

Abstract
Idiopathic pulmonary fibrosis (IPF) is characterized by progressive pulmonary scarring, decline in lung function, and often results in death within 3–5 five years after diagnosis. Fibroblast growth factor (FGF) signaling has been implicated in the pathogenesis of IPF; however, the mechanism through which FGF signaling contributes to pulmonary fibrosis remains unclear. We hypothesized that FGF receptor (FGFR) signaling in fibroblasts is required for the fibrotic response to bleomycin. To test this, mice with mesenchyme-specific tamoxifen-inducible inactivation of FGF receptors 1, 2, and 3 (Col1α2-CreER; TCKO mice) were lineage labeled and administered intratracheal bleomycin. Lungs were collected for histologic analysis, whole lung RNA and protein, and dissociated for flow cytometry and FACS. Bleomycin-treated Col1α2-CreER; TCKO mice have decreased pulmonary fibrosis, collagen production, and fewer α-smooth muscle actin-positive (αSMA+) myofibroblasts compared with controls. Freshly isolated Col1α2-CreER; TCKO mesenchymal cells from bleomycin-treated mice have decreased collagen expression compared with wild type mesenchymal cells. Furthermore, lineage labeled FGFR-deficient fibroblasts have decreased enrichment in fibrotic areas and decreased proliferation. These data identify a cell autonomous requirement for mesenchymal FGFR signaling in the development of pulmonary fibrosis, and for the enrichment of the Col1α2-CreER-positive (Col1α2+) mesenchymal lineage in fibrotic tissue following bleomycin exposure. We conclude that mesenchymal FGF signaling is required for the development of pulmonary fibrosis, and that therapeutic strategies aimed directly at mesenchymal FGF signaling could be beneficial in the treatment of IPF.
Keywords: collagen, fibroblast, fibroblast growth factor (FGF), fibroblast growth factor receptor (FGFR), lung injury, myofibroblast, pulmonary fibrosis, bleomycin
Introduction
Pulmonary fibrosis is characterized by progressive scarring of the lung, with abnormal production of extracellular matrix by activated fibroblasts, or myofibroblasts (1). In idiopathic pulmonary fibrosis (IPF),2 the cause of fibroblast activation and matrix deposition is unknown, although it is suggested to be a result of aberrant healing from an injury of unknown origin (2). IPF affects ∼50,000 patients annually in the United States, and the median survival is 3–5 years after diagnosis (1). Recently approved novel antifibrotic agents Nintedanib (Ofev) and Pirfenidone (Esbriet) decrease the rate of decline in lung function and improve survival in IPF (3, 4); however, overall outcomes remain poor.
Fibroblast growth factors (FGFs) are implicated in the pathogenesis of pulmonary fibrosis. The receptor tyrosine kinase activity of FGF receptors (FGFRs) is inhibited by Nintedanib (5, 6), which decreases bleomycin-induced fibrosis in mice (7, 8). Inhibition of FGF signaling using a soluble ectodomain of FGFR2c decreased TGF-β1 induced primary lung fibroblast proliferation and differentiation in vitro, as well as bleomycin-induced fibrosis in vivo (9). This ectodomain inhibits multiple FGFs known to bind to the IIIc splice variant of FGFR2, including FGFs 1, 2, 4, 6, 8, 9, 17, and 18 (10). A recent study demonstrated altered FGFR1 and FGF1 expression in IPF lungs, and suggested that FGF signaling is critical for fibroblast migration in pulmonary fibrosis (11). Additionally, administration of a specific inhibitor of FGFR1 (NP603) inhibits carbon tetrachloride-induced hepatic fibrosis in rats (12). Although these studies have demonstrated the importance of FGF signaling in the pathogenesis of pulmonary fibrosis, the cellular mechanisms involved remain elusive. In particular, it is not known which cell type(s) are critical targets for FGFs in the pathogenesis of pulmonary fibrosis in vivo.
In primary human lung fibroblasts, administration of TGF-β1 induced FGF receptor expression (13) and FGF2 expression and secretion into culture media (14, 15). Primary lung fibroblasts isolated from rats exposed to peplomycin, a bleomycin-related compound, have increased FGF2 production (16). TGF-β1-induced ERK, JNK, and AP-1 phosphorylation (14, 15), fibroblast differentiation into myofibroblasts, proliferation (17), and FGF2 production (18) are inhibited by FGF2-neutralizing antibodies, suggesting a cooperative mechanism between FGF2 and TGF-β1. These data suggest that FGF signaling in fibroblasts is critical in the pathogenesis of pulmonary fibrosis.
To test whether FGF signaling in lung mesenchyme and fibroblasts is required for the pathogenesis of pulmonary fibrosis in vivo, we have generated mice with a mesenchyme-specific inducible knock-out of FGF receptors 1, 2, and 3 using mice with tamoxifen-inducible Cre recombinase driven by the promoter for procollagen Iα2 (Col1α2-CreER) (19–21). Simultaneous deletion of multiple FGF receptors was performed due to overlapping receptor specificity of many FGFs, including FGF1, FGF2, and FGF9 (22), as well as simultaneous expression of multiple FGFRs in lung mesenchyme in vivo.
In this report we demonstrate that the Col1α2+ lineage is enriched in fibrotic tissue following bleomycin treatment, and that cell-autonomous FGFR signaling is required for this lineage enrichment. Deletion of FGFRs in lung mesenchyme decreases collagen expression and the development of pulmonary fibrosis in response to bleomycin. Our data suggests that inhibition of FGFR signaling pathways in fibroblasts slows the enrichment of lung mesenchyme into injured areas, providing a mechanistic rationale for the use of FGF inhibitors to slow the progression of pulmonary fibrosis.
Results
Gene expression in lung mesenchyme and fibroblasts
Mesenchymal cells were genetically tagged with a membrane-localized green fluorescent protein (GFP) by mating Col1α2-CreER mice with ROSA26mTmG Cre reporter mice (23). Tamoxifen treatment of Col1α2-CreER; ROSA26mTmG/+ mice from postnatal day 21 (P21) to P35 labels peribronchiolar smooth muscle and fibroblasts, perivascular smooth muscle, and interstitial fibroblasts (Fig. 1, A and B). Co-immunofluorescence for GFP and PDGFRα reveals that interstitial fibroblasts targeted by Col1α2-CreER are PDGFRα+ (Fig. 1C). Col1α2+ (GFP+) mesenchymal cells also express periostin and α-smooth muscle actin (αSMA) (Fig. 1, D and E), but not s100a4 (not shown) and epithelial-specific proteins such as pro-surfactant protein C (pro-SPC) (Fig. 1F). In response to bleomycin, lung mesenchymal cells targeted by Col1α2-CreER were enriched in areas of fibrosis as early as 14 days post-bleomycin treatment (Fig. 1, G and H). Col1α2+ cells in areas of fibrosis express αSMA (Fig. 1I), although not all αSMA+ cells in fibrotic areas were GFP+.
Figure 1.

Localization of the Col1α2-CreER lineage and enrichment in fibrotic areas post-bleomycin. Col1α2-CreER; ROSA26mTmG/+ mice were treated with tamoxifen at P21 and lungs were collected for frozen sections and imaged with direct fluorescence microscopy (A and B). Arrows indicate primary sites of Col1α2+ peribronchiolar, perivascular, and interstitial cells. Frozen sections were stained for PDGFRα (C), Periosin (D), αSMA (E), and pro-SPC (F), and microscopy was performed for co-localization with GFP. Tamoxifen-treated Col1α2-CreER; ROSA26mTmG/+ mice were given a single dose of intratracheal bleomycin, and lungs were analyzed after 14 (G) and 21 days (H) for GFP fluorescence. Co-immunofluorescence for αSMA and GFP (I) was performed on 21-day post-bleomycin lung sections.
Col1α2+ cells make up 3.97 ± 0.54% (n = 8) of all single cells from Col1α2-CreER; ROSA26mTmG/+ mice treated with tamoxifen (Fig. 2, A–D), and 32% of CD45, CD31, EpCAM (Lin-negative) cells (Fig. 2G). Very few Col1α2+ cells are found in the Lin-positive population (Fig. 2F). GFP+ cells were collected from tamoxifen-treated mouse lungs by cell sorting, and expression of mesenchymal-specific genes was measured using quantitative RT-PCR and normalized to GAPDH and whole lung expression. Col1α2+ mesenchymal cells were highly enriched for Col1α1, Col1α2, Acta2, Periostin, and PDGFRα (Fig. 2H).
Figure 2.

Isolation and gene expression of Col1α2+ mesenchymal cells. Tamoxifen-treated Col1α2-CreER; ROSA26mTmG/+ mouse lungs were harvested, enzymatically dissociated, and GFP+ cells were identified via flow cytometry. Single cells were identified using forward and side scatter (A and B), and GFP+ cells were identified (D). Analysis of Cre-negative ROSA26mTmG/+ mouse lungs was used to confirm gating (C). Dissociated lungs were stained with BV421-conjugated antibodies against CD31, CD45, and EpCAM (Lin) (E), and abundance of GFP+ cells was measured in both Lin-positive (F) and Lin-negative (G) populations. PE, phycoerythrin. Total RNA was purified from freshly sorted GFP+ cells and analyzed via quantitative RT-PCR for expression of Fgfr1, Col1α1, Col1α2, Acta2, Periostin, Pdgfrα, Fgfr2, Fgfr3, and Fgfr4 (H and I). GFP+ cells were cultured, and RNA was collected from passage number 2 cells for qRT-PCR analysis of Fgfr1, Fgfr2, Fgfr3, and Fgfr4 (J). qRT-PCR data were normalized to Gapdh and expressed as ΔCt. * indicates p < 0.05 compared with whole lung RNA.
