

Abstract
Amplification and/or activation of the c-Myc protooncogene is one of the leading genetic events along hepatocarcinogenesis. The oncogenic potential of c-Myc has been proven experimentally by the finding that its overexpression in the mouse liver triggers tumor formation. However, the molecular mechanism whereby c-Myc exerts its oncogenic activity in the liver remains poorly understood. Here, we demonstrate that the mammalian target of rapamycin complex 1 (mTORC1) cascade is activated and necessary for c-Myc dependent hepatocarcinogenesis. Specifically, we found that ablation of Raptor, the unique member of the mTORC1 complex, strongly inhibits c-Myc liver tumor formation. Also, p70S6K/ribosomal protein S6 (RPS6) and eukaryotic translation initiation factor 4E-binding protein 1/eukaryotic translation initiation factor 4E (4EBP1/eIF4E) signaling cascades downstream of mTORC1 are required for c-Myc-driven tumorigenesis. Intriguingly, microarray expression analysis revealed the upregulation of multiple amino acid transporters, including SLC1A5 and SLC7A6, leading to robust uptake of amino acids, including glutamine, into c-Myc tumor cells. Subsequent functional studies showed that amino acids are critical for activation of mTORC1, as their inhibition suppressed mTORC1 in c-Myc tumor cells. In human HCC specimens, levels of c-Myc directly correlate with those of mTORC1 activation as well as of SLC1A5 and SLC7A6.
Conclusion
Our current study indicates that an intact mTORC1 axis is required for c-Myc-driven hepatocarcinogenesis. Thus, targeting mTOR pathway or amino acid transporters may be an effective and novel therapeutic option for the treatment of HCC with activated c-Myc signaling.
Keywords: Hepatocellular carcinoma, mTOR, Amino acid transporters, RPS6, 4EBP1
Liver cancer is the fifth most common cancer in men and the ninth in women, and the second common cause of cancer death worldwide.1 Both incidence and mortality rates of liver cancer are rising.2 Hepatocellular carcinoma (HCC) is the most common primary liver cancer. Liver resection, liver transplantation, and interventional radiology are still the main therapies for HCC, but they can solely be applied in the early stage of the disease.3 In addition, treatment with the multi-tyrosine kinase inhibitor Sorafenib, the only targeted therapy approved for advanced HCC, provides almost negligible benefits in terms of patients' prognosis.4
Recent advances in the understanding of the molecular pathogenesis of HCC have provided novel biomarkers for disease stratification and targets for treatment.5, 6 Among the putative candidates for targeted therapies in HCC is c-Myc, a well-characterized oncogene.7 c-Myc is a transcriptional factor that contributes to malignant transformation and tumor progression by regulating the expression of a plethora of genes involved in cell growth, metabolism, and carcinogenesis.8 In human HCC, c-Myc is frequently overexpressed and amplified, and its levels directly correlate with poor prognosis.9, 10 The oncogenic potential of c-Myc in the liver is underscored by the finding that its overexpression is sufficient to induce HCC formation in mice.11, 12 However, the molecular mechanisms underlying c-Myc driven hepatocarcinogenesis remain poorly delineated.
Activation of the mammalian target of rapamycin (mTOR) pathway is another critical molecular event in tumorigenesis.13 mTOR is comprised of two distinct complexes, namely complex 1 and complex 2 (mTORC1 and mTORC2).13 mTORC1, with Raptor as its unique and key protein component, is a major regulator of cell growth, metabolism, and survival.13 mTORC1 functions via regulating p70 ribosomal S6 Kinase (p70S6K)/ribosomal protein S6 (RPS6) and eukaryotic translation initiation factor 4E-binding protein 1/eukaryotic translation initiation factor 4E (4EBP1/eIF4E) downstream cascades.13 mTORC2, with Rictor as its unique and key protein component, functions instead via modulating the AGC family kinases, including AKT, PKC and SGKs.14 Similar to c-Myc, the mTOR cascade is often deregulated in human HCC and may represent a novel and promising target for HCC treatment.15 In agreement with this hypothesis, it has been found that over half of HCC cases display an activated mTOR signaling.16 Also, mTOR inhibitors have been shown to effectively inhibit HCC cell growth.17 Furthermore, we recently found that mTOR is essential for hepatocarcinogenesis driven by activated AKT and PIK3CA proto-oncogenes in the mouse.18-20
While both c-Myc and mTOR pathways have been independently implicated in hepatocarcinongenesis, the possible biochemical crosstalk between the two signaling cascades has not been characterized. In the present study, we show that the mTOR pathway is activated along hepatocarcinogenesis driven by c-Myc overexpression in mice. Importantly, ablation of mTORC1 completely inhibited c-Myc HCC development in mice, with both S6K/RSP6 and 4EBP1/eIF4E cascades being necessary for c-Myc induced hepatocarcinogenesis. In addition, we found that c-Myc induces mTORC1 activation through the upregulation of multiple amino acid transporters, including SLC1A5 and SLC7A6, in mouse and human HCC. Altogether, the present findings open the possibility for new therapeutic strategies against HCC, at least for the tumor subset characterized by the activation of the c-Myc oncogene.
Materials and methods
Plasmids and reagents
The plasmids used for the study, including pT3EF1α-c-Myc, pT3EF1α-4EBP1WT, pT3EF1α-4EBP1A4 and pCMV-SB have been described in our previous publications.11, 20 To delete Raptor while co-expressing c-Myc into the mouse liver, shRaptor (against mouse Raptor; Addgene #21399) or shLuc (against Renilla Luciferase) sequences were cloned at the c-terminal of c-Myc to generated pT3EF1α-c-Myc-shLuc and pT3EF1α-c-Myc-shRaptor constructs using standard molecular cloning approach. Rapamycin was purchased from LC Laboratories (Woburn, MA).
Hydrodynamic injection and mouse treatment
FVB/N mice were purchased from Jackson Laboratory (Bar Harbor, ME). AKT2 KO mice were kindly provided by Dr. Morris J. Birnbaum (University of Pennsylvania, USA) and were described in our previous publication.21 Sleeping beauty (SB)-mediated hydrodynamic injections were performed as described.22 In brief, mice were hydrodynamically injected with pT3EF1α-c-Myc (20μg) and pCMV-SB (0.8μg). To determine the requirement of mTORC1 in c-Myc driven liver tumor development, mice were injected with pT3EF1α-c-Myc-shLuc (20μg) or pT3EF1α-c-Myc-shRaptor (20μg) together with pCMV-SB (0.8μg). For blocking the mTORC1 pathway, Rapamycin, which efficiently blocks the p70S6/rpS6 cascade, was intraperitoneally (i.p.) injected in a dose of 6mg/kg/day for 6 days a week starting 3 weeks after hydrodynamic injection of the c-Myc construct. Additional cohorts of mice were injected i.p with vehicle (PEG400 and Tween 80) as the control group. To elucidate the functional role of the 4EBP1/eIF4E pathway, either 40μg of pT3EF1α-4EBP1A4 (the unphosphorylatable form of 4EBP1) or 40μg of pT3EF1α-4EBP1WT (wildtype 4EBP1) were co-injected with pT3EF1α-c-Myc (8ug) together with pCMV-SB (1.96μg). All mice were monitored closely and euthanized as described in the main text. Mice were fed and monitored according to protocols approved by the Committee for Animal Research at the University of California, San Francisco.
Human Tissue Samples
A collection of formalin-fixed, paraffin-embedded HCC (n=88) samples was used in the present study. Sixty-four frozen HCC and corresponding non-tumorous surrounding livers from the same collection were also used. The clinicopathological features of liver cancer patients are summarized in Supplementary Table 1. HCC specimens were collected at the Medical Universities of Greifswald (Greifswald, Germany), Rome La Sapienza (Pietro Valdoni Surgery Department, Rome, Italy), and Sassari (Sassari, Italy). Institutional Review Board approval was obtained at the local Ethical Committee of the Medical Universities of Greifswald, Rome, and Sassari. Informed consent was obtained from all subjects.
