

Abstract
Miniature chromosome maintenance (MCM) proteins play critical roles in DNA replication licensing, initiation and elongation. MCM8, one of the MCM proteins playing a critical role in DNA repairing and recombination, was found to have over-expression and increased DNA copy number in a variety of human malignancies. The gain of MCM8 is associated with aggressive clinical features of several human cancers. Increased expression of MCM8 in prostate cancer is associated with cancer recurrence. Forced expression of MCM8 in RWPE1 cells, the immortalized but non-transformed prostate epithelial cell line, exhibited fast cell growth and transformation, while knocked down of MCM8 in PC3, DU145 and LNCaP cells induced cell growth arrest, and decreased tumor volumes and mortality of severe combined immunodeficiency mice xenografted with PC3 and DU145 cells. MCM8 bound cyclin D1 and activated Rb protein phosphorylation by cyclin-dependent kinase 4 in vitro and in vivo. The cyclin D1/MCM8 interaction is required for Rb phosphorylation and S phase entry in cancer cells. As a result, our study showed that copy number increase and overexpression of MCM8 may play critical roles in human cancer development.
Introduction
Cancer is one of the leading causes of death in the US. It is estimated 1,658,370 new cancer cases diagnosed and 589,430 cancer deaths in the US in 2014(1, 2). The understanding of the mechanisms that lead to the development of these lethal diseases remains incomplete. Prostate cancer is one of the most diagnosed cancers in men in the US. Over 161,000 new cases of prostate cancer were identified in 2016, accounting for approximately 10% of all new cancer cases. Unfortunately, the disease is not always indolent; about 26,730 men are expected to die from prostate cancer this year(3). Despite the high incidence of prostate cancer, the molecular mechanisms that underlie the aggressive behavior of some prostate cancer cases remain unclear. Identification of the molecular events that trigger the aggressive behavior of prostate cancer is crucial in reducing the mortality of this disease.
Comprehensive genomic and transcriptomic analyses of human cancers had revealed numerous numerical or mutational, or epigenomic changes in the cancer genome that may drive gene expression alterations in cancers(4–17). The pattern changes of gene expression and genome abnormalities may provide significant insight into the mechanism of human cancer development. However, the relationship between these abnormalities and behavior of cancers remains unclear. To gain a clear and new understanding of the roles of these genes in the biology of human cancers, experimental analysis of these genes one at a time is required.
In this report, we performed meta-analyses of genome-wide copy number analysis on MCM proteins and led to the identification of MCM8, a critical component of DNA replication licensing gene(18–21), amplified or gain in 17 human malignancies. Similarly, overexpression of MCM8 was found in a wide variety of human malignancies. Over-expression of MCM8 was also identified in prostate cancer cell lines PC3, DU145 and LNCaP. Forced expression in immortalized prostate epithelial cell lines RWPE1 induced transformation, while knocked down of MCM8 induced growth arrest of PC3, DU145, and LNCaP cells. It appears that MCM8 amplification and over-expression underlie the aggressiveness of prostate cancer.
Results
Significant evidence suggests that amplification and overexpression of DNA replication licensing factor have been associated with aggressive human malignancies(22–30). One of these DNA replication licensing factors, MCM7, had been well characterized to drive the aggressive behavior in prostate cancer, colon cancer, lung cancer, ovarian cancer, bladder cancer, liver cancer and gastric cancer(23, 25, 31–38). To investigate whether other DNA replication licensing factors are also implicated in human cancers, we screened 3 data sets of prostate cancer. We found that MCM8, a DNA replication licensing factor involving replication elongation and DNA homologous recombination, were consistently up-regulated in prostate cancer samples in comparison with normal prostate tissues (Table 1). To investigate whether MCM8 is also similarly up-regulated in other human malignancies, microarray data from European Bioinformatics Institute of EMBL were screened for MCM8 expression in a variety of human cancers. The analyses suggest that MCM8 is over-expressed in a large number of human cancers, including, breast cancer, non-small cell lung cancer, liver cancer, medulloblastoma, and glioblastoma multiforme (Table 1). The magnitudes of over-expression range from 2- to 5.2 fold, suggesting a wide significance of MCM8 overexpression in human malignancies.
Table 1.
Human malignancies with MCM8 over-expression
| Malignancy | Fold change | p value |
|---|---|---|
| T-cell acute lymphoblastic leukemia | 5.2 | 1.3 × 10−7 |
| Medulloblastoma | 3.7 | 1.03 × 10−7 |
| Nasopharyngeal carcinoma | 3.5 | 4.77 × 10−9 |
| Glioblastoma Multiforme | 3.25 | 1.32 × 10−7 |
| Pediatric high grade glioma | 3.3 | 3.57 × 10−7 |
| Primitive neuroectodermal tumor | 3.03 | 8.0 × 10−4 |
| Non-small cell lung cancer | 3.0 | 1.3 × 10−19 |
| Adenocarcinoma | 2.64 | 5.3 × 10−10 |
| Squamous carcinoma | 3.4 | 1.4 × 10−16 |
| Adrenocortical carcinoma | 2.64 | 1.65 × 10−4 |
| Hepatocellular carcinoma, HCV related | 2.3 | 0.019 |
| Prostate cancer | ||
| Luo, et al | 2.0 | 0.02 |
| Yu, et al | 2.04 | 0.001 |
| LaTulippe, et al | 1.94 | 7.8 × 10−4 |
| Breast cancer | 2.14 | 1.86 × 10−8 |
| Luminal A | 2.0 | 3.03 × 10−7 |
| Luminal B | 3.03 | 7.6 × 10−8 |
| Triple negative breast cancer | 5.27 | 2.1 × 10−14 |
| Rhabdoid tumor | 2.64 | 1.2 × 10−4 |
To investigate the cause of MCM8 over-expression in prostate cancer, we analyzed 2 prostate cancer copy number data set from University of Pittsburgh and TCGA. The results showed that 19 and 8% of prostate cancer genomes have a gain of MCM8 genome copy, respectively. To examine whether other human malignancies also have similar gain of MCM8 gene copy number, we analyzed additional large sets of Affymetrix SNP 6.0 arrays on 16 other human malignancies including glioblastoma multiforme, breast cancer, colon cancer, pancreatic adenocarcinoma, ovarian cancer, esophageal cancer, liver cancer, lung adenocarcinoma, lung squamous cell carcinoma, sarcoma, diffuse large cell lymphoma, thyroid cancer, rectal adenocarcinoma, acute myeloid leukemia and bladder cancer. The gain of MCM8 gene copy is widespread among human malignancies: ranging from 4.2% in thyroid cancer to 79.7% in rectal adenocarcinoma (Table 2). In some breast and colon cancer samples, up to 16 copies of MCM8 were found in the genomes of cancer cells, suggesting that the gain of MCM8 was the result of amplification of the genome sequence encoding MCM8 in some of these cancers. These analyses suggest that the copy number gain of MCM8 is probably the underlying mechanism of MCM8 overexpression in human malignancies.
Table 2.
Human malignancies with gain of MCM8 gene copy number
| Malignancy | % | Average gains |
|---|---|---|
| Prostate cancer | ||
| Yu et al | 19 | 3.86 |
| TCGA | 8 | 2.76 |
| Bladder cancer (TCGA) | 32 | 2.7 |
| Breast cancer (TCGA) | 40.6 | 3.16 |
| Colon cancer (TCGA | 63.5 | 3.23 |
| Esophageal carcinoma (TCGA) | 48.8 | 2.78 |
| Glioblastoma Multiforme (TCGA) | 36.1 | 2.76 |
| Liver cancer (TCGA) | 30.4 | 2.89 |
| Non-small cell lung cancer | ||
| Adenocarcinoma (TCGA) | 26.1 | 2.75 |
| Squamous cell carcinoma (TCGA) | 32.1 | 2.74 |
| Pancreas adenocarcinoma (TCGA) | 18.4 | 2.7 |
| Head and Neck squamous cell carcinoma (TCGA) | 22.6 | 2.6 |
| Acute myeloid leukemia(TCGA) | 16.9 | 3.2 |
| Ovarian cancer (TCGA) | 61.5 | 3.03 |
| Rectum adenocarcinoma (TCGA) | 79.7 | 3.27 |
| Sarcoma (TCGA) | 31.8 | 2.82 |
| Thyroid carcinoma (TCGA) | 4.2 | 2.86 |
| Uterine corpus endometrial carcinoma (TCGA) | 26.6 | 2.82 |
Analysis of prostate cancer data set from TCGA indicates that gain of MCM8 is associated with higher Gleason’s grade (p=0.0007), more advanced pathological stage (p=0.036) and more likely to have lymph node metastasis (p=0.0025). The gain of MCM8 is also associated with higher level of residual tumors of lung adenocarcinoma (p=0.002), lymph node metastasis (p=0.03) and higher grades (p=0.000098) of esophageal cancer, more advanced stages (p=0.00025) and more frequent lymph node metastasis (p=0.0017) of colon cancer. The gain of MCM8 is also found disproportionately in ovarian cancer (p=0.026) and uterine endometrial carcinoma (p=0.0000024) that were presented as advanced stages of cancer at the time of diagnosis. Urothelial carcinoma with a gain of MCM8 also showed a higher level of aggressiveness by displaying frequent lymph node metastasis and recurrence of cancer after treatment (p=0.049). These results indicate that gain of MCM8 is likely a marker for the aggressiveness of human cancers.
