

Abstract
Obesity and alcohol consumption synergistically promote steatohepatitis, and neutrophil infiltration is believed to be associated with steatosis. However, the underlying mechanisms remain obscure. Peroxisome proliferator-activated receptor-gamma (PPARγ) plays a complex role in lipid metabolism and inflammation, therefore, the purpose of this study was to dissect its role in regulating steatosis and neutrophil infiltration in a clinically relevant mouse steatohepatitis model of 3-month high-fat diet (HFD) feeding plus a binge of ethanol (HFD - plus-binge ethanol). Hepatocyte-specific Pparg disruption reduced liver steatosis, but surprisingly increased hepatic neutrophil infiltration after HFD-plus-binge ethanol. Knockout or knockdown of the PPARγ target gene, fat-specific protein 27 (Fsp27), reduced steatosis without affecting neutrophil infiltration in this model. Moreover, hepatocyte-specific deletion of the Pparg gene but not the Fsp27 gene markedly upregulated hepatic levels of Cxcl1 (a chemokine for neutrophil infiltration) in HFD-plus-binge ethanol-fed mice. In vitro, deletion of the Pparg gene also highly augmented palmitic acid or TNF-α induction of Cxcl1 in mouse hepatocytes. In contrast, activation of PPARγ with a PPARγ agonist attenuated Cxcl1 expression in hepatocytes. Palmitic acid also upregulated IL-8 (a key chemokine for human neutrophil recruitment) expression in human hepatocytes, which was attenuated and enhanced by co-treatment with a PPARγ agonist and antagonist, respectively. Finally, acute ethanol binge markedly attenuated HFD-induced hepatic PPARγ activation, which contributed to the upregulation of hepatic Cxcl1 expression post HFD-plus-bigne ethanol. In conclusion, hepatic PPARγ plays an opposing role in controlling steatosis and neutrophil infiltration, leading to dissociation between steatosis and inflammation. Acute ethanol gavage attenuates hepatic PPARγ activation and subsequently upregulates hepatic CXCL1/IL-8 expression, thereby exacerbating hepatic neutrophil infiltration.
Keywords: PPARgamma, neutrophil, high fat diet, alcohol, CXCL1
Introduction
Excess alcohol consumption and obesity, renders increased risk for the development of steatohepatitis, cirrhosis and hepatocellular carcinoma.1-4 The collaborative relationship between obesity and alcohol on steatohepatitis have been already observed and documented in clinical studies5-7 and in several animal models.8-10 However, the mechanisms underlying the interaction of fat and alcohol on steatohepatitis have not been extensively investigated due to lack of appropriate animal models that reproduce the full spectrum of human steatohepatitis.11 For example, very little overlap of gene expression patterns was found between liver tissues from various mouse models of nonalcoholic fatty liver disease (NAFLD) and patients with different stages of NAFLD.12 Among these various models, only high-fat diet (HFD) feeding associated hepatic gene expression change profiles were more similar to those in human fatty liver than other models.12 HFD feeding for 3 months or longer resulted in severe steatosis and mild elevation of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) but limited inflammation with little neutrophil infiltration. Recently, we developed a mouse model of acute steatohepatitis (HFD-plus-binge ethanol model), in which administration of a single dose of ethanol after 3-month HFD feeding caused marked elevation of serum ALT and AST (reaching to approximately 800 IU/L) and significant hepatic neutrophil infiltration.13 This clinically relevant HFD-plus-binge ethanol model highlights the risk of even episodic binge drinking in obese/overweight individuals that binge drinking could cause significant hepatic neutrophil infiltration and acute steatohepatitis in these individuals. However, how binge drinking/acute ethanol gavage promotes hepatic neutrophil infiltration in HFD-fed mice or in obese individuals are not fully understood. For example, does steatosis promote liver inflammation through neutrophil infiltration?
Peroxisome proliferation-activated receptors (PPAR) have three isoforms (PPARα, PPARβ/δ and PPARγ), which belong to nuclear receptor superfamily of ligand-inducible transcription factors.14 The identified PPARs differ from each other in terms of tissue distribution and ligand specificity, acting as important regulators of many genes involved in adipogenesis, glucose homeostasis, lipid metabolism and inflammation.15 Given the therapeutic potential for type 2 diabetes, PPARγ attracted extensive clinical interest and therefore has been extensively studied16 and tested in clinics for the treatment of NAFLD patients.17-19 However, the results from the treatment of NAFLD by PPARγ agonists have been mixed,20-22 which is partly due to the complex cell- and tissue-specific PPARγ functions. PPARγ is highly expressed in adipose tissues but at very low levels in normal liver.23 Expression of PPARγ is markedly upregulated in fatty livers in animal models24, 25 and in patients with NAFLD.26 Disruption of the Pparg gene in hepatocytes ameliorates steatosis, whereas overexpression promotes the development of fatty liver via the activation of various lipogenic genes, including fat-specific protein 27 (Fsp27),27, 28 and the resultant lipogenesis.23-25, 29 The Fsp27 gene has two forms, Fsp27α and Fsp27β.30 Fsp27α is highly expressed in white adipose tissues, whereas Fsp27β is highly expressed in brown adipose tissues and fatty livers.30 FSP27 is a lipid droplet (LD) protein that plays an important role in LD formation and promotes the development of fatty liver diseases.27, 28, 31, 32 Although the steatogenic function of the PPARγ-FSP27 axis is well documented, its role in hepatic neutrophil infiltration in steatohepatitis remains obscure.
Hepatic neutrophil infiltration is a hallmark of steatohepatitis and is believed to be associated with liver injury and disease progression via generating reactive oxygen species and producing pro-inflammatory mediators.33 Our recent studies showed that blockade of several inflammatory mediators, such as chemokine (C-X-C motif) ligand 1 (CXCL1) and E-selectin, reduced hepatic neutrophil infiltration and ameliorated steatohepatitis in experimental animal model studies, confirming that neutrophil infiltration promoted hepatocyte injury.13, 34 Although the mechanisms underlying neutrophil infiltration in steatohepatitis remain obscure, it is generally believed that steatosis positively correlated with inflammation and liver injury. In the current study, we revealed that hepatic loss of PPARγ ameliorated liver steatosis, but surprisingly aggravated hepatic neutrophil infiltration in mice treated with HFD-plus-binge ethanol. Mechanistically, we demonstrated that PPARγ promoted steatosis via the upregulation of FSP27 and attenuated hepatic neutrophil infiltration via the downregulation of CXCL1/IL-8.
Materials and Methods
Mice
Mice were housed in a temperature-controlled room with a 12-h light/ 12-h dark cycle. C57BL/6J mice, Albumin-Cre mice, and Ppargfl/fl mice on a C57BL/6J background were purchased from the Jackson Laboratory (Bar Harbor, ME). Hepatocyte-specific Pparg knockout (PpargHep-/-) mice were generated through several steps of crossing Albumin-Cre mice and Ppargflox/flox mice in the NIAAA animal facility. Hepatocyte-specific Fsp27 knockout mice (Fsp27Hep-/-) on a C57BL6/N background were generated as reported in our previous study and wild-type (WT) mice were used as controls.27 All mice were cared for in accordance with the guidelines of the National Institutes of Health (NIH), and all animal experimental protocols were reviewed and approved by the National Institute on Alcohol Abuse and Alcoholism (NIAAA) animal care and use committee.