Fgfr1 is expressed in mesenchymal cells and fibroblasts (22). In IPF, expression of FGFRs 1–3 is increased in myofibroblasts (11). To determine the potential for redundant function of Fgfr2, Fgfr3, and Fgfr4 in mesenchymal cells and fibroblasts, we measured Fgfr expression in freshly isolated (non-cultured) Col1α2+ mesenchymal cells. Analysis of Fgfr expression showed that Fgfr1 is the predominant FGF receptor expressed in Col1α2+ cells. Fgfr2, Fgfr3, and Fgfr4 were expressed at ∼16-, 2.15-, and 2.82-fold lower levels than Fgfr1, respectively (Fig. 2I).
When Col1α2+ mesenchymal cells were cultured on collagen-coated dishes, Fgfr1 remained the most abundant FGFR, however, expression of Fgfr2 and Fgfr3 was significantly diminished by passage number 2, and there was little measurable Fgfr4 expression (Fig. 2J). These data indicate that to completely inhibit FGF signaling in lung mesenchyme in vivo, deletion of multiple FGFRs is necessary, as they are capable of compensatory signaling if only Fgfr1 is deleted.
Generation of mesenchyme-specific FGF receptor knock-out mice
Given significant expression of multiple FGF receptors in lung mesenchyme in vivo, we generated mesenchyme-specific knock-out of Fgfrs 1, 2, and 3 using the Col1α2-CreER allele. Col1α2-CreER mice were crossed with Fgfr1f/f; Fgfr2f/f; Fgfr3f/f triple flox mice (Fgfr1/2/3 TFF). Successful generation of Col1α2-CreER; Fgfr1/2/3 TFF mice was confirmed by PCR (Fig. 3A). Treatment of Col1α2-CreER; Fgfr1/2/3 TFF mice with tamoxifen from P21 to P35 generated triple conditional knock-out mice (Col1α2-CreER; TCKO) with significant reduction in Fgfr-1, -2, and -3 expression in freshly sorted Col1α2+ mesenchymal cells (Fig. 3B). Expression of Fgfr4 was unchanged in Col1α2-CreER; TCKO mice (Fig. 3B). Baseline expression, as expressed by ΔCt, of mesenchymal-specific genes such as Col1α1, Col1α2, Col3α1, Fibronectin, Pdgfra, α-smooth muscle actin (Acta2), Periostin (Postn), Lysyl oxidase (Lox), s100a4, and connective tissue growth factor (Ctgf) were also unchanged in freshly sorted Col1α2-CreER; TCKO mesenchyme compared with controls (Fig. 3C). The predominant mesenchymal genes expressed in freshly sorted Col1α2+ cells were Col1α1 and Col3α1 (Fig. 3C).
Figure 3.

Generation of mesenchyme-specific Fgfr1/2/3 triple conditional knock-out mice. Successful generation of Fgfr1f/f; Fgfr2f/f; Fgfr3f/f (Fgfr1/2/3 TFF) mice was confirmed by PCR (A). Tamoxifen-treated Col1α2-CreER; Fgfr1/2/3 TFF; ROSA26mTmG/+ (to generate Col1a2-CreER; TCKO; ROSA26mTmG/+) and Col1α2-CreER; ROSA26mTmG/+ control mouse lungs were harvested, dissociated, and GFP+ cells were collected via cell sorting. Total RNA was purified from freshly sorted GFP+ cells and analyzed via quantitative RT-PCR for Fgfr1, Fgfr2, Fgfr3, and Fgfr4 (B), as well as Col1α1, Col1α2, Col3α1, Fibronectin, Pdgfrα, Postn, Acta2, Lox, s100a4, and Ctgf (C). Data were normalized to Gapdh and expressed as ΔCt. * indicates p < 0.05.
To determine whether lung mesenchyme from Col1α2-CreER; TCKO mice have deficient FGF signaling, sorted GFP+ cells (Col1α2+ lineage) from tamoxifen-treated Col1α2-CreER; TCKO; ROSA26mTmG/+ and Col1α2-CreER; ROSA26mTmG/+ mice were cultured and administered recombinant FGF2 (2 nm) for 48 h. Col1α2-CreER; TCKO mesenchymal cells have decreased expression of FGF-dependent genes Etv4 and Spry2 at baseline and in response to FGF2 (Fig. 4, A and B). Additionally, Etv4 expression was significantly decreased in whole lung RNA and in RNA from freshly sorted GFP+ cells from bleomycin-treated Col1α2-CreER; TCKO; ROSA26mTmG/+ mice compared with controls (Fig. 4, C and D). Western blot analysis from whole-lung protein lysates demonstrated decreased phosphorylation of ERK1/2 in bleomycin-treated Col1α2-CreER; TCKO mice compared with controls (Fig. 5, A and B). Additionally, co-immunofluorescence for phosphorylated ERK1/2 and GFP demonstrated decreased phosphorylation of ERK1/2 overall and in GFP+ cells (Fig. 5, C–J) in bleomycin-treated Col1α2-CreER; TCKO; ROSA26mTmG/+ mice compared with Col1α2-CreER; ROSA26mTmG/+ controls, indicating decreased FGFR-dependent activation of p42/44 MAPK pathways. These data show that mesenchymal cells from Col1α2-CreER; TCKO mice have deficient FGF-induced signaling and gene expression.
Figure 4.
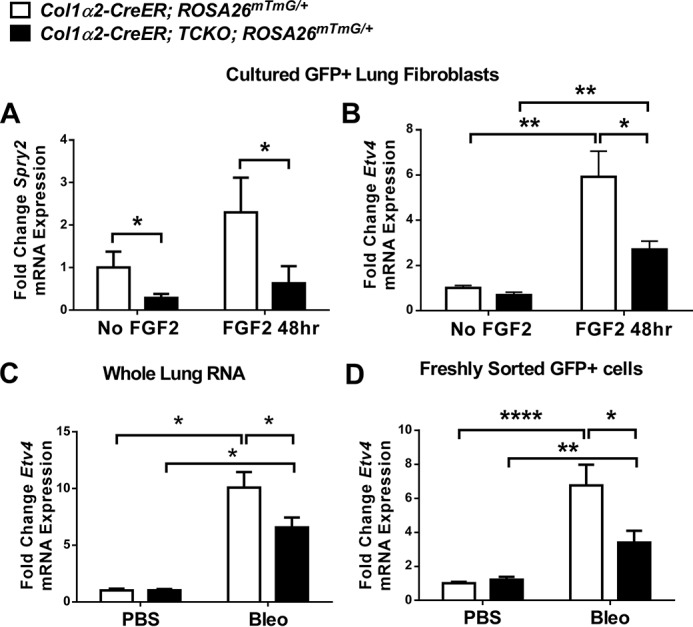
Decreased downstream FGFR signaling in Col1a2-CreER; TCKO mesenchymal cells. Col1α2-CreER; TCKO; ROSA26mTmG/+ and Col1α2-CreER; ROSA26mTmG/+ mouse lungs were harvested, dissociated, and GFP+ cells were collected via FACS and cultured for 2 passages. Cultured mesenchymal cells were treated with recombinant FGF2 (2 nm) for 48 h and total RNA was collected for quantitative RT-PCR analysis of and Spry2 (A) and Etv4 (B). Col1a2-CreER; TCKO; ROSA26mTmG/+ and Col1α2-CreER; ROSA26mTmG/+ mice were treated with intratracheal bleomycin, RNA was collected from whole lungs (C) or GFP+ cells isolated via FACS (D), and analyzed by qRT-PCR for Etv4 expression. Data were normalized to Gapdh and expressed as fold-change from untreated control. * indicates p < 0.05; ** indicates p < 0.005; **** indicates p < 0.0001.
Figure 5.

Decreased ERK1/2 phosphorylation in Col1a2-CreER; TCKO mice. Col1α2-CreER; TCKO and control Fgfr1/2/3 TFF mice were administered a single dose of intratracheal bleomycin (1.2 units/kg) or PBS. Total protein was isolated from whole lungs 21 days post-bleomycin, and analyzed by Western blotting for phosphorylated ERK1/2, total ERK1/2, and tubulin (A). Densitometric analysis was performed on n = 3–4 replicates, and pERK1/2:total ERK1/2 ratios were normalized to tubulin and expressed as fold-change from PBS-treated Fgfr1/2/3 TFF control mice (B). * indicates p < 0.05. C–J, lungs from 21 day post-bleomycin Col1α2-CreER; TCKO; ROSA26mTmG/+ and Col1α2-CreER; ROSA26mTmG/+ mice were inflation-fixed with 4% PFA (20 cm of H2O pressure), and frozen sections were stained for phosphorylated ERK1/2. C and G, confocal images were obtained at ×40 magnification for GFP (green), ERK1/2 (red), and DAPI (blue). Higher magnifications of areas indicated by dotted lines are shown for control (D–F) and TCKO (H–J) mice. Arrows indicate phospho-ERK1/2 positive cells. ns, not significant.