To validate the gain of MCM8 in prostate cancer, qPCR was performed using primers corresponding to intron16/exon17 region on 115 prostate samples, including 19 organ donor prostate tissue samples with ages ranging from 15 to 72, three benign prostate tissues adjacent cancer, and 93 prostate cancer samples. As shown in figure 1A, the gain of MCM8 was not detected in any of the organ donor prostates and benign prostate tissues adjacent to cancer, while 52% (52/93) of the prostate cancer samples were detected with a gain of MCM8. Among those prostate cancer samples from patients that experienced recurrence of the disease after radical prostatectomy, the frequency of gain of MCM8 reached 70.5% (43/49). In contrast, the rate of MCM8 gain was only 26.5% (9/34) for prostate cancers that did not reoccur for at least 5 years after the surgery. These results indicate a strong association between prostate cancer recurrence and the gain of MCM8 (p<0.0001). To visualize the gain of MCM8 in prostate cancer samples, fluorescence in situ hybridization were performed using bacterial artificial chromosome probe corresponding to genome region where MCM8 resides (Figure 1B and C). The results indicate MCM8 gain in 40% (8/20) of the samples examined: 83% (5/6) samples from patients who experienced prostate cancer recurrence had MCM8 gain in their cancer genomes, while only 21% (3/14) indolent cancer samples were positive of the gain of MCM8. Kaplan-Meir analysis based on the status of MCM8 gain showed that patients with MCM8 gain in their cancer genome had PSA-free survival rate of 33.3%, while patients with no MCM8 gain had the 5-year PSA-free survival rate of 74.5% (figure 1D), suggesting that the gain of MCM8 signals a significantly poorer clinical outcomes (p=7.8 × 10−5) for prostate cancer.
Figure 1. The gain of MCM8 gene copy in the genome of prostate cancer.
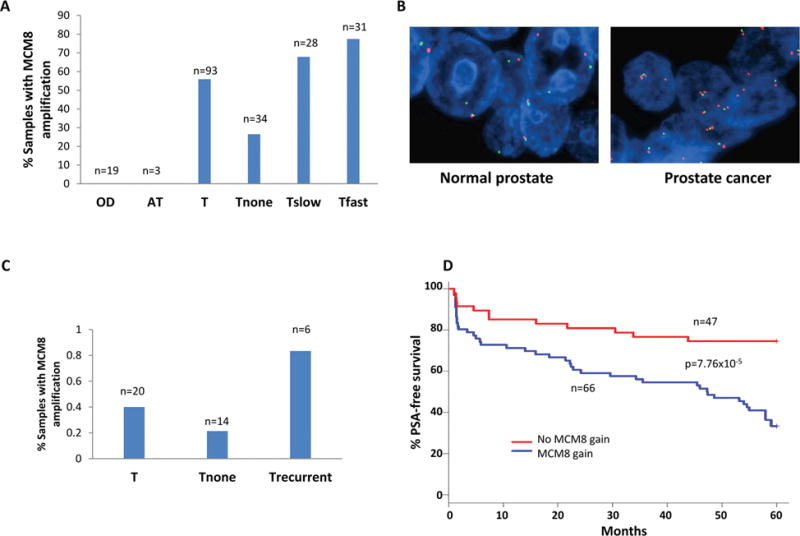
(A) Status of MCM8 gain in organ donor prostate (OD), benign prostate tissue adjacent to cancer (AT), prostate cancer (T). T samples were subdivided into cancers that have no recurrence for at least 5 years after radical prostatectomy (Tnone), cancers that produced recurrences with PSADT≥14 months (Tslow), and cancers that produced recurrences with PSADT≤4 months (Tfast). The gain of MCM8 was determined when copy of MCM8 was estimated to be ≥3 copies per genome relative to β-actin. (B) Representative images of fluorescence in-situ hybridization (FISH) using BAC probes corresponding to MCM8 genome sequence and centromere of chromosome 20. BAC probe for MCM8 was labeled with spectrum orange, while probe for centromere of chromosome 20 was labeled with spectrum green. (C) Prostate cancer samples with MCM8 gain status based on FISH results. (D) Kaplan-Meier analysis of prostate cancer based on the status of MCM8 gain. The analysis was plotted based on the combined results of qPCR and FISH obtained from (A) and (C).
To examine whether MCM8 protein is over-expressed in prostate cancer samples, 533 prostate samples were examined for MCM8 expression through immunostaining using antibodies specific for MCM8 protein. The expression of MCM8 was quantified based on the number of cells positive for MCM8 staining: negative (0), focal positive (0.5), 1–20% cells positive (1), 20–50% cells positive (2), 50–80% cells positive (3) and more than 80% cells positive (4). As shown in figure 2A and B, most organ donor prostate cancers are only focally positive for MCM8 protein with an average score of 0.56. In contrast, a significant number of prostate cancer samples had the expression of MCM8 in more than 20%, with an average of scores of 1.6. The average of MCM8 expression scores for prostate cancer that proved recurrent reached 1.99. Higher MCM8 protein expression score is strongly associated with prostate cancers that had cancer recurrence (p=3.5 × 10−15). To analyze the impact of MCM8 protein expression on PSA-free survival of prostate cancer, Kaplan-Meir analysis was performed based on MCM8 expression levels. As shown in figure 2C, 42.4% patients with MCM8 expression scoring 2 or more survived 5 years without prostate cancer recurrence. In contrast, approximately 90% patients with MCM8 scoring less than 2 survived at least 5 years cancer free. These results indicate that MCM8 overexpression had a significant negative impact on the prostate cancer prognosis (p=9.25 × 10−13).
Figure 2. Overexpression of MCM8 protein in prostate cancer samples.
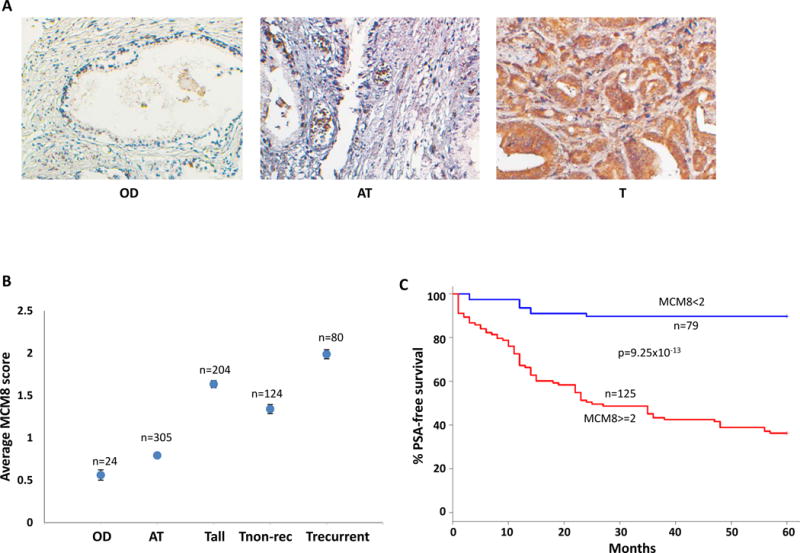
(A) Representative images of immunostaining of MCM8 in an organ donor prostate (OD), benign prostate tissues adjacent to cancer (AT), and prostate cancer (T) samples. (B) Average MCM8 expression scores of OD, AT, prostate cancer (Tall), prostate cancers that produced recurrence (Trecurrent), and prostate cancers that had no recurrence for at least 5 years after radical prostatectomy (Tnon-rec). (C) Kaplan-Meier analysis based on MCM8 expression score ≥2 or <2 on prostate cancers obtained from (B).
To investigate the biological role of MCM8 over-expression in prostate epithelial cells, RWPE1, the immortalized but non-transformed prostate epithelial cells, were transfected with pCDNA4-MCM8-FLAG. Fifteen colonies were screened for inducible expression of MCM8. Two clones were selected for further analysis (RM8#2 and RM8#9). As shown in figure 3A and B, induced expression of MCM8 in RWPE1 cells resulted in an average of 2.5 fold (p<0.01) increase of S-phase and concomitant 32.6% (p<0.01) decrease of G0/G1 phase cells. MCM8 expression increased colony formation by 2.8 fold (p<0.01) and anchorage independent growth by 12.7 fold (p<0.01) of RWPE1 cells (figure 3C and D). Most non-transformed cells grow poorly in anchorage-independent growth assays. Dramatic improvement of anchorage-independent growth of RWPE1 cells induced by MCM8 clearly indicates that RWPE1 cells were transformed. MCM8 appears abundantly expressed in PC3, DU145 and LNCaP cells, in comparison with RWPE1 (figure 3A). To assess the impact of MCM8 overexpression in prostate cancer, prostate cancer cell lines PC3, DU145 and LNCaP were transfected with pSingle-tts-siMCM8 that expresses a shRNA targeting at MCM8 mRNA upon the treatment of doxycycline (5μg/ml). Two clones of each cell lineage were selected for analysis. Knocked down of MCM8 in PC3 (73% for M8P#5 and 71% for M8P#7) by shRNA specific for MCM8 reduced S-phase entry by an average of 51% (p<0.01), and reduced colony formation by over 83% (p<0.01) (figure 3C and D). These biological changes were accompanied by drops of DNA replication licensing of Cdt1 (average 66.7% for M8P#5 and M8P#7), MCM6 (average 59.2%) and MCM7 (average 78.7%), suggesting that down-regulation of MCM8 have significant negative impact on DNA replication licensing. Similar results were also found when DU145 (51% knocked-down for M8D#1 and 75% for M8D#4) and LNCaP (65% for M8L#3 and 63% M8L#5) cells were knocked down with shRNA specific for MCM8: 65% drop (p<0.01) in S-phase cells and 94% drop (p<0.01) in colony formation for DU145, and 44% drop (p<0.01) in S-phase cells and 91% drop (p<0.01) in colony formation for LNCaP cells. Over-expression of MCM8 in non-transforming prostate epithelial cells RWPE1 (RM8#2 and RM8#9, average 2.3 fold) resulted in 2.5 fold increase of S-phase entry in average (figure 3A and B). These were also accompanied by significant increase of Cdt1 (>5 fold), MCM6 (2.2 fold) and MCM7 (>5 fold) licensing. Over-expression of MCM8 is clearly necessary for cancer cell growth and transformation. To investigate the impact of MCM8 overexpression on prostate cancer growth metastasis in vivo, M8P#5 and M8D#1cells were xenografted into the subcutaneous regions of severe combined immunodeficiency mice. These mice were treated with doxycycline to knock down the expression of MCM8 in these cancer cells. As shown in figure 3E–F, mice treated with doxycycline water (5μg/ml) had 74.3% (p<0.01) decrease in tumor volume for M8P#5 cells and 65% (p<0.01) decrease for M8D#1 cells, in comparison with the untreated controls. The mice treated with doxycycline water had 93.8% survival rate. In contrast, only 31% mice xenografted with tumor cells without treatment survived 6 weeks (p=0.00015). These results suggest that MCM8 expression is crucial for cancer growth and aggressiveness.