HFD-plus-binge ethanol model
HFD-plus-binge ethanol model was previously described.13 Briefly, 8- to 12-week-old male mice were fed a HFD (60% kcal% fat; catalog no. D12492; Research Diet, New Brunswick, NJ) for 3 months (3m-HFD-fed mice). After 3m HFD feeding, mice received a single dose of ethanol (5 g/kg body weight as a 53.4% ethanol in water) via gavage at 7:00 AM, and were killed 9 hours later.13 The mice were not subjected to starvation before gavage. After gavage, the mice were kept on a heating pad (37°C) and the diet was removed.
Measurement of serum and liver free fatty acid (FFA) levels
Samples were prepared based on the manufacturer's instruction. Serum and liver FFAs were determined by using a BioVision kit (#K612-100; Milpitas, CA).
Measurement of serum, supernatant and liver CXCL1 protein levels
Samples were collected and prepared according to the manufacturer's instruction. Serum and liver CXCL1 levels, and CXCL1 levels in supernatant from cultured hepatocytes were measured by using an ELISA kit (#SMKC00B, R&D systems, Minneapolis, MN).
Adenovirus Fsp27 shRNA and adenovirus control shRNA
Generation and construction of adenovirus Fsp27 shRNA (Ad-shFsp27) and adenovirus control shRNA (Ad-shRNA) were performed as described previously.27 Briefly, 3m-HFD-fed mice were injected with 109 plaque formation units (Pfu) of Ad-shRNA or Ad-shFsp27 in a total volume of 200 mL PBS via tail vein injection, then the mice were subjected to binge ethanol administration 6 days later.
Statistical Analysis
In this study, all data are shown as the mean ± SEM (n=5-12 in each group). Group comparisons were performed using the unpaired t-test or one-way analysis of variance (ANOVA), followed by Tukey's multiple comparisons as appropriate. P<0.05 was considered statistically significant.
Results
Hepatic Pparg deficiency improves hepatic steatosis but does not affect serum ALT and AST levels following HFD-plus-binge ethanol
Increased hepatic Pparg activation, a typical manifestation of fatty liver disease, promotes steatosis,24 and it is generally believed that reduced hepatic steatosis is correlated with decreased serum ALT and AST levels.35 Thus, we postulated that PpargHep-/- mice had lower steatosis and liver injury than WT mice post HFD-plus-binge ethanol. As expected, compared to WT mice, PpargHep-/- mice had milder liver steatosis and lower hepatic TG levels after HFD-plus-binge ethanol (Fig. 1A and 1B), but surprisingly serum levels of ALT and AST were not reduced in PpargHep-/- mice (Fig. 1C). In addition, body weight gain and liver/body weight ratios were comparable between PpargHep-/- mice and WT mice after HFD-plus-binge ethanol (Supporting Fig.1A and 1B).
Figure 1. Hepatic Pparg deficiency reduces hepatic steatosis, but does not alleviate serum levels of ALT and AST following HFD-plus-binge ethanol.

PpargHep-/- mice and littermate WT controls were fed a HFD diet for 3 months, and then given one binge of ethanol. Blood and liver tissue samples were collected 9 hours post gavage for measurement. (A) Representative H&E staining of liver tissue sections are shown. (B) Serum and liver TG levels. (C) Serum ALT and AST levels. *P<0.05, **P<0.01, ***P<0.001, as indicated.
Hepatic Pparg deficiency increases Cxcl1 expression and neutrophil infiltration following HFD-plus-binge ethanol
In our previous study, we demonstrated that acute inflammation played an important role in liver injury following HFD-plus-binge ethanol.13 Since less steatosis but similar liver injury was found in PpargHep-/- mice compared with WT mice, we postulated that there was more inflammation in PpargHep-/- mice. To test our hypothesis, liver inflammation was examined by performing neutrophil marker myeloperoxidase (MPO) staining. As illustrated in Fig. 2A and 2B, the number of MPO+ neutrophil was comparable in WT and PpargHep-/- mice following HFD feeding without ethanol binge. However, despite milder steatosis, PpargHep-/- mice had a greater degree of hepatic neutrophil infiltration than WT mice after HFD-plus-binge ethanol, which was consistent with the finding that Ly6g (a marker for neutrophils) mRNA levels were higher in PpargHep-/- mice (Fig. 2C). However, the number of Kupffer cells/macrophages as detected by F4/80 staining in the liver was similar between PpargHep-/- mice and WT mice (Supporting Fig. 2A).
Figure 2. Hepatic Pparg deficiency increases Cxcl1 expression and neutrophil infiltration following HFD-plus-ethanol binge.

PpargHep-/- and WT mice were set up for HFD-plus-binge ethanol model. Liver tissues were collected 9 hours post gavage for further analysis. (A) Representative MPO staining is shown (Red arrows indicate MPO+ cells). (B) Quantification of MPO+ cells/per 100× field was determined. (C, D) Total RNA was isolated and subjected to real-time PCR analysis of Ly6g (a marker of neutrophils) and other inflammatory genes. (E) Sera were collected to measure CXCL1 protein levels by using an ELISA kit. (F) Representative CXCL1 protein staining of liver tissue sections are shown (Black arrow indicates positive findings; Bar scale: 100 μm). *P<0.05, **P<0.01, ***P<0.001, as indicated.
To understand the mechanisms by which PpargHep-/- mice had greater hepatic neutrophil infiltration than WT mice after HFD-plus-binge ethanol, hepatic expressions of several chemokines were examined. In HFD feeding alone groups without binge ethanol, hepatic expression of Cxcl1 mRNA level was approximately 5-fold higher in PpargHep-/- mice than WT controls, while hepatic expression of other inflammatory markers, including Cxcl2, Ly6g, IL-1β, IL-6 and P-Selectin, were comparable between WT and PpargHep-/- mice (Fig. 2D). Compared to the HFD feeding alone groups, HFD-plus-binge ethanol highly upregulated hepatic expression of Cxcl1 in both WT and PpargHep-/- mice with much higher levels in PpargHep-/- mice than in WT mice (Fig. 2D). HFD-plus-binge ethanol also upregulated hepatic expression of other inflammatory markers (such as Cxcl2, E-selectin, P-selectin) to a lesser extent, both in WT and PpargHep-/- mice. Expressions of these inflammatory mediators were not higher in PpargHep-/- mice compared to WT mice.
In addition, ELISA analyses confirmed that both hepatic and serum CXCL1 protein levels were much higher in PpargHep-/- mice than WT mice following HFD-plus-binge ethanol (Fig. 2E, supporting Fig. 2B). Moreover, immunohistochemistry staining of CXCL1 also verified that there were more positive CXCL1-stained areas in PpargHep-/- mice compared to WT controls following HFD-plus-binge ethanol (Fig. 2F).
