer 20 % of the molecular layer (9 out of 10 MSA cases in the blue cluster, hypergeometric test, p < 0.001; and 9 out of 10 SCA1 cases in the blue cluster, p < 0.001). The gray cluster is comprised primarily of controls (21 out of 25 controls in the gray cluster, p < 0.001). Interestingly, ET cases and PD cases co-segregated into two clusters: the yellow cluster (11 out of 20 ET cases and 4 out of 10 PD cases, both p < 0.001) and to a lesser extent the orange cluster (5 out of 20 ET cases and 3 out of 10 PD cases, p < 0.01). When compared to the orange cluster, the yellow cluster had higher CF synaptic density (LSD post hoc, p < 0.001) and a lower PC count (p < 0.001), whereas CFs extending into the outer 20 % of the molecular layer and torpedo counts were similar (p = 0.12 and p = 0.22, respectively) (Fig. 5; Table 3). We did not find significant differences in age, sex, or the presence of postural/action tremor between ET cases and PD cases in the yellow and in the orange clusters (Table 4). Interestingly, ET cases that had resting tremor were more likely to be in the orange cluster, although this was not statistically significant (p = 0.16), and a similar trend was also observed in PD cases with resting tremor (p = 0.14). When combining ET and PD, cases in the orange cluster indeed were more often found to have resting tremor than cases in the yellow cluster (p = 0.03) (Table 4).
Fig. 5.
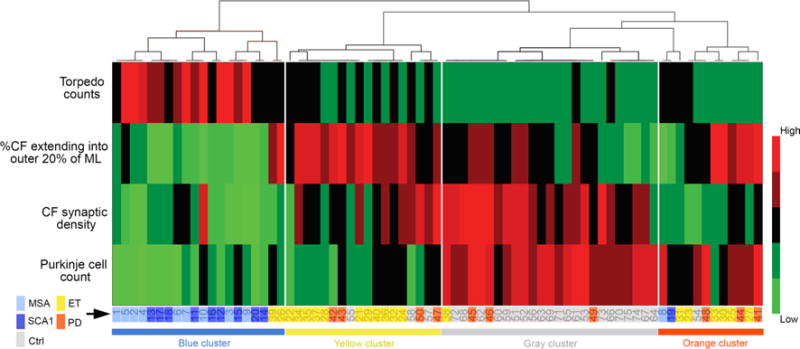
Unsupervised cluster analysis in degenerative movement disorders. We performed unsupervised cluster analysis to assess whether the clinical diagnosis would segregate based on the four pathological features in the cerebellum: [1] CF synaptic density; [2] the percentage of the CFs extending into the outer 20 % of the molecular layer; [3] PC counts; and [4] torpedo counts. Four clusters were identified. The blue cluster consists mainly of MSA cases and SCA1 cases. The gray cluster is composed primarily of controls. ET cases and PD cases segregated both in the yellow cluster and in the orange cluster. Ctrl control, PD Parkinson’s disease, ET essential tremor, MSA multiple system atrophy, SCA1 spinocerebellar ataxia type 1, CF climbing fiber, ML molecular layer
Table 3.