Decreased bleomycin-induced pulmonary fibrosis in mesenchyme-specific FGFR knock-out mice
To test whether deletion of FGF receptors in mesenchyme alters the development of pulmonary fibrosis in response to bleomycin, Col1α2-CreER; TCKO mice and Cre-negative Fgfr1/2/3 TFF controls were treated with tamoxifen (to generate Col1α2-CreER; TCKO and control mice) and subsequently administered a single dose of intratracheal bleomycin (1.2 units/kg). Col1α2-CreER; TCKO mice had attenuated weight loss post-bleomycin (Fig. 6A) and improved survival (Fig. 6B). Histologic analysis revealed significantly decreased bleomycin-induced pulmonary fibrosis in Col1α2-CreER; TCKO mice compared with triple flox/flox (TFF) controls 21 days after bleomycin on H&E-stained sections (Fig. 6D), by Ashcroft scoring (Fig. 6C), and on Masson trichrome-stained sections (Fig. 6F). Quantitative hydroxyproline measurements revealed decreased collagen deposition in Col1α2-CreER; TCKO mice 21 days post-bleomycin (Fig. 6E). There was no difference in histologic fibrosis or hydroxyproline content between Cre-negative TFF controls and mice containing only Col1α2-CreER (Cre controls) in response to bleomycin (not shown).
Figure 6.

Decreased bleomycin-induced fibrosis in Col1a2-CreER; TCKO mice. Col1α2-CreER; TCKO and control Fgfr1/2/3 TFF mice were administered a single dose of intratracheal bleomycin (1.2 units/kg) or PBS. Mice were monitored for weight loss (A) and survival (B) over a 21-day period. * indicates p < 0.05 via two-way analysis of variance. Survival was analyzed using Kaplan-Meier survival analysis, * indicates p < 0.05. Mouse lungs were collected 21 days post-bleomycin for Ashcroft fibrosis scoring (C), H&E staining (D), quantitative hydroxyproline analysis (E), and Masson's trichrome staining (F). To measure the number of αSMA+ myofibroblasts, bleomycin-treated Col1α2-CreER; TCKO and control Col1α2-CreER mouse lungs were collected after 14 days, enzymatically dissociated, fixed, permeabilized, stained for αSMA, and analyzed by flow cytometry (G). * indicates p < 0.05; ** indicates p < 0.005. ns, not significant.
To test whether mesenchyme-specific deletion of FGFRs is required for bleomycin-induced expansion of myofibroblasts, lungs from Col1α2-CreER; TCKO and Col1α2-CreER control mice treated with intratracheal bleomycin were enzymatically dissociated 14 days after bleomycin, fixed, permeabilized, stained with A647-conjugated antibodies against αSMA, and analyzed via flow cytometry. Compared with controls, Col1α2-CreER; TCKO mice had fewer bleomycin-induced αSMA+ cells (Fig. 6G), indicating decreased numbers of myofibroblasts in mice lacking mesenchymal Fgfrs-1, -2, and -3.
To determine whether bleomycin-induced expression of collagen and other pro-fibrotic genes were altered in Col1α2-CreER; TCKO mice, whole lung mRNA was collected 21 days after bleomycin treatment. Quantitative RT-PCR analysis revealed decreased expression of collagen genes Col1α1 and Col3α1 in Col1α2-CreER; TCKO mice compared with TFF controls (Fig. 7, A and B). No significant difference in Col1α2 was measured between bleomycin-treated control and TCKO mice, although control mice, but not TCKO mice, demonstrated a significant increase in Col1α2 expression in response to bleomycin compared with PBS treatment (Fig. 7C). Col1α2-CreER; TCKO mice had decreased bleomycin-induced expression of several pro-fibrotic genes compared with controls, including Ctgf, Lox, and Serpine-1a (Pai1) (Fig. 7, D–F). There was no significant difference in expression of Postn, IL-6, vimentin, or fibronectin in bleomycin-treated Col1α2-CreER; TCKO mice compared with Fgfr1/2/3 TFF controls (Fig. 7, G and H, and data not shown).
Figure 7.

Decreased pro-fibrotic gene expression in Col1a2-CreER; TCKO mice after bleomycin treatment. Col1α2-CreER; TCKO and control Fgfr1/2/3 TFF mice were administered a single dose of intratracheal bleomycin (1.2 units/kg) or PBS and mouse lungs were collected 21 days post-bleomycin for whole lung RNA. Quantitative RT-PCR was performed for Col1α1 (A), Col3α1 (B), Col1α2 (C), Ctgf (D), Lox (E), Pai1 (F), Postn (G), and IL-6 (H). Data were normalized to Gapdh and expressed as fold-change from PBS-treated Fgfr1/2/3 TFF controls. * indicates p < 0.05; ** indicates p < 0.005. ns, not significant.
Reduced collagen expression in fibroblasts lacking Fgfrs
We then tested whether there was cell-autonomous FGFR-dependent gene expression in lung mesenchyme following bleomycin treatment. Col1α2-CreER; TCKO; ROSA26mTmG/+ and Col1α2-CreER; ROSA26mTmG/+ mice were administered intratracheal bleomycin, and lungs were dissociated for cell sorting 21 days post-bleomycin. GFP+ cells were sorted directly into RNA lysis buffer, and analyzed by qRT-PCR. Freshly sorted GFP+ cells from bleomycin-treated Col1α2-CreER; TCKO; ROSA26mTmG/+ mice have decreased Col1a1 expression compared with Col1α2-CreER; ROSA26mTmG/+ controls (Fig. 8A), but unchanged Col3α1, Col1α2, and Ctgf expression (Fig. 8, B–D). GFP+ TCKO cells had decreased Col3α1 expression following PBS treatment (Fig. 8B), and no significant change in Lox and Pai1 expression (Fig. 8, E and F). GFP+ cells from control mice, but not TCKO mice, demonstrated a significant increase in Lox and Pai1 expression in response to bleomycin compared with PBS treatment (Fig. 8, E and F). Additionally, GFP+ cells from Col1α2-CreER; TCKO; ROSA26mTmG/+ mice had significantly decreased bleomycin-induced Postn expression (Fig. 8G), and an attenuated reduction in Pdgfra in response to bleomycin (Fig. 8H).
Figure 8.

Decreased collagen expression in purified Col1a2-CreER; TCKO lung mesenchymal cells. Col1α2-CreER; TCKO; ROSA26mTmG/+ and control Col1α2-CreER; ROSA26mTmG/+ mice were treated with a single dose of intratracheal bleomycin (1.2 units/kg) or PBS. A, lungs were harvested and dissociated 21 days post-bleomycin, and GFP+ cells were collected via cell sorting directly into RNA lysis buffer. RNA purified from GFP+ cells was analyzed by quantitative RT-PCR for Col1α1 (A), Col3α1 (B), Col1α2 (C), Ctgf (D), Lox (E), Pai1 (F), Postn (G), and Pdgfra (H). Data were normalized to Gapdh and expressed as fold-change from PBS-treated controls. * indicates p < 0.05; ** indicates p < 0.005; *** indicates p < 0.001; **** indicates p < 0.0001. ns, not significant.
Freshly sorted GFP+ cells from Col1α2-CreER; TCKO; ROSA26mTmG/+ and Col1α2-CreER; ROSA26mTmG/+ mice were also cultured, serum-starved, and treated with 2 ng/ml of TGF-β1 for 48 h. GFP+ TCKO mesenchymal cells show reduced Col1α1 expression compared with controls in response to TGF-β1 (Fig. 9A), but unchanged phosphorylation of Smad2 (Fig. 9, B and C).
Figure 9.
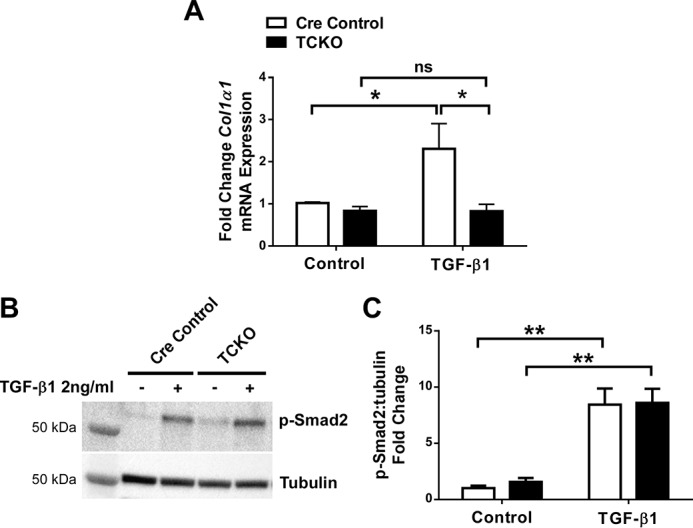
Decreased collagen expression in response to TGF-β1 in cultured Col1α2-CreER; TCKO lung mesenchymal cells. GFP+ cells from uninjured Col1α2-CreER; TCKO; ROSA26mTmG/+ and control Col1α2-CreER; ROSA26mTmG/+ mice were collected by cell sorting and grown in culture. After 2 passages, cells were serum-starved overnight and treated with TGF-β1 (2 ng/ml). RNA was collected from cells 48 h after treatment, and analyzed by quantitative RT-PCR for Col1α1 (A). Data were normalized to Gapdh and expressed as fold-change from PBS-treated controls. B and C, total protein, collected after 1 h of treatment, was analyzed by Western blot analysis for phosphorylated Smad2 and tubulin. Densitometric analysis was performed on n = 6 replicates, and pSmad2 was normalized to tubulin and expressed as fold-change from control (C). * indicates p < 0.05; ** indicates p < 0.005. ns, not significant.
