

Abstract
Recent advances in developing opioid treatments for pain with reduced side effects have focused on the signaling cascades of the μ-opioid receptor (MOR). However, few such signaling targets have been identified for exploitation. To address this need, we explored the role of heat-shock protein 90 (Hsp90) in opioid-induced MOR signaling and pain, which has only been studied in four previous articles. First, in four cell models of MOR signaling, we found that Hsp90 inhibition for 24 h with the inhibitor 17-N-allylamino-17-demethoxygeldanamycin (17-AAG) had different effects on protein expression and opioid signaling in each line, suggesting that cell models may not be reliable for predicting pharmacology with this protein. We thus developed an in vivo model using CD-1 mice with an intracerebroventricular injection of 17-AAG for 24 h. We found that Hsp90 inhibition strongly blocked morphine-induced anti-nociception in models of post-surgical and HIV neuropathic pain but only slightly blocked anti-nociception in a naive tail-flick model, while enhancing morphine-induced precipitated withdrawal. Seeking a mechanism for these changes, we found that Hsp90 inhibition blocks ERK MAPK activation in the periaqueductal gray and caudal brain stem. We tested these signaling changes by inhibiting ERK in the above-mentioned pain models and found that ERK inhibition could account for all of the changes in anti-nociception induced by Hsp90 inhibition. Taken together, these findings suggest that Hsp90 promotes opioid-induced anti-nociception by an ERK mechanism in mouse brain and that Hsp90 could be a future target for improving the therapeutic index of opioid drugs.
Keywords: brain, extracellular-signal-regulated kinase (ERK), heat-shock protein 90 (Hsp90), opiate opioid, pain, 17-AAG, anti-nociception, μ-opioid receptor
Introduction
Chronic pain is a serious public health problem in the United States, with an estimated ∼100 million affected people and an annual economic cost of $600 billion (1). Moderate to severe cancer and non-cancer chronic pain seriously degrades patient quality of life and interferes with daily activities (2). There are multiple drug classes for the treatment of chronic pain, including tricyclic antidepressants and gabapentinoids, but often the only efficacious option for serious pain is opioid drugs such as morphine or fentanyl. Although efficacious, opioid drugs induce a constellation of side effects, including abuse liability, tolerance, constipation, and respiratory depression (3, 4). These limitations have spurred a search for new analgesic drugs with a reduction or elimination of side effects, which has persisted for more than 100 years with little success (5).
Recent advances in the understanding of signal transduction downstream of the μ-opioid receptor (MOR)2 have suggested new approaches for drug discovery. Notably, the ubiquitous signaling regulator β-arrestin2 (βarr2) has been found to reduce anti-nociception (6) and promote opioid side effects (4, 7), leading to the development of biased ligands that activate MOR without recruiting βarr2, one of which is currently in clinical trials (8–11). Although promising, this signal transduction-focused approach is limited by the availability of signaling targets to exploit; some targets such as Raf-1 (12) and RGS proteins (13) have been shown to regulate opioid side effects, but they have not been exploited for drug discovery to our knowledge, and few other such targets are known.
To address this need and improve our chances of developing new opioid therapies, we investigated the ubiquitous and crucial signaling regulator Hsp90 as a potential regulator of opioid signaling and behavioral responses. Hsp90 is a chaperone protein that prevents protein aggregation and degradation. However, it can also facilitate the activation of signal transduction molecules, including kinases, by promoting a conformation that is ideal for activation (14, 15). Hsp90, present in all cell types, makes up 2–3% of total cell protein content and up to 4–5% during heat stress. The activity of Hsp90 is regulated and directed by co-chaperone proteins, including Hop/STIP1 and Cdc37, which are more selective by tissue and signaling type than Hsp90 itself (14, 16). Because of this selectivity, co-chaperone proteins may represent future targets for modulation that could decrease the side effects of modulating Hsp90 itself, as has been done for Hsp70 (17).
Hsp90 has only been studied in two pain models to date. First, it has been found that Hsp90 inhibition reverses diabetes-induced mechanical hypoalgesia (18). Second, it has been shown that Hsp90 inhibition reverses chronic constriction injury (CCI)-induced mechanical allodynia and potentiates morphine anti-nociception in this model, whereas Hsp90 activation has the opposite effect when combined with lipopolysaccharide treatment (19). Only two studies have examined the role of Hsp90 specifically in MOR signal transduction and opioid response. One study found in an in vitro model that Hsp90 inhibition reduces adenylyl cyclase superactivation, a cellular marker of dependence (20). An article by the Devi group found that Hsp90 is up-regulated by chronic morphine treatment in the presynaptic terminal and that administration of the Hsp90 inhibitor geldanamycin in the periphery immediately prior to naloxone precipitation reduces the somatic signs of withdrawal (21). This limited evidence from the literature suggests that Hsp90 has a role in regulating MOR signal transduction and opioid behavioral responses. However, in the CCI and dependence in vivo studies (19, 21) the Hsp90 inhibitor was administered 3 h or less prior to measuring pain or withdrawal, which may not be a good model for chronic pain treatment.