Pathological features of all cases and controls grouped by clustering
| Variables | Blue cluster | Yellow cluster | Gray cluster | Orange cluster | p value |
|---|---|---|---|---|---|
| n | 20 | 18 | 25 | 12 | |
| Diagnosisc | <0.001a | ||||
| Controls | 0 (0.0 %) | 3 (16.7 %) | 21 (84.0 %) | 1 (8.3 %) | |
| PD cases | 0 (0.0 %) | 4 (22.2 %) | 3 (12.0 %) | 3 (25.0 %) | |
| ET cases | 2 (10.0 %) | 11 (61.1 %) | 1 (4.0 %) | 6 (50.0 %) | |
| MSA cases | 9 (45.0 %) | 0 (0.0 %) | 0 (0.0 %) | 1 (8.3 %) | |
| SCA1 cases | 9 (45.0 %) | 0 (0.0 %) | 0 (0.0 %) | 1 (8.3 %) | |
| Purkinje cell counts | 5.7 ± 2.2 Median = 5.4 | 7.9 ± 1.3 Median = 8.1 | 12.0 ± 1.5 Median = 11.7 | 10.7 ± 1.8 Median = 10.1 | <0.001b |
| Torpedo counts | 77.6 ± 67.6 Median = 50.5 | 12.7 ± 7.7 Median = 15.0 | 5.1 ± 7.7 Median = 3.0 | 9.6 ± 6.50 Median = 8.5 | <0.001b |
| CF synaptic density (puncta/100 μm) | 16.9 ± 3.3 Median = 17.0 | 21.9 ± 3.1 Median = 22.00 | 24.8 ± 3.7 Median = 24.00 | 18.5 ± 1.2 Median = 18.00 | <0.001b |
| Percentage of CF extending into outer 20 % ML | 9.7 ± 9.4 Median = 6.0 | 25.9 ± 4.9 Median = 26.0 | 16.5 ± 5.6 Median = 17.0 | 23.1 ± 12.1 Median = 22.8 | <0.001b |
Values represent mean ± standard deviation or number (percentage), and for variables with non-normal distribution, the median is reported as well
CF climbing fiber, ET essential tremor, ML molecular layer, MSA multiple system atrophy, PD Parkinson’s disease, SCA1 spinocerebellar ataxia type 1, VGlut2 vesicular glutamate transporter
Chi-square test
Kruskal–Wallis test
The percentages indicate column percentages
Table 4.
Clinical features of essential tremor and Parkinson’s disease cases grouped by yellow and orange clusters
| Variables | ET cases
|
p value | |
|---|---|---|---|
| Yellow cluster | Orange cluster | ||
| n | 11 | 6 | |
| Age at death (years) | 84.0 ± 6.9 | 86.7 ± 5.5 | 0.43a |
| Sex | 0.30b | ||
| Men | 6 (54.5 %) | 1 (16.7 %) | |
| Women | 5 (45.5 %) | 5 (83.3 %) | |
| Rest tremor | 0.16b | ||
| No | 8 (72.7 %) | 2 (33.3 %) | |
| Yes | 3 (27.3 %) | 4 (66.7 %) | |
| Action/postural tremor | |||
| No | 0 (0 %) | 0 (0 %) | |
| Yes | 11 (100 %) | 6 (100 %) | |
|
| |||
| PD cases
|
p value | ||
| Yellow cluster | Orange cluster | ||
|
| |||
| n | 4 | 3 | |
| Age at death (years) | 74.0 ± 5.4 | 79.33 ± 2.1 | 0.14a |
| Sex | 1.00b | ||
| Men | 3 (75.0 %) | 3 (100.0 %) | |
| Women | 1 (25.0 %) | 0 (0.0 %) | |
| Rest tremor | 0.14b | ||
| No | 3 (75.0 %) | 0 (0.0 %) | |
| Yes | 1 (25.0 %) | 3 (100.0 %) | |
| Action/postural tremor | 1.00b | ||
| No | 2 (50.0 %) | 1 (33.3 %) | |
| Yes | 2 (50.0 %) | 2 (66.7 %) | |
|
| |||
| Combined group (ET and PD cases) |
p value | ||
| Yellow cluster | Orange cluster | ||
|
| |||
| n | 15 | 9 | |
| Age at death (years) | 81.3 ± 7.8 | 84.2 ± 5.8 | 0.31a |
| Sex | 0.68b | ||
| Men | 9 (60.0 %) | 4 (44.4 %) | |
| Women | 6 (40.0 %) | 5 (55.6 %) | |
| Rest tremor | 0.03b | ||
| No | 11 (73.3 %) | 2 (22.2 %) | |
| Yes | 4 (26.7 %) | 7 (77.8 %) | |
| Action/postural tremor | 1.00b | ||
| No | 2 (50.0 %) | 1 (33.3 %) | |
| Yes | 13 (50.0 %) | 7 (66.7 %) | |
Values represent mean ± standard deviation or number
ET essential tremor, PD Parkinson’s disease
Two independent samples t test
Chi-square test
Discussion
Our study systematically investigated CF pathological features more broadly, by extending our studies of ET to other cerebellar degenerative disorders as well as disorders in which cerebellum, as part of a larger tremor loop, has been postulated to be involved. Interestingly, our study demonstrates that these disorders seem to differ with respect to the pattern of CF synaptic pathology they exhibit. Specifically, PD cases and ET cases had more CFs extending into the PF territory, but ET cases showed additional complex branching and elongation of CFs in the PF territory along with a reduction of CF synaptic density. On the other hand, MSA cases and SCA1 cases had much regressed CFs in the molecular layer and decreased CF synaptic density, whereas some MSA cases additionally had thickened CF collaterals parallel to PC layers. We further investigated combined pathological features of CFs and PCs across the diagnostic groups with an unsupervised cluster analysis and found that ET cases and PD co-segregated into two clusters, whereas MSA cases and SCA1 cases clustered together, separate from controls. Overall, our studies demonstrated differences across degenerative movement disorders in terms of their CF and PC pathologies (Table 5).
