

Abstract
Human immunodeficiency virus (HIV) has a number of circulating recombinant forms that are the product of recombination between different HIV subtypes. The first circulating recombinant form of HIV-1 to be identified was CRF01_AE, which originated in Central Africa and is now most prevalent in Southeast and East Asia. In this study, we investigated the timescale, evolutionary history, and population genetics of the HIV-1 CRF01_AE strains primarily responsible for the epidemic in Asia. A further aim of our study was to define and standardize the nomenclature and provide well-characterized reference sequences for the phylogenetic transmission clusters of CRF01_AE. We analysed a data set of 334 near-complete genome sequences from various risk groups, sampled between 1990 and 2011 from nine countries. Phylogenetic analyses of these sequences were performed using maximum likelihood and Bayesian methods. Our study confirms that the diversity of HIV-1 CRF01_AE originated in Central Africa in the mid-1970s, was introduced into Thailand between 1979 and 1982, and began expanding there shortly afterwards (1982–1984). Subsequently, multiple clusters significantly contributed to China’s HIV epidemic. A Bayesian skyline plot revealed the rapid expansion of CRF01_AE in China around 1999–2000. We identified at least eight different clusters of HIV-1 CRF01_AE formed by rapid expansion into different risk groups and geographic regions in China since the late 1980s.
Introduction
Human immunodeficiency virus (HIV) has undergone multiple cross-species transmissions from nonhuman primates into humans, producing two major types1: HIV-1 and HIV-2. The globally circulating strains of HIV-1 are extremely diverse, as a result of high rates of mutation, recombination, and replication2–6. Group M, the most common group of HIV-1, is responsible for the large majority of AIDS cases across the world. It is further classified into nine subtypes (A–D, F–H, J, and K) and four sub-subtypes (A1, A2, F1, and F2), as well as a number of circulating recombinant forms (CRFs) with various unique recombinant forms (URFs)7, 8.
The first CRF of HIV-1 to be identified was CRF01_AE, initially named “subtype E”. It represents a putative recombinant between subtypes A and E, but a parental (non-recombinant) subtype E has not been found9, 10. Although CRF01_AE contains a subtype E vif, vpr, env, nef, and long terminal repeat (LTR), most or all of the remaining genome derives from subtype A. Although the “subtype E” segments in this CRF should be referred to as “U” (unclassified) according to the recommended nomenclature for HIV-18, the historical “subtype E” designation has been retained to refer to the putative non-A regions in this CRF.
CRF01_AE is most prevalent in Thailand and neighboring countries in Southeast and East Asia. It originated in Central Africa and has been found among mid-1980s samples from the Democratic Republic of Congo11. However, the earliest known strains of CRF01_AE were first identified in samples from northern Thailand in 1989 among female commercial sex workers12–14. CRF01_AE then spread into various risk groups in Thailand and neighboring regions15–19. It is also a component of at least 16 CRFs identified in Africa and Asia (http://www.hiv.lanl.gov).
Viral transmission events can be investigated using phylogenetic analyses of HIV sequences isolated from different patients. An analysis of 33 near-complete genomes found that CRF01_AE in Vietnam formed at least three phylogenetic transmission clusters from founder strains being introduced into new locations and risk groups17. Another study identified at least three phylogenetic transmission clusters of strains that most likely contributed to the CRF01_AE epidemic in Hong Kong20. A recent analysis of 1957 CRF01_AE gag p17 sequences, collected between 1990 and 2010 from 15 different countries, identified 27 phylogenetic transmission clusters21. A more comprehensive study used a statistical phylogeographic analysis of 2736 CRF01_AE partial pol sequences to uncover global patterns of dispersal22. Other phylogenetic studies have shown that the CRF01_AE epidemic in China was driven by multiple independent clusters introduced in the 1990s18, 23, 24. Despite this research into CRF01_AE, we still have an incomplete understanding of the distinct clusters circulating in the Asian region.
To obtain a more comprehensive picture of the spatiotemporal dynamics of the HIV-1 CRF01_AE epidemic in Asia, we analysed a data set of 334 near-complete genomes of CRF01_AE sampled from 1990 to 2011 from nine countries. We used phylogenetic, molecular clock, and Bayesian skyline analyses to explore the origin of CRF01_AE transmission clusters and to estimate the timeline and demographic history of each of the clusters. We also suggest the use of consistent and standardized nomenclatural criteria for the transmission clusters and provide a set of 10 well-characterized reference sequences.
Materials and Methods
Sample selection and sequence data
Based on the results of an HIV molecular epidemiology survey conducted between 2010 and 2011 of various risk groups in Jilin province, China25, we obtained four new near-complete genome sequences of CRF01_AE (552–9636 nt relative to HXB2) from plasma virus RNA as previously described18, 26–28. These sequences, from one heterosexual and three men who have sex with men (MSM), were named JL100034, JL100038, JL110010, and JL110056, respectively (GenBank accession numbers KP860667–KP860670).
All available near-complete genome sequences of CRF01_AE (one per patient) with known sampling dates and geographic information were retrieved from the Los Alamos National Laboratory (LANL) HIV Sequence Database (http://www.hiv.lanl.gov). HIV BLAST was used to identify closely related CRF01_AE sequences in the HIV-1 database29. Sequence quality was analysed using the Quality Control tool on the LANL site, whereas the genotype assignment of all sequences was confirmed using RIP v.3.030. Hypermutation analysis was performed using Hypermut v2.031. A total of 330 sequences of CRF01_AE were combined with the four newly generated sequences to form this data set (Tables 1, 2 and 3 and S1).
Table 1.