In our current studies, we tested the role of Hsp90 in MOR signaling and pain using the selective inhibitor 17-AAG (22) on a 24-h treatment regimen to more closely model chronic pain treatment. The results from both in vitro and in vivo experiments indicated strong impacts of Hsp90 inhibition on opioid-induced signaling and pain behaviors, showing that Hsp90 regulates pain behavior through an ERK mechanism. These findings suggest that Hsp90 could be a target for promoting analgesia and reducing the side effects of opioid therapy.
Results
Hsp90 inhibition in cell models of MOR signaling
We first sought to explore the role of Hsp90 in regulating MOR signaling in four in vitro models. These include Chinese hamster ovary (CHO), human embryonic kidney (HEK), and human bone osteosarcoma (U2OS) cell lines stably expressing the human MOR (see “Experimental procedures” for cell line details) and the human neuroblastoma cell line SH-SY5Y, which endogenously expresses the MOR and δ-opioid receptor (DOR) (23). All cells were treated with 1 μm 17-AAG for 24 h and then stimulated with the MOR full agonist DAMGO ([d-Ala2, N-MePhe4, Gly2-ol]-enkephalin) for 10 min; signaling protein expression and signal transduction were analyzed by Western blotting.
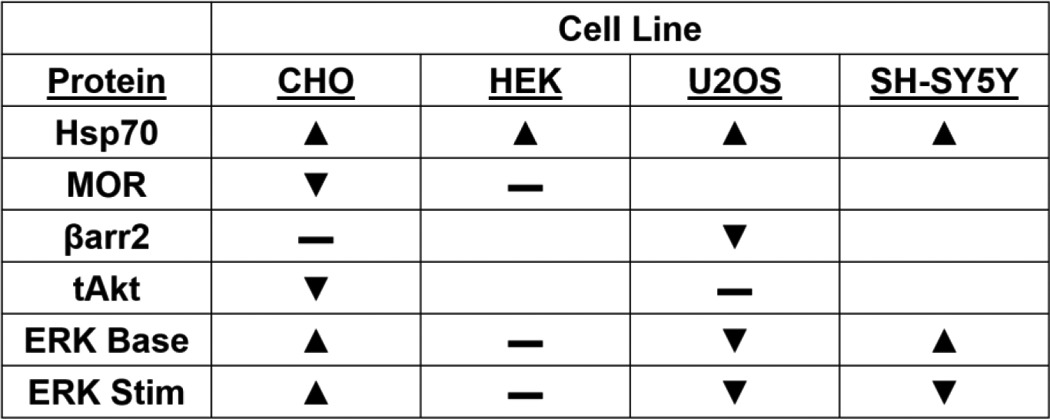
For signaling protein expression changes, we found that Hsp70 was induced in all four cell lines, which is a canonical effect of this class of Hsp90 inhibitors and serves as a marker of Hsp90 inhibition (Fig. 1, A and B) (24, 25). We also found that Hsp90 inhibition altered the protein expression levels of the MOR itself and of the signaling regulator βarr2 and the kinase Akt. Unlike Hsp70, however, the regulation of these protein levels differed considerably by cell line; the MOR was decreased in CHO cells but unaltered in HEK, βarr2 levels were decreased in U2OS but not CHO cells, and Akt levels were decreased in CHO cells but not U2OS (Fig. 1B).
Figure 1.