Table 5.
Patterned climbing fiber pathological features in degenerative movement disorders
| Control | Parkinson’s disease | Essential tremor | Multiple system atrophy | Spinocerebellar ataxia type 1 | |
|---|---|---|---|---|---|
| Climbing fiber synaptic density | − | − | ↓ | ↓↓ | ↓↓ |
| Percentage of climbing fibers extending into outer 20 % of the molecular layer | − | ↑ | ↑ | ↓ | ↓ |
| Climbing fiber length in the outer 20 % of the molecular layer | − | ↑ | ↑↑ | ↓ | ↓ |
| Number of climbing fiber synapses in the outer 20 % of the molecular layer | − | ↑ | ↑↑ | ↓ | ↓ |
| Number of climbing fiber branches in the outer 20 % of the molecular layer | − | ↑ | ↑↑ | ↓ | − |
| Percentage of climbing fiber synapses on the thin Purkinje cell dendritic branchlets | − | ↑ | ↑↑ | N/A | N/A |
| Climbing fiber collaterals | − | − | − | ↑ | − |
| Purkinje cell counts | − | − | ↓ | ↓↓ | ↓↓ |
| Torpedo counts | − | ↑ | ↑ | ↑↑ | ↑↑ |
N/A not available due to the fragmentation of Purkinje cell dendrites of multiple system atrophy cases and spinocerebellar ataxia type 1 case
It has been proposed that ET is a cerebellar degenerative disorder given the progressive nature of the disease and associated neurodegenerative changes in the cerebellum [32]. The observation of decreased CF synaptic density in ET, as is seen in cerebellar degenerative disorders, lends an additional support to the degenerative model of ET. We found a marked reduction in the numbers of CFs in the cerebellar cortex of MSA and SCA1 cases, possibly secondary to inferior olivary nucleus pathology [50, 51]. However, in contrast with MSA and SCA1, we did not find a dramatic PC dendritic fragmentation in ET, indicating a relatively milder degree of degeneration. Nonetheless, the decrease in CF synaptic density could represent an early phase of cerebellar degeneration.
The cerebellum in PD is an under-studied area, and the cerebellothalamic pathway has been implicated in PD resting tremor and other motor symptoms. It has been postulated that nigro-striatal dysfunction can lead to the generation of parkinsonian tremor and the cerebellum can play an important role in modulating the tremor frequency and intensity [23, 58]. Since the olivo-cerebellar pathway has long been thought to be involved in the tremor loops, the CF pathology in PD cases could be important in modulating tremor presentation. On the other hand, CF pathology could also be a consequence of long-standing tremor-related rhythmic loops involving the CFs, since CF synaptic distributions on PC dendrites could be dynamically regulated in response to excessive or deficient CF activity [8–10].
Interestingly, ET cases had increased complexity of CF synapses in the outer molecular layer as compared to PD and controls, highlighting the uniqueness of this pathological feature in ET. Cerebellar degenerative disorders, such as MSA and SCA1, have much regressed CF distributions, very different from ET and PD, supporting the notion that tremor might require a relatively intact cerebellar circuitry for the propagation of the rhythmic oscillatory activity in the brain.