The above data indicated that acute binge ethanol induced much higher levels of hepatic expression of Ly6g (neutrophil marker) and Cxcl1 mRNAs in HFD-fed PpargHep-/- mice than in HFD-fed WT mice. However, acute binge ethanol induced comparable levels of hepatic Ly6g and Cxcl1 mRNA expression in chow-fed WT and PpargHep-/- mice (supporting Fig. 2C). The reason why PPARγ has no effect on liver inflammation in chow-fed mice after acute ethanol binge is probably because chow-fed mice have very low hepatic PPARγ activation (lower nuclear PPARγ) in contrast to highly activated hepatic PPARγ in HFD-fed mice (reference,24 also see results below in the current study).
Fsp27 is the downstream signal of Pparg-induced steatosis, but does not affect hepatic neutrophil infiltration in mice with HFD-plus-binge ethanol
The above data suggest that PPARγ promotes steatosis but inhibits neutrophil infiltration-induced inflammation. To understand the underlying mechanisms, we examined hepatic expression of Fsp27, a downstream target gene of PPARγ,28 to see whether Fsp27 mediates both the steatogenic and anti-inflammatory effect of PPARγ. Fsp27 exists two isoforms of Fsp27 gene, including Fsp27a and Fsp27b.30 In our previous study, Fsp27 was reported to be highly up-regulated in mice on chronic-plus-binge ethanol model, and up-regulation of Fsp27 contributed to alcohol-induced liver injury.27 Here we found that HFD- plus-binge ethanol induced much higher levels of hepatic Fsp27a and Fsp27b mRNA in mice than HFD feeding alone, and that both Fsp27a and Fsp27b mRNA levels were markedly reduced in PpargHep-/- mice compared to WT mice after HFD feeding or HFD-plus-binge ethanol (Fig. 3A). In addition, mRNA encoding hepatic Cidea were also reduced, but mRNA encoding hepatic Cideb were not reduced in PpargHep-/- mice compared with WT mice (Fig. 3A).
Figure 3. Hepatic Fsp27, a Pparg downstream target gene, contributes to the pro-steatotic effect of PPARγ in mice following HFD-plus-binge ethanol.
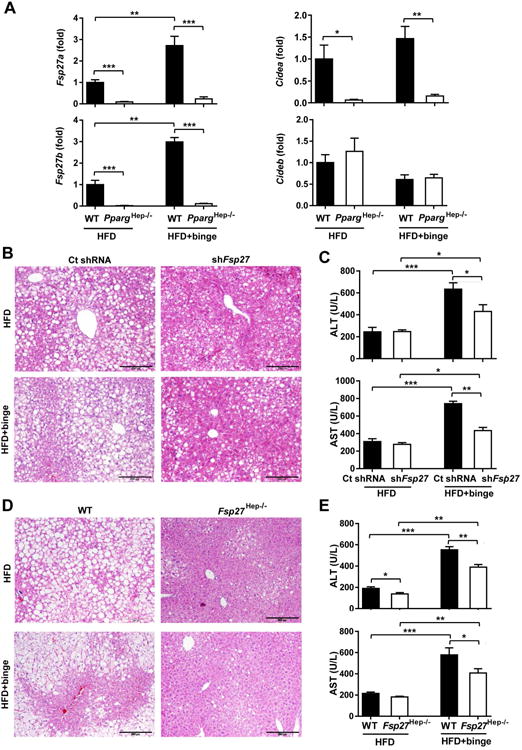
(A) PpargHep-/- and WT mice were set up for HFD-plus-binge ethanol model. Liver tissue samples were collected, and Cidea, Cideb and Fsp27 mRNA expressions were determined by real-time PCR. (B-C) Male C57BL/6N mice were fed a HFD for 3 months, and administered with Ad-control-shRNA (109Pfu) or Ad-shFsp27 (109Pfu) for the final 6 days. Ethanol (5g/kg) was given by oral gavage 9h before killing. Liver tissues and sera were collected for measurement. Liver histology was analyzed and serum ALT and AST levels were analyzed. (D-E) Fsp27Hep-/- mice and WT mice were set up for HFD-plus-binge ethanol model. Liver histology is shown, and serum ALT and AST level were analyzed. *P<0.05, **P<0.01, ***P<0.001, as indicated.
In order to test the pro-steatotic function of FSP27, Fsp27shRNA adenovirus was used to knock down Fsp27 gene expression. There were no changes in HFD consumption and body weight in mice 3 days or 6 days after injecting with control shRNA and Fsp27shRNA adenovirus (Supporting Fig. 3A). Both hepatic Fsp27a and Fsp27b mRNAs were lowered by Fsp27shRNA adenovirus injection, thus confirming hepatic Fsp27 expression was reduced (Supporting Fig. 3B).
In both the HFD feeding alone and HFD-plus-binge ethanol groups, injection of Fsp27shRNA adenovirus reduced liver steatosis (Fig. 3B) and ameliorated hepatic TG levels (Supporting Fig. 3C). Interestingly, compared to the shRNA control, injection of Fsp27shRNA reduced serum ALT and AST levels in HFD-plus-binge ethanol groups but not in HFD feeding alone groups (Fig. 3C).
Furthermore, genetic disruption of Fsp27 did not affect body weight (Supporting Fig. 3D) but led to reduced steatosis in both HFD and HFD-plus-binge ethanol groups (Fig. 3D). Both serum ALT and AST levels were markedly reduced in Fsp27Hep-/- mice compared to WT mice in HFD-plus-binge ethanol groups, whereas only ALT levels were reduced in Fsp27Hep-/- mice in HFD alone groups (Fig. 3E). Thus, Fsp27 is responsible for the pro-steatotic function of hepatic PPARγ in this HFD-plus-binge ethanol model.
In order to investigate the anti-inflammatory function of Fsp27 in HFD-plus-binge ethanol model, we tested neutrophil infiltration by performing MPO staining and real-time PCR analysis of Ly6g mRNA in mice with hepatic Fsp27 knockdown and Fsp27 disruption. As illustrated in Fig. 4A-D, neither Fsp27 knockdown nor Fsp27 deletion altered the number of infiltrated neutrophil and hepatic expression of Ly6g in HFD feeding alone or in HFD-plus-binge ethanol groups, except for a mild increase in the number of infiltrated neutrophil following Fsp27 knockdown in HFD feeding alone. Moreover, hepatic expression of Cxcl1 mRNA was not altered by hepatic Fsp27 knockdown or Fsp27 disruption (Fig. 4E).
Figure 4. Hepatic Fsp27 deletion does not affect neutrophil infiltration following HFD-plus-binge ethanol.