Hsp90 inhibition has multivariate effects on protein expression and signal transduction in vitro. Four different cell models of MOR signal transduction were treated with 1 μm 17-AAG or vehicle for 24 h, serum-starved (see “Experimental procedures”), and treated with vehicle, 10 μm DAMGO, or 10 μm LPA (a non-opioid GPCR endogenous agonist) for 10 min. Results were measured by Western blot, and analyzed as reported under “Experimental procedures.” The sample size reported in each experiment graph represents an independent technical replicate consisting of two or more averaged replicate wells counted as n = 1. All data are reported as the mean ± S.D. A, representative Western blots for each target are shown, with vehicle (Veh) or 17-AAG (AAG) treatment. The cell line from which each representative blot was taken is indicated below the target name. The numbers with arrows on the left side of each image represents the position of the nearest molecular weight markers. These images also indicate that GAPDH and tERK were not altered by 17-AAG treatment (data not shown). B, the expression levels of four separate signaling proteins normalized to GAPDH and vehicle control were analyzed in each cell line as labeled. *, ***, and ****, p < 0.05, 0.001, and 0.0001, respectively, versus same cell line vehicle control by unpaired 2-tailed t test. Only Hsp70 shows consistent regulation across cell lines with Hsp90 inhibition. C, signal transduction via ERK phosphorylation is reported for each cell line in a separate graph. The pERK signal was normalized to tERK and, in the case of CHO, was normalized further to MOR levels as they were decreased. The pERK/tERK signal was further normalized to the vehicle:vehicle group. * and **, p < 0.05 and 0.01 versus same pretreatment group (vehicle, 17-AAG) vehicle control; #, ##, ###, and ####, p < 0.05, 0.01, 0.001, and 0.0001, respectively, versus vehicle:vehicle group; both sets by two-way ANOVA with Fisher's least significant difference post-hoc test. Each line showed a different alteration in the ERK signaling pattern after Hsp90 inhibition, and both DAMGO and LPA showed similar behavior.
This heterogeneity continued when we analyzed the phosphorylation and activation of ERK (pERK) in these cells in response to DAMGO after Hsp90 inhibition (total ERK levels (tERK) were not altered by Hsp90 inhibition (Fig. 1A)). In CHO cells, the per-unit ERK activation was strongly increased by Hsp90 inhibition in response to DAMGO and the endogenous GPCR agonist lysophosphatidic acid (LPA) (Fig. 1C). However, in HEK cells the pattern of ERK activation was not changed by Hsp90 inhibition, whereas it was decreased in U2OS cells. Lastly, the SH-SY5Y cells displayed an unusual ERK activation pattern whereby the unstimulated baseline was strongly increased but ERK was not stimulated above that elevated baseline by either DAMGO or LPA. As summarized in Table 1, Hsp90 inhibition showed heterogeneous results in different cell lines, which suggested that in vitro models would not be reliable in studying the role of Hsp90 in the opioid system and would necessitate the use of relevant in vivo models for the study of this protein.
Table 1.
Summary of the effects of Hsp90 inhibition in vitro
The results of Fig. 1 are summarized here. Stim, stimulation; triangle, increased; inverted triangle, decreased; —, no change; blank, not investigated.
Hsp90 inhibition in mouse models of opioid-induced behavior
For the in vivo studies, mice were treated with 17-AAG by the i.c.v. route to isolate the role of Hsp90 specifically in the brain, with a 24-h recovery time as described above, to more closely model chronic pain treatment. The i.c.v. route results in higher concentrations of injected drug molecules around the wall of the ventricles and associated areas, which have high levels of opioid receptors and are known to be important in producing various effects of opioid drugs such as morphine (e.g. anti-nociception, physical dependence, etc.). However, we could not find in the literature that 17-AAG had been administered by the i.c.v. route, although Hsp70 is up-regulated in the brain in response to systemic 17-AAG (25). We thus performed a dose-response study of i.c.v. 17-AAG in male CD-1 mice, with a readout of Hsp70 up-regulation by Western blotting (Fig. 2). We found robust dose-dependent Hsp70 up-regulation in the periaqueductal gray (PAG), a region high in MOR expression and critical for pain regulation (26), with an approximate A50 of 0.084 nmol. Based on these results, we chose the approximate A90 dose of 0.5 nmol for further studies, which reduced the risk of off-target effects from using the highest dose tested (5 nmol).
Figure 2.