Mesenchyme-specific FGFR KO mice demonstrate decreased enrichment of the Col1α2+ lineage in fibrotic areas
We observed fewer GFP+ cells in bleomycin-treated Col1α2-CreER; TCKO; ROSA26mTmG/+ lungs, as seen in Fig. 5, G–J, and hypothesized that the mechanism by which Col1α2-CreER; TCKO mice have decreased fibrosis and myofibroblasts after bleomycin injury is through impaired lineage expansion of Col1α2+ mesenchyme into areas of fibrosis. To test this, Col1α2-CreER; TCKO; ROSA26mTmG/+ and Col1α2-CreER; ROSA26mTmG/+ control mice were provided a 4-week washout period post-tamoxifen, and administered intratracheal PBS or bleomycin. The Col1α2+ lineage was initially evaluated using GFP immunofluorescence and co-localization with αSMA 21 days post-bleomycin (Fig. 10, A–H). In bleomycin-treated Col1α2-CreER; TCKO; ROSA26mTmG/+ mice, there were fewer GFP+ cells in fibrotic areas, and fewer αSMA+ cells throughout the lungs compared with controls (Fig. 10, A–E, also see Figs. 5, C–J, and 6G). To better determine αSMA+ expression in GFP+ TCKO mesenchymal cells, dissociated lungs from bleomycin-treated Col1α2-CreER; TCKO; ROSA26mTmG/+ and Col1α2-CreER; ROSA26mTmG/+ mice were stained with A647-conjugated anti-αSMA antibodies and analyzed by flow cytometry. PBS-treated TCKO mice had unaltered abundance of αSMA+ cells in both GFP+ and GFP− populations (Fig. 10, I and J). Following bleomycin treatment, there were fewer αSMA+, GFP− cells in TCKO mice but unchanged αSMA+, GFP+ cells in TCKO lungs compared with controls (Fig. 10, I and J), suggesting an indirect effect of FGFR signaling on αSMA expression following bleomycin.
Figure 10.

Altered αSMA expression in Col1a2-CreER; TCKO mice following bleomycin treatment. Col1α2-CreER; TCKO; ROSA26mTmG/+ and control Col1α2-CreER; ROSA26mTmG/+ mice were administered a single dose of intratracheal bleomycin (1.2 units/kg) or PBS. (A–H) Sections from lungs collected 21 days post-bleomycin were stained for αSMA and GFP. A and E, confocal images were obtained at ×20 magnification for GFP (green), αSMA (red), and DAPI (blue). Higher magnifications of areas indicated by dotted lines are shown for control (B–D) and TCKO (F–H) mice. Arrows indicate αSMA+ cells. I and J, lungs were dissociated, fixed, permeabilized, stained with BV421-conjugated antibodies against CD45, CD31, and EpAM (Lin) as well as A647-conjugated anti-αSMA antibodies, and analyzed by flow cytometry. Lin-negative cells were analyzed for expression of GFP and αSMA, and the percentage of Lin-negative cells that are αSMA+, GFP- (I) and αSMA+, GFP+ (J) are shown. * indicates p < 0.05. ns, not significant.
To quantify the abundance of GFP-labeled mesenchymal cells in Col1α2-CreER; TCKO; ROSA26mTmG/+ mouse lungs compared with controls, GFP immunohistochemistry in areas with similar amounts of fibrosis were compared. The Col1α2+ lineage had decreased enrichment in areas of fibrosis following bleomycin in TCKO mice (Fig. 11, A–C). Additionally, the overall abundance of GFP+ cells, as a percentage of total lung single cells detected using flow cytometry, was decreased in TCKO mice compared with controls (Fig. 11D). The overall abundance of GFP+ cells did not significantly increase following bleomycin compared with PBS controls (not shown), consistent with prior reports (24). We then assessed proliferation in response to bleomycin in TCKO mice by EdU labeling prior to lung collection, enzymatic dissociation, and cell sorting. Col1α2-CreER; TCKO mice have decreased numbers of EdU+ cells within the overall single cell population and in the Lin-neg mesenchymal population (Fig. 11, E and F), demonstrating decreased proliferation in mesenchyme after bleomycin-induced injury. We then measured growth in cultures of Lin-neg, GFP+ cells isolated from Col1α2-CreER; TCKO; ROSA26mTmG/+ and control Col1α2-CreER; ROSA26mTmG/+ mice 21 days after bleomycin. Compared with controls, Col1α2-CreER; TCKO; ROSA26mTmG/+ mesenchymal cells demonstrate decreased growth over a 9-day period (Fig. 11G). This demonstrates that FGFR signaling is required for proliferation and expansion of a pre-existing mesenchymal population that contributes to bleomycin-induced pulmonary fibrosis.
Figure 11.

Decreased proliferation and enrichment of Col1α2+ lineage in fibrotic areas in bleomycin-treated Col1a2-CreER; TCKO mice. Col1α2-CreER; TCKO; ROSA26mTmG/+ and control Col1α2-CreER; ROSA26mTmG/+ mice were administered a single dose of intratracheal bleomycin (1.2 units/kg) and lungs were collected 21 days post-bleomycin. Immunohistochemistry for GFP was performed (A and B) and the percentage of total fibrotic areas that were GFP+ was analyzed using ImageJ (C). Whole lungs were dissociated and the percentage of total live single cells that were GFP+ following bleomycin treatment were analyzed by flow cytometry (D). Proliferation was measured by injecting mice with EdU (50 mg/kg i.p.) prior to collection, and dissociated lungs were stained for CD45, CD31, and EpCam (Lin), fixed, permeabilized, and then stained for EdU incorporation. Stained cells were measured by flow cytometry, and EdU incorporation within all single cells (D) or Lin-negative cells (E) was analyzed. GFP+ cells from bleomycin-treated Col1α2-CreER; TCKO; ROSA26mTmG/+ and control Col1α2-CreER; ROSA26mTmG/+ mice were sorted into 96-well plates (2,000 cells/well) and cultured for 48 h, 5 days, 7 days, and 9 days prior to measurement of total cellular DNA using a CyQuant assay (F). * indicates p < 0.05; ** indicates p < 0.005. ns, not significant.
Discussion
In this report, we demonstrate that FGFRs-1, -2, and -3 in Col1α2+ lung mesenchyme and fibroblasts are required for bleomycin-induced pulmonary fibrosis and collagen production. Furthermore, we showed that FGFR signaling is required for bleomycin-induced collagen expression in freshly isolated fibroblasts. Additionally, intact FGFR signaling in lung mesenchyme is required for lineage-specific expansion of fibrotic tissue in response to bleomycin.
FGF signaling has been implicated in the pathogenesis of pulmonary fibrosis, as non-selective inhibition of receptor tyrosine kinases (7) and nonspecific inhibition of FGF receptors (9, 25) decrease bleomycin-induced pulmonary fibrosis in rodents. FGFR signaling (in addition to VEGFR and PDGFR) is inhibited by Nintedanib, which decreases the rate of decline in lung function in IPF patients (3, 4). The mechanism by which inhibition of multiple receptor tyrosine kinases decreases the progression of pulmonary fibrosis remains unclear. Nintedanib reduces fibronectin and collagen expression, induces autophagy, and inhibits TGFβ signaling in IPF lung fibroblasts (26). Our data demonstrates that there is a cell-autonomous role for FGFR signaling in pulmonary fibrosis that promotes expansion of lung fibroblasts into injured areas within the lung. A recent report by MacKenzie et al. (11) described increased migration of fibroblasts in response to FGF1, further supporting our findings that FGF signaling is required for fibroblast expansion within fibrotic areas.
Our studies use bleomycin as a model agent to induce fibrosis. One percent of patients treated with bleomycin develop pulmonary fibrosis (27). In mice, bleomycin causes alveolar epithelial injury, inflammation, and fibroblast proliferation and differentiation into myofibroblasts. Although bleomycin does not cause lesions characteristic for IPF (fibroblastic foci and honeycombing) in the mouse lung, bleomycin remains a well established and useful model to study underlying mechanisms of alveolar epithelial cell injury and subsequent fibroblast activation and fibrosis (27, 28, 30, 31).