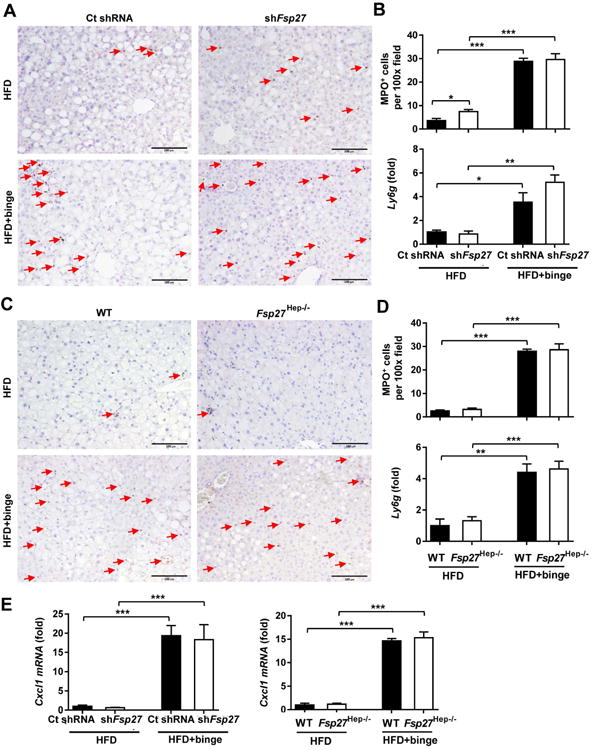
(A, B) Male C57BL/6N mice were fed a HFD for 3 months, and administered with Ad-control-shRNA (109Pfu) or Ad-shFsp27 (109Pfu) for the final 6 days. Ethanol (5g/kg) was given by oral gavage 9 hours before killing. Liver tissues and sera were collected for measurement. Representative MPO staining is shown in panel A. Quantification of MPO+ cells/per 100× field and real-time PCR analysis of Ly6g are shown in panel B. (C-D) Fsp27Hep-/- mice and WT mice were set up for HFD-plus-binge ethanol model. Liver tissue samples were collected. Representative MPO staining is shown in panel C. Quantification of MPO+ cells/per 100× field and real-time PCR analysis of Ly6g mRNA are shown in panel D. (E) Real-time PCR analysis of Cxcl1 mRNA in liver samples from panels A and C. Red arrows in panels A and C indicate MPO+ cells. *P<0.05, **P<0.01, ***P<0.001, as indicated.
The above data suggest that FSP27 does not play a role in inducing hepatic neutrophil infiltration and Cxcl1 mRNA expression in HFD-fed mice even when hepatic Fsp27 mRNA expression is highly elevated. Because FSP27 is expressed at very low levels in the livers from chow-fed mice,27 FSP27 very unlikely affects hepatic Cxcl1 mRNA expression and neutrophil infiltration in these mice.
Hepatic Pparg deficiency reduces liver fibrogenic response/fibrosis in HFD-plus-binge ethanol
Liver steatosis and inflammation are associated with the development of fibrosis.36-39 Previous studies reported an anti-inflammatory and anti-fibrogenic properties of PPARγ in non-parenchymal liver cells.40 Thus, we wondered whether hepatic PPARγ also affected liver fibrogenic response/fibrosis in post HFD-plus-binge ethanol. Compared to WT mice, PpargHep-/- mice had fewer areas of Sirius red staining following HFD feeding or HFD-plus-binge ethanol (Fig. 5A-C). In addition, Tgfb, Col1a1, Col3a1, Col4a1 and Col12a1 mRNAs encoding fibrogenic markers, were reduced in PpargHep-/- mice compared to WT mice post HFD-plus-binge ethanol.
Figure 5. Hepatic Pparg deficiency attenuates liver fibrosis following HFD-plus-binge ethanol.

PpargHep-/- and WT mice were set up for HFD-plus-binge ethanol model. Liver tissue samples were collected. (A) Representative Sirius red staining of liver tissue sections is shown. (B) Quantification of fibrotic area/per 100× field was determined. (C) Real-time PCR analyses of fibrosis-related genes. *P<0.05, **P<0.01, ***P<0.001, as indicated.
As PPARγ in hepatocyte oppositely regulates steatosis and inflammation as demonstrated above, it remains a question whether steatosis dominantly modulates fibrosis instead of inflammation in the HFD-plus-binge ethanol model. To answer this question, we further tested liver fibrosis by performing hepatic Fsp27 knockdown and Fsp27 knockout. As shown in Supporting Fig. 4A-B, Sirius red staining revealed that less fibrosis lesions were found in the Fsp27 knockdown and the Fsp27 knockout groups, after both HFD feeding and HFD-plus-binge ethanol challenge. Because knockdown/knockout of Fsp27 reduced steatosis but not inflammation, the data from Supporting Fig. 4 suggest that steatosis not inflammation plays a predominate role in promoting liver fibrosis in this HFD-plus-binge ethanol model.
Interaction of PPARγ and NF-κB regulates Cxcl1 expression in hepatocytes
To understand the mechanisms by which activation of PPARγ inhibits the upregulation of Cxcl1 in the liver, we isolated primary hepatocytes and treated them with different regimens of palmitic acid (PA) and/or troglitazone (a PPARγ agonist). Treatment with PA markedly upregulated Cxcl1 mRNA expression in hepatocytes (Fig. 6A). Interestingly, PA-mediated upregulation of Cxcl1 mRNA was markedly enhanced in hepatocytes from PpargHep-/- mice (12-fold induction in Pparg knockout hepatocytes vs. a 2-fold change in WT hepatocytes). In addition, treatment with TNF-α also upregulated Cxcl1 mRNA expression in isolated hepatocytes with much higher levels in Pparg knockout hepatocytes than WT hepatocytes (Fig. 6B). In contrast, activation of PPARγ by treatment with troglitazone attenuated TNF-α-mediated upregulation of Cxcl1 mRNA and CXCL1 protein in hepatocytes (Fig. 6C). Surprisingly, treatment with troglitazone did not affect PA-mediated upregulation of Cxcl1 mRNA in hepatocytes (Supporting Fig. 5). Moreover, we have previously demonstrated that NF-κB activation is involved in upregulation of Cxcl1 mRNA in WT hepatocytes.13 Here we found that treatment with the NF-κB inhibitor Bay also markedly reduced PA-mediated induction of Cxcl1 mRNA in Pparg knockout hepatocytes (Fig. 6D).
Figure 6. Interaction between PPARγ and NF-κB modulates FFAs-induced upregulation of Cxcl1 in hepatocytes.

(A, B) Primary hepatocytes from PpargHep-/- mice and WT mice were isolated and stimulated with palmitic acid (PA, 0.3mM) or TNF-α (10ng/ml). Hepatocytes were collected and Cxcl1 mRNA levels were determined by real-time qPCR. (C) Primary hepatocytes from C57BL/6N mice were isolated and stimulated with troglitazone (Tro, a PPARγ agonist) and TNF-α (10ng/ml) for 12 hours, then cells were harvested and Cxcl1 mRNA levels were determined by real-time PCR. (D) Primary hepatocytes from PpargHep-/- mice and WT mice were isolated and stimulated by PA with pre-treatment by different doses of Bay-7085 (an NF-kB inhibitor) for 30 minutes. Cxcl1 mRNA expression was determined by real-time PCR. (E) HepG2 cells were stimulated by PA with PPARγ agonist (Tro) or antagonist (GW9662) pretreatment. Cells were collected after the treatment at different time points, and IL-8 mRNA levels were determined by real-time PCR. *P<0.05, **P<0.01, ***P<0.001, as indicated.