We used an unsupervised cluster analysis based on CF and PC pathologies and found that the cases and controls could all be categorized into four clusters. Interestingly, ET cases and PD cases co-segregated into two distinct clusters. Clinically, cases in one cluster (designated as orange cluster: higher PC counts, lower torpedo counts, and lower CF synaptic density) were more likely to have resting tremor than the other cluster (designated as yellow cluster: lower PC counts, higher torpedo counts, and higher CF synaptic density) (Fig. 5; Table 3). These results showed the clinicopathological heterogeneity of ET and PD. Based on these findings, it is possible that PCs and their intact axons are crucial to propagate the oscillation rhythm required for resting tremor. Further research is mandated to explore this possibility.
CF synaptic distributions are important to control PC physiology and normal cerebellar function. In our study, tremor disorders have abnormal CFs distributed in the PF territories. These abnormal CF synapses might contribute to tremor presentation as a mouse model with such abnormal CF synaptic pathology has 10 Hz tremor in the eyes, which falls into ET frequency [59]. In addition, abnormal CF synapses in the PF territories strongly correlate with tremor severity in ET cases, further supporting the notion that these synaptic pathologies can directly link to tremor [31, 41]. The regressed CF pathology might also contribute to cerebellar ataxia, as several animal models of different genetic mutations of SCAs have regressed CF distribution in the molecular layer, disturbed PC physiology, and ataxia phenotypes [3, 13, 19, 53]. However, animal models of SCAs might not completely recapitulate human diseases. For example, we did not observe abnormal CFs surrounding the PC soma in human SCA1 cases, but this neuropathological feature has been well-described in the SCA1 mouse models [13]. Therefore, studying postmortem human brains in the genetic disorders still remains crucial to understand the disease pathogenesis.
We additionally observed unique CF distributions that run parallel to the PC layer in MSA cases. These fibers could be the collateral sprouts of CFs. In the setting of rapid PC degeneration, CFs lose their primary innervation targets and form synapses with the neighboring PCs [20]. However, in MSA, there is an extensive PC loss (Table 1); therefore, many CFs without target PCs might have this unique distribution to search for the remaining PCs. Interestingly, SCA1 cases had similar PC loss as MSA cases, (Table 1), but SCA1 cases did not have parallel distributions of CFs, suggesting that other unique feature of MSA, such as α-synuclein, aggregates in the oligodendrocytes might be important.
The major limitation in the current study is the modest sample size, but nonetheless, we were able to detect numerous case–control differences across different diagnostic groups. Future studies will focus on the CF pathology in a larger sample size of PD and degenerative ataxias with different clinical presentations. For example, an analysis of CF pathology within MSA cases who are primarily parkinsonian vs. primarily ataxic would be of interest; such an analysis was not possible in our sample. In addition, differences in CF pathology were observed at the group level, but this could not necessarily be translated to individual levels due to the significant overlap across diagnostic groups. Other pathological features of the cerebellum are likely to play roles in these disorders. Future studies will also focus on the comparison of cerebellar pathology in neurodegenerative disorders with and without cerebellar involvement; for example, frontotemporal dementia cases with C9Orf72 repeat expansions have more cerebellar involvement when compared to those without C9Orf72 repeat expansions [4].
In conclusion, we studied several aspects of CF pathology in tremor and cerebellar degenerative disorders and also in controls. Our study provides the first insight to CF-PC synapses during aging and in different disease states. Each degenerative movement disorder demonstrated its patterned CF and PC pathological features, which might be the result of the respective degenerative processes. Further studies on how these abnormal CF and PC pathologies can lead to tremor and/or ataxia will be crucial to gain an understanding of these disorders. In addition, therapies targeting abnormal CF synapses could be a promising strategy.
Acknowledgments
Dr. Arnulf H. Koeppen, Veterans Affairs Medical Center, Albany, New York, USA provided the rabbit polyclonal anti-VGluT2 antibody and the ten specimens of SCA1. We also thank the patients and families for the donation of brains and NYBB for processing of autopsy tissue.
Funding Dr. Kuo has received funding from the National Institutes of Health: NINDS #K08 NS083738 (principal investigator), and the Louis V. Gerstner Jr. Scholar Award, PD Foundation, and International ET Foundation, and NIEHS Pilot Grant ES009089 (principal investigator). Dr. Louis has received research support from the National Institutes of Health: NINDS #R01 NS042859 (principal investigator), NINDS #R01 NS39422 (principal investigator), NINDS #R01 NS086736 (principal investigator), NINDS #R01 NS073872 (principal investigator), NINDS #R01 NS085136 (principal investigator), and NINDS #R01 NS088257 (principal investigator). He has also received support from the Claire O’Neil ET Research Fund (Yale University). Dr. Faust has received funding from the National Institutes of Health: NINDS #R01 NS04289 (co-investigator), NINDS #R21 NS077094 (principal investigator), NINDS #R01 NS085136 (principal investigator), and NINDS #R01 NS088257 (principal investigator).