Col1α2-CreER mice have been used previously to inactivate genes in lung mesenchyme that are important for the development of pulmonary fibrosis, including Tert (32), smoothened (33), TgfβR1 (19), Notch1 (34), and CEBPβ (20). It is important to note that this allele targets multiple mesenchymal populations, and it is difficult to assess which population within the Col1α2+ lineage directly contributes to fibrotic areas in the lung. Furthermore, we have found that cultured lung fibroblasts generated from enzymatically dissociated Col1α2-CreER; ROSA26mTmG/+ whole lungs, without enrichment for GFP+ cells via cell sorting, results in a heterogeneous population of cells, with only 50–60% of the cells being GFP+ (not shown). Incorporating the ROSA26mT/mG Cre reporter allele with the Col1α2-CreER allele allows for direct analysis of lineage labeled and genetically manipulated mesenchymal cells both in vivo and in vitro.
This study underscores the importance of studying fibroblasts in vivo, as we have found that Fgfr expression changes significantly once lung fibroblasts are placed in culture, even after a single passage. We demonstrate significant expression of all Fgfrs in freshly isolated lung mesenchyme; however, only Fgfr1 is expressed when isolated fibroblasts are cultured. Low levels of Fgfr3 and Fgfr4 expression in cultured lung fibroblasts has previously been reported (35). FGFRs 1–4 were also found to be expressed in myofibroblasts of fibroblastic foci in human IPF lung biopsies (11). The mechanism(s) responsible for this change in Fgfr expression is not known; however, altered cellular phenotypes and gene expression as a result of culture conditions, including substrate stiffness and coating of culture dishes have been well described (36).
Simultaneous deletion of multiple FGF receptors was performed due to 1) overlapping receptor specificity of many FGFs to FGFRs 1–3, including FGF1, FGF2, and FGF9 (22), and 2) simultaneous expression of multiple Fgfrs in lung mesenchyme in vivo (Fig. 2). Although mesenchymal cells with deletion of FGFRs 1–3 demonstrate a functional deficiency of FGF signaling, it remains possible that FGFR4 could further contribute to lung fibroblast pathophysiology. Targeting Fgfr4 in lung mesenchyme and fibroblasts is an area of future study. Additionally, the individual roles of FGFR1, FGFR2, and FGFR3 in lung mesenchyme following injury and in the development of pulmonary fibrosis is not known, and future studies will be needed to determine the relative contribution of individual FGFRs. Mice expressing a soluble dominant-negative FGFR2 in late gestation, but not postnatally, causes airspace enlargement and emphysema (37). FGFR3 may represent a significant FGFR in adult lung pathophysiology. FGFR3 and FGFR4 have been shown to be critical for early postnatal alveologenesis (35, 38), and mice with combined deficiencies of Fgfr3 and Fgfr4 have excessive elastic fiber deposition; however, their importance in lung injury and pulmonary fibrosis are not well described. Recent reports have also implicated FGFR3 in the pathogenesis of lung cancer (39–41).
Although mesenchyme-specific FGFR signaling is important for bleomycin-induced pulmonary fibrosis, the endogenous FGF ligand(s) that mediate this pathology remains unclear. FGF1 and FGF2 are capable of activating most FGF receptors, including FGFR1, the mesenchyme-specific FGFR2c spice variant, and FGFR3 (10, 22). Inhibition of FGFR2c ligands by expressing a soluble FGFR2c ectodomain (25) decreases bleomycin-induced fibrosis, and whereas this implicates FGF signaling in the pathogenesis of fibrosis, it does not point to a single ligand, as FGFR2c interacts with multiple ligands, including FGF1, FGF2, FGF8, FGF9, and FGF18 (10). We recently reported that Fgf2 knock-out mice have preserved fibrosis in response to bleomycin (42), which suggests that other FGF ligands are either required for bleomycin-induced fibrosis or compensate for the absence of FGF2. A combined knock-out of FGF1 and FGF2 has decreased carbon tetrachloride-induced hepatic fibrosis (43), but the response of these mice to intratracheal bleomycin is not known. FGF1 is increased in IPF lungs, and is capable of increasing fibroblast migration in vitro (11). FGF9 and FGF18 are increased in IPF, stimulate fibroblast migration, and decrease fibroblast apoptosis (44, 45). Inhibition of endogenous FGFR2b ligands (FGF7 and FGF10) does not alter bleomycin-induced pulmonary fibrosis (46), suggesting that these ligands are not essential for fibrosis. As the FGF ligand(s) critical for bleomycin-induced fibrosis remain unclear, further examination of the critical FGFRs are likely to provide insight to the ligands involved, as FGF-FGFR specificity is well described (22). Furthermore, pharmacologic targeting of FGFRs may be more efficacious than targeting specific FGF ligands, particularly given overlap in ligand-receptor interactions and as multiple FGF ligands are likely involved in fibrosis. Furthermore, targeting FGFR tyrosine kinases may potentially be done in a cell-type specific manner. Continued studies using Col1α2-CreER to inactivate individual Fgfrs will determine which receptor is most critical for bleomycin-induced pulmonary fibrosis, and will allow for improved therapeutics targeted against fibroblasts contributing to fibrotic diseases.
FGF signaling in other cell types may also contribute to the development of fibrosis. A recent report suggested that endothelial FGFR1 signaling is required for hepatic fibrosis (47), as endothelial-specific deletion of FGFR1 lessened liver fibrosis in response to bile duct ligation. The role of endothelium and angiogenesis, and in particular endothelial-specific FGF signaling, in pulmonary fibrosis is not well understood (48) and warrants further study.
We show that inactivation of FGFRs in mesenchymal cells decreases collagen expression in response to bleomycin and TGF-β1. This appears to be a cell-autonomous effect, as collagen expression is altered in isolated Cre-expressing mesenchymal cells following bleomycin treatment. These data also support prior reports where FGF2 signaling is required for TGF-β1 induced collagen expression in lung fibroblasts (14, 15, 17). Other studies have demonstrated an inhibitory effect of FGFs on pulmonary fibrosis and TGF-β1-induced myofibroblast differentiation (44, 45, 49–52); however, those studies use exogenous or overexpressed FGF ligands and no study to date has demonstrated an inhibitory role of endogenous FGF ligands on pulmonary fibrosis in vivo.
Our data also suggests that deletion of FGF receptors leads to a cell non-autonomous effect on mesenchymal cells that are not targeted by Col1α2-CreER. Deletion of FGFRs in the Col1α2+ lineage leads to alteration in the abundance of GFP-negative, αSMA+ cells as well as expression of Col3α1, Ctgf, Lox, and Pai1 in whole lung, but not GFP+ (Col1α2+ lineage) mesenchymal cells. Future studies will investigate the factor(s) involved in this interaction, as well as the cell types involved. It should be noted that our experiments inactivated FGFRs prior to lung injury, and future studies will also address whether inactivation of FGFRs in the Col1α2+ lineage post-bleomycin has similar cell autonomous and non-autonomous effects. Additional studies are needed to determine the mechanistic role of FGFRs in multiple phases of fibroblast activation following injury and in response to pro-fibrotic growth factors such as TGF-β1, including migration, metabolism, differentiation into myofibroblasts, and recovery from injury.
In summary, this study provides direct evidence that deletion of FGFRs in lung mesenchyme in adult mice decreases pulmonary fibrosis, reduces fibroblast-specific collagen expression, and decreases fibroblast enrichment in fibrotic areas in response to bleomycin. This study provides a mechanistic rationale for the use of FGF inhibitors to slow the progression of pulmonary fibrosis. Improved understanding of the contributions of FGF and other growth factor pathways to the development of fibrosis remains important, and future studies are needed to determine fibroblast-specific, FGFR-dependent pathways amenable to therapeutic intervention for improved therapy in IPF and other fibrotic diseases.
Experimental procedures
Animal care and use
Mice were housed in a pathogen-free barrier facility and handled in accordance with standard protocols, animal welfare regulations, and the NIH guide for the Care and Use of Laboratory Animals. All procedures complied with the standards for the care and use of laboratory animals as stated in the Guide or the Care and Use of Laboratory Animals (NIH publication No. 85-23, revised 1996), and all protocols were approved by the Animal Studies Committee at Washington University School of Medicine and at the University of Chicago.
Experiments were carried out using Col1α2-CreER mice (53) mated to mice containing floxed alleles for FGF receptors 1, 2, and 3 (Fgfr1f/f; Fgfr2f/f; Fgfr3f/f) (54, 55) referred to as TFF. To maintain this line of mice, male Col1α2-CreER; Fgfr1f/f; Fgfr2f/f; Fgfr3f/f mice were mated to female Fgfr1f/f; Fgfr2f/f; Fgfr3f/f; ROSA26mTmG/mTmG mice. Cre-negative TFF littermates and Cre+ mice without floxed Fgfr alleles were used as controls. Mice containing the Cre reporter allele and the ROSA26mTmG allele allow for identification of targeted fibroblasts after treatment with tamoxifen. After treatment with tamoxifen, these mice are referred to as triple conditional knock-out mice (Col1α2-CreER; TCKO). All mice were maintained in a C57BL/6J;129X1 mixed background.