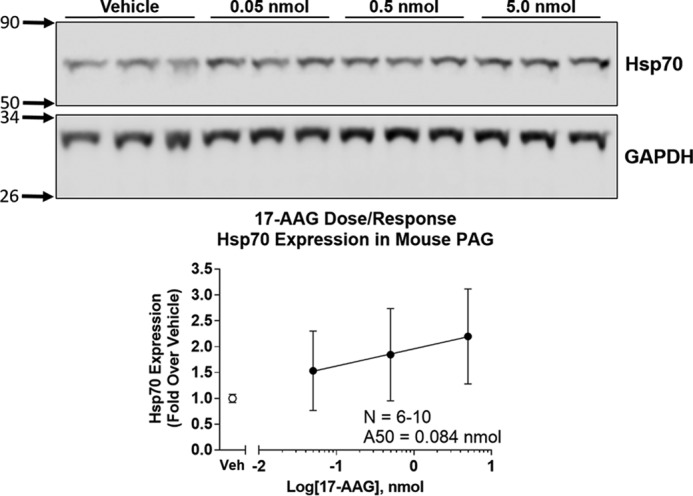
Hsp90 inhibitor (17-AAG) dose response in mouse brain. Mice were injected i.c.v. with a dose response of 17-AAG or an equalized vehicle and allowed to recover for 24 h. The mice were sacrificed, and the PAG dissected, removed, and analyzed by Western blotting. Representative blots of Hsp70 and GAPDH are shown with treatment and molecular weight markers indicated. The Hsp70 signal is normalized to GAPDH and further to vehicle control. GAPDH was not altered by 17-AAG treatment. The data were reported as the mean ± S.D. Linear regression was performed on the data points, and the A50 (0.084 nmol) was calculated as described under “Experimental procedures.” The reported sample size is for experiments performed on individual mice (biological replicates) over two technical replicates on different days with different mouse cohorts by the same experimenter. Based on this experiment, 0.5 nmol of 17-AAG was chosen for further experiments.
We then tested the effects of Hsp90 inhibition on opioid response in several pain and dependence/withdrawal models. We first established a clinically relevant post-surgical pain model by paw incision (27) with concurrent 17-AAG injection and a 24-h recovery. Pain response and allodynia were measured by mechanical stimulation with Von Frey filaments (28). The animals showed a robust allodynia post-surgery that was not altered by 17-AAG treatment (Fig. 3A). The vehicle-treated animals showed a robust time-dependent anti-nociception in response to systemic morphine (1, 3.2, and 10 mg/kg s.c.), which was strongly blocked by 17-AAG treatment (area under the curve (AUC) reduction of 70.2, 81.5, and 67.0% at 1, 3.2, and 10 mg/kg with 17-AAG) (Fig. 3A and Table 2). The AUC values from individual mice at the three dosages were used to construct dose-response curves (Fig. 3B). Analysis of these curves showed an A50 of 3.98 mg/kg for vehicle-treated mice and an A50 of 522.2 mg/kg for 17-AAG-treated mice, a 131.2-fold shift (Table 2). We observed strongly reduced but still present anti-nociception in 17-AAG-treated mice at 10 mg/kg morphine but not at 1 and 3.2 mg/kg, suggesting that this shift represents a change in potency (not efficacy) of the opioid system in this pain model with 17-AAG treatment.
Figure 3.

Hsp90 inhibition decreases opioid-induced anti-nociception. All data are reported as the mean ± S.D. A, post-surgical pain was induced by paw-incision surgery, and mechanical thresholds were measured with Von Frey filaments as described under “Experimental procedures.” Vehicle or 17-AAG (0.5 nmol) was injected i.c.v. concurrent with surgery. After 24 h, morphine was injected (1, 3.2, or 10 mg/kg s.c.), and mechanical thresholds were measured over a time course. Each dose is reported in a separate graph. *, **, ***, and **** (same with # and $), p < 0.05, 0.01, 0.001, and 0.0001, respectively, versus same time point by two-way ANOVA with Fisher's least significant difference post-hoc test. AUC analysis results are summarized in Table 2. The reported sample size of individual mice includes two to three technical replicates for each dose on different days by the same experimenter. B, the AUC values from each individual mouse in A were used to construct dose-response curves, and linear regression analysis was performed to calculate the A50 values (see “Experimental procedures” and results in Table 2). Vehicle = 3.98 mg/kg and 17-AAG = 522.2 mg/kg, a 131.2-fold shift. The results suggest a strong decrease in potency for the system. C, 52 °C tail-flick baselines were determined as reported under “Experimental procedures” prior to and 24 h post-i.c.v. injection of vehicle or 0.5 nmol of 17-AAG. DAMGO (0.01, 0.032, or 0.1 nmol) was then injected i.c.v., and tail-flick latencies were determined over a time course. * and ** = p < 0.05 and 0.01, respectively, versus 0.1 nmol time point; #, p < 0.05 versus 0.032 nmol time point; both by two-way ANOVA with Fisher's least significant difference post-hoc test. AUC analysis results are summarized in Table 2. The reported sample size of individual mice includes three technical replicates for each dose from two different experimenters at different institutions; each replicate showed very similar results. D, dose-response curves were created and analyzed from C using the same method as described in B. The results are summarized in Table 2. Vehicle = 0.095 nmol and 17-AAG = 0.251 nmol, a 2.64-fold shift. The results suggest a modest decrease in efficacy for the system. E, HIV peripheral neuropathy was established using the method outlined under “Experimental procedures.” This figure demonstrates progressive, severe, mechanical allodynia, measured as described in A, over a 21-day treatment period. Both hind paws were measured, and the injection days were noted (Inj). The reported sample size of individual mice includes every HIV neuropathy mouse used in this study combined with six technical replicates by the same experimenter on different days. F, HIV neuropathy was established as described in E, with pre-gp120, day 19, and day 21 post-i.c.v. injection baselines noted. On day 20, vehicle or 17-AAG was injected i.c.v. as above followed by 10 mg/kg morphine s.c. on day 21. 17-AAG had no effect on pain baselines but abolished (AUC reduction of 97.6% (Table 2)) morphine anti-nociception. * and ****, p < 0.05 and 0.0001 versus same time point by two-way ANOVA with Fisher's least significant difference post-hoc test. The reported sample size of individual mice includes two technical replicates performed on different days by the same experimenter. Both hind paws showed extremely similar results, and thus only the data from the left paw is shown (data not shown).