Geographic source, sampling year, and risk factor for HIV-1 CRF01_AE strains analysed in the present study.
| Geographic source | Sampling year | na | Risk factorb | |||||
|---|---|---|---|---|---|---|---|---|
| Hetero | IDU | MSM | MTCT | ST | n/a | |||
| China | 1997–2011 | 154 (4) | 56 (1) | 21 | 56 (3) | 3 | 9 | 9 |
| Vietnam | 1997–1998 | 33 | 17 | 16 | ||||
| Afghanistan | 2007 | 1 | 1 | |||||
| Central African Republic | 1990 | 3 | 1 | 2 | ||||
| Hong Kong | 2004 | 1 | 1 | |||||
| Indonesia | 1993 | 1 | 1 | |||||
| Japan | 1993–2000 | 2 | 2 | |||||
| Thailand | 1990–2009 | 134 | 14 | 30 | 1 | 89 | ||
| United States | 1998–2005 | 5 | 4 | 1 | ||||
| Total | 334 (4) | 95 (1) | 67 | 56 (3) | 4 | 9 | 103 | |
aThe numbers of HIV-1 CRF01_AE sequences newly reported in this study are shown in parentheses.
bRisk group: Hetero, heterosexual; IDU, injecting drug user; MSM, men who have sex with men; MTCT, mother-to-child transmission; ST, sexual transmission, unspecified type; n/a, not available.
Table 2.
Classification and risk factor of distinct HIV-1 CRF01_AE clusters analysed in the present study.
| CRF01_AE cluster | na | Risk factorb | |||||
|---|---|---|---|---|---|---|---|
| Hetero | IDU | MSM | MTCT | ST | n/a | ||
| CRF01_1AE | 40 (1) | 24 (1) | 14 | 2 | |||
| CRF01_2AE | 26 | 8 | 18 | ||||
| CRF01_3AE | 3 | 1 | 1 | 1 | |||
| CRF01_4AE | 25 | 1 | 1 | 21 | 1 | 1 | |
| CRF01_5AE | 37 (3) | 2 | 34 (3) | 1 | |||
| CRF01_6AE | 4 | 4 | |||||
| CRF01_7AE | 3 | 3 | |||||
| CRF01_8AE | 5 | 3 | 1 | 1 | |||
| CRF01_9AE | 3 | 3 | |||||
| CRF01_10AE | 5 | 5 | |||||
| Ungrouped | 183 | 49 | 25 | 1 | 4 | 7 | 97 |
| Total | 334 (4) | 95 (1) | 67 | 56 (3) | 4 | 9 | 103 |
aNumbers of HIV-1 CRF01_AE sequences newly reported in our study are shown in parentheses.
bRisk group: Hetero, heterosexual; IDU, injecting drug user; MSM, men who have sex with men; MTCT, mother-to-child transmission; ST, sexual transmission, unspecified type; n/a, not available.
Table 3.
Classification and sampling year of distinct HIV-1 CRF01_AE clusters analysed in the present study.
| CRF01_AE cluster | na | Sampling year | ||||
|---|---|---|---|---|---|---|
| 1990–1994 | 1995–1999 | 2000–2004 | 2005–2009 | 2010–2011 | ||
| CRF01_1AE | 40 (1) | 38 | 2 (1) | |||
| CRF01_2AE | 26 | 19 | 7 | |||
| CRF01_3AE | 3 | 3 | ||||
| CRF01_4AE | 25 | 9 | 16 | |||
| CRF01_5AE | 37 (3) | 21 | 16 (3) | |||
| CRF01_6AE | 4 | 4 | ||||
| CRF01_7AE | 3 | 3 | ||||
| CRF01_8AE | 5 | 5 | ||||
| CRF01_9AE | 3 | 2 | 1 | |||
| CRF01_10AE | 5 | 3 | 2 | |||
| Ungrouped | 183 | |||||
| Total | 334 (4) | 24 | 3 | 90 | 34 (4) | |
aNumbers of HIV-1 CRF01_AE sequences newly reported in our study are shown in parentheses.
An initial alignment of all 334 sequences was performed using Gene Cutter from the LANL site and then adjusted manually in BioEdit v7.0.9.032. If gaps were inserted unambiguously and the alignment columns contained gaps in more than 50% of the sequences, they were removed using Gap Strip/Squeeze v2.1.0 on the LANL site.
The combined data set of 334 sequences includes samples from various risk groups: heterosexuals (Hetero); injecting drug users (IDUs); men who have sex with men (MSM); mother-to-child transmission (MTCT); sexual transmission with unspecified type (ST); and unknown risk. The samples are drawn from broad geographical regions: 13 provinces in China; Afghanistan; Central African Republic; Hong Kong; Indonesia; Japan; Thailand; United States; and five provinces in Vietnam. As listed in Tables 1 and S1, 154 were obtained from various risk groups in 13 provinces across China between 1997 and 2011 and 180 were previously reported from 8 other countries between 1990 and 2009. The main risk groups are unknown risk (30.84%), Hetero (28.44%), IDUs (20.06%), and MSM (16.77%). The samples are primarily from China (46.11%), Thailand (40.12%), and Vietnam (9.88%).
The study was approved by the institutional review board of the National Center for AIDS/STD Control and Prevention, China CDC. A written informed consent, as well as a socio-demographic questionnaire, was obtained for each of the four new near-complete genome sequences. All methods were performed in accordance with the relevant guidelines and regulations.
Phylogenetic analyses
To study the amount of evolutionary information contained in the data set, a likelihood-mapping analysis33 was performed using TREE-PUZZLE v5.334 by analysing 10000 randomly chosen quartets for the entire tree. For each sequence quartet, three unrooted tree topologies are possible. For a random sample of quartets, the likelihoods for the three possible topologies are reported as dots in an equilateral triangle. The distribution of points in different sections of this triangle indicates the tree-likeness of the data: the three corners represent fully resolved tree topologies, indicating the presence of tree-like phylogenetic signal; the center represents the sets of points where all three trees are equally supported, indicating a lack of phylogenetic signal; and the three areas on the sides indicate support for conflicting tree topologies. To infer the phylogeny, we used a maximum-likelihood approach with the GTR + G model in RAxML v8.0.935. Support for the inferred relationships was evaluated by a bootstrap analysis with 1000 replicates.