Footnotes
Conflict of interest The authors report no conflicts of interest.
References
- 1.Apps R, Hawkes R. Cerebellar cortical organization: a one-map hypothesis. Nat Rev Neurosci. 2009;10:670–681. doi: 10.1038/nrn2698. [DOI] [PubMed] [Google Scholar]
- 2.Babij R, Lee M, Cortes E, Vonsattel JPG, Faust PL, Louis ED. Purkinje cell axonal anatomy: quantifying morphometric changes in essential tremor versus control brains. Brain. 2013;136:3051–3061. doi: 10.1093/brain/awt238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barnes JA, Ebner BA, Duvick LA, Gao W, Chen G, Orr HT, Ebner TJ. Abnormalities in the climbing fiber-Purkinje cell circuitry contribute to neuronal dysfunction in ATXN1[82Q] mice. J Neurosci. 2011;31:12778–12789. doi: 10.1523/JNEUROSCI.2579-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bocchetta M, Cardoso MJ, Cash DM, Ourselin S, Warren JD, Rohrer JD. Patterns of regional cerebellar atrophy in genetic frontotemporal dementia. Neuroimage Clin. 2016;11:287–290. doi: 10.1016/j.nicl.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Braak H, Braak E. Diagnostic criteria for neuropathologic assessment of Alzheimer’s disease. Neurobiol Aging. 1997;18:S85–S88. doi: 10.1016/s0197-4580(97)00062-6. [DOI] [PubMed] [Google Scholar]
- 7.Braak H, Rüb U, Jansen Steur ENH, Del Tredici K, de Vos RAI. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology. 2005;64:1404–1410. doi: 10.1212/01.WNL.0000158422.41380.82. [DOI] [PubMed] [Google Scholar]
- 8.Cesa R, Scelfo B, Strata P. Activity-dependent presynaptic and postsynaptic structural plasticity in the mature cerebellum. J Neurosci. 2007;27:4603–4611. doi: 10.1523/JNEUROSCI.5617-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cesa R, Strata P. Axonal and synaptic remodeling in the mature cerebellar cortex. Prog Brain Res. 2005;148:45–56. doi: 10.1016/S0079-6123(04)48005-4. [DOI] [PubMed] [Google Scholar]
- 10.Cesa R, Strata P. Axonal competition in the synaptic wiring of the cerebellar cortex during development and in the mature cerebellum. Neuroscience. 2009;162:624–632. doi: 10.1016/j.neuroscience.2009.02.061. [DOI] [PubMed] [Google Scholar]
- 11.Cheng MM, Tang G, Kuo SH. Harmaline-induced tremor in mice: videotape documentation and open questions about the model. Tremor Other Hyperkinet Mov (N Y) 2013:3. doi: 10.7916/D8H993W3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choe M, Cortes E, Vonsattel JPG, Kuo SH, Faust PL, Louis ED. Purkinje cell loss in essential tremor: random sampling quantification and nearest neighbor analysis. Mov Disord. 2016;31:393–401. doi: 10.1002/mds.26490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebner BA, Ingram MA, Barnes JA, Duvick LA, Frisch JL, Clark HB, Zoghbi HY, Ebner TJ, Orr HT. Purkinje cell ataxin-1 modulates climbing fiber synaptic input in developing and adult mouse cerebellum. J Neurosci. 2013;33:5806–5820. doi: 10.1523/JNEUROSCI.6311-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eccles JC, Llinás R, Sasaki K. The excitatory synaptic action of climbing fibres on the Purkinje cells of the cerebellum. J Physiol (Lond) 1966;182:268–296. doi: 10.1113/jphysiol.1966.sp007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erickson-Davis CR, Faust PL, Vonsattel JPG, Gupta S, Honig LS, Louis ED. Hairy baskets associated with degenerative Purkinje cell changes in essential tremor. J Neuropathol Exp Neurol. 2010;69:262–271. doi: 10.1097/NEN.0b013e3181d1ad04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fanciulli A, Wenning GK. Multiple-system atrophy. NEJM. 2015;372:249–263. doi: 10.1056/NEJMra1311488. [DOI] [PubMed] [Google Scholar]