Table 2.
Summary of in vivo anti-nociception assays
The relevant results of Figs. 3 and 6 are summarized here. ΔAUC, change in AUC percentage, either increased (+) or decreased (−) with treatment. The A50 values are derived from the dose-response curves shown in Fig. 3. Veh, vehicle.
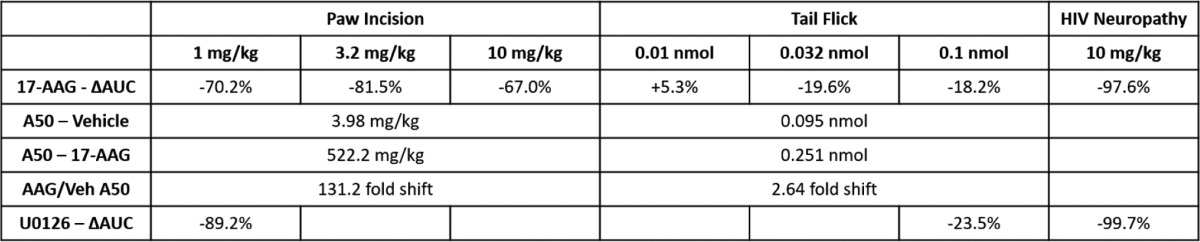
Seeking to determine whether this effect of 17-AAG was consistent across pain models, we next tested the effects of the inhibitor in a model of thermal tail-flick anti-nociception in naive mice treated with i.c.v. DAMGO. Tail-flick baselines were not altered by 17-AAG treatment, whereas both vehicle- and 17-AAG-treated mice displayed robust anti-nociception in response to 0.01, 0.032, and 0.1 nmol of DAMGO i.c.v. (Fig. 3C). 17-AAG treatment reduced the extent of the anti-nociception but by far less than in the paw-incision model, with an AUC increase of 5.3% at 0.01 nmol and a reduction of 19.6 and 18.2% at 0.032 and 0.1 nmol (Fig. 3C and Table 2 compared with a 67.0–81.5% reduction with paw incision). As described above, the AUC values of the individual mice were used to construct dose-response curves (Fig. 3D). Analysis of these curves revealed an A50 of 0.095 nmol for vehicle-treated mice and an A50 of 0.251 nmol for 17-AAG-treated mice; this was a 2.64-fold shift, far less than the 131.2-fold shift for paw incision (Table 2). Because the magnitude of the anti-nociception was equal for both vehicle- and 17-AAG-treated mice at the low dose of 0.01 nmol, we concluded that 17-AAG treatment reduces the efficacy (not potency) of the opioid system in this pain model.
Both the paw-incision and tail-flick models represent acute pain, with durations of 24 h or less. To investigate the role of Hsp90 in chronic pain anti-nociception, we established a mouse model of HIV peripheral neuropathy (29). The pain state was induced by intrathecal injection of HIV gp120 protein at days 1, 3, and 5, which induced a progressive and strong mechanical allodynia over a 3-week treatment period (Fig. 3E). We injected 17-AAG on day 20 as described above and measured mechanical allodynia as for the paw-incision experiments on day 21 in response to 10 mg/kg morphine. As for the other two pain models, 17-AAG treatment did not alter the pain baseline. However, 17-AAG treatment completely abolished anti-nociception in response to 10 mg/kg morphine s.c. (AUC reduction of 97.6%) (Fig. 3F and Table 2). This result demonstrates that Hsp90 has a strong role in chronic pain anti-nociception, likely stronger than in acute pain. Dose-response experiments could not be completed in this model, as the motor-activating effects of the next half-log dose of morphine (32 mg/kg) were too strong for us to reliably measure mechanical thresholds.