Figure 3. MCM8 is critical to the transformation of prostate cancer.
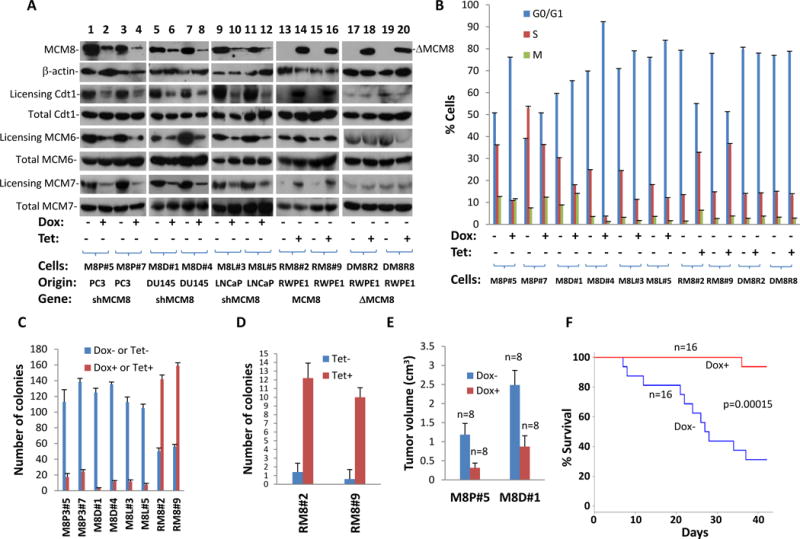
(A) Immunoblotting of MCM8 and β-actin of protein extracts from PC3 (M8P#5 and M8P#7), DU145 (M8D#1 and M8D#4) and LNCaP (M8L#3 and M8L#5) cell transformed with pSingle-tts-shMCM8, treated with or without 5 μg/ml doxycycline. RWPE1 cells (RM8#2 and RM8#9) transformed with pCDNA4-MCM8-FLAG/pCDNA6-TO, or with pCDNA4-ΔMCM8del261-290-FLAG/pCDNA6-TO (DM8R2 and DM8R8), and treated with or without tetracycline (1 μg/ml) were similarly blotted with antibodies specific for MCM8 and β-actin. DNA licensing of Cdt, MCM6, and MCM7 was shown. (B) BrdU labeling and cell cycle analysis of the replicate samples from (A). Triplicate experiments were performed. Standard deviation is indicated. (C) Colony formation analyses of the replicate samples from (A). Triplicate experiments were performed. Standard deviation is indicated. (D) Anchorage-independent growth of RWPE1 cells transformed with pCDNA4-MCM8-FLAG and pCDNA6. Replicate samples from (A) were analyzed. Triplicate experiments were performed. Standard deviation is indicated. (E) Knocked down of MCM8 expression reduced tumor volume of SCID mice xenografted M8P#5 and M8D#9 cells. Standard deviation is indicated. (F) Knocked down of MCM8 expression reduced mortality of SCID mice xenografted with M8P#5 and M8D#9 cells.
To investigate the mechanism of MCM8 mediated cancer growth and transformation, Yeast two-hybrid cDNA library from human prostate was screened for MCM8 interacting protein by pBD-MCM8full that expresses bait domain-MCM8 fusion protein (BD-MCM8). Thirty-one colonies were found to grow in most stringent selective medium and positive for β-galactosidase. Eleven unique clones were identified. One of these clones contains a cDNA encoding cyclin D1 (CCND1). To validate these results, pAD-CCND1 and pBD-MCM8 were co-transfected into Yeast AH109 cells. Co-transfection of pBD-MCM8 (full length) and pAD-CCND1 showed positive galactosidase activity and grew in most stringent selective medium, while all negative controls were negative, confirming the initial Yeast Two-hybrid screening results (Figure 4A). To test the binding between MCM8 and cyclin D1 in vivo, co-immunoprecipitations of MCM8 and cyclin D1 were performed on the protein extracts of PC3 and RWPE1 cells. As shown in Figure 4B, co-immunoprecipitation of MCM8 and cyclin D1 was readily apparent. To visualize whether MCM8 and cyclin D1 co-localize in the nucleus, double immunofluorescence staining using antibodies against MCM8 and cyclin D1 were performed in RWPE1 cells. As demonstrated in figure 4C, MCM8 and a significant amount of cyclin D1 were colocalized in the nuclei. Similar colocalization results were obtained with PC3 cells (data not shown).
Figure 4. MCM8 interacts with cyclin D1.
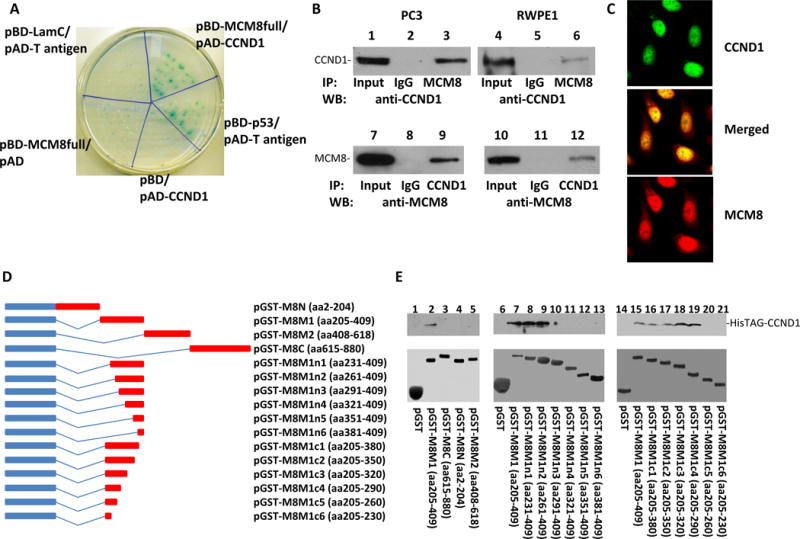
(A) Yeast Two-hybrid screening identified that MCM8 bound cyclin D1. Co-transformants of pBD-MCM8full with pAD-CCND1 on SD agar plate with high stringent nutrient selection (SD-leu-Trp-His-Ade) are shown. Co-transfection of pBD-p53 and pAD-T-antigen is the positive control, while co-transfection of pBD-LamC and pAD-T-antigen, pBD-MCM8full, and pAD, pBD and pAD-CCND1 are the negative controls. (B) Co-immunoprecipitations of MCM8 (lanes 3 and 6) or cyclin D1 (lanes 9 and 12) using antibodies specific for MCM8 or cyclin D1 were performed on protein extracts from PC3 (left panels) and RWPE1 (right panels). The immunoprecipitates were blotted with the indicated antibodies. (C) Immunofluorescence staining of RWPE-1 cells with an antibody against cyclin D1 recognized by FITC- conjugated secondary antibodies for mouse and antibodies specifically against MCM8 recognized by TRITC conjugated secondary antibodies for rabbit. (D) Mapping binding motif of cyclin D1 on MCM8. Left: Schematic diagram of fusion proteins of a series of MCM8 deletion mutants with glutathione-S-transferase (GST) protein. Right: Binding assays of GST or GST-MCM8 deletion mutants with HisTAG-cyclin D1 purified from E.coli. The bound cyclin D1 was blotted with anti-cyclin D1 antibodies. Top panel: Immunoblots with antibodies specific for cyclin D1; Bottom panel: Coomassie staining of the fusion proteins.