Finally, IL-8 is a functional homologue of mouse Cxcl1 and its expression in human hepatocytes is upregulated by PAs,41 playing a critical role in promoting neutrophil infiltration in human steatohepatitis. Thus, we determined whether PPARγ also regulates IL-8 expression in human hepatoma HepG2 cells. Treatment with PA induced IL8 mRNA in HepG2 cells. Co-treatment with the PPARγ agonist troglitazone inhibited PA-mediated upregulation of CXCL1 mRNA, whereas co-treatment with the PPARγ antagonist GW9662 enhanced it (Fig. 6E).
HFD feeding activates hepatic PPARγ, which is attenuated by a binge of ethanol
Several murine models of obesity and diabetes developed fatty liver with enhanced levels of PPARγ expression.42 Previous studies also showed that Pparg mRNA level was downregulated in response to the combination of acute ethanol and lipopolysaccharide (LPS).43 To further clarify the modulation of hepatic PPARγ in response to HFD-plus-binge ethanol, we examined nuclear PPARγ staining in this model. Very few PPARγ positive nuclei of hepatocytes were detected in chow- and chow-plus-binge ethanol-fed mice (Fig. 7A). However, a significant number of PPARγ-stained nuclei of hepatocytes were found in mouse livers from HFD and HFD-plus-binge ethanol with a lower number in the HFD-plus-binge ethanol group compared to HFD feeding group. Western blot analyses confirmed that expression of nuclear PPARγ protein (activated PPARγ) in hepatocytes was increased by 12 folds in HFD-fed mice as compared with chow-fed mice, but was significantly suppressed in the HFD-plus-binge ethanol-fed mice compared to that in HFD alone-fed mice (Fig. 7B-C). In addition, hepatic expression of total PPARγ protein was also higher in HFD-fed mice than in chow-fed mice, and that acute ethanol binge markedly downregulated hepatic expression of PPARγ protein in HFD-fed mice (Fig. 7B-C). Moreover, hepatic expression of Pparg mRNA was much higher in HFD-fed mice than in chow-fed mice, and such expression was markedly attenuated in mice following HFD-plus-binge ethanol (Fig. 7D).
Figure 7. Acute ethanol binge suppresses hepatic Pparg activation and expression in HFD-fed mice.
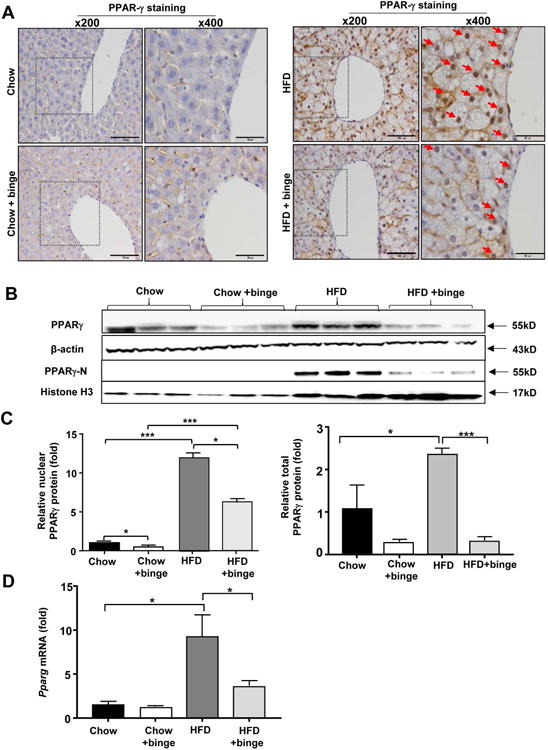
C57BL/6J male mice were divided into 4 groups, 2 groups were fed a chow diet and other 2 groups were fed a HFD diet for 3 months. On the last day of 3m-feeding, mice were given maltose or ethanol by oral gavage 9 hours before killing (Chow, Chow+binge, HFD, HFD+binge). Liver tissues were collected for further analysis. (A) Representative PPARγ nuclear staining is shown (Red arrows indicate positive staining of hepatocyte nucleus). (B-C) Total hepatic protein and hepatic nuclear protein were extracted and subjected to Western blot analysis of total PPARγ and nuclear PPARγ protein. Representative blots are shown in panel B. Quantification of total PPARγ/β-actin and nuclear PPARγ/histone H3 is shown in panel C. (D) Real-time PCR analysis of Pparg mRNA was performed. *P<0.05, **P<0.01, ***P<0.001, as indicated.
Discussion
By using a clinically relevant mouse steatohepatitis model induced by HFD-plus-binge ethanol, mechanistic insights into the role of steatosis and inflammation in liver disease were investigated leading to several novel findings. First, hepatic PPARγ promoted steatosis via Fsp27 upregulation while attenuating neutrophil infiltration via CXCL1 suppression. Second, acute ethanol binge inhibits PPARγ activation, which is partially responsible for acute ethanol induced upregulation of hepatic CXCL1. Third, acute ethanol gavage induces hepatic neutrophil infiltration in HFD-fed mice independent of steatosis. Finally, PPARγ agonists also inhibit IL-8 expression in human hepatocytes. These findings were used to generate a working model depicting the interaction of fat and acute ethanol in promoting steatohepatitis (Fig. 8).
Figure 8. The dichotomy role of PPARγ underlying the synergistic effect of HFD and acute ethanol binge on steatohepatitis.
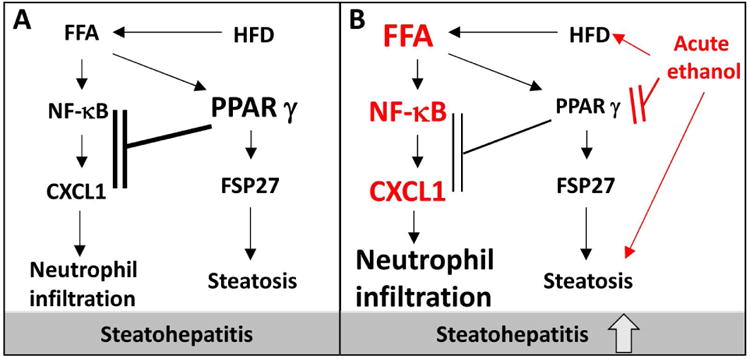
(A) Post HFD feeding, hepatic PPARγ becomes activated and upregulates hepatic expression of FSP27, thereby promoting steatosis. PPARγ also inhibits hepatic CXCL1 expression via the inhibition of NF-κB, and subsequently reduces hepatic neutrophil infiltration. (B) Acute ethanol binge inhibits PPARγ activation and subsequently diminishes the inhibitory effect of PPARγ on the NF-κB/CXCL1 axis, thus leading CXCL1 upregulation and massive hepatic neutrophil infiltration in HFD-fed mice. Acute ethanol also elevates FFA levels to further enhance the NF-κB/CXCL1 pathway.