We next tested the effects of Hsp90 inhibition on the opioid side effects of dependence and withdrawal. This was performed first in an acute model of dependence and withdrawal, with 17-AAG treatment for 20 h as described above, a single 100 mg/kg s.c. injection of morphine with a 4-h recovery, and the precipitation of withdrawal with 10 mg/kg naloxone i.p. (Fig. 4A). We also tested 17-AAG in a model of chronic dependence and withdrawal, with an escalating dosage protocol over 4 days (see “Experimental procedures”) with daily re-injection of 17-AAG, and precipitation of withdrawal on day 4 with 30 mg/kg naloxone i.p. (Fig. 4B). In both models of dependence and withdrawal, 17-AAG treatment significantly increased the production of withdrawal-associated feces and urine without altering jumping behavior. The above results suggest that Hsp90 inhibition with 17-AAG worsens the opioid therapeutic index by decreasing anti-nociception and increasing side effects.
Figure 4.

Hsp90 inhibition increases opioid-induced withdrawal symptoms. Acute (A) and chronic (B) morphine dependence and withdrawal paradigms were established with concurrent vehicle and 17-AAG (0.5 nmol) i.c.v. injections as described under “Experimental procedures.” Withdrawal was precipitated with 10 mg/kg (acute) or 30 mg/kg (chronic) naloxone i.p. with behaviors recorded for 20 min. Jumps and urine/feces weight are reported for each paradigm as the mean ± S.D. *, p < 0.05 versus the same measure vehicle control by unpaired two-tailed t test; n = 24–29 mice/group for each paradigm; three technical replicates were used for each paradigm by the same experimenter using different mouse cohorts on different days. 17-AAG treatment increased urine and feces weight, but not jumps, in both paradigms. C, mice were exposed to three 3-min training sessions on a Rotarod device followed by vehicle or 0.5 nmol of 17-AAG injected i.c.v. and a 24-h recovery. Another 3-min trial was then performed with acceleration from 4 to 16 rpm and no further treatment. The sample size reported for individual mice consisted of one technical replicate. 17-AAG had no effect on Rotarod performance (p > 0.05). Data are reported as the mean ± S.D.
Lastly, to determine whether the above results were due to off-target motor, sedative, or similar effects, we measured the effect of 17-AAG treatment on performance in the rotarod test (30). Both 17-AAG and vehicle-treated mice had unaltered performance in this test, suggesting that the above results are specific to the opioid system and are not off-target (Fig. 4C).
Hsp90 inhibition alters protein expression and signal transduction in the brain
In an effort to explore the mechanism of Hsp90 regulation of the above-described opioid-induced behaviors, we measured brain protein expression and ERK and Akt signal transduction in the PAG and caudal brain stem (pons and medulla) by Western blotting after 24 h of 17-AAG treatment and 0.1 nmol of DAMGO administered i.c.v. for 10 min. As expected, we found up-regulation of Hsp70 in both brain regions in response to 17-AAG (Fig. 5, A–C). Contrary to some of our in vitro findings shown in Fig. 1B, we did not observe changes in MOR, βarr2, or STAT3. Importantly for our analysis, 17-AAG treatment did not change the protein expression levels of GAPDH, tERK, or tAkt (Fig. 5, A–C, and data not shown).
Figure 5.