- 17.Fox CA, Barnard JW. A quantitative study of the Purkinje cell dendritic branchlets and their relationship to afferent fibres. J Anat. 1957;91:299–313. [PMC free article] [PubMed] [Google Scholar]
- 18.Fremeau RT, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron. 2001;31:247–260. doi: 10.1016/s0896-6273(01)00344-0. [DOI] [PubMed] [Google Scholar]
- 19.Furrer SA, Waldherr SM, Mohanachandran MS, Baughn TD, Nguyen K-T, Sopher BL, Damian VA, Garden GA, La Spada AR. Reduction of mutant ataxin-7 expression restores motor function and prevents cerebellar synaptic reorganization in a conditional mouse model of SCA7. Hum Mol Genet. 2013;22:890–903. doi: 10.1093/hmg/dds495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grasselli G, Strata P. Structural plasticity of climbing fibers and the growth-associated protein GAP-43. Front Neural Circuits. 2013;7:25. doi: 10.3389/fncir.2013.00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grodd W, Hülsmann E, Lotze M, Wildgruber D, Erb M. Sensorimotor mapping of the human cerebellum: fMRI evidence of somatotopic organization. Hum Brain Mapp. 2001;13:55–73. doi: 10.1002/hbm.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Handforth A. Harmaline tremor: underlying mechanisms in a potential animal model of essential tremor. Tremor Other Hyperkinet Mov (N Y) 2012:2. doi: 10.7916/D8TD9W2P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helmich RC, Toni I, Deuschl G, Bloem BR. The pathophysiology of essential tremor and Parkinson’s tremor. Curr Neurol Neurosci Rep. 2013;13:378. doi: 10.1007/s11910-013-0378-8. [DOI] [PubMed] [Google Scholar]
- 24.Ito M, Yamaguchi K, Nagao S, Yamazaki T. Long-term depression as a model of cerebellar plasticity. Prog Brain Res. 2014;210:1–30. doi: 10.1016/B978-0-444-63356-9.00001-7. [DOI] [PubMed] [Google Scholar]
- 25.Koeppen AH. The pathogenesis of spinocerebellar ataxia. Cerebellum. 2005;4:62–73. doi: 10.1080/14734220510007950. [DOI] [PubMed] [Google Scholar]
- 26.Koeppen AH, Davis AN, Morral JA. The cerebellar component of Friedreich’s ataxia. Acta Neuropathol. 2011;122:323–330. doi: 10.1007/s00401-011-0844-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koeppen AH, Ramirez RL, Bjork ST, Bauer P, Feustel PJ. The reciprocal cerebellar circuitry in human hereditary ataxia. Cerebellum. 2013;12:493–503. doi: 10.1007/s12311-013-0456-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuo SH, Lin CY, Wang J, Liou JY, Pan MK, Louis RJ, Wu WP, Gutierrez J, Louis ED, Faust PL. Deep brain stimulation and climbing fiber synaptic pathology in essential tremor. Ann Neurol. 2016 doi: 10.1002/ana.24728. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kuo SH, Tang G, Louis ED, Ma K, Babji R, Balatbat M, Cortes E, Vonsattel J-PG, Yamamoto A, Sulzer D, Faust PL. Lingo-1 expression is increased in essential tremor cerebellum and is present in the basket cell pinceau. Acta Neuropathol. 2013;125:879–889. doi: 10.1007/s00401-013-1108-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuo SH, Erickson-Davis C, Gillman A, Faust PL, Vonsattel JPG, Louis ED. Increased number of heterotopic Purkinje cells in essential tremor. J Neurol Neurosurg Psychiatry. 2011;82:1038–1040. doi: 10.1136/jnnp.2010.213330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin CY, Louis ED, Faust PL, Koeppen AH, Vonsattel JPG, Kuo SH. Abnormal climbing fibre-Purkinje cell synaptic connections in the essential tremor cerebellum. Brain. 