Bleomycin-induced lung fibrosis
Mice were fed tamoxifen chow for 14 days starting at 3 weeks of age, and subsequently fed regular chow. Adult mice between 8 and 10 weeks of age were sedated, orally intubated, and administered a single dose of intratracheal bleomycin (1.2 units/kg) (Sigma) in sterile phosphate-buffered saline (PBS) or PBS alone. Mice were monitored daily and weight was measured twice weekly. Mice with >25% weight loss or significant respiratory distress were humanely euthanized and treated as a death. At various times after treatment, mice were euthanized with an overdose of a mixture containing ketamine and xylazine, and exsanguinated by cutting the abdominal aorta. Lungs were then slowly perfused with 5 ml of PBS via the right ventricle. The trachea was dissected and cannulated, and lungs were fixed via intra-tracheal inflation with 10% phosphate-buffered formalin at a pressure of 20 cm H2O for 10 min, followed by immersion in fresh fixative for 24 h at room temperature. Samples were then dehydrated in ethanol and xylene, embedded in paraffin, cut in 5-μm sections, and stained with hematoxylin and eosin (H&E) and Masson's trichrome.
Fibrosis scoring
Ashcroft scoring was performed as described previously (29). Briefly, all lobes were sectioned on a single slide, and 20 non-overlapping trichrome-stained ×10 fields were scored per sample, and the average score of all fields indicates the Ashcroft score of a single sample. The scoring scale was as follows: 0 = no abnormalities, 1 = slight thickening of alveolar membranes, 2 = small areas of fibrosis (<10%), 3 = 10–20% fibrotic area, 4 = 20–40% fibrotic area, 5 = 40–60% fibrosis, 6 = 60–80% fibrosis, 7 = >80% fibrosis, and 8 = complete fibrosis.
Whole slide scanning
Digital scanning of whole slides was performed using a Nanozoomer 2.0 HT digital slide scanner (Hamamatsu, Bridgewater, NJ), available through the Washington University Hope Center Alafi Neuroimaging Lab (NIH Shared Instrumentation Grant (S10 RR027552)), or using an Aperio ScanScope XT through the University of Chicago Human Tissue Resource Center.
Hydroxyproline assay
Left lung samples were transferred into glass tubes and hydrolyzed with 200 μl of 6 n HCl at 110 °C for 48 h. The hydrolyzed samples were evaporated to dryness to remove excess HCl, reconstituted with 400 μl of H2O, and filtered in 1.5-ml centrifuge tubes equipped with a 0.45-μm semipermeable membrane filter. Samples were added to a 96-well microplate, chloramine T solution was added and the plate incubated at room temperature for 20 min. 100 μl of Erlich's reagent was added to each well and the plate was incubated at 65 °C for 18 min. This method gives an orange-red color that is linear up to 6 μg of hydroxyproline. A550 nm was obtained, and compared with a hydroxyproline standard curve.
Immunohistochemistry and immunofluorescence
Five-μm sections were prepared from paraffin-embedded tissues, and deparaffinized with xylene. Endogenous peroxidase was inhibited by incubation with 3% H2O2 in methanol, and sections were then re-hydrated. Antigen retrieval was performed using a pressure cooker and citrate buffer (pH 6.0). Samples were incubated in PBS, and blocked in PBS + 0.2% Triton X-100 + 0.1% BSA + 5% serum. Primary antibodies for GFP (GFP-1010, Aves Inc, Tigard, OR), αSMA (M0851, Dako North America, Carpinteria, CA), PDGFRa (sc-338, Santa Cruz Biotechnology, Dallas, TX), pro-SPC (ab90716, Abcam, Cambridge, UK), s100a4 (ab27957, Abcam), and periostin (ab14041, Abcam) were added to blocking buffer and slides were incubated overnight at 4 °C. Slides were washed with PBS, and for immunohistochemistry studies a biotin-conjugated secondary antibody was added, followed by streptavidin-HRP. Colorimetric reaction was performed using DAB staining (Vector Labs, Burlingame, CA), and sections were counterstained with hematoxylin. Immunofluorescent imaging was performed using a Zeiss Apotome or a Marianas Tokogawa spinning disk confocal microscope, and image processing was performed using Zeiss Axioplan and ImageJ (NIH, Bethesda, MD).
Quantification of GFP immunohistochemistry
Paraffin sections from PBS- and bleomycin-treated mice were deparaffinized and stained with an anti-GFP antibody and colorimetric reaction was performed using DAB staining. 10–15 non-overlapping ×10 fields containing areas of tissue fibrosis were analyzed using ImageJ software to determine the percentage of total fibrotic area that was stained for GFP.
RNA isolation and quantitative real-time PCR
RNA was isolated from whole lungs or whole left lungs via homogenization in TRIzol, and subsequent purification using RNeasy spin columns (74104, Qiagen, Valencia, CA), with on-column DNA digest, per the manufacturer's instructions. RNA concentration was determined utilizing a Nanodrop spectrophotometer. cDNA was made using the Bio-Rad iScript Reverse Transcription Supermix for RT-qPCR kit (170-8841, Bio-Rad). Quantitative RT-PCR was performed on an Applied Biosystems StepOne thermocycler using ABI TaqMan® Fast Advaned Master Mix (number 4444557, Applied Biosystems, Foster City, CA) and TaqMan® gene expression assays. All samples were normalized to Gapdh and then scaled relative to controls using the standard ΔCt method. Data are reported as ΔCt or fold-change relative to wild type PBS-treated mice.
Protein extraction and Western blotting
Protein was extracted from whole left lung or cultured fibroblasts via homogenization in RIPA buffer with freshly added 2% Protease Inhibitor Mixture (P8340, Sigma) and Phosphatase Inhibitor Mixture I and II (P2850 and P5726, Sigma). Protein concentration was determined utilizing a Pierce BCA assay kit (23225, Thermo Scientific, Rockford, IL). Total protein (50–100 μg) was separated on 4–12% polyacrylamide gels (Bio-Rad) and transferred to PVDF membranes using a Trans-Blot Turbo transfer system (Bio-Rad). Membranes were blocked for 1 h at room temperature with gentle shaking in TBST (50 mm Tris, pH 7.4, 150 mm NaCl, 0.1% Tween 20) containing 5% nonfat milk or 5% BSA, and then probed with primary antibodies against phospho-ERK1/2 (number 4376, Cell Signaling Technology, Danvers, MA), ERK1/2 (Cell Signaling number 9102), tubulin (Abcam number ab6046), and phospho-Smad2 (Cell Signaling number 3101) overnight at 4 °C. After three rinses in TBST, membranes were incubated for 1 h at room temperature in horseradish peroxidase-linked secondary antibodies in TBST with 5% nonfat milk, rinsed again in TBST, and developed using SuperSignal West Femto Substrate (34096, Thermo Scientific). Protein bands were quantified using Image Lab (Bio-Rad), normalized to tubulin, and scaled relative to control samples set at a value of 1.
Flow cytometry and cell sorting
Mice were euthanized with an overdose of a mixture containing ketamine and xylazine, and exsanguinated by cutting the abdominal aorta. Lungs were then slowly perfused with 5 ml of cold PBS via the right ventricle. The trachea was dissected and cannulated, and lungs were inflated with DMEM digest media containing LiberaseTM (Roche Diagnostics), hyaluronidase (Sigma), elastase (Worthington Biochemical, Lakewood, NJ), and DNase (Sigma). Inflated lungs were transferred to digest media and placed on ice until all mice were dissected. Lungs were then minced with razor blades and incubated in digest medium at 37 °C for 30 min with agitation. Digest enzymes were inactivated with DMEM with 10% FBS, and samples were filtered with a 100-μm strainer. Red blood cells were then lysed with ACK buffer (Thermo Scientific) and cells were resuspended in FACS buffer (PBS + 3% FBS + 1 mm EDTA). Cells were stained with BV-421-conjugated antibodies against CD45, CD31, and CD326 (EpCAM) (BioLegend, San Diego, CA). GFP+ cells were sorted using a Sony Synergy (Washington University) or FACSAria II (University of Chicago), using a 100-μm diameter nozzle. For culture, cells were sorted directly into complete media containing 10% FBS. For RNA isolation, between 50,000 and 100,000 cells were sorted directly into RLT lysis buffer with β-mercaptoethanol, and RNA was purified using an RNeasy Plus Micro Kit (Qiagen).
For detection of αSMA+ myofibroblasts via flow cytometry, dissociated cells were stained with BV-421-conjugated antibodies against CD45, CD31, and CD326 (EpCAM), and subsequently fixed with 4% paraformaldehyde. Fixed cells were washed with PBS + 1% BSA + 0.1% sodium azide and permeabilized using the BD cytofix/cytoperm kit (BD Biosciences). Fixed cells were stained with A647-conjugated anti-αSMA antibodies (number NBP2–34522AF647, Novus Biologicals, Littleton, CO), washed, and measured using a BD LSR II flow cytometer. Data were analyzed using FlowJo software.
Lung fibroblast isolation and culture
GFP-labeled cells were sorted directly into culture media, and subsequently cultured on collagen-coated plates in DMEM with 10% FBS, penicillin/streptomycin, fungizone, l-glutamine, and HEPES. Cells were grown in a humidified incubator at 37 °C with 5% CO2. Media was replaced every other day, and cultured fibroblasts were used for experiments between passages 2 and 5.