Hsp90 inhibition induces protein expression and signaling changes in mouse brain. Mice were injected i.c.v. with vehicle or 0.5 nmol of 17-AAG, allowed to recover for 24 h, and then injected i.c.v. with vehicle (Veh) or 0.1 nmol of DAMGO for 10 min. Brains were extracted and analyzed by Western blot analysis as reported under “Experimental procedures.” All data are reported as the mean ± S.D. A, representative blots from each protein target are shown, with the treatment and molecular weight markers indicated as described in the legend for Fig. 1. PAG (B) and caudal brain stem (pons and medulla) (C) protein expression changes are indicated. The signal for each target is normalized to GAPDH and further normalized to the vehicle control for each. MOR and βarr2 in the PAG were measured by immunoprecipitation (see “Experimental procedures”). *, p < 0.05 versus same target vehicle control by unpaired 2-tailed t test. The reported sample size of the individual mice consists of two technical replicates performed by two different experimenters at different institutions. Only Hsp70 expression was altered by 17-AAG treatment in both brain regions, contrary to some of the results reported in the legend for Fig. 1. Importantly, GAPDH, tERK, and tAkt protein levels were not changed (see A; GAPDH and tAkt quantified data not shown). PAG (D) and caudal brain stem (E) ERK signaling changes are indicated. pERK was normalized to the tERK signal and further normalized to the vehicle:vehicle group. **, ***, and ****, p < 0.01, 0.001, and 0.0001, respectively, versus vehicle:vehicle group by Fisher's least significant difference post-hoc test. The reported sample size of individual mice for both D and E consisted of two technical replicates each performed by different experimenters at different institutions. Hsp90 inhibition increased the ERK signaling baseline, and stimulation above that baseline by DAMGO was completely lost in both brain regions. PAG (F) and caudal brain stem (G) Akt signaling changes are indicated. Akt was analyzed as described for ERK from the same samples and with the same technical replicates. * and **, p < 0.05 and 0.01 versus vehicle:vehicle group by Fisher's least significant difference post-hoc test. Hsp90 inhibition induced a very similar, albeit less robust pattern in Akt as in ERK. H, in a control experiment, ERK signaling was assessed in mice injected i.c.v. with the highest dose of 17-AAG tested in the dose response (5 nmol). PAG ERK signaling in these mice was assessed as described in D and E. *, p < 0.05 versus vehicle:vehicle group by Fisher's least significant difference post-hoc test. 5 nmol of 17-AAG induces the same ERK pattern as seen in D and E with no increased magnitude of the effect, suggesting that 0.5 nmol has a maximal effect on brain signaling, justifying the use of the lower dose. The reported sample size of individual mice consisted of one technical replicate.
Both brain regions also displayed an unusual pattern of ERK signaling in response to 17-AAG whereby the pERK baseline was increased to the same level as DAMGO stimulation in the vehicle-treated brains, but stimulation above that baseline in response to DAMGO was completely lost (Fig. 5, D and E). This was a similar pattern of signaling as observed in SH-SY5Y cells treated with 17-AAG (Fig. 1C). We also examined pAkt levels in both regions and found a pattern similar to but less robust than the ERK signaling pattern (Fig. 5, F and G).
As a control, we also examined ERK activation in mice treated with 5 nmol of 17-AAG, the highest dose tested in our dose-response experiment shown in Fig. 2. The ERK pattern observed in this experiment was identical to that observed with 0.5 nmol of 17-AAG, with no increase in the magnitude of signaling, further justifying the choice of 0.5 nmol of 17-AAG for our studies (Fig. 5H).
Determining the mechanism of Hsp90 regulation of anti-nociception
In our signaling studies of mouse brain, we determined that Hsp70 expression and ERK signaling were altered by 17-AAG treatment. We thus tested these two proteins as mechanisms for Hsp90 regulation of anti-nociception. First, we tested the role of Hsp70 by combining Hsp90 inhibition by 17-AAG with Hsp70 inhibition by pifithrin-μ, a selective Hsp70 inhibitor. 17-AAG and 50 μg of pifithrin-μ, a previously established i.c.v. dose of this inhibitor (31), were co-injected i.c.v. with a 24-h recovery (along with single drug and vehicle controls). The mice were then i.c.v. injected with 0.1 nmol of DAMGO i.c.v., and a tail-flick assay performed as described above. We found that pifithrin-μ had no effect on the tail-flick response, with indistinguishable vehicle and pifithrin groups (AUC of 100 and 100.5%), and indistinguishable 17-AAG and 17-AAG/pifithrin groups (AUC of 76.3 and 78.7%) (Fig. 6A). This finding suggests that up-regulated Hsp70 is not responsible for the anti-nociception changes we observed.
Figure 6.