2014;137:3149–3159. doi: 10.1093/brain/awu281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Louis ED. Essential tremors: a family of neurodegenerative disorders? Arch Neurol. 2009;66:1202–1208. doi: 10.1001/archneurol.2009.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Louis ED. From neurons to neuron neighborhoods: the rewiring of the cerebellar cortex in essential tremor. Cerebellum. 2014;13:501–512. doi: 10.1007/s12311-013-0545-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Louis ED, Babij R, Cortes E, Vonsattel JPG, Faust PL. The inferior olivary nucleus: a postmortem study of essential tremor cases versus controls. Mov Disord. 2013;28:779–786. doi: 10.1002/mds.25400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Louis ED, Babij R, Lee M, Cortes E, Vonsattel JPG. Quantification of cerebellar hemispheric purkinje cell linear density: 32 ET cases versus 16 controls. Mov Disord. 2013;28:1854–1859. doi: 10.1002/mds.25629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Louis ED, Babij R, Ma K, Cortes E, Vonsattel JPG. Essential tremor followed by progressive supranuclear palsy: postmortem reports of 11 patients. J Neuropathol Exp Neurol. 2012;72:8–17. doi: 10.1097/NEN.0b013e31827ae56e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louis ED, Lee M, Babij R, Ma K, Cortes E, Vonsattel JPG, Faust PL. Reduced Purkinje cell dendritic arborization and loss of dendritic spines in essential tremor. Brain. 2014;137:3142–3148. doi: 10.1093/brain/awu314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Louis ED, Mazzoni P, Ma KJ, Moskowitz CB, Lawton A, Garber A, Vonsattel JPG. Essential tremor with ubiquitinated intranuclear inclusions and cerebellar degeneration. Clin Neuropathol. 2012;31:119–126. doi: 10.5414/NP300414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Louis ED, Erickson-Davis C, Pahwa R, Lyons KE, Garber A, Moskowitz CB, Lawton A, Faust PL, Vonsattel JPG. Essential tremor with ubiquitinated Purkinje cell intranuclear inclusions. Acta Neuropathol. 2010;119:375–377. doi: 10.1007/s00401-010-0641-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Louis ED, Faust PL, Vonsattel JPG, Honig LS, Rajput A, Robinson CA, Rajput A, Pahwa R, Lyons KE, Ross GW, Borden S, Moskowitz CB, Lawton A, Hernandez N. Neuropathological changes in essential tremor: 33 cases compared with 21 controls. Brain. 2007;130:3297–3307. doi: 10.1093/brain/awm266. [DOI] [PubMed] [Google Scholar]
- 41.Louis RJ, Lin CY, Faust PL, Koeppen AH, Kuo SH. Climbing fiber synaptic changes correlate with clinical features in essential tremor. Neurology. 2015;84:2284–2286. doi: 10.1212/WNL.0000000000001636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McKay BE, Engbers JDT, Mehaffey WH, Gordon GRJ, Molineux ML, Bains JS, Turner RW. Climbing fiber discharge regulates cerebellar functions by controlling the intrinsic characteristics of purkinje cell output. J Neurophysiol. 2007;97:2590–2604. doi: 10.1152/jn.00627.2006. [DOI] [PubMed] [Google Scholar]
- 43.McKeith IG, Dickson DW, Lowe J, Emre M, O’Brien JT, Feldman H, Cummings J, Duda JE, Lippa C, Perry EK, Aarsland D, Arai H, Ballard CG, Boeve B, Burn DJ, Costa D, Del Ser T, Dubois B, Galasko D, Gauthier S, Goetz CG, Gomez-Tortosa E, Halliday G, Hansen LA, Hardy J, Iwatsubo T, Kalaria RN, Kaufer D, Kenny RA, Korczyn A, Kosaka K, Lee VMY, Lees A, Litvan I, Londos E, Lopez OL, Minoshima S, Mizuno Y, Molina JA, Mukaetova-Ladinska EB, Pasquier F, Perry RH, Schulz JB, Trojanowski JQ, Yamada M, Consortium on DLB Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 44.Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: a commentary. Neurobiol Aging. 1997;18:S91–S94. doi: 10.1016/s0197-4580(97)00058-4. [DOI] [PubMed] [Google Scholar]