To validate the interaction between MCM8 and cyclin D1 in vitro, MCM8 coding region was segmented into 4 regions: MCM8N (aa2-204), MCM8M1 (aa205-409), MCM8M2 (aa409-618), MCM8C (aa615-880). These cDNA fragments were constructed into pGEX-5X-3 to create GST-MCM8N, GST-MCM8M1, GST-MCM8M2 and GST-MCM8C fusion proteins. These constructs were transfected into E.coli strain BL21. Each of these fusion proteins was induced to express with Isopropyl β-D-1-thiogalactopyranoside (IPTG), purified and assayed for their binding activity with recombinant HisTAG-cyclin D1 purified from bacterial protein extract. The results of the binding assays indicate that GST-MCM8M1 (aa408-618) bound with cyclin D1 in the cell-free system (figure 4D and E), while GST-MCM8N, GST-MCM8M2, and GST-MCM8C were negative in the binding assays. These results indicate that the cyclin D1 binding motif is located in the region of amino acids 205–409 in MCM8 protein. The interaction between MCM8 and cyclin D1 is direct. No “bridge protein” is required in their interaction in vitro. A series of deletion mutants of GST-MCM8M1 were assayed to identify the motif that is required for interaction with cyclin D1. A stretch of 30 amino acid sequence located in 261–290 of MCM8 was found crucial for MCM8 binding with cyclin D1 because the fusion proteins with deletion of this sequence did not bind with cyclin D1, while all proteins containing this sequence bound with cyclin D1 (figure 4D and E).
Cyclin D1 is a binding protein and co-factor for cyclin-dependent kinase 4 (CDK4)(39), and plays a crucial role in promoting cell cycle entry to S phase through phosphorylation of retinoblastoma (Rb) protein(40). Interestingly, in vitro binding assays indicate that the presence of MCM8 produced a multi-protein complex that includes CDK4, cyclin D1and MCM8 (figure 5A). To investigate whether binding of MCM8 with cyclin D1 has an impact on the kinase activity of CDK4, protein kinase assays were performed on recombinant CDK4/cyclin D1 protein with or without MCM8, using recombinant Rb as a substrate. As shown in figure 5, the presence of recombinant GST-MCM8 enhanced kinase activity of CDK4 and reduced the Km value by 6.8 fold (455 μM vs. 3100 μM) in comparison with cyclin D1/CDK4 alone. These experiments suggest that MCM8 is a cofactor for enhancing the kinase activity of CDK4. In contrast, such enhancement of CDK4 kinase activity was abrogated when the cyclin D1 interaction motif was deleted from GST-MCM8 (GST-dMCM8, figure 5B). The peptide corresponding to aa261-290 of MCM8 blocked the kinase enhancement effect of MCM8 on CDK4/cyclin D1 complex (figure 5B). Co-immunoprecipitation analysis showed that MCM8 formed a large complex with cyclin D1, CDK4, p21 and p27 in PC3 cells (figure 5C), suggesting complex nature of these interactions. To investigate the impact of MCM8 on Rb phosphorylation in vivo, MCM8 mRNA was knocked down by induction MCM8 shRNA with doxycycline in M8P#5 (PC3) and M8D#1 (DU145) cells. As shown in figure 6A, a decrease of MCM8 protein (>90% for M8P#5 and 82% for M8D#1) level accompanied by a reduction of Rb phosphorylation in both cell types. The in vitro CDK4 kinase activity also showed similar decreases when MCM8 was knocked down in these cell lines (figure 6B). These experiments suggest that the presence of MCM8 is essential for Rb phosphorylation. To examine whether binding of MCM8 with cyclin D1 is required for CDK4 kinase activation, a mutant MCM8 that contains only 31 amino acids from MCM8 (aa1,261-290) and FLAG-TAG sequence was ligated into pCDNA4 to create pCDNA4-CBM-FLAG (cyclin D1 binding motif-FLAG) to interfere the binding between MCM8 and cyclin D1. The mutant was cotransfected with pCDNA6 into PC3 cells. Two clones were selected for further analysis. As shown in figure 6A, induction of mutant MCM8 effectively decreased the phosphorylation level of Rb, in comparison with uninduced controls. Similar decreases were also identified in the in vitro kinase assays (figure 6B), while MCM8 mutant that contains no cyclin D1 binding motif had no impact on Rb phosphorylation (figure 6A–B), DNA replication licensing and cell cycle S-phase entry (figure 3B). These analyses support that MCM8/cyclin D1 interaction is crucial for CDK4 kinase activation. It appears that DNA replication licensing in vivo is also dependent on MCM8/cyclin D1 interaction. Interestingly, over-expression of cyclin D1 partially reversed the inhibitory effect induced by MCM8 knockdown (58–65% for M8P#5 and 41–67% for M8D#1) in cell cycle analysis (figure 6C and D), probably due to forcing more CDK4/cyclin D1 complex formation due to a larger quantity of cyclin D1. In addition, copy number analysis of TCGA data suggests that most samples with MCM8 gain contain at least one intact Rb copy (Supplemental Table 1), supporting a clinical significance of MCM8/cyclin D1/CDK4/Rb signaling.
Figure 5. MCM8 enhanced CDK4 kinase activity.
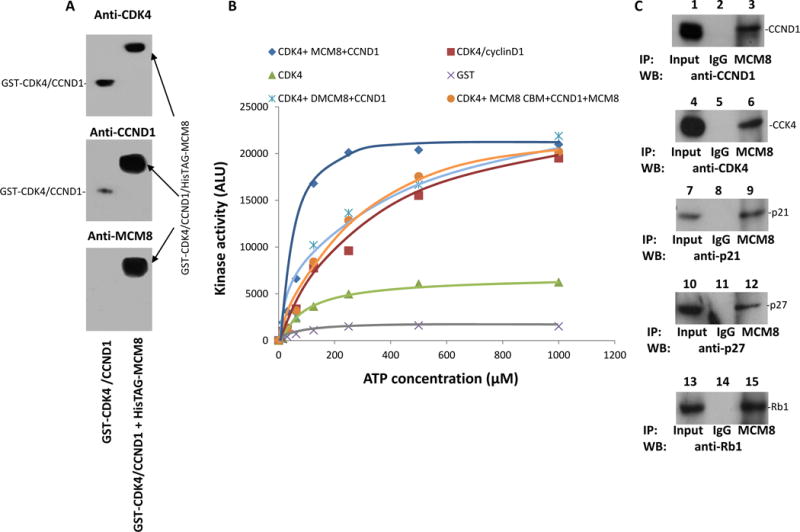
(A) Complex formation of GST-CDK4/cyclin D1/HisTAG-MCM8. The binding assays were performed on E.coli produced GST-CDK4 with GST-CCND1 (lane 1) or GST-CDK4 with GST-CCND1 and HisTAG-MCM8 (lane 2). The GST-CDK4/cyclin D1/HisTAG-MCM8 complex was purified from HisTAG column, resolved in 6% non-denaturing gel, and immunoblotted with antibodies specific for CDK4, cyclin D1 or MCM8. (B) One hundred nanograms of GST-CDK4/CCND1 (from Sf9 cells) were incubated with various concentrations of ATP as indicated and 50 ng of Rb, in the absence or presence of MCM8 or ΔMCM8delaa261-290 (dMCM8) or ΔMCM8aa261-290 (MCM8 cyclin D1 binding motif –MCM8 CBM) plus MCM8 for 30 min at 37°C using ATP-GLO™ kinase assay kit. CDK4 and GST were used as controls. Rb phosphorylation quantified after immunoprecipitation using antibodies specific for Rb protein. (C) MCM8 co-immunoprecipitated with p27, p21, cyclin D1 and CDK4 in PC3 cells.
Figure 6. MCM8 is required for Rb phosphorylation in vivo.
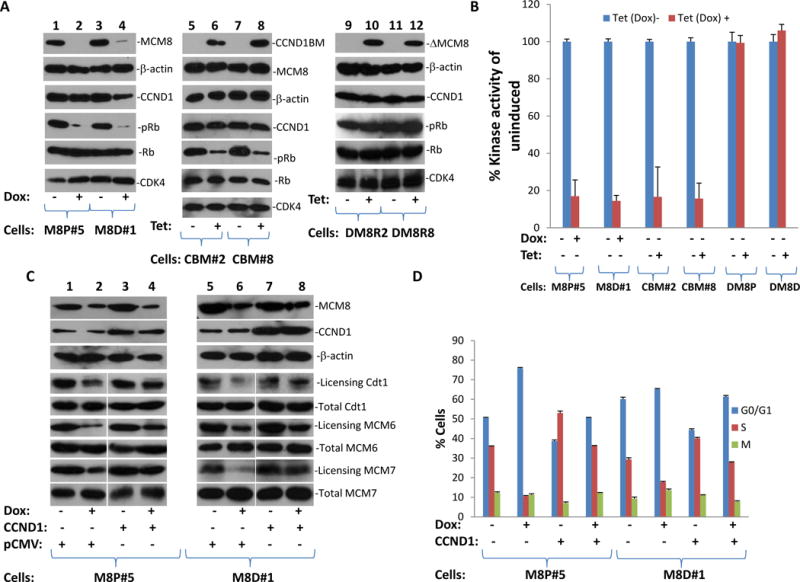
(A) Knocked down of MCM8 abrogated Rb phosphorylation in M8P#5 and M8D#1 cells (left panel). Expression of mutant MCM8 that interferes with cyclin D1/MCM8 binding decreased Rb phosphorylation in PC3 (CBM#2 and CBM#8) cells transformed with pCDNA4-CBM-FLAG/pCDNA6 (cyclin D1 binding motif-FLAG) (middle panel), while expression of MCM8 mutant that lacks cyclin D1 binding motif had no impact on Rb phosphorylation. (B) In vitro kinase assays of immunoprecipitates of CDK4 from replicate samples of (A) using GST-Rb as substrate. Triplicate experiments were performed. Standard deviation is indicated. (C) Immunoblot analysis of cyclin D1, MCM8 and β-actin in M8P#5 and M8D#1 cells transfected with pCMVscript (pCMV) or pCMV-CCND1 (CCND1). DNA licensing of Cdt, MCM6, and MCM7 was shown. (D) BrdU labeling and cell cycle analyses of replicate samples from (C). Triplicate experiments were performed. Standard deviation is indicated.