In vivo proliferation assay
Prior to collection, mice were given an intraperitoneal injection of EdU (50 mg/kg, Thermo Fisher). Mouse lungs were enzymatically dissociated as described above, and cells were stained with BV-421-conjugated antibodies against CD45, CD31, and CD326 (EpCAM) (BioLegend). Cells were then fixed, permeabilized, and stained for EdU using the Click-iT EdU assay (Thermo Fisher) according to the manufacturer's protocol. Flow cytometry was performed using a BD LSR II cytometer, and data analyzed using FlowJo.
In vitro proliferation assay
Mouse lungs were dissociated and stained with BV-421-conjugated antibodies against CD45, CD31, and CD326 (EpCAM) (BioLegend) as described, dead cells were excluded using 7-aminoactinomycin staining, and 2000 GFP+ cells were directly sorted into individual wells of 96-well collagen-coated plates containing complete media using a FACSAria II sorter. Cells were grown as described above. At specified times, plates were washed with PBS and placed at −80 °C. DNA content was measured using a CyQuant assay per manufacturer protocol.
Statistical analysis
Significant differences in mean values were calculated using paired Student's t tests or one-way analysis of variance. Survival analysis was performed using Kaplan-Meier analysis. A p value of less than 0.05 was considered to be significant.
Author contributions
R. D. G. participated in the design of the study, performed all animal experiments and most molecular analyses, performed statistical analysis, and drafted the manuscript. L. L. participated in animal care and performed quantitative RT-PCR assays. C. S. participated in animal experiments and in the design of the study. S. J. D. performed immunohistochemistry and immunofluorescence and participated in animal experiments. H. Y. K. performed quantitative RT-PCR assays and participated in animal experiments. L. C. developed FGFR3 flox mice used in this study. D. M. O. participated in the design of the study, and helped draft the manuscript. All authors read and approved the final manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants HL111190 and T32HL007317, American Heart Association Grant 14FTF19840029, Washington University Hope Center Alafi Neuroimaging Lab, and a National Institutes of Health Shared Instrumentation Grant S10 RR027552. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IPF
- idiopathic pulmonary fibrosis
- TCKO
- triple conditional knockout
- αSMA
- α smooth muscle actin
- PDGFRα
- platelet-derived growth factor receptor α
- TFF
- triple flox/flox
- P21
- postnatal day 21
- EdU
- 5-ethynyl-2-deoxyuridine
- qRT
- quantitative RT
- pro-SPC
- pro-surfactant protein C.
References
- 1. King T. E. Jr., Pardo A., and Selman M. (2011) Idiopathic pulmonary fibrosis. Lancet 378, 1949–1961 [DOI] [PubMed] [Google Scholar]
- 2. Blackwell T. S., Tager A. M., Borok Z., Moore B. B., Schwartz D. A., Anstrom K. J., Bar-Joseph Z., Bitterman P., Blackburn M. R., Bradford W., Brown K. K., Chapman H. A., Collard H. R., Cosgrove G. P., Deterding R., et al. (2014) Future directions in idiopathic pulmonary fibrosis research: an NHLBI workshop report. Am. J. Respir. Crit. Care Med. 189, 214–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Richeldi L., du Bois R. M., Raghu G., Azuma A., Brown K. K., Costabel U., Cottin V., Flaherty K. R., Hansell D. M., Inoue Y., Kim D. S., Kolb M., Nicholson A. G., Noble P. W., Selman M., et al. (2014) Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2071–2082 [DOI] [PubMed] [Google Scholar]
- 4. King T. E. Jr., Bradford W. Z., Castro-Bernardini S., Fagan E. A., Glaspole I., Glassberg M. K., Gorina E., Hopkins P. M., Kardatzke D., Lancaster L., Lederer D. J., Nathan S. D., Pereira C. A., Sahn S. A., Sussman R., et al. (2014) A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 370, 2083–2092 [DOI] [PubMed] [Google Scholar]
- 5. Schaefer C. J., Ruhrmund D. W., Pan L., Seiwert S. D., and Kossen K. (2011) Antifibrotic activities of pirfenidone in animal models. Eur. Respir. Rev. 20, 85–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Oku H., Shimizu T., Kawabata T., Nagira M., Hikita I., Ueyama A., Matsushima S., Torii M., and Arimura A. (2008) Antifibrotic action of pirfenidone and prednisolone: different effects on pulmonary cytokines and growth factors in bleomycin-induced murine pulmonary fibrosis. Eur. J. Pharmacol. 590, 400–408 [DOI] [PubMed] [Google Scholar]
- 7. Wollin L., Maillet I., Quesniaux V., Holweg A., and Ryffel B. (2014) Antifibrotic and anti-inflammatory activity of the tyrosine kinase inhibitor nintedanib in experimental models of lung fibrosis. J. Pharmacol. Exp. Ther. 349, 209–220 [DOI] [PubMed] [Google Scholar]
- 8. Chaudhary N. I., Roth G. J., Hilberg F., Müller-Quernheim J., Prasse A., Zissel G., Schnapp A., and Park J. E. (2007) Inhibition of PDGF, VEGF and FGF signalling attenuates fibrosis. Eur. Respir. J. 29, 976–985 [DOI] [PubMed] [Google Scholar]
- 9. Yu Z. H., Wang D. D., Zhou Z. Y., He S. L., Chen A. A., and Wang J. (2012) Mutant soluble ectodomain of fibroblast growth factor receptor-2 IIIc attenuates bleomycin-induced pulmonary fibrosis in mice. Biol. Pharm. Bull. 35, 731–736 [DOI] [PubMed] [Google Scholar]
- 10. Zhang X., Ibrahimi O. A., Olsen S. K., Umemori H., Mohammadi M., and Ornitz D. M. (2006) Receptor specificity of the fibroblast growth factor family: the complete mammalian FGF family. J. Biol. Chem. 281, 15694–15700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. MacKenzie B., Korfei M., Henneke I., Sibinska Z., Tian X., Hezel S., Dilai S., Wasnick R., Schneider B., Wilhelm J., El Agha E., Klepetko W., Seeger W., Schermuly R., Günther A., and Bellusci S. (2015) Increased FGF1-FGFRc expression in idiopathic pulmonary fibrosis. Respir. Res. 16, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lin N., Chen S., Pan W., Xu L., Hu K., and Xu R. (2011) NP603, a novel and potent inhibitor of FGFR1 tyrosine kinase, inhibits hepatic stellate cell proliferation and ameliorates hepatic fibrosis in rats. Am. J. Physiol. Cell Physiol. 301, C469–C477 [DOI] [PubMed] [Google Scholar]
- 13. Thannickal V. J., Aldweib K. D., Rajan T., and Fanburg B. L. (1998) Upregulated expression of fibroblast growth factor (FGF) receptors by transforming growth factor-β1 (TGF-β1) mediates enhanced mitogenic responses to FGFs in cultured human lung fibroblasts. Biochem. Biophys. Res. Commun. 251, 437–441 [DOI] [PubMed] [Google Scholar]
- 14. Finlay G. A., Thannickal V. J., Fanburg B. L., and Paulson K. E. (2000) Transforming growth factor-β1-induced activation of the ERK pathway/activator protein-1 in human lung fibroblasts requires the autocrine induction of basic fibroblast growth factor. J. Biol. Chem. 275, 27650–27656 [DOI] [PubMed] [Google Scholar]
- 15. Khalil N., Xu Y. D., O'Connor R., and Duronio V. (2005) Proliferation of pulmonary interstitial fibroblasts is mediated by transforming growth factor-β1-induced release of extracellular fibroblast growth factor-2 and phosphorylation of p38 MAPK and JNK. J. Biol. Chem. 280, 43000–43009 [DOI] [PubMed] [Google Scholar]
- 16. Osaki T., Yoneda K., Tatemoto Y., Yamamoto T., Yokoyama T., and Enzan H. (2001) Peplomycin, a bleomycin derivative, induces myofibroblasts in pulmonary fibrosis. Int. J. Exp. Pathol. 82, 231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xiao L., Du Y., Shen Y., He Y., Zhao H., and Li Z. (2012) TGF-β1 induced fibroblast proliferation is mediated by the FGF-2/ERK pathway. Front. Biosci. 17, 2667–2674 [DOI] [PubMed] [Google Scholar]
- 18. Strutz F., Zeisberg M., Renziehausen A., Raschke B., Becker V., van Kooten C., and Müller G. (2001) TGF-β1 induces proliferation in human renal fibroblasts via induction of basic fibroblast growth factor (FGF-2). Kidney Int. 59, 579–592 [DOI] [PubMed] [Google Scholar]
- 19. Hoyles R. K., Derrett-Smith E. C., Khan K., Shiwen X., Howat S. L., Wells A. U., Abraham D. J., and Denton C. P. (2011) An essential role for resident fibroblasts in experimental lung fibrosis is defined by lineage-specific deletion of high-affinity type II transforming growth factor β receptor. Am. J. Respir. Crit. Care Med. 183, 249–261 [DOI] [PubMed] [Google Scholar]
- 20. Hu B., Wu Z., Nakashima T., and Phan S. H. (2012) Mesenchymal-specific deletion of C/EBPβ suppresses pulmonary fibrosis. Am. J. Pathol. 180, 2257–2267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parapuram S. K., Thompson K., Tsang M., Hutchenreuther J., Bekking C., Liu S., and Leask A. (2015) Loss of PTEN expression by mouse fibroblasts results in lung fibrosis through a CCN2-dependent mechanism. Matrix Biol. 43, 35–41 [DOI] [PubMed] [Google Scholar]