Strategies for identifying and defining transmission clusters differ between studies. Here we identify them on the basis of within-cluster genetic distance (cut-off of 6%) and bootstrap support (cut-off of 99%) for groupings with more than two sequences, as implemented in Cluster Picker v1.2.336. Genetic distances between and within clusters were calculated in MEGA v7.1.1837 using the maximum composite likelihood38 with 1000 bootstrap replicates. Rate variation among sites was modelled with a gamma distribution. A plot of genetic distances between clusters was generated using the pheatmap package in R. In addition, we used the web-based tool Evolview v239 to visualize and annotate the phylogenetic tree with geographic location, phylogenetic cluster, and risk group.
To investigate the temporal signal in the data set, analyses of the correlation between root-to-tip genetic distance and year of sampling were performed on the maximum-likelihood tree using the program TempEst v1.540. We also estimated the evolutionary rate for the data set using least-squares dating in LSD v0.241. We then used a Bayesian phylogenetic approach for joint estimation of the ages of each of the 10 CRF01_AE clusters and the demographic history of all of the strains. This was done by analysing the 334 sequences using a GTR + G substitution model with an uncorrelated lognormal relaxed-clock model42 and a Bayesian skyline coalescent tree prior43 in BEAST v1.8.244. The molecular clock was calibrated using the sampling dates of the sequences. Posterior distributions of parameters, including the tree, were estimated using Markov chain Monte Carlo (MCMC) sampling. The MCMC was run for 500 million steps, with the first 10% removed as burn-in. Samples were drawn every 50,000 steps. Convergence and sufficient sampling were evaluated by calculating the effective sample sizes of the parameters using Tracer v1.5 (http://beast.bio.ed.ac.uk/software/tracer). Trees were summarized as maximum clade credibility (MCC) trees using TreeAnnotator (part of the BEAST package) and visualized in FigTree v1.4.3.
We wished to test the hypothesis that a tip with a given discrete trait (geographic location or risk group) is more likely to share that discrete trait with a neighboring tip than would be expected by chance. For each discrete trait in our data set, we calculated the association index (AI), Fitch parsimony score (PS), and monophyletic clade size (MC) statistics using Bayesian Tip-Significance Testing (BaTS) software version 1.045. AI and PS scores indicate migration events between trait values, and MC scores indicate the number of taxa in the largest clade monophyletic for that trait value. Therefore, low AI and PS scores and high MC scores indicate a strong trait association.
To accommodate the uncertainty in the phylogenetic estimate, we used the posterior set of trees from the Bayesian phylogenetic analysis described above. The topological robustness of this sample of trees was determined by comparing it with the null distribution of trees obtained from 10,000 bootstrap replicates of discrete characters. The P-value is then calculated as the proportion of trees from the null distribution for which the value of the statistic is equal to, or more extreme than, the median estimate from the posterior sample of trees. We reject the null hypothesis for significance levels of 0.001, 0.001, and 0.05 for AI, PS, and MC statistics, respectively.
Results
Likelihood mapping and phylogeny of HIV-1 CRF01_AE strains
The phylogenetic signal from the data set was investigated by likelihood-mapping analysis33. Our likelihood-mapping analysis revealed that the quartets from the data set were primarily distributed in the corners (92.2%) rather than the sides (7.5%) or center (0.3%) of the triangle, indicating a strong tree-like phylogenetic signal (Supplementary Figure S1).
The phylogeny of HIV-1 CRF01_AE strains, inferred using maximum likelihood, indicates the presence of 10 transmission clusters (Figs 1 and 2 and S2 and Tables 2 and 3 and S1). Cluster names were based on our previous numbering system and with the addition of new clusters in this study18. Sequences from Cluster 1 (designated as CRF01_1AE; n = 40) were found among Hetero (n = 24), IDU (n = 14), and unknown risk (n = 2) patients in eight provinces of China. Cluster 2 sequences (CRF01_2AE; n = 17) were found among Hetero (n = 8) and IDU (n = 18) patients in three provinces of China and five provinces of Vietnam. Sequences from Cluster 3 (CRF01_3AE; n = 30) were collected from Hetero (n = 1), IDU (n = 1), and unknown risk (n = 1) patients in three provinces of China. The risk groups associated with these three clusters were primarily Hetero and IDUs.
Figure 1.
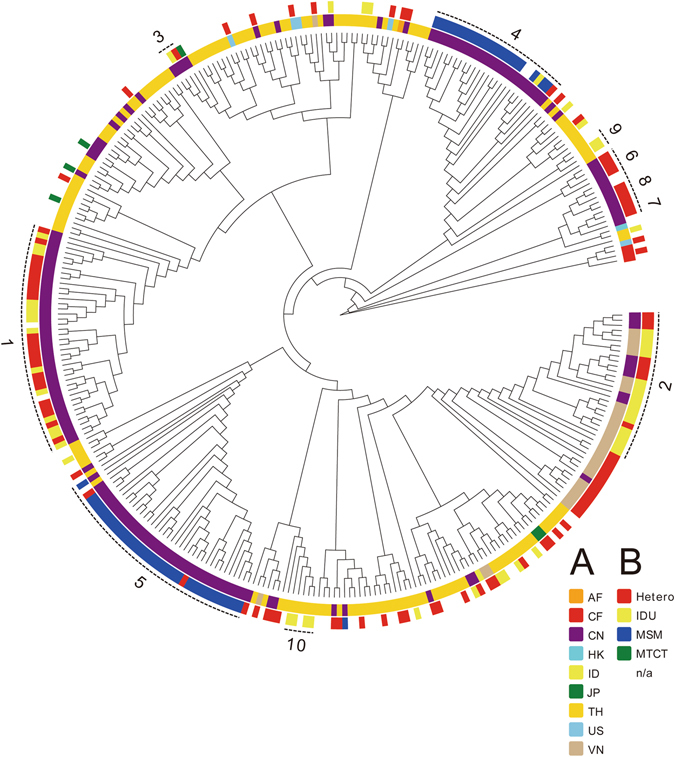
Maximum-likelihood phylogeny of HIV-1 CRF01_AE strains. Maximum-likelihood phylogeny of near-complete genome sequences of HIV-1 CRF01_AE. The two circles of colored cells show geographic location (inner circle, A) and risk group (outer circle, B).