Statistical analysis
GraphPad Prism version 6.0 (GraphPad Software Inc., La Jolla, CA) was used to evaluate statistical significance by Tukey–Kramer, Student's t and Mann–Whitney tests and linear regression analyses. Overall survival was estimated according to Kaplan–Meier and Log-rank (Mantel–Cox) test. Univariate and multivariate Cox analysis was used to estimate hazard ratios for risk factors using STATA 9 statistical software (Stata Corporation, College Station, TX, USA). Values of P<0.05 were considered significant. Data are expressed as mean ± SEM for each group. Two-tailed unpaired t test was used to compare the differences between two groups.
A detailed description of Materials and Methods is supplied as Supporting Material.
Results
The mTORC1 pathway is activated and required for c-Myc dependent hepatocarcinogenesis
In accordance with previous data,12, 23 hydrodynamic gene delivery of the c-Myc oncogene into the mouse liver resulted in lethal burden of liver cancer by ∼6 weeks post injection (Fig. 1). To investigate the possible crosstalk between c-Myc and mTOR pathways in hepatocarcinogenesis, we evaluated the status of mTOR in c-Myc mouse tumor tissues. Of note, we found that downstream effectors of mTORC1 (p-RPS6 and p-4EBP1) as well as of mTORC2 (p-AKTS473) were upregulated in c-Myc tumor samples (Fig. 1B, C), thus implying that the mTOR cascade is activated along c-Myc hepatocarcinogenesis.
Figure 1. The mTOR pathway is activated in HCC from c-Myc mice.
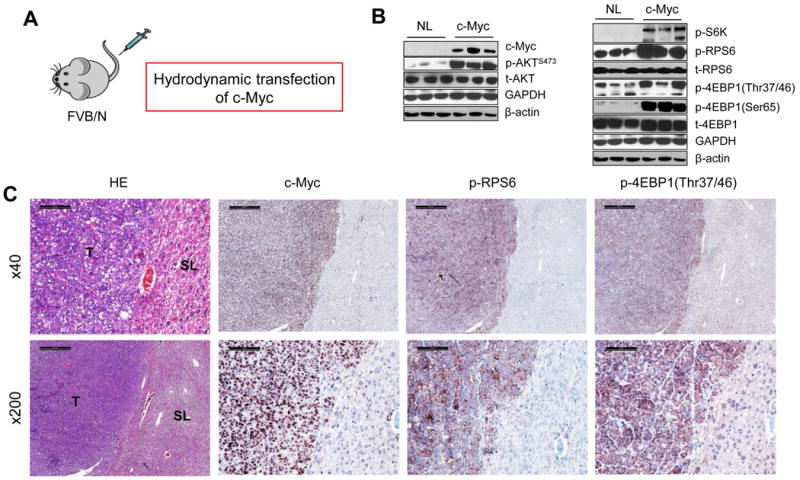
(A) Schematic representation of hydrodynamic gene delivery of the c-Myc protooncogene in the mouse. (B) Representative Western blotting from normal (NL) and c-Myc liver tissues. (C) Representative immunohistochemistry showing strong upregulation of c-Myc, phosphorylated/activated (p-)RPS6 and phosphorylated/inactivated (p-)4EBP1 in tumor lesions (T) when compared with surrounding non-neoplastic livers (SL) from c-Myc injected liver tissue. Upper level: scale bar=500μm; Lower level: scale bar=100μm.
Raptor is the unique and essential subunit of mTORC1.13 To investigate whether mTORC1 is required for c-Myc driven hepatocarcinogenesis, we applied miR-30 based shRNA to silence Raptor while co-expressing c-Myc in the mouse liver (Fig. 2A). In brief, the shRNA sequence against mouse Raptor or Renilla Luciferase18 was cloned downstream of c-Myc in a pT3-EF1α vector (c-Myc-shRaptor or c-Myc-shLuc); subsequently, the plasmids were hydrodynamically injected into mice. Studies from our and other labs have demonstrated that shRaptor sequence is effective in silencing Raptor expression24 in mouse cells, including in mouse liver cells.18 As expected, all c-Myc-shLuc injected mice developed lethal tumor burden and required to be euthanized within 6-7 weeks post-injection. In striking contrast, none of c-Myc-shRaptor injected mice developed HCC up to 12.3 weeks post injection (Fig. 2B, C). Histological evaluation confirmed the lack of neoplastic lesions in c-Myc-shRaptor livers (Fig. 2D). Altogether, the present results indicate that mTORC1 is activated and required for c-Myc induced liver tumor formation in mice.
Figure 2. mTORC1 is required for c-Myc driven HCC formation.
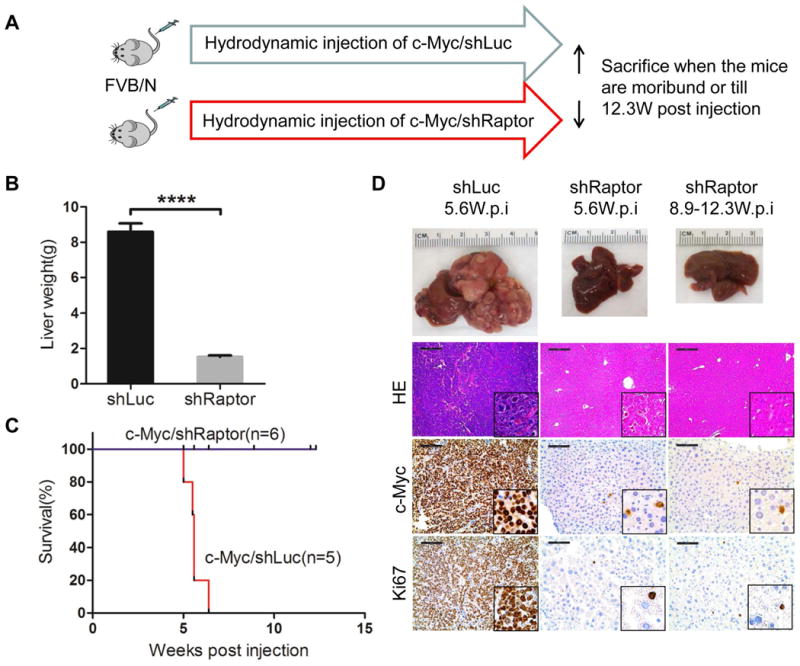
(A) Study design. (B) Liver weight of c-Myc/shLuc (shLuc) and c-Myc/shRaptor (shRaptor) mice. Data are presented as mean ± SEM. ****P<0.0001. (C) Survival curve. (D) Gross images of livers, Hematoxylin & eosin (HE), c-Myc and Ki67 staining in shLuc and shRaptor mouse liver at specific weeks post injection (W.p.i). Magnifications: 100× (HE), scale bar=200μm; 200× (c-Myc and Ki67), scale bar=100μm; 400× (insets).