- 45.Nitschke MF, Kleinschmidt A, Wessel K, Frahm J. Soma-totopic motor representation in the human anterior cerebellum. A high-resolution functional MRI study. Brain. 1996;3:1023–1029. doi: 10.1093/brain/119.3.1023. [DOI] [PubMed] [Google Scholar]
- 46.Palay SL, Chan-Palay V. Cerebellar cortex: cytology and organization. Springer; Berlin: 1974. [Google Scholar]
- 47.Pan JJ, Lee M, Honig LS, Vonsattel JPG, Faust PL, Louis ED. Alzheimer’s-related changes in non-demented essential tremor patients vs. controls: links between tau and tremor? Parkinsonism Relat Disord. 2014;20:655–658. doi: 10.1016/j.parkreldis.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pool M, Thiemann J, Bar-Or A, Fournier AE. Neurite T-racer: a novel ImageJ plugin for automated quantification of neurite outgrowth. J Neurosci Methods. 2008;168:134–139. doi: 10.1016/j.jneumeth.2007.08.029. [DOI] [PubMed] [Google Scholar]
- 49.Rajput AH, Robinson CA, Rajput ML, Robinson SL, Rajput A. Essential tremor is not dependent upon cerebellar Purkinje cell loss. Parkinsonism Relat Disord. 2012;18:626–628. doi: 10.1016/j.parkreldis.2012.01.013. [DOI] [PubMed] [Google Scholar]
- 50.Ramirez EP, Vonsattel JPG. Neuropathologic changes of multiple system atrophy and diffuse lewy body disease. Semin Neurol. 2014;34:210–216. doi: 10.1055/s-0034-1381732. [DOI] [PubMed] [Google Scholar]
- 51.Seidel K, Siswanto S, Brunt ERP, den Dunnen W, Korf HW, Rüb U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol. 2012;124:1–21. doi: 10.1007/s00401-012-1000-x. [DOI] [PubMed] [Google Scholar]
- 52.Sharifi S, Nederveen AJ, Booij J, van Rootselaar A-F. Neuroimaging essentials in essential tremor: a systematic review. Neuroimage Clin. 2014;5:217–231. doi: 10.1016/j.nicl.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shuvaev AN, Horiuchi H, Seki T, Goenawan H, Irie T, Iizuka A, Sakai N, Hirai H. Mutant PKCγ in spinocerebellar ataxia type 14 disrupts synapse elimination and long-term depression in Purkinje cells in vivo. J Neurosci. 2011;31:14324–14334. doi: 10.1523/JNEUROSCI.5530-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Symanski C, Shill HA, Dugger B, Hentz JG, Adler CH, Jacobson SA, Driver-Dunckley E, Beach TG. Essential tremor is not associated with cerebellar Purkinje cell loss. Mov Disord. 2014;29:496–500. doi: 10.1002/mds.25845. [DOI] [PubMed] [Google Scholar]
- 55.Thach WT. Somatosensory receptive fields of single units in cat cerebellar cortex. J Neurophysiol. 1967;30:675–696. doi: 10.1152/jn.1967.30.4.675. [DOI] [PubMed] [Google Scholar]
- 56.Vonsattel JPG, Del Amaya MP, Keller CE. Twenty-first century brain banking. Processing brains for research: the Columbia University methods. Acta Neuropathol. 2008;115:509–532. doi: 10.1007/s00401-007-0311-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Watanabe M. Molecular mechanisms governing competitive synaptic wiring in cerebellar Purkinje cells. Tohoku J Exp Med. 2008;214:175–190. doi: 10.1620/tjem.214.175. [DOI] [PubMed] [Google Scholar]
- 58.Wu T, Hallett M. The cerebellum in Parkinson’s disease. Brain. 2013;136:696–709. doi: 10.1093/brain/aws360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yoshida T, Katoh A, Ohtsuki G, Mishina M, Hirano T. Oscillating Purkinje neuron activity causing involuntary eye movement in a mutant mouse deficient in the glutamate receptor delta2 subunit. J Neurosci. 2004;24:2440–2448. doi: 10.1523/JNEUROSCI.0783-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]