- 22. Ornitz D. M., and Itoh N. (2015) The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol. 4, 215–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muzumdar M. D., Tasic B., Miyamichi K., Li L., and Luo L. (2007) A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605 [DOI] [PubMed] [Google Scholar]
- 24. Tsukui T., Ueha S., Abe J., Hashimoto S., Shichino S., Shimaoka T., Shand F. H., Arakawa Y., Oshima K., Hattori M., Inagaki Y., Tomura M., and Matsushima K. (2013) Qualitative rather than quantitative changes are hallmarks of fibroblasts in bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 183, 758–773 [DOI] [PubMed] [Google Scholar]
- 25. Ju W., Zhihong Y., Zhiyou Z., Qin H., Dingding W., Li S., Baowei Z., Xing W., Ying H., and An H. (2012) Inhibition of α-SMA by the ectodomain of FGFR2c attenuates lung fibrosis. Mol. Med. 18, 992–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rangarajan S., Kurundkar A., Kurundkar D., Bernard K., Sanders Y. Y., Ding Q., Antony V. B., Zhang J., Zmijewski J., and Thannickal V. J. (2016) Novel mechanisms for the antifibrotic action of nintedanib. Am. J. Respir. Cell Mol. Biol. 54, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Moeller A., Ask K., Warburton D., Gauldie J., and Kolb M. (2008) The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 40, 362–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mouratis M. A., and Aidinis V. (2011) Modeling pulmonary fibrosis with bleomycin. Curr. Opin. Pulm. Med. 17, 355–361 [DOI] [PubMed] [Google Scholar]
- 29. Ashcroft T., Simpson J. M., and Timbrell V. (1988) Simple method of estimating severity of pulmonary fibrosis on a numerical scale. J. Clin. Pathol. 41, 467–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Matute-Bello G., Frevert C. W., and Martin T. R. (2008) Animal models of acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 295, L379–L399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Moore B. B., and Hogaboam C. M. (2008) Murine models of pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 294, L152–L160 [DOI] [PubMed] [Google Scholar]
- 32. Liu T., Yu H., Ding L., Wu Z., Gonzalez De Los Santos F., Liu J., Ullenbruch M., Hu B., Martins V., and Phan S. H. (2015) Conditional knockout of telomerase reverse transcriptase in mesenchymal cells impairs mouse pulmonary fibrosis. PloS One 10, e0142547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hu B., Liu J., Wu Z., Liu T., Ullenbruch M. R., Ding L., Henke C. A., Bitterman P. B., and Phan S. H. (2015) Reemergence of hedgehog mediates epithelial-mesenchymal crosstalk in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 52, 418–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hu B., Wu Z., Bai D., Liu T., Ullenbruch M. R., and Phan S. H. (2015) Mesenchymal deficiency of Notch1 attenuates bleomycin-induced pulmonary fibrosis. Am. J. Pathol. 185, 3066–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Srisuma S., Bhattacharya S., Simon D. M., Solleti S. K., Tyagi S., Starcher B., and Mariani T. J. (2010) Fibroblast growth factor receptors control epithelial-mesenchymal interactions necessary for alveolar elastogenesis. Am. J. Respir. Crit. Care Med. 181, 838–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tschumperlin D. J. (2015) Matrix, mesenchyme, and mechanotransduction. Ann. Am. Thorac. Soc. 12, S24–S29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hokuto I., Perl A. K., and Whitsett J. A. (2003) Prenatal, but not postnatal, inhibition of fibroblast growth factor receptor signaling causes emphysema. J. Biol. Chem. 278, 415–421 [DOI] [PubMed] [Google Scholar]
- 38. Weinstein M., Xu X., Ohyama K., and Deng C. X. (1998) FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 125, 3615–3623 [DOI] [PubMed] [Google Scholar]
- 39. Yin Y., Betsuyaku T., Garbow J. R., Miao J., Govindan R., and Ornitz D. M. (2013) Rapid induction of lung adenocarcinoma by fibroblast growth factor 9 signaling through FGF receptor 3. Cancer Res. 73, 5730–5741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yin Y., Ren X., Smith C., Guo Q., Malabunga M., Guernah I., Zhang Y., Shen J., Sun H., Chehab N., Loizos N., Ludwig D. L., and Ornitz D. M. (2016) Inhibition of FGF Receptor 3-dependent lung adenocarcinoma with a human monoclonal antibody. Dis. Model. Mech. 9, 563–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Arai D., Hegab A. E., Soejima K., Kuroda A., Ishioka K., Yasuda H., Naoki K., Kagawa S., Hamamoto J., Yin Y., Ornitz D. M., and Betsuyaku T. (2015) Characterization of the cell of origin and propagation potential of the fibroblast growth factor 9-induced mouse model of lung adenocarcinoma. J. Pathol. 235, 593–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Guzy R. D., Stoilov I., Elton T. J., Mecham R. P., and Ornitz D. M. (2015) Fibroblast growth factor 2 is required for epithelial recovery, but not for pulmonary fibrosis, in response to bleomycin. Am. J. Respir. Cell Mol. Biol. 52, 116–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yu C., Wang F., Jin C., Huang X., Miller D. L., Basilico C., and McKeehan W. L. (2003) Role of fibroblast growth factor type 1 and 2 in carbon tetrachloride-induced hepatic injury and fibrogenesis. Am. J. Pathol. 163, 1653–1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Coffey E., Newman D. R., and Sannes P. L. (2013) Expression of fibroblast growth factor 9 in normal human lung and idiopathic pulmonary fibrosis. J. Histochem. Cytochem. 61, 671–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Joannes A., Brayer S., Besnard V., Marchal-Sommé J., Jaillet M., Mordant P., Mal H., Borie R., Crestani B., and Mailleux A. A. (2016) FGF9 and FGF18 in idiopathic pulmonary fibrosis promote survival and migration and inhibit myofibroblast differentiation of human lung fibroblasts in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 310, L615–L629 [DOI] [PubMed] [Google Scholar]
- 46. MacKenzie B., Henneke I., Hezel S., Al Alam D., El Agha E., Chao C. M., Quantius J., Wilhelm J., Jones M., Goth K., Li X., Seeger W., Königshoff M., Herold S., Rizvanov A. A., Günther A., and Bellusci S. (2015) Attenuating endogenous Fgfr2b ligands during bleomycin-induced lung fibrosis does not compromise murine lung repair. Am. J. Physiol. Lung Cell. Mol. Physiol. 308, L1014–L1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ding B. S., Cao Z., Lis R., Nolan D. J., Guo P., Simons M., Penfold M. E., Shido K., Rabbany S. Y., and Rafii S. (2014) Divergent angiocrine signals from vascular niche balance liver regeneration and fibrosis. Nature 505, 97–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hanumegowda C., Farkas L., and Kolb M. (2012) Angiogenesis in pulmonary fibrosis: too much or not enough? Chest 142, 200–207 [DOI] [PubMed] [Google Scholar]
- 49. Shimbori C., Bellaye P. S., Xia J., Gauldie J., Ask K., Ramos C., Becerril C., Pardo A., Selman M., and Kolb M. (2016) Fibroblast growth factor-1 attenuates TGF-β1-induced lung fibrosis. J. Pathol. 240, 197–210 [DOI] [PubMed] [Google Scholar]
- 50. Ramos C., Montaño M., Becerril C., Cisneros-Lira J., Barrera L., Ruíz V., Pardo A., and Selman M. (2006) Acidic fibroblast growth factor decreases α-smooth muscle actin expression and induces apoptosis in human normal lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 291, L871–L879 [DOI] [PubMed] [Google Scholar]
- 51. Gupte V. V., Ramasamy S. K., Reddy R., Lee J., Weinreb P. H., Violette S. M., Guenther A., Warburton D., Driscoll B., Minoo P., and Bellusci S. (2009) Overexpression of fibroblast growth factor-10 during both inflammatory and fibrotic phases attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Crit. Care Med. 180, 424–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sugahara K., Iyama K., Kuroda M. J., and Sano K. (1998) Double intratracheal instillation of keratinocyte growth factor prevents bleomycin-induced lung fibrosis in rats. J. Pathol. 186, 90–98 [DOI] [PubMed] [Google Scholar]
- 53. Zheng B., Zhang Z., Black C. M., de Crombrugghe B., and Denton C. P. (2002) Ligand-dependent genetic recombination in fibroblasts: a potentially powerful technique for investigating gene function in fibrosis. Am. J. Pathol. 160, 1609–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jacob A. L., Smith C., Partanen J., and Ornitz D. M. (2006) Fibroblast growth factor receptor 1 signaling in the osteo-chondrogenic cell lineage regulates sequential steps of osteoblast maturation. Dev. Biol. 296, 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu K., Xu J., Liu Z., Sosic D., Shao J., Olson E. N., Towler D. A., and Ornitz D. M. (2003) Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063–3074 [DOI] [PubMed] [Google Scholar]