Rapamycin treatment effectively inhibits c-Myc induced hepatocarcinogenesis in the early stage
mTORC1 functions via p70S6K/RPS6 and 4EBP1/eIF4E cascades.25 First, we assessed whether p70S6K/RPS6 is a key mTORC1 downstream effector in c-Myc tumor development. Our and other investigations have demonstrated that Rapamycin is a partial mTORC1 inhibitor, as it effectively inhibits the p70S6K/RPS6 axis without affecting 4EBP1/eIF4E signaling.20 Thus, we subjected c-Myc mice to Rapamycin treatment. Specifically, c-Myc was hydrodynamically injected into mice. Three weeks post injection, when the tumors start to emerge,11 c-Myc mice were treated with Rapamycin or vehicle for additional 3 weeks (Fig. 3A). While all vehicle treated mice became moribund due to high tumor burden, none of the Rapamycin treated mice developed a palpable liver mass. Upon dissection, only small tumor nodules were visible on the liver of Rapamycin treated mice (Fig. 3C), leading to a significantly lower liver weight when compared with vehicle treated mice (Fig. 3B). Histologically, the small lesions from Rapamycin treated mice were identical to those from untreated or vehicle treated mice (Fig. 3C). Immunostaining demonstrated the expression of c-Myc in the tumor lesions and tumor cells were proliferating, as revealed by Ki67 staining (Fig. 3D). At the molecular level, as previously reported,26 Rapamycin treatment induced the inhibition of p70S6K/pRPS6, with limited effects on p-4EBP1/eIF4E (Fig. 3E). We next determined whether Rapamycin is effective in inhibiting c-Myc HCC cell growth in vitro. We used HCC3-4 and HCC4-4 mouse liver tumor cell lines, which were derived from c-Myc transgenic mice.27 We found that Rapamycin effectively suppressed the growth of HCC3-4 and HCC4-4 with IC50 around ∼25μM (Sup. Fig. 1A). Consistently, Rapamycin treatment strongly inhibited p70S6K/pRPS6 in the two Myc HCC cell lines (Sup. Fig. 1B).
Figure 3. Rapamycin treatment effectively inhibits c-Myc induced hepatocarcinogenesis.
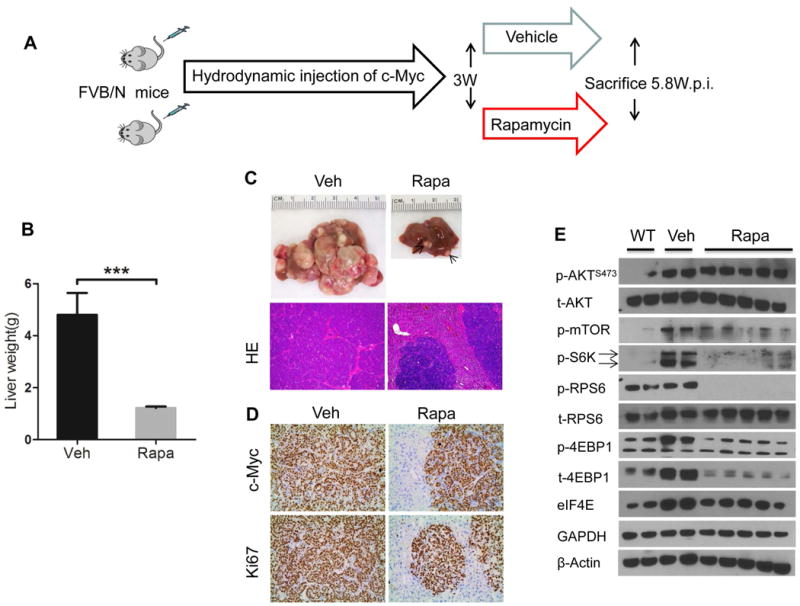
(A) Study design. (B) Liver weight of c-Myc injected mice treated with vehicle (Veh) and Rapamycin (Rapa) for 2.8 weeks. Data are presented as mean ± SEM. ***P<0.001. (C) Gross and HE images in Veh and Rapa-treated mouse livers. The arrow indicates a small tumor nodule in a Rapa-treated mouse liver. (D) c-Myc and Ki67 staining in Veh and Rapa mouse livers. Magnifications:100× (HE); 200× (c-Myc and Ki67). (E) Representative Western blotting from wild type (WT), Veh, and Rapa liver tissues. Arrows at the left side of the blots indicate the correct band for the indicated proteins.
In summary, Rapamycin effectively inhibits c-Myc induced hepatocarcinogenesis via inhibition of the p70S6K/pRPS6 axis.
Inhibition of 4EBP1/eIF4E delays but does not suppress c-Myc induced hepatocarcinogenesis
Next, we investigated whether the 4EBP1/eIF4E cascade is required for c-Myc induced hepatocarcinogenesis. Thus, we co-injected c-Myc with 4EBP1A4, the unphosphorylatable form of 4EBP120 (c-Myc/4EBP1A4), or 4EBP1 wild-type (c-Myc/4EBP1WT) into mice. It is important to note that 4EBP1WT can still be phosphorylated by the endogenous mTORC1 complex, preventing it from binding to eIF4E. Therefore, 4EBP1WT remains inactive although overexpressed. In contrast, the 4EBP1A4 mutant form, with T37, T46, S65 and T70 being replaced by Alanine, cannot be phosphorylated and, thus, continuously binds to eIF4E.28 4EBP1A4 functions to inhibit eIF4E mediated CAP dependent translation downstream of mTORC1.28 We found that, by 6.7 weeks post injection, all c-Myc/4EBP1WT mice developed lethal burden of liver tumors. In contrast, all c-Myc/4EBP1A4 mice appeared to be healthy with no palpable abdominal mass at this time point. Upon dissection, small few nodules could be found in the liver of c-Myc/4EBP1A4 mice. Large tumors were instead detected in c-Myc/4EBP1A4 mice by 10 to 14.2 weeks post injection (Fig. 4A-C).
Figure 4. Inhibition of 4EBP1/eIF4E delays c-Myc induced hepatocarcinogenesis.
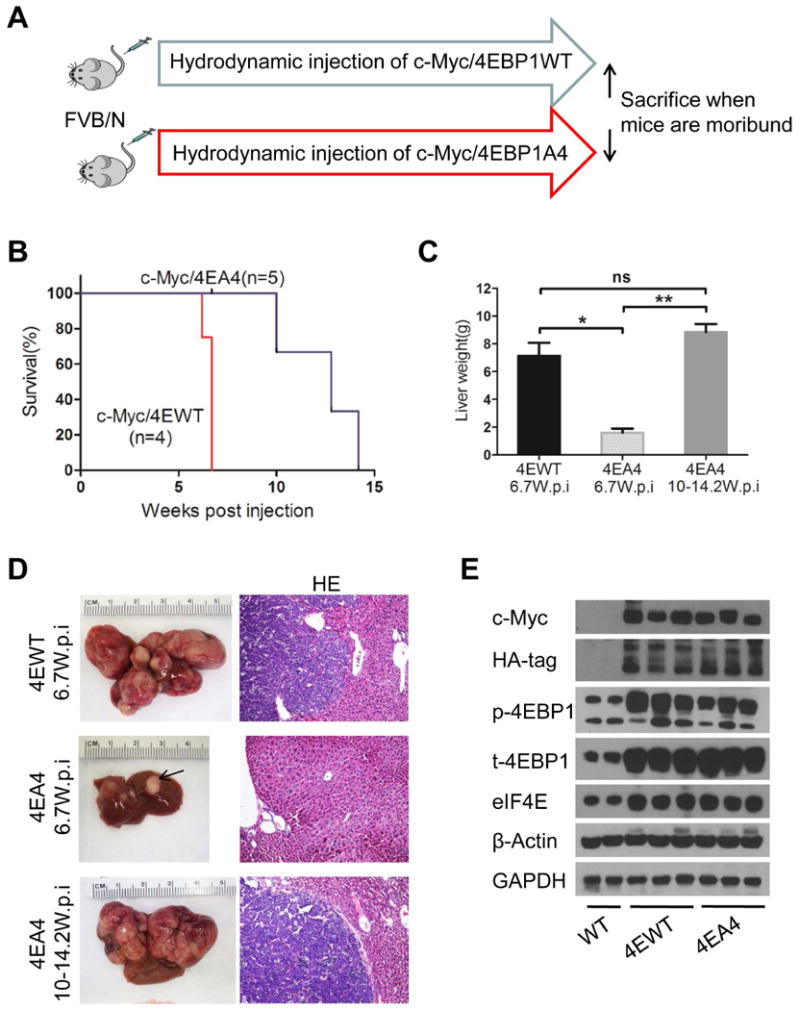
(A) Study design. (B) Survival curve. (C) Liver weight of c-Myc/4EBP1WT (4EWT) and c-Myc/4EBP1A4 (4EA4) mice. Data are presented as mean ± SEM.*P<0.05; **P<0.01; ns, not significant. (D) Gross and HE images of 4EWT and 4EA4 mouse livers. Magnifications: 100× (HE). (E) Representative Western blotting from WT, 4EWT and 4EA4 liver tissues.