The role of hepatic PPARγ in the development of fatty liver has been well documented in several animal models of fatty liver induced by HFD feeding,24 chronic ethanol feeding,25 or genetic deletion of leptin.23 Given all of these models are associated with little liver inflammation (eg. neutrophil infiltration), the effect of hepatic PPARγ in liver inflammation has not been investigated and still remains obscure. In the current study, we used a recently developed mouse model of HFD-plus-binge ethanol, which induces significant hepatic neutrophil infiltration,13 to study the role of PPARγ in modulating steatosis and neutrophil recruitment in the liver. The most striking finding was that although steatosis was reduced, hepatic neutrophil infiltration was markedly exacerbated in PpargHep-/- mice after HFD-plus-binge ethanol, suggesting that hepatic PPARγ acts an anti-inflammatory pathway via the inhibition of hepatic neutrophil infiltration. We have previously demonstrated that hepatic CXCL1, a key chemokine to promote neutrophil infiltration, was upregulated by more than 30-fold after HFD-plus-binge ethanol, and played an important role in hepatic neutrophil infiltration in this model.13 Fascinatingly, in the current study, we found that hepatic and serum levels of CXCL1 protein were further elevated in PpargHep-/- mice compared to those in WT mice (approximately 2500 pg/ml in serum from PpargHep-/- mice vs 1200 pg/ml in WT) after HFD-plus-binge ethanol, indicating that hepatic PPARγ played a critical role in inhibiting CXCL1 protein production in hepatocytes, thereby limiting hepatic neutrophil infiltration. The inhibitory effect of PPARγ on Cxcl1 mRNA expression in hepatocytes were also confirmed in cultured primary hepatocytes, as disruption of the Pparg gene in hepatocytes upregulated FA- or TNF-α-induced Cxcl1 mRNA, while activation of PPARγ by treatment with the PPARγ agonist troglitazone decreased TNF-α-induced Cxcl1 mRNA levels. Interestingly, activation of PPARγ by troglitazone did not affect PA-mediated Cxcl1 mRNA expression in hepatocytes. The reason why troglitazone treatment does not affect PA-mediated upregulation of hepatic Cxcl1 mRNA is probably because PA itself is a strong ligand for PPARγ activation 44 and thus may diminish the effect of PPARγ agonist treatment.
Mechanistically, we also demonstrated that inhibition of NF-κB abolished PA-mediated upregulation of Cxcl1 mRNA in PpargHep-/- hepatocytes, suggesting that PPARγ attenuates hepatic Cxcl1 mRNA levels via an NF-κB-dependent mechanism. Several previous studies suggested that PPARγ inhibited the expression of pro-inflammatory genes by interfering with NF-κB DNA binding.45-49 This mechanism likely also explains the PPARγ inhibition of Cxcl1 gene expression in hepatocytes via the inhibition of FFA activation of NF-κB. In addition, hepatic and serum FFA levels were comparable in PpargHep-/- mice compared to WT mice following HFD-plus-binge ethanol (Supporting Fig. 6A-B). Taken together, these data suggest that upregulated hepatic Cxcl1 mRNA expression in PpargHep-/- mice is due to enhanced FFA signaling (through activation of NF-κB) in hepatocytes, and is not due to altered FFA levels compared to WT mice.
Liver fibrosis is always associated with steatosis and inflammation. Hepatic steatosis generally promotes liver fibrogenesis by producing reactive oxygen species and danger associated molecule patterns (DAMPs), while inflammation plays a complex role in promoting or suppressing liver fibrogenesis.37, 38 Interestingly, compared to WT mice, PpargHep-/- mice had reduced liver steatosis and fibrosis although these mice had enhanced liver inflammation post HFD-plus-binge ethanol. These results suggest that hepatic PPARγ promotes liver fibrosis via the stimulation of hepatic steatosis, while the anti-inflammatory effects of hepatic PPARγ are less important for liver fibrosis in the HFD-plus-binge ethanol model.
Hepatic PPARγ was upregulated and activated in mice following HFD feeding 24, 25 or chronic ethanol feeding.25 Here we found that hepatic PPARγ was also highly upregulated and activated after HFD feeding, but surprisingly, this upregulation and activation were attenuated by an additional ethanol binge challenge. As a result, the attenuation of hepatic PPARγ likely diminished the inhibitory effect of PPARγ on hepatic neutrophil infiltration, resulting in highly elevated hepatic CXCL1 level and hepatic neutrophil infiltration observed in HFD-plus-binge ethanol-fed mice compared to HFD alone-fed mice. In addition, HFD feeding or acute ethanol gavage alone induced fat accumulation in the liver, but HFD-plus-binge ethanol did not further exacerbate steatosis compared to HFD feeding alone.13 One possible explanation with regard to underlying mechanisms may be that in addition to promote steatosis, acute ethanol binge also reduced the steatogenic effect of PPARγ via the inhibition of PPARγ activation, resulting in comparable levels of steatosis in HFD-plus-binge ethanol and HFD feeding groups. Another interesting observation was that acute ethanol binge downregulated hepatic expression of PPARγ protein and mRNA in HFD-fed mice as demonstrated in the current study, but chronic ethanol feeding upregulated hepatic Pparg expression as reported in a previous study.25 However, the mechanisms by which hepatic PPARγ is oppositely regulated by acute and chronic ethanol feeding remain unknown, and further studies are required to clarify.
Hepatic neutrophil infiltration, a hallmark of steatohepatitis, is generally believed to be associated with the degree of steatosis.33 In the current study, we provided evidence suggesting that hepatic neutrophil infiltration is independent of steatosis in HFD-plus-binge ethanol model. First, PpargHep-/- mice had significantly reduced steatosis but had increased hepatic neutrophil infiltration than WT mice post HFD-plus-binge ethanol. Second, inhibition of the PPARγ downstream target gene Fsp27 also markedly improved steatosis but did not affect hepatic neutrophil infiltration and hepatic Cxcl1 mRNA expression in mice fed with HFD-plus-binge ethanol.
Another important finding from the current study is that activation of PPARγ not only inhibits PA-mediated induction of CXCL1 in mouse hepatocytes but also suppresses PA stimulation of IL-8, a functional homologue of mouse CXCL1, in human hepatocytes. PA induction of IL-8 in human hepatocytes likely plays an important role in promoting neutrophil infiltration and disease progression in human nonalcoholic steatohepatitis (NASH).41 Thus, treatment with PPARγ agonists may be beneficial for human NASH via the inhibition of IL-8 production in hepatocytes.
Clinical implications of this study
The complex effects of PPARγ on hepatic steatosis, fibrosis, and neutrophil infiltration compromise the potential use of PPARγ aa a therapeutic target for the treatment of NASH.19 The PPARγ agonist pioglitazone, a prescription drug for type 2 diabetes, should be cautiously used for the treatment of NASH because activation of hepatic PPARγ while beneficial for the inhibition of neutrophil infiltration, could exacerbate steatosis and fibrosis. Furthermore, the PPARγ-NF-κB-CXCL1/IL-8 axis identified in the current study yields new insights into the development of potential treatment of neutrophil-mediated steatohepatitis reflecting the synergistic effect of obesity and alcohol consumption in hepatic neutrophil infiltration.