Discussion
MCM8 is one of the DNA replication licensing factors participating in DNA replication initiation and elongation(41–43). Recent studies suggest that MCM8 is recruited to the DNA repair site to facilitate DNA homologous recombination and double-strand breaks(44, 45). Genome instability was identified in mice that are deficient in MCM8(46). Mutations of MCM8 resulted in human gonadal failure(47). To our knowledge, this is the first report showing that widespread gain and overexpression of MCM8 are present in human malignancies. Since the gain of MCM8 is associated with aggressive characteristics of several human cancers, it implies that the gain of MCM8 may play significant roles in driving the cancer metastasis. Most aggressive cancer genomes demonstrate extensive chromosome rearrangement, deletion, and amplification of genes critical to cell survival, growth and migration. It is conceivable that overexpression of MCM8 may facilitate and exacerbate the process of chromosome rearrangement.
Cyclin D1/CDK4 complex has been a critical target by many proteins that regulate cell cycle progression, including p27(48, 49), p16(50), p21(51), Hsp90(52), ccdc37(53), PCNA(54), MyoD(55), etc. MCM8 probably contains versatile functions beyond DNA replication licensing and repair. It is possible that MCM8 serves as one of the assembly factors in building CDK4/cyclin D1 complex since the presence of MCM8 enhanced the recruitment of CDK4/cyclin D1 complex. In addition, our analysis showed that MCM8 is crucial in CDK4 activation by significantly lowering the substrate threshold required for CDK4 kinase activation. Phosphorylation of Rb correlates with the level of MCM8. Indeed, forced expression of MCM8 in cancer cell lines resulted in dramatic increase of cell entry into S phase in non-transformed cells, while knocked down of MCM8 generates growth arrest of cancer cells. These results indicate that MCM8 is essential for cell S phase entry. Over-expression of MCM8 may produce two consequences: Promoting cell growth through increasing phosphorylation and inactivation of Rb molecule and promoting DNA recombination that produces abnormal chromosome rearrangements and mutations. As a result, abnormal expression of MCM8 could be one of the fundamental causes that initiate the carcinogenic process.
The underlying cause of MCM8 overexpression in human cancers is probably the gain of MCM8 gene copy in the cancer genome. For most cases, the increase of copy number of MCM8 appears moderate, estimated 3–4 copies per cancer genome. However, in some samples of breast and colon cancers, the copy number reached 16 copies per genome, clearly indicating amplification in the region of MCM8. The expression of MCM8 is inhibited during S, G2 and early M phase to prevent additional DNA synthesis(56). Terminally differentiated cells do not express replication complex proteins. Amplification or gain of MCM8 may weaken the control of its expression. The continuing presence of MCM8 throughout the cell cycle may induce improper DNA synthesis that leads to genome instability of cancer cells, in addition to increased recruitment of larger proportion of cell population into the proliferation cycle. The association of MCM8 gain with some of the aggressive features of human cancers may indicate the significance of MCM8 in driving the behavior of human cancers. Overexpression of MCM8 gene in prostate immortalized epithelial cells inducing marked transformation underlies the important role of this gene in cancer development. The significance of the discovery of MCM8 gain and over-expression in human malignancies not only lies at a new insight gained towards understanding the carcinogenic process of human cancers but also at identifying a new base to develop potential therapeutic intervention to treat cancers that over-express MCM8 protein.
Materials and Methods
Materials and cell lines
All cell lines, including PC3, Du145, RWPE1, and LNCaP were purchased from American Type Cell Culture (Manassas, VA). PC3 cells were cultured with F12K medium supplemented with 10% fetal bovine serum (InVitrogen, Carlsbad, CA). DU145 cells were cultured with modified Eagle medium supplemented with 10% fetal bovine serum (InVitrogen). LNCaP cells were cultured with RPMI 1640 supplemented with 10% fetal bovine serum. RWPE1 cells were cultured with K-SFM supplemented with recombinant human Epidermal Growth Factor (rhEGF) and Bovine Pituitary Extract (BPE). The genomes of these cell lines were tested for a short tandem repeat (STR) DNA profile on eight different loci (CSF1PO, D13S317, D16S539, D5S818, D7S820, THO1, TPOX, and vWA) of the genomes by PCR using the primer sets for CSF1PO, D13S317, D16S539, D5S818, D7S820, TH01, TPOX, vWA recommended by ATCC on April 22, 2016 (latest). These cell lines were authenticated because the STR profiles of the cell lines perfectly matched those published by ATCC. All antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), including: Antibodies for MCM6 (sc-55577), MCM7 (sc-9966), MCM8 (sc-47117), Cdt1 (sc-28262), CCND1 (sc-20044), p27 (sc-1641), p21 (sc-6246), CDK4 (sc-136241), Rb (sc-102), pRb (sc-271930) and β-actin (sc-47778).
Sample preparation
Fresh prostate cancer tissues were prepared as described previously(11, 12). Tumor samples were microdissected to 70% purity for qPCR analysis. Formalin-fixed and paraffin-embedded (FFPE) tissues were used in FISH assays. One hundred thirty-three samples including 93 prostate cancers, three benign prostate tissues adjacent to cancer and 19 normal prostate samples from healthy organ donors were examined. The construction of microarrays of prostate tissues was previously described(25). The procedure of tissue collection, de-identification, and experimental protocols were approved by institutional review board of University of Pittsburgh.
Fluorescence In-situ Hybridization and SYBR-green real-time quantitation PCR
The similar procedure was previously described[12–14]. The FISH probe was prepared by combining 7 ml of SpectrumOrange-labeled Bacterial artificial chromosome (BAC) sequence containing MCM8 (RP11-1203216, InVitrogen, Inc, Grand Island, NY) /50% formamide with 1 ml of direct-labeled CEP20 SpectrumGreen (Vysis, Downers Grove, IL). The cutoff for a gain of MCM8 is an average of at least 2.5 copies per genome (for detailed procedure, please see Ren et al. (25)). For quantitative RT-PCR, QuantiTect SYBR Green RT-PCR Kit (Qiagen, CA) was used, and the reaction was carried out in an Eppendorf Realplex Mastercycler machine (Thermo-Fisher, Inc Waltham, MA). The procedure was similar to those previously described(25). The assays were performed with 100 ng genomic DNA for all of the experimental and control samples. MCM8 amplification was performed with primers CTATGGCAGTGCTACATTGGG/CCTGTTGCTCATTCCAGAACC for 45 cycles of the following program: 94°C for 10 s, 62°C for 5 s, and 72°C for 10 s. A separate quantification of β-actin DNA copy was also performed in parallel with MCM8 analysis using the following primers TCTTTGCACTTTCTGCATGTCCCC/GTCCATCACGATGCCAGTGGTAC.
Plasmid construction
For construction of pBD-MCM8full fusion protein, a mutagenic primer set (5′- TGTGGCCTATAATCATATGAATGGAGAGTATAGAGGC -3′ and 5′- TTCATTTCAAGCAAAGTCGACTCTGCAATAAACCCAGGAGGC- 3′) was used. PCR was performed on the cDNA template from the donor prostate (Clontech) using the following conditions: 94 °C for 1 min, followed by 35 cycles of 94 °C for 30 s, 68 °C for 3 min, and a final 3 min extension step at 68 °C. The PCR product was restricted with Nde1 and Sal1 (New England Biolabs, MA), gel purified, and ligated into a similarly digested pGBKT7 vector. The fusion protein contained 840 amino acids from the MCM8.
For the construction of pGST-MCM8N, a GST fusion protein, a mutagenic primer set (5′- AGGAATTCCCGGGTCGACAATGGAGAGTATAGAGGC -3′ and 5′- AGTCACGATGCG GCCGCCAAAGGCTCATAGTTGTACAC -3′) was used. PCR was performed using these primers under the following conditions: 94 °C for 1 min, followed by 35 cycles of 94 °C for 30 s, 68 °C for 3 min, and a final 10 min extension step at 68 °C. The PCR product was then ligated into a pCR2.1 TA cloning vector (Invitrogen). A similar strategy was used for constructing pGST-MCM8M1, pGST-MCM8M2 and pGST-MCM8C using the following pair of primers: AGGAATTCCCGGGTCGACACACAGCTCAAGAATGTCAGA/AGTCACGATGCGGCCGCCCGAGTTGACAATGAGTTTAAAC for pGST-MCM8M1, AGGAATTCCCGGGTCGACtCGCTTTGCCCTGTCATTTTTG/AGTCACGATGCGGCCGCCTCTCTGCTTTCCAGCTCTTATTG for pGST-MCM8M2, and AGGAATTCCCGGGTCGACGGAAAGCAGAGAACCATTAGC/AGTCACGATGCGGCCGCCTCTGCAATAAACCCAGGAGGC for pGST-MCM8C. The inserts of these plasmids were retrieved with Sal1 and Not1 and were ligated into a similarly restricted pGEX-5x-3 vector. A series of deletions, including 5′ or 3′ deletions, of pGST-MCM8M1 were then constructed using the primer sets listed in Table 3. The procedures for constructing these mutants are similar to that described for pGST-MCM8N. E. coli BL21 cells were transformed with these mutant constructs for induction of recombinant protein expression.