Histological evaluation demonstrated that liver tumor nodules from c-Myc/4EBP1A4 mice were similar to those developed in c-Myc/4EBP1WT and c-Myc-only injected mice (Fig. 4D). At the molecular level, ectopically injected HA-tagged 4EBP1WT and 4EBP1A4 constructs could be readily detected within the tumor lesions (Fig. 4E).
Thus, our study indicates that 4EBP1/eIF4E is a key downstream effector of mTORC1 whose inhibition delays, but cannot completely block, c-Myc induced hepatocarcinogenesis.
As neither Rapamycin nor 4EBP1A4 was able to completely prevent c-Myc tumorigenesis in vivo, we inhibited the two cascades simultaneously by injecting the mice with c-Myc/4EBP1A4 plasmid and treating them with Rapamycin, starting 4 weeks post hydrodynamic injection (Fig. 5A). Noticeably, combined 4EBP1A4 and Rapamycin treatment completely inhibited c-Myc driven hepatocarcinogenesis (Fig. 5B-D), thus recapitulating the effects of Raptor knockdown in c-Myc overexpressing livers (Fig. 2).
Figure 5. Combined treatment with 4EBP1A4 transfection and Rapamycin administration supresses c-Myc induced hepatocarcinogenesis.
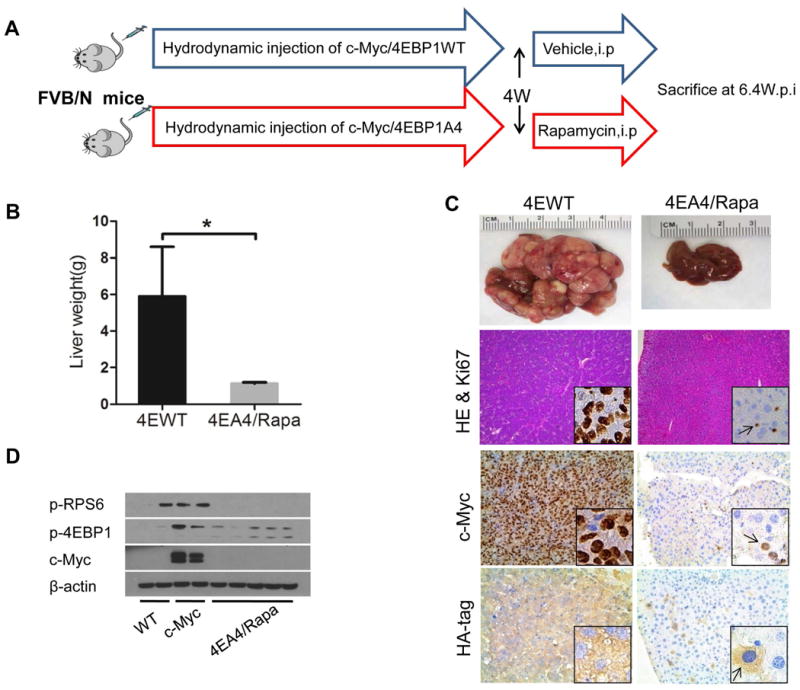
(A) Study design. (B) Liver weight of c-Myc/4EBP1WT and c-Myc/4EBP1A4 mice treated with vehicle (4EWT) or Rapamycin (4EA4/Rapa) for 2.4 weeks. Data are presented as mean ± SEM. *P<0.05. (C) Gross images of livers, HE and Ki67 (insets) staining, c-Myc and HA-tag staining in 4EWT and 4EA4/Rapa mouse liver. Arrow indicates positive staining cells. Magnifications:100× (HE); 200× (c-Myc and HA-tag); 400× (Ki67 and insets). (D) Representative western blotting in WT, c-Myc and 4EA4/Rapa liver tissues.
c-Myc driven hepatocarcinogenesis is independent of AKT2, the major AKT isoform in the liver
mTORC1 is well-characterized as being activated by the insulin/PI3K/AKT pathway in the liver. Thus, we investigated whether c-Myc induced hepatocarcinogenesis depends on AKT2, the major AKT isoform downstream of insulin signaling in the liver. In a previous study, we showed that loss of AKT2 completely abolishes the oncogenic potential of activated PIK3CA in mice.21 To determine the requirement of AKT2 for c-Myc dependent hepatocarcinogenesis, we injected c-Myc into AKT2-/- mice or control AKT2+/+ littermates (Sup. Fig. 2A). Surprisingly, we found that in both cohorts of mice, liver tumor developed at the same rate and latency. Indeed, by ∼6 weeks post injection, all c-Myc injected AKT2-/- or control AKT2+/+ mice developed lethal burden of liver tumors (Sup. Fig. 2B). Histologically, tumors developed in the AKT2-/- or AKT2+/+ genetic background were undistinguishable (Sup. Fig. 2C). At the molecular level, Western blotting demonstrated that AKT2 was successfully knocked out in AKT2-/- c-Myc liver tumor tissues (Sup. Fig. 2D). In addition, p-AKT(S473) levels were higher in AKT2+/+ c-Myc tumor tissues than in AKT2-/- c-Myc corresponding lesions. Furthermore, p-RPS6 and p-4EBP1 expression was similar in AKT2-/- and AKT2+/+mouse liver lesions.
Altogether, our study strongly suggests that c-Myc induced mTORC1 activation and tumorigenesis is independent of AKT2.
Amino acids are major factors promoting mTORC1 activation along c-Myc hepatocarcinogenesis
To further explore the mechanisms whereby mTORC1 becomes activated following c-Myc overexpression, we performed transcriptomic analysis of normal liver and c-Myc liver tumor tissues. Intriguingly, we found that multiple members of the amino acid transporter families (SLC1, 7, 36 and 38 families) were up-regulated in c-Myc liver tumors (Fig. 6A). Based on these preliminary findings, we next determined the expression of SLC family of transporters in HCC3-4 and HCC4-4 mouse liver tumor cell lines using qRT-PCR. Of note, we found that SLC1A5, SLC7A6, SLC7A1, SLC7A3, SLC7A5, SLC36A2 and SLC1A1 mRNAs were markedly higher in HCC3-4 and HCC4-4 liver tumor cells than in primary hepatocytes (*P<0.05,**P<0.01, ***P<0.001) (Fig. 6B and Sup. Fig. 3). Based on mRNA expression patterns, SLC1A5 and SLC7A6 appear to be the major SLC family of amino acid transporters in c-Myc liver tumor cells. Western blotting confirmed that protein levels of SLC1A5 and SLC7A6 were significantly up-regulated in HCC3-4 and HCC4-4 c-Myc liver tumor cell lines when compared with primary hepatocytes (Fig. 6C).
Figure 6. Amino acids transporters are the major factors responsible for amino acid uptake along c-Myc driven tumorigenesis.