Figure. 2.

Geographic distribution of HIV-1 CRF01_AE clusters identified in the present study. The geographic distribution of HIV-1 CRF01_AE clusters is shown at the (A) country level, and at the provincial level for (B) China and (C) Vietnam. Each CRF01_AE cluster identified in this study is color-coded, as shown on the left. Maps were obtained from Craft MAP website (http://www.craftmap.box-i.net/).
Cluster 4 sequences (CRF01_4AE; n = 25) were found in seven provinces of China, whereas Cluster 5 sequences (CRF01_5AE; n = 37) were collected from four provinces of China and from Thailand. The predominant risk group associated with Clusters 4 and 5 was MSM. The four sequences from Cluster 6 (CRF01_6AE) were all collected from Hetero patients in Fujian province, China. Cluster 7 (CRF01_7AE) included only three sequences collected from Hetero patients in Yunnan province, China. Cluster 8 sequences (CRF01_8AE; n = 5) were found among Hetero (n = 3), ST (n = 1), and unknown risk (n = 1) patients in two Chinese provinces. Cluster 9 (CRF01_9AE; n = 3) only included sequences collected only from IDUs in Thailand. Cluster 10 (n = 5) included sequences collected only from IDUs in Thailand, with the short branches in the tree implying that these were recent transmissions.
The remaining 183 sequences (designated as Ungrouped) were scattered throughout the main CRF01_AE clade and had been sampled in China (n = 29), Vietnam (n = 16), Afghanistan (n = 1), Hong Kong (n = 1), Indonesia (n = 1), Japan (n = 2), Thailand (n = 125), United States (n = 5), and Central African Republic (n = 3). Of the 56 MSM sequences in our analysis, all but one were found within either CRF01_4AE (n = 21) or CRF01_5AE (n = 34) and all originated from China.
Genetic diversity and demographic analysis
We estimated the genetic diversity within and between each of the 10 HIV-1 CRF01_AE clusters (Supplementary Figure S4). The smallest genetic distance separated Clusters 2 and 9 (4.4%), whereas the largest was between Clusters 4 and 8 (7.6%).
A plot of root-to-tip genetic distance against year of sampling indicated a strong temporal signal with no clear outlier sequences (correlation coefficient = 0.91; slope = 4.74 × 10−3), reflecting a relatively clocklike pattern of molecular evolution (Fig. 3). The estimated evolutionary rate for the data set using least-squares dating was 4.60 × 10−3 substitutions per site per year. In our Bayesian phylogenetic analysis, we estimated a substitution rate of 4.70 × 10−3 substitutions per site per year (95% credibility interval: 4.46 × 10−3–4.92 × 10−3). The rate estimates from the three methods are in close agreement, as expected when there is low rate variation across branches and a low degree of age clustering among the tips46.
Figure 3.
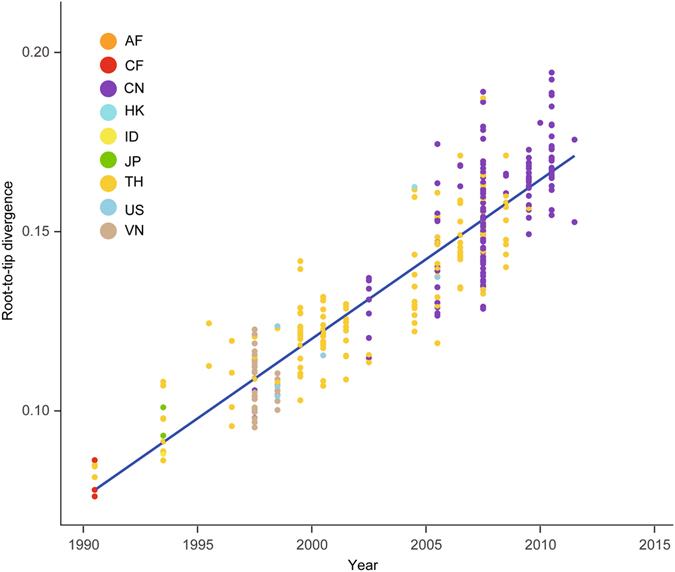
Regression of the root-to-tip genetic distance against year of sampling for 334 HIV-1 CRF01_AE sequences. Genetic distances are based on the tree in Supplementary Figure S2. Colors indicate different sampling locations.
The age of each CRF01_AE cluster was also estimated in the analysis (Supplementary Table S2). The first divergences between the sequences from Central African Republic and Thailand were estimated to have occurred in 1974 (95% credibility interval: 1972–1977) and 1981 (95% credibility interval: 1980–1983), respectively. These estimates are consistent with those obtained by Feng et al.18.
We further investigated the past population dynamics of CRF01_AE using a Bayesian skyline plot, which depicts the changes in effective population size over time43. The effective population size seems to have experienced a complex dynamic, characterized by two phases of exponential growth (1985–1988 and 1999–2000) separated by a periods of constant or declining population size (Fig. 4). The estimates of the phylogenetic relationships among the CRF01_AE sequences using Bayesian coalescent framework were consistent with those inferred using maximum likelihood (Fig. 5).