Table 3.
Primer sequences for pGST-MCM8 mutants
| Plasmid name | Sequence |
|---|---|
| pGST-MCM81n1 | aggaattcccgggtcgacataaagcctctttgcaccaag/agtcacgatgcggccgcccgagttgacaatgagtttaaac |
| pGST-MCM81n2 | aggaattcccgggtcgaccccacaaagtgtcctgtgcct/agtcacgatgcggccgcccgagttgacaatgagtttaaac |
| pGST-MCM81n3 | aggaattcccgggtcgacatcaaaatccaggaattgatg/agtcacgatgcggccgcccgagttgacaatgagtttaaac |
| pGST-MCM81n4 | aggaattcccgggtcgacgatagctgtgtcccgggagac/agtcacgatgcggccgcccgagttgacaatgagtttaaac |
| pGST-MCM81n5 | aggaattcccgggtcgactgtatgttccttttgtatatt/agtcacgatgcggccgcccgagttgacaatgagtttaaac |
| pGST-MCM81n6 | aggaattcccgggtcgacatgttgatggagttctcactt/agtcacgatgcggccgcccgagttgacaatgagtttaaac |
| pGST-MCM81c1 | aggaattcccgggtcgacacacagctcaagaatgtcaga/agtcacgatgcggccgctccatgcttacacccatcctc |
| pGST-MCM81c2 | aggaattcccgggtcgacacacagctcaagaatgtcaga/agtcacgatgcggccgccttgtcattcttatttcgagaac |
| pGST-MCM81c3 | aggaattcccgggtcgacacacagctcaagaatgtcaga/agtcacgatgcggccgccacaagatcatgaacaagctc |
| pGST-MCM81c4 | aggaattcccgggtcgacacacagctcaagaatgtcaga/agtcacgatgcggccgctgactgccagtccatcgtaac |
| pGST-MCM81c5 | aggaattcccgggtcgacacacagctcaagaatgtcaga/agtcacgatgcggccgcaagactgtattttccatctgg |
| pGST-MCM81c6 | aggaattcccgggtcgacacacagctcaagaatgtcaga/agtcacgatgcggccgcattactgacacgaaccactgtc |
Construction of MCM8 expressing cell lines
MCM8 cDNA was obtained by extended long PCR (Roche Applied Science, NJ) using primers (AGAAGCTTCCAGTTGTGAACTAGGAGAGC/AGCTCGAGCATAGTTTGAAGCTGGTAAACTTTTGGGC) on cDNA templates from donor prostate tissue. The PCR product was digested with HindIII and XhoI and ligated into a similarly digested pCDNA4-FLAG vector. This construct and pCDNA6 were transfected into RWPE1 cells, the immortalized prostate epithelial cell line with low MCM8 expression, using superfect™ kit (InVitrogen, CA). The transformed cells were cultured with 100 μg/ml zeocin and 5 μg/ml blasticidin. Colonies were selected for MCM8-FLAG expression in the presence of (5 μg/ml) tetracycline.
To construct the vector that expresses shRNA specific for MCM8, the following oligonucleotides were used: 5′-tcgagctctcaattccaaccttgctcgtgccgaagcacgagcaaggttggaattgagaga-3′/5′-agcttctctcaattccaaccttgctcgtgcttcggcacgagcaaggttggaattgagagc-3′. These primers were annealed and ligated into a pSingle-tts-shRNA vector. The constructs were then transfected into PC3, LNCaP, and DU145 cells to generate cells that are doxycycline-inducible for the suppression of MCM8 expression.
Immunoblot detection of MCM8, chromatin association and co-immunoprecipitation
The procedure of MCM8 immunoblotting is similar to previously described(25). The primary mouse anti-MCM8 antibody was obtained from Santa Cruz Biotechnology, Santa Cruz, CA. The MCM8 expression was detected with the ECL system (Amersham Life Science) according to the manufacturer’s protocols. Co-immunoprecipitation and chromatin association analyses were performed similarly to those previously described(57).
Cell Cycle Analysis
The similar procedure was described previously(58–61). Briefly, cells were cultured to 40%-50% confluence. These cells were synchronized with medium containing no serum for 24 h to G0 phase. The cells were then stimulated with 10% fetal bovine serum for 4 h and labeled with 5-Bromo-2-DeoxyUridine (10 μM). The cells were then harvested and stained with propidium iodide /tritonX-100. BrdU labeling and nuclei were quantified using a FACSCalibur Automated Benchtop Flow Cytometer (Becton Dickinson, MA), and analyzed by WinMDI 2.9 program.
Immunohistochemistry and tissue array analysis
The clinical features of these cases were tabulated in Table 4. The procedure was described previously (62, 63). The expression of MCM8 was graded as 0 (0%), 0.5 (focal positive), 1 (<20%), 2 (20–50%), 3 (50–80%) or 4 (>80%) based on % cells positive. The scores were the averages of 2 independent observers.
Table 4.
Clinical and Pathological characteristics of Prostate Cancer Samples
| Pathological stage | Number | Gleason scores | Recurrent | *PC free* | Undetermined outcome+ |
|---|---|---|---|---|---|
| T2a | 5 | 5–7 | 3/7 | 2/7 | 2/7 |
| T2b | 115 | 6–10 | 45/115 | 69/115 | 1/115 |
| T2c | 2 | 5,7 | 1/2 | 1/2 | 0/2 |
| T3a | 23 | 6–8 | 11/23 | 12/23 | 0/23 |
| T3b | 14 | 7–9 | 7/14 | 7/14 | 0/14 |
| T3c | 1 | 7 | 0/1 | 1/1 | 0/1 |
| T4 | 3 | 8 | 0/3 | 3/3 | 0/3 |
- clinical follow-up for 5 years;
- No adequate clinical follow-up.
Tumor growth and spontaneous metastasis
Power analysis were performed based on the assumption that 75% mice xenografted with M8P#5 and M8D#1 without doxycycline treatment may die in 6 weeks, while mice treated with doxycycline may have a mortality of 27% in the same period. Thus, a total of 32 mice (3 weeks old) were used. All male mice severe combined immune-deficient (SCID) mice (Taconic, Germantown, NY) were chosen due to the relevance of prostate cancer. No randomization was performed due to inbred nature of the SCID mice. These mice were subcutaneously implanted at the abdominal flank with ~1 × 107 viable cells. The detailed procedure of xenograft tumor monitoring was described previously(25). All procedures were approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
TCGA data analysis
Data were downloaded from The Cancer Genome Atlas (TCGA) (https://tcga-data.nci.nih.gov/tcga/dataAccessMatrix.htm). Hg19 CNV (SNP array, BI Genome-wide SNP 6) level 3 tumor sample data were collected for multiple cancer types: BLCA (bladder urothelial carcinoma), BRCA (breast invasive carcinoma), COAD (colon adenocarcinoma), ESCA (esophageal carcinoma), GBM (glioblastoma multiforme), LIHC (liver hepatocellular carcinoma), LUAD (lung adenocarcinoma), LUSC (lung squamous cell carcinoma), PAAD (pancreatic adenocarcinoma), PRAD (prostate adenocarcinoma), LAML (acute myeloid leukemia), OV (ovarian serous cystadenocarcinoma), THCA (thyroid carcinoma), UCEC (uterine corpus endometrial carcinoma), SARC (sarcoma), READ (rectum adenocarcinoma), DLBC (lymphoid neoplasm diffuse large B-cell lymphoma), HNSC (head and neck squamous cell carcinoma). CNV segments overlapped with the MCM8 gene were defined to be the MCM8 related CNVs. Fisher’s exact test was applied to test whether amplification (with segment mean equal to or greater than 0.2) or non-amplification (with segment mean smaller than 0.2) status of MCM8 related CNVs was associated with the clinical information. Clinical information chosen included: ajcc_pathologic_tumor_stage (Stage I, II VS III, IV), ajcc_metastasis_pathologic_pm (M0 VS M1), lymph_nodes_examined_he_count (0 VS others), tumor_grade (G1, G2 VS others), residual_tumor (R0 VS others), pathologic_N (N0 VS others), pathologic_T (T2 VS higher), clinical_stage (Stage I, II VS others), gleason_score (for PRAD, <=7 VS higher), aggressive (new_tumor_event_dx_indicator is “Yes”, lymph_nodes_examined_he_count is non-zero, lymph_nodes_examined is not 1 VS new_tumor_event_dx_indicator is “No”, lymph_nodes_examined_he_count is non-zero, lymph_nodes_examined is not 0 or 1). For lymph_nodes_examined, Wilcoxon rank sum test was applied. These association p-values were adjusted by the Benjamini-Hochberg method for multiple comparisons.
Survival analysis
The samples were divided into A (red), and B (blue) group and Kaplan-Meier survival curves were plotted. Groups were segregated based on MCM8 gain or non-gain status (figure 1) or MCM8 expression score≥2 or <2 status (figure 2) or treated with or without doxycycline (figure 3). The p-value by the log-rank test was calculated to indicate the significant level of the difference between these two groups.
Yeast Two-Hybrid library screening
The Yeast competent cell preparation and the procedure of Yeast two-hybrid cDNA library screening were described previously(64). Fresh AH109 competent cells (100 μl) were co-transfected with pBD-MCM8full (0.5 μg) and 0.5 μg DNA from prostate Yeast Two-Hybrid cDNA library constructed in pACT2. The transformants were plated on the high stringency selection plate (SD-Ade/-His/-Leu/-Trp). The grown colonies were then assayed for β-galactosidase as described previously (64). Thirty-one transformants were found positive for β-galactosidase and grown in most stringent selection condition. Through restriction enzyme cutting pattern analysis, 11 transformants were found unique. One of these transformants was identified as a clone containing cyclin D1 cDNA in frame with the activation domain of GAL4.