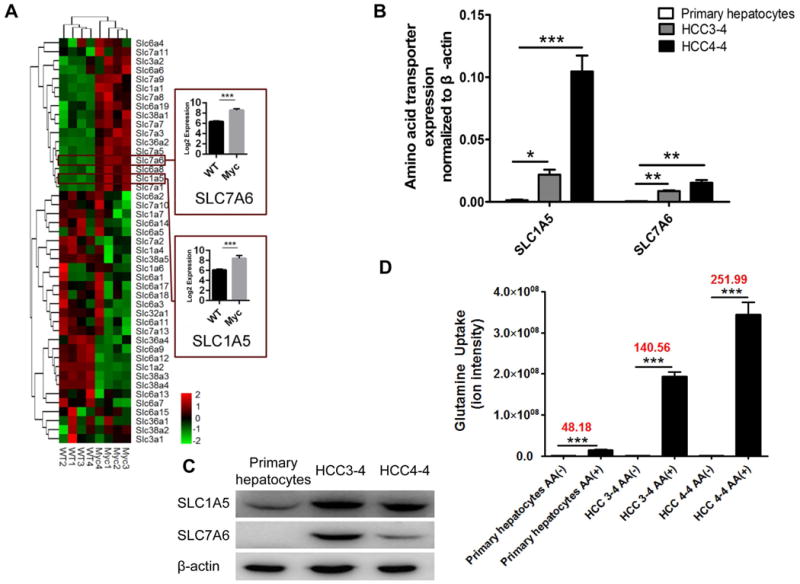
(A) Expression of SLC family members of transporters in normal liver and c-Myc liver tumors analyzed by microarray analysis. (B) Panel of amino acid transporter mRNAs was analyzed by RT-PCR in primary hepatocytes and HCC3-4/HCC4-4 c-Myc liver tumor cell lines. (C) Western blotting of SLC1A5 and SLC7A6 in primary hepatocytes and HCC3-4/HCC4-4 c-Myc liver tumor cells. (D) Glutamine uptake rates in primary hepatocytes and HCC3-4/HCC4-4 cells. (B)&(D) Data are presented as mean ± SEM. *P<0.05; **P<0.01; ***P<0.001.
Subsequently, we investigated whether upregulation of amino acid transporters leads to increased amino acid uptake into c-Myc liver tumor cells. For this purpose, we analyzed the uptake rate of 20 amino acids in primary mouse hepatocytes and c-Myc liver tumor cells. We found that among the 20 amino acids, glutamine showed the most drastic increase with high amounts taken up by primary hepatocytes, HCC3-4 and HCC4-4 compared with their controls without adding amino acids (Fig. 6D and Sup. Fig. 4). Also, increased uptake of isoleucine, tyrosine, methionine, threonine, and alanine was detected (Sup. Fig. 4)
Amino acids are known to be able to activate mTORC1.29, 30 Thus, we tested the hypothesis that amino acids are the major regulators of mTORC1 in c-Myc tumors. For this purpose, we evaluated the effects of amino acids on mTOR activity in c-Myc HCC cell lines. Noticeably, we found that deprivation of amino acids in the culture medium strongly inhibited mTOR activity in HCC4-3 and HCC4-4 cells, as determined by p-4EBP1, p-RPS6, and p-P70S6K expression (Fig. 7A). Adding amino acids back to the culture medium rescued the expression of p-4EBP1, p-RPS6, and p-P70S6K in c-Myc cells (Fig. 7A). Similar results were obtained in 6 human HCC cell lines, which display high levels of c-Myc (Sup. Fig. 5). Next, we determined whether SLC1A5 and SLC7A6 are the major amino acid transporters regulating mTORC1 signaling in c-Myc tumor cells. Strikingly, efficient knockdown of SLC1A5 and SLC7A6 expression (Fig. 7B) resulted in a decreased uptake of glutamine (Fig. 7D) and reduced mTORC1 activity (Fig. 7B) in c-Myc liver tumor cell lines. Subsequently, treatment with L-γ-glutamyl-p-nitroanilide (GPNA), a glutamine analogue and inhibitor of amino acid transporters, strongly inhibited glutamine uptake into c-Myc HCC cell lines. Once again, the inhibition of amino acid transporters was paralleled by a decline of mTORC1 activity in HCC3-4 and HCC4-4 cells (Fig. 7C).
Figure 7. Amino acids and their transporters are major determinants of mTORC1 activation in c-Myc liver tumor cells.
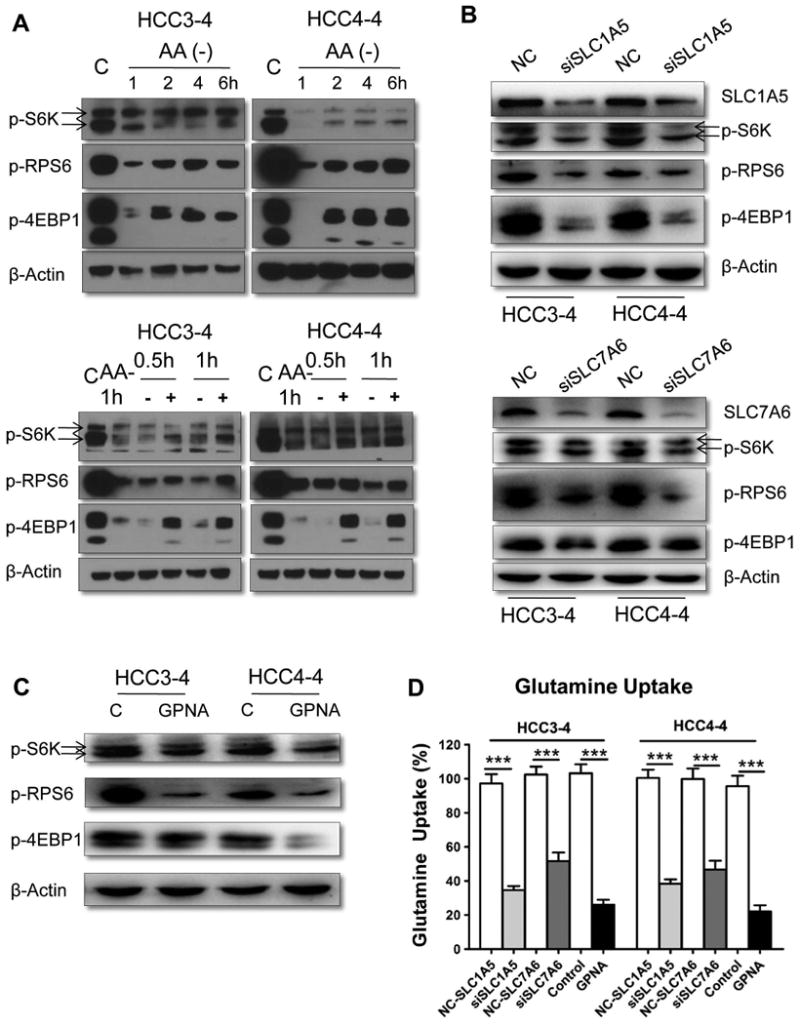
(A) Amino acid deprivation inhibited mTORC1 activation in HCC3-4 and HCC4-4 cells (upper-level). Amino acid addback partially restored mTORC1 activity (lower-level). (B) Silencing of SLC1A5 (upper-level) or SLC7A6 (lower-level) reduces mTORC1 signaling. (C) GPNA, a SLC1A5 inhibitor, strongly inhibits mTORC1 activation in HCC3-4 and HCC4-4 cells. (D) Glutamine uptake rates in HCC3-4 and HCC4-4 cells. Data are presented as mean ± SEM. ***P<0.001.