Figure 4.
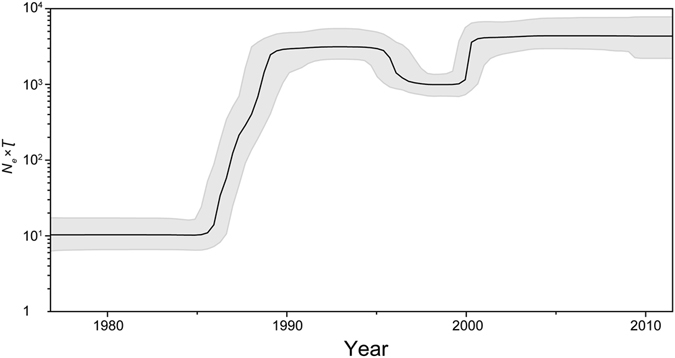
Bayesian skyline demographic reconstruction of HIV-1 CRF01_AE. The vertical axis shows the effective number of infections (N e) multiplied by mean viral generation time (τ). The solid line and shaded region represent the median and 95% credibility interval of N eτ through time.
Figure 5.

Maximum-clade-credibility tree estimated from near-complete genome sequences of HIV-1 CRF01_AE. Sequence names include accession number, geographic location, and year of sampling. Only internal nodes with posterior probability support >0.5 are shown with white, grey, and black circles.
Phylogenetic association with geographic location and risk group
Based on the AI and PS statistics, we rejected the null hypothesis of no association between the selected trait (geographic location or risk group) and the phylogeny (P < 0.001; Tables 4 and 5). For the MC statistic, we also rejected the null hypothesis of no association between geographic location and the structure of the phylogeny (P < 0.05), with the exception of the MC (ID), MC (HK), and MC (AF) statistics because of insufficient sample sizes from these geographic locations (n = 1; Table 4). For the risk group, the MC (Hetero), MC (MSM), and MC (n/a) statistics rejected the null hypothesis of no association with the structure of the phylogeny (P < 0.05; Table 5). However, the MC (IDU) statistic was not significantly larger than expected by chance (P = 0.192), and the MC (MTCT) and MC (ST) statistics were not different from those expected by chance. The results of our detailed analysis of geographic location are summarized in Supplementary Tables S3, S4, and S5.
Table 4.
Statistical analysis of geographic location of CRF01_AE sequences.
| Statistic | No. of sequences | Observed mean (95% CI) | Null mean (95% CI) | P-value |
|---|---|---|---|---|
| AI | 10.9 (10.0, 11.8) | 23.9 (21.7, 26.1) | <0.001* | |
| PS | 82.5 (80.0, 85.0) | 139.7 (132.3, 147.1) | <0.001* | |
| MC (AF) | 1 | 1.0 (1.0, 1.0) | 1.0 (1.0, 1.0) | N/A |
| MC (CF) | 3 | 1.8 (1.0, 2.0) | 1.0 (1.0, 1.0) | 0.005* |
| MC (CN) | 154 | 10.0 (10.0, 10.0) | 4.5 (3.1, 6.1) | 0.001* |
| MC (HK) | 1 | 1.0 (1.0, 1.0) | 1.0 (1.0, 1.0) | N/A |
| MC (ID) | 1 | 1.0 (1.0, 1.0) | 1.0 (1.0, 1.0) | N/A |
| MC (JP) | 2 | 2.0 (2.0, 2.0) | 1.0 (1.0, 1.0) | 0.002* |
| MC (TH) | 134 | 40.0 (40.0, 40.0) | 3.9 (3.0, 5.5) | 0.001* |
| MC (US) | 5 | 2.0 (2.0, 2.0) | 1.0 (1.0, 1.0) | 0.008* |
| MC (VN) | 33 | 5.0 (5.0, 5.0) | 1.7 (1.0, 2.3) | 0.001* |
AI, association index.
PS, parsimony score.
MC, monophyletic clade statistic.
95% CI, 95% credbility interval.
*Statistically significant (P < 0.05).
N/A, not available because of the observed 95% CI contains the null 95% CI.
Table 5.
Statistical analysis of risk group for CRF01_AE sequences.
| Statistic | No. of sequences | Observed mean (95% CI) | Null mean (95% CI) | P-value |
|---|---|---|---|---|
| AI | 20.1 (19.2, 21.1) | 29.7 (27.7, 31.7) | <0.001* | |
| PS | 133.2 (130.0, 136.0) | 184.4 (176.8, 191.7) | <0.001* | |
| MC (Hetero) | 56 | 5.0 (5.0, 5.0) | 3.0 (2.0, 4.1) | 0.017* |
| MC (IDU) | 21 | 3.0 (3.0, 3.0) | 2.4 (2.0, 3.1) | 0.192 |
| MC (MSM) | 56 | 5.0 (5.0, 5.0) | 2.2 (1.8, 3.0) | 0.002* |
| MC (MTCT) | 3 | 1.0 (1.0, 1.0) | 1.0 (1.0, 1.0) | N/A |
| MC (ST) | 9 | 1.0 (1.0, 1.0) | 1.0 (1.0, 1.0) | N/A |
| MC (n/a) | 9 | 40.0 (40.0, 40.0) | 3.1 (2.1, 4.5) | 0.001* |
AI, association index.
PS, parsimony score.
MC, monophyletic clade statistic.
95% CI, 95% credibility interval.
*Statistically significant (P < 0.05).
Hetero, heterosexual.
IDU, injecting drug user.
MSM, men who have sex with men.
MTCT, mother-to-child transmission.
ST, sexual transmission, unspecified type.
n/a, not available.