In vitro kinase assay
GST-CDK4 was purchased from Abcam, Inc (Cambridge, MA), while GST-CDK4/cyclin D1 was from Signal Chem, Inc (Richmond, Canada). For GST protein, E. coli. harboring GST or GST-CDK4 were grown overnight in room temperature. The recombinant proteins were induced with 1 mM IPTG for 4 hours. GST was purified by glutathione column and diluted to 1 ng/ul with 1X kinase assay buffer provided by the manufacturer (Cell Signaling, Inc, Danvers, MA). This was followed by combining 25 μl GST or GST-CDK4 with 1 ng substrate (GST-Rb) with or without 25 ng of cyclin D1, or HisTAG-MCM8, or HisTAG- ΔMCM8del261-290 or 2 ng of HisTAG-MCM8CBM in 1xkinase reaction buffer. The solutions were incubated at 37°C for 60 minutes. The reactions were terminated by adding 25 μl of 2N NaOH stop solution to each reaction well. The kinase activities were quantified using the kit and protocols of ADP-Glo™ Kinase Assay from Promega, Inc, Madison, WI.
For in vitro MCM8/cyclin D1/CDK4 binding assay, the following condition was used: 25 ng of each of HisTAG-MCM8, GST-cyclinD1 and GST-CDK4 produced from E.coli. were incubated with 150 mM NaCl, 25 mM MOPS, pH 7.2, 12.5 mM MgCl2, 12.5 mM β-glycerol phosphate at 37°C for 2 h. The products were then resolved in 6% non-denaturing polyacrylamide gel electrophoresis.
Supplementary Material
Acknowledgments
This work was supported by grants from National Cancer Institute (RO1 CA098249) to JHL and a grant from University of Pittsburgh Cancer Institute.
Footnotes
Conflict of interest: All authors in this study declare no conflict of interest.
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc).
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA: a cancer journal for clinicians. 2008;58(2):71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA: a cancer journal for clinicians. 2009;59(4):225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RG, Barzi A, et al. Colorectal cancer statistics, 2017. CA: a cancer journal for clinicians. 2017 doi: 10.3322/caac.21395. [DOI] [PubMed] [Google Scholar]
- 4.Network TCGAR. The Molecular Taxonomy of Primary Prostate Cancer. Cell. 2015;163(4):1011–25. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, Ellrott K, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nature genetics. 2013;45(10):1113–20. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nature genetics. 2013;45(10):1127–33. doi: 10.1038/ng.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, et al. Pan-cancer patterns of somatic copy number alteration. Nature genetics. 2013;45(10):1134–40. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu YP, Ding Y, Chen R, Liao SG, Ren BG, Michalopoulos A, et al. Whole-Genome Methylation Sequencing Reveals Distinct Impact of Differential Methylations on Gene Transcription in Prostate Cancer. The American journal of pathology. 2013 doi: 10.1016/j.ajpath.2013.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Luo JH, Ding Y, Chen R, Michalopoulos G, Nelson J, Tseng G, et al. Genome-wide methylation analysis of prostate tissues reveals global methylation patterns of prostate cancer. The American journal of pathology. 2013;182(6):2028–36. doi: 10.1016/j.ajpath.2013.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu YP, Song C, Tseng G, Ren BG, Laframboise W, Michalopoulos G, et al. Genome abnormalities precede prostate cancer and predict clinical relapse. The American journal of pathology. 2012;180(6):2240–8. doi: 10.1016/j.ajpath.2012.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu YP, Landsittel D, Jing L, Nelson J, Ren B, Liu L, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J Clin Oncol. 2004;22(14):2790–9. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 12.Luo JH, Yu YP, Cieply K, Lin F, Deflavia P, Dhir R, et al. Gene expression analysis of prostate cancers. Molecular carcinogenesis. 2002;33(1):25–35. doi: 10.1002/mc.10018. [DOI] [PubMed] [Google Scholar]
- 13.Luo JH. Gene expression alterations in human prostate cancer. Drugs Today (Barc) 2002;38(10):713–9. doi: 10.1358/dot.2002.38.10.704653. [DOI] [PubMed] [Google Scholar]
- 14.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, et al. Increased methylation variation in epigenetic domains across cancer types. Nature genetics. 2011;43(8):768–75. doi: 10.1038/ng.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feber A, Wilson GA, Zhang L, Presneau N, Idowu B, Down TA, et al. Comparative methylome analysis of benign and malignant peripheral nerve sheath tumors. Genome research. 2011;21(4):515–24. doi: 10.1101/gr.109678.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vallot C, Stransky N, Bernard-Pierrot I, Herault A, Zucman-Rossi J, Chapeaublanc E, et al. A novel epigenetic phenotype associated with the most aggressive pathway of bladder tumor progression. Journal of the National Cancer Institute. 2011;103(1):47–60. doi: 10.1093/jnci/djq470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu YP, Paranjpe S, Nelson J, Finkelstein S, Ren B, Kokkinakis D, et al. High throughput screening of methylation status of genes in prostate cancer using an oligonucleotide methylation array. Carcinogenesis. 2005;26(2):471–9. doi: 10.1093/carcin/bgh310. [DOI] [PubMed] [Google Scholar]
- 18.Edwards MC, Tutter AV, Cvetic C, Gilbert CH, Prokhorova TA, Walter JC. MCM2–7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J Biol Chem. 2002;277(36):33049–57. doi: 10.1074/jbc.M204438200. [DOI] [PubMed] [Google Scholar]
- 19.Blow JJ, Hodgson B. Replication licensing–defining the proliferative state? Trends in cell biology. 2002;12(2):72–8. doi: 10.1016/s0962-8924(01)02203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Labib K, Kearsey SE, Diffley JF. MCM2-7 proteins are essential components of prereplicative complexes that accumulate cooperatively in the nucleus during G1-phase and are required to establish, but not maintain, the S-phase checkpoint. Mol Biol Cell. 2001;12(11):3658–67. doi: 10.1091/mbc.12.11.3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu ZH, Xu H, Leno GH. DNA replication in quiescent cell nuclei: regulation by the nuclear envelope and chromatin structure. Mol Biol Cell. 1999;10(12):4091–106. doi: 10.1091/mbc.10.12.4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Honeycutt KA, Chen Z, Koster MI, Miers M, Nuchtern J, Hicks J, et al. Deregulated minichromosomal maintenance protein MCM7 contributes to oncogene driven tumorigenesis. Oncogene. 2006;25(29):4027–32. doi: 10.1038/sj.onc.1209435. [DOI] [PubMed] [Google Scholar]
- 23.Luo JH. Oncogenic activity of MCM7 transforming cluster. World journal of clinical oncology. 2011;2(2):120–4. doi: 10.5306/wjco.v2.i2.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laitinen S, Martikainen PM, Tolonen T, Isola J, Tammela TL, Visakorpi T. EZH2, Ki-67 and MCM7 are prognostic markers in prostatectomy treated patients. International journal of cancer. 2008;122(3):595–602. doi: 10.1002/ijc.23145. [DOI] [PubMed] [Google Scholar]
- 25.Ren B, Yu G, Tseng GC, Cieply K, Gavel T, Nelson J, et al. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene. 2006;25(7):1090–8. doi: 10.1038/sj.onc.1209134. [DOI] [PubMed] [Google Scholar]
- 26.Shohet JM, Hicks MJ, Plon SE, Burlingame SM, Stuart S, Chen SY, et al. Minichromosome maintenance protein MCM7 is a direct target of the MYCN transcription factor in neuroblastoma. Cancer research. 2002;62(4):1123–8. [PubMed] [Google Scholar]
- 27.Padmanabhan V, Callas P, Philips G, Trainer TD, Beatty BG. DNA replication regulation protein Mcm7 as a marker of proliferation in prostate cancer. Journal of clinical pathology. 2004;57(10):1057–62. doi: 10.1136/jcp.2004.016436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Facoetti A, Ranza E, Benericetti E, Ceroni M, Tedeschi F, Nano R. Minichromosome maintenance protein 7: a reliable tool for glioblastoma proliferation index. Anticancer research. 2006;26(2A):1071–5. [PubMed] [Google Scholar]
- 29.Feng CJ, Li HJ, Li JN, Lu YJ, Liao GQ. Expression of Mcm7 and Cdc6 in oral squamous cell carcinoma and precancerous lesions. Anticancer research. 2008;28(6A):3763–9. [PubMed] [Google Scholar]
- 30.Freeman A, Morris LS, Mills AD, Stoeber K, Laskey RA, Williams GH, et al. Minichromosome maintenance proteins as biological markers of dysplasia and malignancy. Clin Cancer Res. 1999;5(8):2121–32. [PubMed] [Google Scholar]