Based on this body of evidence, we investigated whether SLC1A5 and SLC7A6 are direct transcriptional targets of c-Myc in liver tumors. Towards this goal we analyzed the public ChIP-seq data (GSE76078) from the liver specific c-Myc transgenic mouse model.31 Importantly, ChIP-seq data show that Myc binds SLC1A5 and SLC7A6 in the promoter-region together with RNA polymerase II holoenzyme (RNAPII) as well as active histone markers H3K4me3 and H3K27ac in the c-Myc transgenic liver tumor samples (Sup. Fig. 6A). Next, we examined the effects of c-Myc silencing on the expression of SLC1A5 and SLC7A6. qRT-PCR analysis confirmed that the siRNA used against human c-Myc significantly reduced the expressions levels of c-Myc (Sup. Fig. 7A), as well as SLC1A5 and SLC7A6 in HCC3-4 and HCC4-4 cells (Sup. Fig. 7B). Knockdown of c-Myc also decreased the uptakes of glutamine in HCC3-4 and HCC4-4 cells compared with that from control siRNA transfected cells (Sup. Fig. 7C). Finally, to verify the public ChIP-seq results, we performed ChIP–qPCR to examine the occupancy of c-Myc to the promoter regions of SLC1A5 and SLC7A6 genes. Consistently, anti-c-Myc ChIP showed significant enrichments over the SLC1A5 and SLC7A6 promoter regions in both HCC3-4 and HCC4-4 cells when compared with those of cell immuneprecipitated with normal IgG antibody as the negative controls (Sup. Fig. 6B).
Altogether, the present data indicate that c-Myc directly upregulates the expression of SLC1A5 and SLC7A6 transporters, thus resulting in increased uptake of amino acids, which in turn leads to mTORC1 activation and eventual liver tumor formation (Sup. Fig. 8).
Levels of c-Myc correlate with those of mTORC1 activation and SLC1A5/SLC7A6 in human HCC patients
Finally, we evaluated the relationship of c-Myc with mTORC1 cascade as well as SLC1A5 and SLC7A6 genes in a collection of human HCC specimens. First, levels of c-Myc, p-RPS6, and p-4EBP1 were determined in a collection of human HCC specimens (n = 88) by immunohistochemistry (Fig. 8A). Higher immunoreactivity for c-Myc, p-RPS6, and p-4EBP1 proteins was detected in 56.8%, 54.5%, and 47.7% of HCC specimens, respectively, when compared with surrounding non-tumorous liver tissues. Importantly, 38 HCC specimens showing c-Myc upregulation also exhibited elevated levels of p-RPS6 and p-4EBP1. The Pearson correlation coefficient between c-Myc and p-RPS6 was 0.524 (p<0.0001), whereas between c-Myc and p-4EBP1 was 0.626 (p<0.00001) (Fig. 8B). In addition, all HCC samples exhibiting c-Myc amplification (21/88; 23.9%) displayed elevated immunoreactivity for p-RPS6 and p-4EBP1. Subsequently, we evaluated the mRNA levels of c-Myc in the HCC samples whose clinicopathological data were available (n=64; Supplementary Table 1). We found that c-Myc mRNA levels were significantly higher in HCC with poorer prognosis when compared with those with better prognosis. Similarly, mTORC1 activity was highest in HCC specimens with unfavorable outcome. Of note, a highly significant correlation between c-Myc mRNA levels and mTORC1 activity was detected in the HCC collection (P <0.0001; Fig. 8C). Subsequently, we have evaluated the relationship between c-Myc and SLC1A5/SLC7A6 mRNA levels. Noticeably, expression levels of SLC1A5 and SLC7A6 were weakly but statistically significantly correlated with those of c-Myc (P=0.0002 and P=0.0017, respectively; Sup. Fig. 9A). This weak but statistical significant correlation between c-Myc and SLC1A5/SLC7A6 mRNA expression was validated using TCGA data set (Sup. Fig. 9B). Furthermore, high expression of c-Myc, SLC1A5, and SLC7A6 correlated with lower HCC survival rate and this association remained significant after multivariate Cox regression analysis (p<0.01; Supplementary Table 2), suggesting that they are independent prognostic factors for HCC. Kaplan-Meier analysis confirmed this finding for SLC1A5 (p=0.004) and SLC7A6 (p<0.0001) (Sup. Fig. 10). No relationship between the mRNA levels of c-Myc, SLC1A5, and SLC7A6 and other clinicopathological features of the patients, including age, gender, etiology, presence of cirrhosis, tumor size, and tumor grade were detected (Supplementary Table 2).
Figure 8. mTORC1, SLC1A5 and SLC7A6 are associated with poor prognosis and c-Myc levels in human HCC.
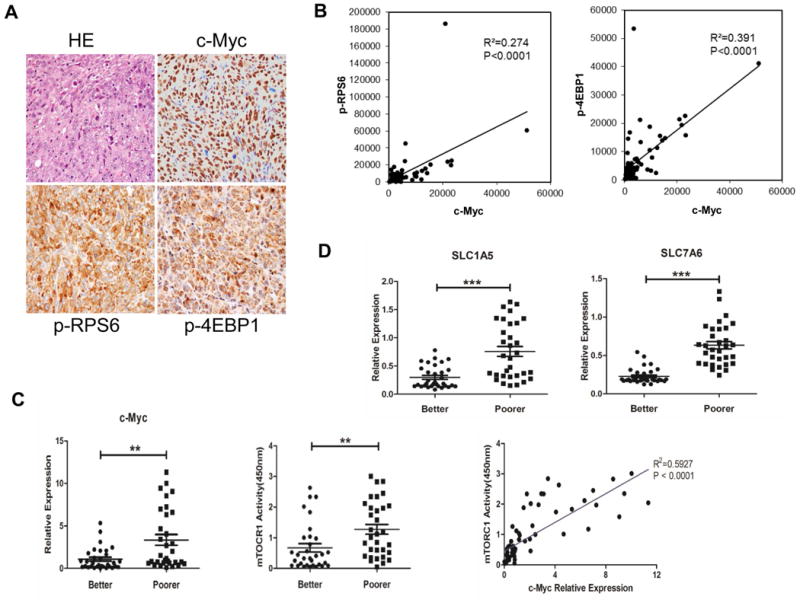
(A) Representative immunohistochemistry of a HCC displaying concomitant strong expression of c-Myc and mTORC1 activation (as indicated by p-RPS6 and p-4EBP1 staining). (B) The Pearson correlation coefficient between c-Myc and p-RPS6, p-4EBP1 are respectively 0.524 (p<0.0001, two-tailed, R2=0.274) and 0.626 (p<0.0001, two-tailed, R2=0.391). (C) Levels of c-Myc mRNA and mTORC1 activity were assessed in human HCC characterized by better and poorer prognosis by quantitative real-time RT-PCR and ELISA assay, respectively. ** P <0.01. mTORC1 activity directly correlates with c-Myc mRNA levels in the collection of human HCC samples. (D) Levels of SLC1A5 and SLC7A6 were assessed in human HCC characterized by better and poorer prognosis by quantitative real-time RT-PCR. *** P <0.001.
Altogether, the present data indicate that human HCC with high c-Myc levels are characterized by elevated activity of mTORC1 and upregulation of SLC1A5 and SLC7A6 gene expression.
Discussion
Both c-Myc and mTOR pathways have been implicated in tumorigenesis, and the biochemical crosstalk between the two pathways has been found to play a critical role during tumor growth. For instance, it has been shown that c-Myc directly upregulates the transcription of eIF4E protooncogene, whose activity is tightly regulated by phosphorylated 4EBP1 downstream of mTORC1.32 In addition, c-Myc has been identified as a direct target of mTOR,33 and 4EBP1/eIF4E modulates the translation levels of c-Myc.34 Also, using a c-Myc driven mouse model of B cell lymphoma, Pourdehnad et al showed that mTOR-dependent 4EBP1/eIF4E axis confers survival to c-Myc lymphoma cells.35 However, whether mTOR and c-Myc cascades functionally interact to promote hepatocarcinogenesis has not been elucidated to date.