N/A, not available because of the observed 95% CI contains the null 95% CI.
Discussion
Phylodynamics of HIV-1 CRF01_AE
The most prevalent genetic type of HIV-1 in Asia is CRF01_AE. Our evolutionary analyses, based on all of the available near-complete genome sequences of CRF01_AE that included country of origin and year of sampling, revealed the presence of 10 independent clusters. These strongly supported clusters represent founder variants that led to substantial viral spread, most likely into a highly active, high-risk group of HIV-naïve individuals. Population-level transmission depends on the probabilities of transmission and the structure of the social/sexual networks into which a founder virus enters47–50. Subsequent transmissions of a founder virus might remain limited to its original transmission network, but can eventually move outside the network and spread regionally, nationally, or even globally.
The basal divergences within HIV-1 CRF01_AE involved the samples not only from Hetero patients collected from the Central African Republic in 1990, but also from a Hetero patient in US collected in 1998. It was previously proposed that HIV-1 CRF01_AE outbreaks in Thailand were directly seeded by the HIV-1 CRF01_AE strains of African origin9, 12, 13. We identified 10 independent clusters within the CRF01_AE pandemic, including eight detected in China, and the origins of these clusters date from the late 1980s to the late 1990s. The sequences within some of the clusters were quite dispersed and were identified in as many as eight Chinese provinces. These results support a scenario of multiple CRF01_AE founder viruses that were introduced into epidemiologically linked, high-risk groups in China. As these founder viruses spread within transmission/social/sexual networks, they became the ancestors of each of these independent clusters. Further sampling might reveal the presence of additional HIV-1 CRF01_AE clusters. As more sequences are characterized within other countries, more local, regional, national or global clusters are likely to emerge, presenting a challenge to HIV nomenclature.
Our Bayesian skyline plot analysis revealed that a bottleneck occurred in the second half of the 1990s. This is consistent with demographic data on the decline of HIV prevalence among female commercial sex workers and male sexually transmitted disease patients in Thailand during this period (Supplementary Figure S5). This was most likely a result of the implementation of effective HIV-control measures in Thailand beginning in the late 1980s, including the 100% Condom Program51–53. The second period of population growth around 2000 coincided with China’s first explosive travel to Thailand during the late 1990s to early 2000s (Supplementary Table S6)54. Therefore, it is tempting to speculate that China’s “free travel” policy provided an opportunity for the establishment, spatial dissemination, and epidemic growth of multiple clusters of CRF01_AE strains from Thailand to China. Furthermore, we found that geographic locations and risk groups are indeed having a significant influence on the complex transmission dynamics of CRF01_AE. The phylogeny of CRF01_AE is likely to have been structured by geography and risk-group traits, especially for China, Thailand, and Vietnam, and for those in the MSM risk group.
HIV-1 nomenclature
The official nomenclature of HIV-1 includes groups M, N, O, and P, each of which represents a single zoonotic transmission event. Subtypes within the HIV-1 M group were formed by epidemiological factors. The circulating recombinant forms include viruses such as CRF01_AE, CRF02_AG, and CRF04_cpx, which are recombinant viruses with some parts of their genomes clustering with more than one subtype. The official nomenclature also includes some “sub-subtypes” such as A1, A2, F1, and F2, each of which is nearly as distant from each other as subtype B is from subtype D.
There have also been many unofficial designations for local strains and subclades within subtypes, such as the “B-prime” or “Thai-B” and “A3” and “A4” viruses. Identifying subclades within a subtype without standards to name them can lead to a great deal of confusion. For example, Feng et al.18 reported seven distinct phylogenetic clusters of CRF01_AE strains from China. However, their “Cluster 2” was the same as “Cluster 3” first identified among IDUs in northern Vietnam and the nearby Chinese province of Guangxi17, and was also called “IMC-1” by Shiino et al.55. The best way to standardize the nomenclature of these new phylogenetic clusters is to provide well-characterized reference sequences and to employ the same cluster-identification strategies.
We propose that the 10 HIV-1 CRF01_AE clusters be designated as CRF01_1AE through CRF01_10AE. These clusters are labeled with numbers rather than letters placed before the suffix “AE”. We are proposing that as new clusters AE are identified, reference sequences should be made available in a public HIV database and that the authors use the next available number. Therefore, we are suggesting a method by which HIV-1 CRF01_AE cluster nomenclature will provide a consistent and standardized method to name newly identified transmission-derived clusters among all subtypes and CRFs. We are also providing well-characterized near-complete genome sequences of CRF01_AE as reference sequences by selecting the sequence that had the deepest branch in each of the 10 currently identified CRF01_AE clusters (Supplementary Table S7).
New CRF01_AE cluster reference sequences should include the nomenclature, the representative sequence name, the accession number, the year of sampling, the country of sampling (origin), associated publication(s), and any other demographic information. The more HIV samples that are sequenced and characterized within countries and regions, the more unique clusters that are likely to be identified. This will present challenges to nomenclature and our ability to refer to these variants in a consistent and standardized way.
Electronic supplementary material
Acknowledgements
This study was supported by the National Science and Technology Major Project for Infectious Diseases Control and Prevention (2012ZX10001-002 and 2012ZX10001-008), National Natural Science Foundation of China (81361120407), NIH Foundation (R01AI094562), and SKLID Development Grant (2012SKLID103).