Supplementary Material
Acknowledgments
This work was supported by the intramural program of NIAAA, NIH.
Abbreviations
- AH
alcoholic hepatitis
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CXCL1
(C-X-C motif) ligand 1
- FFAs
free fatty acids
- FSP27
fat-specific protein 27
- HFD
high-fat diet
- HFD-plus-ethanol binge
Feeding mice with an HFD for 3 months followed by a binge of ethanol
- KO
knockout
- LD
lipid droplet
- LPS
lipopolysaccharide
- m
months
- MPO
myeloperoxidase
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PA
palmitic acid
- PPARγ
peroxisome proliferation-activated receptor γ. TG, triglyceride
- TG
triglyceride
- TNF
tumor necrosis factor
- Tro
troglitazone
- WT
wild-type
Footnotes
No conflicts of interest exist for any of the authors.
Author names in bold designate shared co-first authorship.
References
- 1.Hart CL, Morrison DS, Batty GD, Mitchell RJ, Davey Smith G. Effect of body mass index and alcohol consumption on liver disease: analysis of data from two prospective cohort studies. BMJ. 2010;340:c1240. doi: 10.1136/bmj.c1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Naveau S, Giraud V, Borotto E, Aubert A, Capron F, Chaput JC. Excess weight risk factor for alcoholic liver disease. Hepatology. 1997;25:108–11. doi: 10.1002/hep.510250120. [DOI] [PubMed] [Google Scholar]
- 3.Ruhl CE, Everhart JE. Joint effects of body weight and alcohol on elevated serum alanine aminotransferase in the United States population. Clin Gastroenterol Hepatol. 2005;3:1260–8. doi: 10.1016/s1542-3565(05)00743-3. [DOI] [PubMed] [Google Scholar]
- 4.Diehl AM. Obesity and alcoholic liver disease. Alcohol. 2004;34:81–7. doi: 10.1016/j.alcohol.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 5.Alatalo PI, Koivisto HM, Hietala JP, Puukka KS, Bloigu R, Niemela OJ. Effect of moderate alcohol consumption on liver enzymes increases with increasing body mass index. Am J Clin Nutr. 2008;88:1097–103. doi: 10.1093/ajcn/88.4.1097. [DOI] [PubMed] [Google Scholar]
- 6.Shen Z, Li Y, Yu C, Shen Y, Xu L, Xu C, et al. A cohort study of the effect of alcohol consumption and obesity on serum liver enzyme levels. Eur J Gastroenterol Hepatol. 2010;22:820–5. doi: 10.1097/MEG.0b013e3283328b86. [DOI] [PubMed] [Google Scholar]
- 7.Loomba R, Bettencourt R, Barrett-Connor E. Synergistic association between alcohol intake and body mass index with serum alanine and aspartate aminotransferase levels in older adults: the Rancho Bernardo Study. Aliment Pharmacol Ther. 2009;30:1137–49. doi: 10.1111/j.1365-2036.2009.04141.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gabele E, Dostert K, Dorn C, Patsenker E, Stickel F, Hellerbrand C. A new model of interactive effects of alcohol and high-fat diet on hepatic fibrosis. Alcohol Clin Exp Res. 2011;35:1361–7. doi: 10.1111/j.1530-0277.2011.01472.x. [DOI] [PubMed] [Google Scholar]
- 9.Xu J, Lai KK, Verlinsky A, Lugea A, French SW, Cooper MP, et al. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J Hepatol. 2011;55:673–82. doi: 10.1016/j.jhep.2010.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Demori I, Voci A, Fugassa E, Burlando B. Combined effects of high-fat diet and ethanol induce oxidative stress in rat liver. Alcohol. 2006;40:185–91. doi: 10.1016/j.alcohol.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 11.Maher JJ. Modeling fatty liver disease in animals: Is there an optimal approach, and is the effort worthwhile? Hepatology. 2016 doi: 10.1002/hep.28823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, et al. Comparison of Gene Expression Patterns Between Mouse Models of Nonalcoholic Fatty Liver Disease and Liver Tissues From Patients. Gastroenterology. 2016;151:513–525 e0. doi: 10.1053/j.gastro.2016.05.051. [DOI] [PubMed] [Google Scholar]
- 13.Chang B, Xu MJ, Zhou Z, Cai Y, Li M, Wang W, et al. Short- or long-term high-fat diet feeding plus acute ethanol binge synergistically induce acute liver injury in mice: An important role for CXCL1. Hepatology. 2015;62:1070–85. doi: 10.1002/hep.27921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002;53:409–35. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 15.Janani C, Ranjitha Kumari BD. PPAR gamma gene--a review. Diabetes Metab Syndr. 2015;9:46–50. doi: 10.1016/j.dsx.2014.09.015. [DOI] [PubMed] [Google Scholar]
- 16.Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, et al. PPARgamma signaling and metabolism: the good, the bad and the future. Nat Med. 2013;19:557–66. doi: 10.1038/nm.3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polyzos SA, Bugianesi E, Kountouras J, Mantzoros CS. Nonalcoholic fatty liver disease: Updates on associations with the metabolic syndrome and lipid profile and effects of treatment with PPAR-gamma agonists. Metabolism. 2016 doi: 10.1016/j.metabol.2016.08.001. [DOI] [PubMed] [Google Scholar]
- 18.Musso G, Cassader M, Paschetta E, Gambino R. Pioglitazone for advanced fibrosis in NASH: New evidence, new challenges. Hepatology. 2016 doi: 10.1002/hep.28960. [DOI] [PubMed] [Google Scholar]
- 19.Tailleux A, Wouters K, Staels B. Roles of PPARs in NAFLD: potential therapeutic targets. Biochim Biophys Acta. 2012;1821:809–18. doi: 10.1016/j.bbalip.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 20.Lutchman G, Modi A, Kleiner DE, Promrat K, Heller T, Ghany M, et al. The effects of discontinuing pioglitazone in patients with nonalcoholic steatohepatitis. Hepatology. 2007;46:424–9. doi: 10.1002/hep.21661. [DOI] [PubMed] [Google Scholar]
- 21.Ratziu V, Charlotte F, Bernhardt C, Giral P, Halbron M, Lenaour G, et al. Long-term efficacy of rosiglitazone in nonalcoholic steatohepatitis: results of the fatty liver improvement by rosiglitazone therapy (FLIRT 2) extension trial. Hepatology. 2010;51:445–53. doi: 10.1002/hep.23270. [DOI] [PubMed] [Google Scholar]
- 22.Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, et al. Long-Term Pioglitazone Treatment for Patients With Nonalcoholic Steatohepatitis and Prediabetes or Type 2 Diabetes Mellitus: A Randomized Trial. Ann Intern Med. 2016;165:305–15. doi: 10.7326/M15-1774. [DOI] [PubMed] [Google Scholar]
- 23.Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O, Ward JM, et al. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest. 2003;111:737–47. doi: 10.1172/JCI17223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moran-Salvador E, Lopez-Parra M, Garcia-Alonso V, Titos E, Martinez-Clemente M, Gonzalez-Periz A, et al. Role for PPARgamma in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J. 2011;25:2538–50. doi: 10.1096/fj.10-173716. [DOI] [PubMed] [Google Scholar]