In this study, we show that the mTOR pathway is induced in c-Myc liver tumor cells and that activated mTORC1 is required for c-Myc tumor development in vivo. In addition, using pharmacological and genetic approaches, we demonstrated that both p70S6K/RPS6 and 4EBP1/eIF4E cascades downstream of mTORC1 have key roles along c-Myc dependent tumorigenesis. Furthermore, a strong association between c-Myc levels and mTORC1 activity was detected in a collection of human HCC specimens, thus indicating that the functional crosstalk between c-Myc and mTORC1 is conserved across species. Our study therefore provides, for the first time, a critical link between the two oncogenic pathways in hepatocarcinogenesis. The signaling and metabolic pathways downstream of p70S6K/RPS6 and 4EBP1/eIF4E clearly require further investigator. Previous studies have shown that fatty acid synthase (FASN) and its mediated de novo lipogenesis is a major metabolic pathway downstream of p70S6K/RPS6 during tumor development.36, 37 In accordance with these findings, preliminary data from our group indicate that levels of FASN and acetyl-CoA carboxylase (ACC) lipogenic proteins are upregulated in c-Myc liver tumor samples (Che L, unpublished results). Further studies are ongoing to determine whether de-regulated fatty acid metabolism pathway is the major event downstream of the p70S6K/RPS6 cascade in c-Myc mice.
Further analysis showed that c-Myc upregulates the expression of multiple amino acid transporters, including SLC1A5 and SLC7A6. They belong to the solute carrier (SLC) group of membrane transport proteins, which include over 300 members.38 These transporters facilitate the transport of large numbers of substrates across the cell membrane and have critical roles in various physiological and pathological processes.38 Expression of amino acid transporters have been reported to be upregulated in human cancer cells and their increased facilitates the uptake of amino acids that are required for tumor cell growth.39 Among all the amino acid transporters, SLC1A5 is the most studied. For instance, recent studies showed that SLC1A5 controls glutamine uptake and proliferation in breast cancer cells,40 and targeting SLC1A5 efficiently decreases the growth of non-small cell lung cancer.41 The functional contribution of SLC7A6 during tumorigenesis is not well characterized. A study suggests that SCL7A6 is an androgen-responsive gene that is required for the growth of metastatic castration-resistant prostate cancer.42 Reports on the role of amino acid transporters in HCC and the mechanisms whereby they contribute to hepatocarcinogenesis are scanty. Here, we show that c-Myc directly upregulates SLC1A5 and SLC7A6, leading to the increased uptake of amino acids to the cells and activation of the mTOR cascade. Therefore, SLC1A5 and SLC7A6 are the major amino acid transporters and the determinant of mTOR activity along c-Myc driven liver tumorigenesis. In our study, we show that a weak but statistically significant correlation between c-Myc mRNA expression with SLC1A5 and SLC7A6 in human HCCs exists using both our sample set and the TCGA data set (Sup. Fig. 9). There are several possible reasons for the weak association. First, it is most likely that SLC1A5 and SLC7A6 expression is regulated by multiple factors during hepatocarcinogenesis, and c-Myc is only one of these factors. For instance, serine/threonine kinases SGK1 and SGK3 were found to induce SLC1A5 expression.43 Second, mRNA expression may not accurately reflect the eventual protein levels and, thus, the activity of the gene. Concerning c-Myc, it has been clearly demonstrated that the protein stability can be modulated via proteolysis as well as phosphorylation.44 It is clearly important to further study the correlation between c-Myc and SLC1A5/SLC7A6 at protein as well as posttranslational modification levels in human HCC samples.
The present study has several important translational implications. First, our data support the use of mTOR inhibitors for the treatment of HCC with high levels of c-Myc. Recent clinical studies showed that treatment with Everolimus, an mTORC1 inhibitor, did not improve the overall survival in patients with advanced HCC.45 However, a biomarker-based selection of patients with HCC who may truly benefit from mTOR inhibition was not conducted in the clinical trial. It is plausible to hypothesize that the use of c-Myc expression as a biomarker of mTOR activation may enrich HCC patients who may benefit from treatment with mTOR inhibitors. Another important issue that might explain the failure of the aforementioned clinical trial is the fact that Everolimus is a partial mTORC1 inhibitor, as it only inhibits the p70S6K/RPS6 cascade, but does not affect the 4EBP1/eIF4E axis. Based on the present and our previous study,20 the 4EBP1/eIF4E branch of mTORC1 also plays a critical role in hepatocarcinogenesis, and the lack of 4EBP1/eIF4E inhibition might strongly limit Everolimus efficacy in HCC patients. Therefore, the use of pan mTOR inhibitors, such as MLN0128 or AZD8055, should be tested in HCC patients.
In addition to mTOR, SLC membrane transporters have been identified as valid druggable targets,38, 46 as small molecules or antibodies may block the membrane transporters and, thus, inhibit the uptake of substrates. Many FDA approved drugs function primarily through the inhibition of SLC transporters, such as diuretic drugs that target SLC12 and neuropsychiatric drugs targeting SLC6.38 Many efforts have been made to develop drugs that can target amino acid transporters for cancer treatment.39 Among them, several drugs, including Benzylserine and GPNA, have been shown to efficiently inhibit SLC1A5.39 Obviously, additional studies are required to develop more efficient drugs targeting amino acid transporters as well as to determine whether these drugs can efficiently inhibit tumor growth in vivo. Nevertheless, our study strongly supports targeting amino acid transporters as a therapeutic strategy at least in the subset of HCC with elevated c-Myc expression. In this regard, the c-Myc mouse might represent an excellent preclinical model to assess the antineoplastic activity of these drugs for the treatment of HCC.
Supplementary Material
Acknowledgments
We would like to thank Dr. Morris J. Birnbaum from University of Pennsylvania for providing AKT2 KO mice and Dr. Dean W. Felsher of Stanford University for HCC3-4 and HCC4-4 mouse c-Myc liver tumor cell lines.
Financial Support: This work is supported by NIH grants R01CA136606 and R21CA198490 to XC; P30DK026743 for UCSF Liver Center; National Natural Science Foundation of China (No. 81470839), National 1000-Talent Program (No. 20141770980), and Tsinghua University Initiative Scientific Research Program (No. 20161080086) to LC; National Natural Science Foundation of China (No. 81602424) to JH; China Scholarship Council (CSC) (No. 201506270117) to Pin Liu; and the Deutsche Forschungsgemeinschaft DFG (grant number RI2695/1-1) to SR.
List of Abbreviations
- eIF4E
eukaryotic translation initiation factor 4E
- FASN
fatty acid synthase
- GPNA
L-γ-glutamyl-p-nitroanilide
- HCC
Hepatocellular carcinoma
- mTOR
mammalian target of rapamycin
- mTORC1
mammalian target of rapamycin complex 1
- mTORC2
mammalian target of rapamycin complex 2
- p70S6K
p70 ribosomal S6 Kinase
- RPS6
ribosomal protein S6
- 4EBP1
eukaryotic translation initiation factor 4E-binding protein 1
Footnotes
Conflict of interests: nothing to disclose.