Author Contributions
X.L., M.K. and Y.S. conceived and designed the study. X.L., H.Z., L.L, Y.F., and M.K. performed the experiments and analyzed the data. X.L., M.K. and S.Y.W.H. drafted the manuscript. X.L., Y.F., M.K., S.Y.W.H. and Y.S. interpreted data and provided critical review. All authors reviewed and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Xingguang Li, Haizhou Liu and Lu Liu contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-03820-8
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Sharp PM, Hahn BH. Origins of HIV and the AIDS pandemic. Cold Spring Harbor Perspectives in Medicine. 2011;1:a006841. doi: 10.1101/cshperspect.a006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Robertson DL, Sharp PM, McCutchan FE, Hahn BH. Recombination in HIV-1. Nature. 1995;374:124–126. doi: 10.1038/374124b0. [DOI] [PubMed] [Google Scholar]
- 3.Saksena NK, et al. Coinfection and genetic recombination between HIV-1 strains: possible biological implications in Australia and South East Asia. Annals of the Academy of Medicine, Singapore. 1997;26:121–127. [PubMed] [Google Scholar]
- 4.Moutouh L, Corbeil J, Richman DD. Recombination leads to the rapid emergence of HIV-1 dually resistant mutants under selective drug pressure. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:6106–6111. doi: 10.1073/pnas.93.12.6106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coffin JM. Structure, replication, and recombination of retrovirus genomes: some unifying hypotheses. Journal of General Virology. 1979;42:1–26. doi: 10.1099/0022-1317-42-1-1. [DOI] [PubMed] [Google Scholar]
- 6.Hemelaar J. The origin and diversity of the HIV-1 pandemic. Trends in Molecular Medicine. 2012;18:182–192. doi: 10.1016/j.molmed.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Louwagie J, et al. Genetic diversity of the envelope glycoprotein from human immunodeficiency virus type 1 isolates of African origin. Journal of Virology. 1995;69:263–271. doi: 10.1128/jvi.69.1.263-271.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Robertson DL, et al. HIV-1 nomenclature proposal. Science. 2000;288:55–56. doi: 10.1126/science.288.5463.55d. [DOI] [PubMed] [Google Scholar]
- 9.Gao F, et al. The heterosexual human immunodeficiency virus type 1 epidemic in Thailand is caused by an intersubtype (A/E) recombinant of African origin. Journal of Virology. 1996;70:7013–7029. doi: 10.1128/jvi.70.10.7013-7029.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson JP, et al. Testing the hypothesis of a recombinant origin of human immunodeficiency virus type 1 subtype E. Journal of Virology. 2000;74:10752–10765. doi: 10.1128/JVI.74.22.10752-10765.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalish ML, et al. Recombinant viruses and early global HIV-1 epidemic. Emerging Infectious Diseases. 2004;10:1227–1234. doi: 10.3201/eid1007.030904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCutchan FE, et al. Genetic variants of HIV-1 in Thailand. AIDS Research and Human Retroviruses. 1992;8:1887–1895. doi: 10.1089/aid.1992.8.1887. [DOI] [PubMed] [Google Scholar]
- 13.Carr JK, et al. Full-length sequence and mosaic structure of a human immunodeficiency virus type 1 isolate from Thailand. Journal of Virology. 1996;70:5935–5943. doi: 10.1128/jvi.70.9.5935-5943.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson KE, et al. Risk factors for HIV infection among young adult men in northern Thailand. JAMA. 1993;270:955–960. doi: 10.1001/jama.1993.03510080059032. [DOI] [PubMed] [Google Scholar]
- 15.Ou CY, et al. Wide distribution of two subtypes of HIV-1 in Thailand. AIDS Research and Human Retroviruses. 1992;8:1471–1472. doi: 10.1089/aid.1992.8.1471. [DOI] [PubMed] [Google Scholar]
- 16.Kilmarx PH, et al. Explosive spread and effective control of human immunodeficiency virus in northernmost Thailand: the epidemic in Chiang Rai province, 1988-99. AIDS. 2000;14:2731–2740. doi: 10.1097/00002030-200012010-00013. [DOI] [PubMed] [Google Scholar]
- 17.Liao H, et al. Phylodynamic analysis of the dissemination of HIV-1 CRF01_AE in Vietnam. Virology. 2009;391:51–56. doi: 10.1016/j.virol.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 18.Feng Y, et al. The rapidly expanding CRF01_AE epidemic in China is driven by multiple lineages of HIV-1 viruses introduced in the 1990s. AIDS. 2013;27:1793–1802. doi: 10.1097/QAD.0b013e328360db2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCutchan FE, et al. Diversity of the envelope glycoprotein among human immunodeficiency virus type 1 isolates of clade E from Asia and Africa. Journal of Virology. 1996;70:3331–3338. doi: 10.1128/jvi.70.6.3331-3338.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen JH, et al. Molecular epidemiological study of HIV-1 CRF01_AE transmission in Hong Kong. Journal of acquired immune deficiency syndromes. 2009;51:530–535. doi: 10.1097/QAI.0b013e3181aac516. [DOI] [PubMed] [Google Scholar]
- 21.Abubakar YF, Meng Z, Zhang X, Xu J. Multiple independent introductions of HIV-1 CRF01_AE identified in China: what are the implications for prevention? PLOS ONE. 2013;8:e80487. doi: 10.1371/journal.pone.0080487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Angelis K, et al. Global Dispersal Pattern of HIV Type 1 Subtype CRF01_AE: A genetic trace of human mobility related to heterosexual sexual activities centralized in Southeast Asia. The Journal of Infectious Diseases. 2015;211:1735–1744. doi: 10.1093/infdis/jiu666. [DOI] [PubMed] [Google Scholar]