- 25.Zhang W, Sun Q, Zhong W, Sun X, Zhou Z. Hepatic Peroxisome Proliferator-Activated Receptor Gamma Signaling Contributes to Alcohol-Induced Hepatic Steatosis and Inflammation in Mice. Alcohol Clin Exp Res. 2016;40:988–99. doi: 10.1111/acer.13049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pettinelli P, Videla LA. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab. 2011;96:1424–30. doi: 10.1210/jc.2010-2129. [DOI] [PubMed] [Google Scholar]
- 27.Xu MJ, Cai Y, Wang H, Altamirano J, Chang B, Bertola A, et al. Fat-Specific Protein 27/CIDEC Promotes Development of Alcoholic Steatohepatitis in Mice and Humans. Gastroenterology. 2015;149:1030–1041 e6. doi: 10.1053/j.gastro.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsusue K, Kusakabe T, Noguchi T, Takiguchi S, Suzuki T, Yamano S, et al. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metab. 2008;7:302–11. doi: 10.1016/j.cmet.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, et al. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J Biol Chem. 2003;278:498–505. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 30.Xu X, Park JG, So JS, Lee AH. Transcriptional activation of Fsp27 by the liver-enriched transcription factor CREBH promotes lipid droplet growth and hepatic steatosis. Hepatology. 2015;61:857–69. doi: 10.1002/hep.27371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liangpunsakul S, Gao B. Alcohol and fat promote steatohepatitis: a critical role for fat-specific protein 27/CIDEC. J Investig Med. 2016;64:1078–81. doi: 10.1136/jim-2016-000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langhi C, Baldan A. CIDEC/FSP27 is regulated by peroxisome proliferator-activated receptor alpha and plays a critical role in fasting- and diet-induced hepatosteatosis. Hepatology. 2015;61:1227–38. doi: 10.1002/hep.27607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao B, Tsukamoto H. Inflammation in Alcoholic and Nonalcoholic Fatty Liver Disease: Friend or Foe? Gastroenterology. 2016;150:1704–9. doi: 10.1053/j.gastro.2016.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bertola A, Park O, Gao B. Chronic plus binge ethanol feeding synergistically induces neutrophil infiltration and liver injury in mice: a critical role for E-selectin. Hepatology. 2013;58:1814–23. doi: 10.1002/hep.26419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belfort R, Harrison SA, Brown K, Darland C, Finch J, Hardies J, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med. 2006;355:2297–307. doi: 10.1056/NEJMoa060326. [DOI] [PubMed] [Google Scholar]
- 36.Leandro G, Mangia A, Hui J, Fabris P, Rubbia-Brandt L, Colloredo G, et al. Relationship between steatosis, inflammation, and fibrosis in chronic hepatitis C: a meta-analysis of individual patient data. Gastroenterology. 2006;130:1636–42. doi: 10.1053/j.gastro.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 37.Seki E, Schwabe RF. Hepatic inflammation and fibrosis: functional links and key pathways. Hepatology. 2015;61:1066–79. doi: 10.1002/hep.27332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koyama Y, Brenner DA. Liver inflammation and fibrosis. J Clin Invest. 2017;127:55–64. doi: 10.1172/JCI88881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson MW, Harmon C, O'Farrelly C. Liver immunology and its role in inflammation and homeostasis. Cell Mol Immunol. 2016;13:267–76. doi: 10.1038/cmi.2016.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moran-Salvador E, Titos E, Rius B, Gonzalez-Periz A, Garcia-Alonso V, Lopez-Vicario C, et al. Cell-specific PPARgamma deficiency establishes anti-inflammatory and anti-fibrogenic properties for this nuclear receptor in non-parenchymal liver cells. J Hepatol. 2013;59:1045–53. doi: 10.1016/j.jhep.2013.06.023. [DOI] [PubMed] [Google Scholar]
- 41.Joshi-Barve S, Barve SS, Amancherla K, Gobejishvili L, Hill D, Cave M, et al. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology. 2007;46:823–30. doi: 10.1002/hep.21752. [DOI] [PubMed] [Google Scholar]
- 42.Inoue M, Ohtake T, Motomura W, Takahashi N, Hosoki Y, Miyoshi S, et al. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun. 2005;336:215–22. doi: 10.1016/j.bbrc.2005.08.070. [DOI] [PubMed] [Google Scholar]
- 43.Ohata M, Suzuki H, Sakamoto K, Hashimoto K, Nakajima H, Yamauchi M, et al. Pioglitazone prevents acute liver injury induced by ethanol and lipopolysaccharide through the suppression of tumor necrosis factor-alpha. Alcohol Clin Exp Res. 2004;28:139S–144S. doi: 10.1097/01.alc.0000134412.38510.f7. [DOI] [PubMed] [Google Scholar]
- 44.Sauma L, Stenkula KG, Kjolhede P, Stralfors P, Soderstrom M, Nystrom FH. PPAR-gamma response element activity in intact primary human adipocytes: effects of fatty acids. Nutrition. 2006;22:60–8. doi: 10.1016/j.nut.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 45.Romics L, Jr, Kodys K, Dolganiuc A, Graham L, Velayudham A, Mandrekar P, et al. Diverse regulation of NF-kappaB and peroxisome proliferator-activated receptors in murine nonalcoholic fatty liver. Hepatology. 2004;40:376–85. doi: 10.1002/hep.20304. [DOI] [PubMed] [Google Scholar]
- 46.Wang N, Verna L, Chen NG, Chen J, Li H, Forman BM, et al. Constitutive activation of peroxisome proliferator-activated receptor-gamma suppresses pro-inflammatory adhesion molecules in human vascular endothelial cells. J Biol Chem. 2002;277:34176–81. doi: 10.1074/jbc.M203436200. [DOI] [PubMed] [Google Scholar]
- 47.Pascual G, Fong AL, Ogawa S, Gamliel A, Li AC, Perissi V, et al. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-gamma. Nature. 2005;437:759–63. doi: 10.1038/nature03988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pascual G, Sullivan AL, Ogawa S, Gamliel A, Perissi V, Rosenfeld MG, et al. Anti-inflammatory and antidiabetic roles of PPARgamma. Novartis Found Symp. 2007;286:183–96. doi: 10.1002/9780470985571.ch16. discussion 196-203. [DOI] [PubMed] [Google Scholar]
- 49.Scirpo R, Fiorotto R, Villani A, Amenduni M, Spirli C, Strazzabosco M. Stimulation of nuclear receptor peroxisome proliferator-activated receptor-gamma limits NF-kappaB-dependent inflammation in mouse cystic fibrosis biliary epithelium. Hepatology. 2015;62:1551–62. doi: 10.1002/hep.28000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.