References
- 1.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Ryerson AB, Eheman CR, Altekruse SF, Ward JW, Jemal A, Sherman RL, Henley SJ, et al. Annual Report to the Nation on the Status of Cancer, 1975-2012, featuring the increasing incidence of liver cancer. Cancer. 2016 doi: 10.1002/cncr.29936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pascual S, Herrera I, Irurzun J. New advances in hepatocellular carcinoma. World J Hepatol. 2016;8:421–438. doi: 10.4254/wjh.v8.i9.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 5.Llovet JM, Villanueva A, Lachenmayer A, Finn RS. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat Rev Clin Oncol. 2015;12:408–424. doi: 10.1038/nrclinonc.2015.103. [DOI] [PubMed] [Google Scholar]
- 6.Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology. 2015;149:1226–1239.e1224. doi: 10.1053/j.gastro.2015.05.061. [DOI] [PubMed] [Google Scholar]
- 7.Gabay M, Li Y, Felsher DW. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb Perspect Med. 2014;4 doi: 10.1101/cshperspect.a014241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dang CV. MYC on the path to cancer. Cell. 2012;149:22–35. doi: 10.1016/j.cell.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Abou-Elella A, Gramlich T, Fritsch C, Gansler T. c-myc amplification in hepatocellular carcinoma predicts unfavorable prognosis. Mod Pathol. 1996;9:95–98. [PubMed] [Google Scholar]
- 10.Peng SY, Lai PL, Hsu HC. Amplification of the c-myc gene in human hepatocellular carcinoma: biologic significance. J Formos Med Assoc. 1993;92:866–870. [PubMed] [Google Scholar]
- 11.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- 12.Chow EK, Fan LL, Chen X, Bishop JM. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. Hepatology. 2012;56:1331–1341. doi: 10.1002/hep.25776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaubitz C, Prouteau M, Kusmider B, Loewith R. TORC2 Structure and Function. Trends Biochem Sci. 2016;41:532–545. doi: 10.1016/j.tibs.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60:855–865. doi: 10.1016/j.jhep.2013.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, Tovar V, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972–1983. 1983.e1971–1911. doi: 10.1053/j.gastro.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kirstein MM, Boukouris AE, Pothiraju D, Buitrago-Molina LE, Marhenke S, Schutt J, Orlik J, et al. Activity of the mTOR inhibitor RAD001, the dual mTOR and PI3-kinase inhibitor BEZ235 and the PI3-kinase inhibitor BKM120 in hepatocellular carcinoma. Liver Int. 2013;33:780–793. doi: 10.1111/liv.12126. [DOI] [PubMed] [Google Scholar]
- 18.Hu J, Che L, Li L, Pilo MG, Cigliano A, Ribback S, Li X, et al. Co-activation of AKT and c-Met triggers rapid hepatocellular carcinoma development via the mTORC1/FASN pathway in mice. Sci Rep. 2016;6:20484. doi: 10.1038/srep20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang C, Che L, Hu J, Zhang S, Jiang L, Latte G, Demartis MI, et al. Activated mutant forms of PIK3CA cooperate with RasV12 or c-Met to induce liver tumor formation in mice via AKT2/mTORC1 cascade. Liver Int. 2015 doi: 10.1111/liv.13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Cigliano A, Jiang L, Li X, Fan B, Pilo MG, Liu Y, et al. 4EBP1/eIF4E and p70S6K/RPS6 axes play critical and distinct roles in hepatocarcinogenesis driven by AKT and N-Ras proto-oncogenes in mice. Hepatology. 2015;61:200–213. doi: 10.1002/hep.27396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang C, Che L, Hu J, Zhang S, Jiang L, Latte G, Demartis MI, et al. Activated mutant forms of PIK3CA cooperate with RasV12 or c-Met to induce liver tumour formation in mice via AKT2/mTORC1 cascade. Liver Int. 2016;36:1176–1186. doi: 10.1111/liv.13055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol. 2014;184:912–923. doi: 10.1016/j.ajpath.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Juric V, Ruffell B, Evason KJ, Hu J, Che L, Wang L, Chen X, et al. Monocytes promote liver carcinogenesis in an oncogene-specific manner. J Hepatol. 2016;64:881–890. doi: 10.1016/j.jhep.2015.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choo AY, Yoon SO, Kim SG, Roux PP, Blenis J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc Natl Acad Sci U S A. 2008;105:17414–17419. doi: 10.1073/pnas.0809136105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao Z, Fan-Minogue H, Bellovin DI, Yevtodiyenko A, Arzeno J, Yang Q, Gambhir SS, et al. MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase. Cancer Res. 2011;71:2286–2297. doi: 10.1158/0008-5472.CAN-10-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249–261. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14:133–139. doi: 10.1038/nrm3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24:400–406. doi: 10.1016/j.tcb.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kress TR, Pellanda P, Pellegrinet L, Bianchi V, Nicoli P, Doni M, Recordati C, et al. Identification of MYC-Dependent Transcriptional Programs in Oncogene-Addicted Liver Tumors. Cancer Res. 2016;76:3463–3472. doi: 10.1158/0008-5472.CAN-16-0316. [DOI] [PubMed] [Google Scholar]
- 32.Lin CJ, Cencic R, Mills JR, Robert F, Pelletier J. c-Myc and eIF4F are components of a feedforward loop that links transcription and translation. Cancer Res. 2008;68:5326–5334. doi: 10.1158/0008-5472.CAN-07-5876. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt EV, Ravitz MJ, Chen L, Lynch M. Growth controls connect: interactions between c-myc and the tuberous sclerosis complex-mTOR pathway. Cell Cycle. 2009;8:1344–1351. doi: 10.4161/cc.8.9.8215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu G, Lou Z, Gupta M. The long non-coding RNA GAS5 cooperates with the eukaryotic translation initiation factor 4E to regulate c-Myc translation. PLoS One. 2014;9:e107016. doi: 10.1371/journal.pone.0107016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci U S A. 2013;110:11988–11993. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Calvisi DF, Wang C, Ho C, Ladu S, Lee SA, Mattu S, Destefanis G, et al. Increased lipogenesis, induced by AKT-mTORC1-RPS6 signaling, promotes development of human hepatocellular carcinoma. Gastroenterology. 2011;140:1071–1083. doi: 10.1053/j.gastro.2010.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamming DW, Sabatini DM. A Central role for mTOR in lipid homeostasis. Cell Metab. 2013;18:465–469. doi: 10.1016/j.cmet.2013.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lin L, Yee SW, Kim RB, Giacomini KM. SLC transporters as therapeutic targets: emerging opportunities. Nat Rev Drug Discov. 2015;14:543–560. doi: 10.1038/nrd4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bhutia YD, Babu E, Ramachandran S, Ganapathy V. Amino Acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res. 2015;75:1782–1788. doi: 10.1158/0008-5472.CAN-14-3745. [DOI] [PubMed] [Google Scholar]
- 40.van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, Ritchie W, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene. 2016;35:3201–3208. doi: 10.1038/onc.2015.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hassanein M, Qian J, Hoeksema MD, Wang J, Jacobovitz M, Ji X, Harris FT, et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int J Cancer. 2015;137:1587–1597. doi: 10.1002/ijc.29535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang Q, Tiffen J, Bailey CG, Lehman ML, Ritchie W, Fazli L, Metierre C, et al. Targeting amino acid transport in metastatic castration-resistant prostate cancer: effects on cell cycle, cell growth, and tumor development. J Natl Cancer Inst. 2013;105:1463–1473. doi: 10.1093/jnci/djt241. [DOI] [PubMed] [Google Scholar]
- 43.Palmada M, Speil A, Jeyaraj S, Bohmer C, Lang F. The serine/threonine kinases SGK1, 3 and PKB stimulate the amino acid transporter ASCT2. Biochem Biophys Res Commun. 2005;331:272–277. doi: 10.1016/j.bbrc.2005.03.159. [DOI] [PubMed] [Google Scholar]
- 44.Thomas LR, Tansey WP. Proteolytic control of the oncoprotein transcription factor Myc. Adv Cancer Res. 2011;110:77–106. doi: 10.1016/B978-0-12-386469-7.00004-9. [DOI] [PubMed] [Google Scholar]
- 45.Zhu AX, Kudo M, Assenat E, Cattan S, Kang YK, Lim HY, Poon RT, et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. Jama. 2014;312:57–67. doi: 10.1001/jama.2014.7189. [DOI] [PubMed] [Google Scholar]
- 46.McCracken AN, Edinger AL. Nutrient transporters: the Achilles' heel of anabolism. Trends Endocrinol Metab. 2013;24:200–208. doi: 10.1016/j.tem.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.