- 23.An M, et al. Reconstituting the epidemic history of HIV strain CRF01_AE among men who have sex with men (MSM) in Liaoning, northeastern China: implications for the expanding epidemic among MSM in China. Journal of Virology. 2012;86:12402–12406. doi: 10.1128/JVI.00262-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ye J, et al. Phylogenetic and temporal dynamics of human immunodeficiency virus type 1 CRF01_AE in China. PLOS ONE. 2013;8:e54238. doi: 10.1371/journal.pone.0054238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X, et al. Molecular epidemiology of HIV-1 in Jilin Province, Northeastern China: Emergence of a new CRF07_BC transmission cluster and intersubtype recombinants. PLOS ONE. 2014;9:e110738. doi: 10.1371/journal.pone.0110738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rousseau CM, et al. Large-scale amplification, cloning and sequencing of near full-length HIV-1 subtype C genomes. Journal of Virological Methods. 2006;136:118–125. doi: 10.1016/j.jviromet.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 27.Li X, et al. Near full-length genome identification of a novel HIV-1 recombinant form (CRF01_AE/B’/C) among heterosexuals in Jilin, China. AIDS Research and Human Retroviruses. 2014;30:695–700. doi: 10.1089/aid.2013.0278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, et al. Near full-length genome sequence of a novel HIV type 1 second-generation recombinant form (CRF01_AE/CRF07_BC) identified among men who have sex with men in Jilin, China. AIDS Research and Human Retroviruses. 2013;29:1604–1608. doi: 10.1089/aid.2013.0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karlin S, Altschul SF. Applications and statistics for multiple high-scoring segments in molecular sequences. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:5873–5877. doi: 10.1073/pnas.90.12.5873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Siepel AC, Halpern AL, Macken C, Korber BT. A computer program designed to screen rapidly for HIV type 1 intersubtype recombinant sequences. AIDS Research and Human Retroviruses. 1995;11:1413–1416. doi: 10.1089/aid.1995.11.1413. [DOI] [PubMed] [Google Scholar]
- 31.Rose PP, Korber BT. Detecting hypermutations in viral sequences with an emphasis on G – >A hypermutation. Bioinformatics. 2000;16:400–401. doi: 10.1093/bioinformatics/16.4.400. [DOI] [PubMed] [Google Scholar]
- 32.Hall, T. A. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series41, 95–98, doi:citeulike-article-id:691774 (1999).
- 33.Strimmer K, von Haeseler A. Likelihood-mapping: a simple method to visualize phylogenetic content of a sequence alignment. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:6815–6819. doi: 10.1073/pnas.94.13.6815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18:502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- 35.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ragonnet-Cronin M, et al. Automated analysis of phylogenetic clusters. BMC Bioinformatics. 2013;14:317. doi: 10.1186/1471-2105-14-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kumar S, Stecher G, Tamura K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:11030–11035. doi: 10.1073/pnas.0404206101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.He Z, et al. Evolview v2: an online visualization and management tool for customized and annotated phylogenetic trees. Nucleic Acids Research. 2016;44:W236–241. doi: 10.1093/nar/gkw370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen) Virus Evolution. 2016;2:vew007. doi: 10.1093/ve/vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.To TH, Jung M, Lycett S, Gascuel O. Fast dating using least-squares criteria and algorithms. Systematic Biology. 2016;65:82–97. doi: 10.1093/sysbio/syv068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Drummond AJ, Ho SYW, Phillips MJ, Rambaut A. Relaxed phylogenetics and dating with confidence. PLOS Biology. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Bayesian coalescent inference of past population dynamics from molecular sequences. Molecular Biology and Evolution. 2005;22:1185–1192. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 44.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Molecular Biology and Evolution. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parker J, Rambaut A, Pybus OG. Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infection, Genetics and Evolution. 2008;8:239–246. doi: 10.1016/j.meegid.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 46.Duchene S, Geoghegan JL, Holmes EC, Ho SYW. Estimating evolutionary rates using time-structured data: a general comparison of phylogenetic methods. Bioinformatics. 2016;32:3375–3379. doi: 10.1093/bioinformatics/btw005. [DOI] [PubMed] [Google Scholar]
- 47.Bengtsson L, Lu X, Liljeros F, Thanh HH, Thorson A. Strong propensity for HIV transmission among men who have sex with men in Vietnam: behavioural data and sexual network modelling. BMJ Open. 2014;4:e003526. doi: 10.1136/bmjopen-2013-003526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Young AM, Jonas AB, Mullins UL, Halgin DS, Havens JR. Network structure and the risk for HIV transmission among rural drug users. AIDS and Behaviour. 2013;17:2341–2351. doi: 10.1007/s10461-012-0371-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rothenberg RB, et al. Social network dynamics and HIV transmission. AIDS. 1998;12:1529–1536. doi: 10.1097/00002030-199812000-00016. [DOI] [PubMed] [Google Scholar]
- 50.De P, Singh AE, Wong T, Yacoub W, Jolly AM. Sexual network analysis of a gonorrhoea outbreak. Sexually Transmitted Infections. 2004;80:280–285. doi: 10.1136/sti.2003.007187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rojanapithayakorn W, Hanenberg R. The 100% condom program in Thailand. AIDS. 1996;10:1–7. doi: 10.1097/00002030-199601000-00001. [DOI] [PubMed] [Google Scholar]
- 52.Celentano DD, et al. Risk factors for HIV-1 seroconversion among young men in northern Thailand. JAMA. 1996;275:122–127. doi: 10.1001/jama.1996.03530260036028. [DOI] [PubMed] [Google Scholar]
- 53.Hanenberg RS, Rojanapithayakorn W, Kunasol P, Sokal DC. Impact of Thailand’s HIV-control programme as indicated by the decline of sexually transmitted diseases. Lancet. 1994;344:243–245. doi: 10.1016/S0140-6736(94)93004-X. [DOI] [PubMed] [Google Scholar]
- 54.Administration, C. N. T. The Yearbook of China Tourism Statistics. (China Tourism Publishing House, 1987–2014).
- 55.Shiino T, et al. Phylodynamic analysis reveals CRF01_AE dissemination between Japan and neighboring Asian countries and the role of intravenous drug use in transmission. PLOS ONE. 2014;9:e102633. doi: 10.1371/journal.pone.0102633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.