- 31.Iizuka N, Tsunedomi R, Tamesa T, Okada T, Sakamoto K, Hamaguchi T, et al. Involvement of c-myc-regulated genes in hepatocellular carcinoma related to genotype-C hepatitis B virus. J Cancer Res Clin Oncol. 2006;132(7):473–81. doi: 10.1007/s00432-006-0094-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Dekken H, Tilanus HW, Hop WC, Dinjens WN, Wink JC, Vissers KJ, et al. Array comparative genomic hybridization, expression array, and protein analysis of critical regions on chromosome arms 1q, 7q, and 8p in adenocarcinomas of the gastroesophageal junction. Cancer genetics and cytogenetics. 2009;189(1):37–42. doi: 10.1016/j.cancergencyto.2008.08.018. [DOI] [PubMed] [Google Scholar]
- 33.Nishihara K, Shomori K, Fujioka S, Tokuyasu N, Inaba A, Osaki M, et al. Minichromosome maintenance protein 7 in colorectal cancer: implication of prognostic significance. International journal of oncology. 2008;33(2):245–51. [PubMed] [Google Scholar]
- 34.Fujioka S, Shomori K, Nishihara K, Yamaga K, Nosaka K, Araki K, et al. Expression of minichromosome maintenance 7 (MCM7) in small lung adenocarcinomas (pT1): Prognostic implication. Lung cancer (Amsterdam, Netherlands) 2009;65(2):223–9. doi: 10.1016/j.lungcan.2008.11.007. [DOI] [PubMed] [Google Scholar]
- 35.Ota T, Clayton AC, Minot DM, Shridhar V, Hartmann LC, Gilks CB, et al. Minichromosome maintenance protein 7 as a potential prognostic factor for progression-free survival in high-grade serous carcinomas of the ovary. Mod Pathol. 2011;24(2):277–87. doi: 10.1038/modpathol.2010.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fristrup N, Birkenkamp-Demtroder K, Reinert T, Sanchez-Carbayo M, Segersten U, Malmstrom PU, et al. Multicenter validation of cyclin D1, MCM7, TRIM29, and UBE2C as prognostic protein markers in non-muscle-invasive bladder cancer. The American journal of pathology. 2013;182(2):339–49. doi: 10.1016/j.ajpath.2012.10.017. [DOI] [PubMed] [Google Scholar]
- 37.Zhou YM, Zhang XF, Cao L, Li B, Sui CJ, Li YM, et al. MCM7 expression predicts post-operative prognosis for hepatocellular carcinoma. Liver Int. 2012;32(10):1505–9. doi: 10.1111/j.1478-3231.2012.02846.x. [DOI] [PubMed] [Google Scholar]
- 38.Kang W, Tong JH, Chan AW, Cheng AS, Yu J, To K. MCM7 serves as a prognostic marker in diffuse-type gastric adenocarcinoma and siRNA-mediated knockdown suppresses its oncogenic function. Oncology reports. 31(5):2071–8. doi: 10.3892/or.2014.3094. [DOI] [PubMed] [Google Scholar]
- 39.Matsushime H, Ewen ME, Strom DK, Kato JY, Hanks SK, Roussel MF, et al. Identification and properties of an atypical catalytic subunit (p34PSK-J3/cdk4) for mammalian D type G1 cyclins. Cell. 1992;71(2):323–34. doi: 10.1016/0092-8674(92)90360-o. [DOI] [PubMed] [Google Scholar]
- 40.Kato J, Matsushime H, Hiebert SW, Ewen ME, Sherr CJ. Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes & development. 1993;7(3):331–42. doi: 10.1101/gad.7.3.331. [DOI] [PubMed] [Google Scholar]
- 41.Maiorano D, Cuvier O, Danis E, Mechali M. MCM8 is an MCM2–7-related protein that functions as a DNA helicase during replication elongation and not initiation. Cell. 2005;120(3):315–28. doi: 10.1016/j.cell.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 42.Volkening M, Hoffmann I. Involvement of human MCM8 in prereplication complex assembly by recruiting hcdc6 to chromatin. Molecular and cellular biology. 2005;25(4):1560–8. doi: 10.1128/MCB.25.4.1560-1568.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gozuacik D, Chami M, Lagorce D, Faivre J, Murakami Y, Poch O, et al. Identification and functional characterization of a new member of the human Mcm protein family: hMcm8. Nucleic acids research. 2003;31(2):570–9. doi: 10.1093/nar/gkg136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee KY, Im JS, Shibata E, Park J, Handa N, Kowalczykowski SC, et al. MCM8-9 complex promotes resection of double-strand break ends by MRE11-RAD50-NBS1 complex. Nature communications. 6:7744. doi: 10.1038/ncomms8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park J, Long DT, Lee KY, Abbas T, Shibata E, Negishi M, et al. The MCM8-MCM9 complex promotes RAD51 recruitment at DNA damage sites to facilitate homologous recombination. Molecular and cellular biology. 33(8):1632–44. doi: 10.1128/MCB.01503-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lutzmann M, Grey C, Traver S, Ganier O, Maya-Mendoza A, Ranisavljevic N, et al. MCM8- and MCM9-deficient mice reveal gametogenesis defects and genome instability due to impaired homologous recombination. Molecular cell. 47(4):523–34. doi: 10.1016/j.molcel.2012.05.048. [DOI] [PubMed] [Google Scholar]
- 47.AlAsiri S, Basit S, Wood-Trageser MA, Yatsenko SA, Jeffries EP, Surti U, et al. Exome sequencing reveals MCM8 mutation underlies ovarian failure and chromosomal instability. The Journal of clinical investigation. 125(1):258–62. doi: 10.1172/JCI78473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kato JY, Matsuoka M, Polyak K, Massague J, Sherr CJ. Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin-dependent kinase 4 activation. Cell. 1994;79(3):487–96. doi: 10.1016/0092-8674(94)90257-7. [DOI] [PubMed] [Google Scholar]
- 49.Toyoshima H, Hunter T. p27, a novel inhibitor of G1 cyclin-Cdk protein kinase activity, is related to p21. Cell. 1994;78(1):67–74. doi: 10.1016/0092-8674(94)90573-8. [DOI] [PubMed] [Google Scholar]
- 50.Xiong Y, Zhang H, Beach D. Subunit rearrangement of the cyclin-dependent kinases is associated with cellular transformation. Genes Dev. 1993;7(8):1572–83. doi: 10.1101/gad.7.8.1572. [DOI] [PubMed] [Google Scholar]
- 51.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. p21 is a universal inhibitor of cyclin kinases. Nature. 1993;366(6456):701–4. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 52.Stepanova L, Leng X, Parker SB, Harper JW. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 1996;10(12):1491–502. doi: 10.1101/gad.10.12.1491. [DOI] [PubMed] [Google Scholar]
- 53.Dai K, Kobayashi R, Beach D. Physical interaction of mammalian CDC37 with CDK4. J Biol Chem. 1996;271(36):22030–4. doi: 10.1074/jbc.271.36.22030. [DOI] [PubMed] [Google Scholar]
- 54.Xiong Y, Zhang H, Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992;71(3):505–14. doi: 10.1016/0092-8674(92)90518-h. [DOI] [PubMed] [Google Scholar]
- 55.Zhang JM, Wei Q, Zhao X, Paterson BM. Coupling of the cell cycle and myogenesis through the cyclin D1-dependent interaction of MyoD with cdk4. EMBO J. 1999;18(4):926–33. doi: 10.1093/emboj/18.4.926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kearsey SE, Maiorano D, Holmes EC, Todorov IT. The role of MCM proteins in the cell cycle control of genome duplication. Bioessays. 1996;18(3):183–90. doi: 10.1002/bies.950180305. [DOI] [PubMed] [Google Scholar]
- 57.Shi YK, Yu YP, Zhu ZH, Han YC, Ren B, Nelson JB, et al. MCM7 Interacts with Androgen Receptor. The American journal of pathology. 2008;173(6):1758–67. doi: 10.2353/ajpath.2008.080363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Han YC, Zheng ZL, Zuo ZH, Yu YP, Chen R, Tseng GC, et al. Metallothionein 1 h tumour suppressor activity in prostate cancer is mediated by euchromatin methyltransferase 1. The Journal of pathology. 2013;230(2):184–93. doi: 10.1002/path.4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang H, Luo K, Tan LZ, Ren BG, Gu LQ, Michalopoulos G, et al. p53-induced gene 3 mediates cell death induced by glutathione peroxidase 3. The Journal of biological chemistry. 2012;287(20):16890–902. doi: 10.1074/jbc.M111.322636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Han YC, Yu YP, Nelson J, Wu C, Wang H, Michalopoulos GK, et al. Interaction of integrin-linked kinase and miniature chromosome maintenance 7-mediating integrin {alpha}7 induced cell growth suppression. Cancer research. 2010;70(11):4375–84. doi: 10.1158/0008-5472.CAN-09-4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ren B, Yu YP, Tseng GC, Wu C, Chen K, Rao UN, et al. Analysis of integrin alpha7 mutations in prostate cancer, liver cancer, glioblastoma multiforme, and leiomyosarcoma. Journal of the National Cancer Institute. 2007;99(11):868–80. doi: 10.1093/jnci/djk199. [DOI] [PubMed] [Google Scholar]
- 62.Jing L, Liu L, Yu YP, Dhir R, Acquafondada M, Landsittel D, et al. Expression of myopodin induces suppression of tumor growth and metastasis. The American journal of pathology. 2004;164(5):1799–806. doi: 10.1016/S0002-9440(10)63738-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu YP, Tseng GC, Luo JH. Inactivation of myopodin expression associated with prostate cancer relapse. Urology. 2006;68(3):578–82. doi: 10.1016/j.urology.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 64.Yu YP, Luo JH. Myopodin-mediated suppression of prostate cancer cell migration involves interaction with zyxin. Cancer research. 2006;66(15):7414–9. doi: 10.1158/0008-5472.CAN-06-0227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.