ERK inhibition recapitulates the effects of Hsp90 inhibition on anti-nociception. Tail-flick, HIV neuropathy/Von Frey, and paw-incision/Von Frey tests were performed as described in the legend for Fig. 3 and under “Experimental procedures.” Data are reported as the mean ± S.D. A, vehicle or 17-AAG (0.5 nmol) combined with pifithrin-μ (50 μg), as well as single injection controls, were injected i.c.v. with a 24-h recovery period. 0.1 nmol of DAMGO was then injected i.c.v., and tail-flick latencies were determined in a time course. ** and ***, p < 0.01 and 0.001 for vehicle versus pifithrin/17-AAG; #, ##, and ###, p < 0.05, 0.01, and 0.001, respectively, for pifithrin versus pifithrin/17-AAG group; both at the same time point using a two-way ANOVA with Fisher's least significant difference post-hoc test. AUC analysis revealed: 17-AAG, 23.7% decrease; pifithrin, 0.5% increase; pifithrin/17-AAG, 21.3% decrease. Pifithrin had no effect alone or in combination with 17-AAG on tail-flick anti-nociception. The sample size of individual mice reported consisted of two technical replicates by the same experimenter on different days. B, vehicle or 5 μg of U0126 was injected i.c.v. with a 15-min treatment period followed by 0.1 nmol of i.c.v. DAMGO and a tail-flick time course. *, p < 0.05 versus same time point via two-way ANOVA with a Fisher's least significant difference post-hoc test. AUC analysis revealed a decrease of 23.5% with U0126, very similar to the AUC values for 17-AAG treatment (Table 2). The sample size of individual mice reported consisted of two technical replicates performed on different days by the same experimenter. C, paw-incision surgery and pre- and post-surgery mechanical withdrawal thresholds were performed in mice. 5 μg of U0126 or vehicle control was injected i.c.v. with a 15-min treatment time. Morphine (1 mg/kg s.c.) was then injected, and withdrawal thresholds were determined over a time course. ** and ****, p < 0.01 and 0.0001 versus same time point via two-way ANOVA with a Fisher's Least Significant Difference post-hoc test. AUC analysis revealed that U0126 reduced anti-nociception by 89.2%, very similarly to the reduction caused by 17-AAG above (Table 2). The sample size of individual mice reported consisted of 2 technical replicates performed by the same experimenter on different days. D, HIV neuropathy was induced as in Fig. 3 and under “Experimental procedures.” On day 21, U0126 or vehicle control was i.c.v. injected as above with a 15 min treatment time. This was followed by 10 mg/kg morphine s.c., and mechanical thresholds were measured in a time course. *, **** = p < 0.05, 0.0001 versus same time point by 2 Way ANOVA with Fisher's least significant difference post-hoc test. AUC analysis revealed that U0126 reduced morphine anti-nociception by 99.7%, very similar to the value for 17-AAG (Table 2). The sample size of individual mice reported consisted of three technical replicates performed by the same experimenter on different days. Both hind paws showed very similar results; only the left paw is reported here (data not shown).
For ERK signaling, we reasoned that the loss of DAMGO stimulation seen with 17-AAG treatment (Fig. 5, D and E) was more critical for acute anti-nociception than the elevated baseline. We thus modeled the change in ERK signaling seen with 17-AAG by i.c.v. injection of 5 μg of U0126, a selective ERK inhibitor used at this i.c.v. dose previously (32). Vehicle or U0126 was i.c.v. injected with a 15-min treatment time followed by 0.1 nmol of DAMGO i.c.v. and a tail-flick assay as described above. We observed that U0126 reduced tail-flick anti-nociception with a pattern very similar to 17-AAG (Fig. 6B and Table 2) and a very similar AUC reduction of 23.5% (versus 18.2% for 17-AAG in Fig. 3C). These results suggest that the loss of ERK stimulation with 17-AAG treatment is the mechanism for the reduction of tail-flick anti-nociception seen with 17-AAG treatment.
We then extended these results to the paw-incision model to determine whether ERK signaling would explain the results there as well. We performed paw-incision surgery as described above, except without the 17-AAG i.c.v. injection. After the 24-h recovery time, the animals displayed significant mechanical allodynia as measured by Von Frey filaments. The mice were then i.c.v. injected with 5 μg of U0126 (or vehicle control) with a 15-min treatment time followed by 1 mg/kg morphine s.c. We observed that the vehicle-treated mice displayed robust time-dependent anti-nociception, as expected. We also observed that U0126 blocked nearly all morphine anti-nociception in this model, with an AUC reduction of 89.2% (Fig. 6C and Table 2). Again, the U0126 results showed a similar pattern to that of 17-AAG (results from Fig. 3A), with similar AUC reductions (89.2% for U0126 versus 70.2% for 17-AAG).
We also tested the role of ERK in our HIV peripheral neuropathy model. Peripheral neuropathy was established as described above with a 21-day treatment period. On day 21, the mice received vehicle or U0126 i.c.v. as described above, with a 15-min treatment time followed by 10 mg/kg morphine s.c. Again, we found that ERK inhibition strongly blocked morphine anti-nociception in this model, with an AUC reduction of 99.7% (Fig. 6D and Table 2). This was again very similar to the 17-AAG treatment AUC reduction value of 97.6% in this model (Table 2). Taken together, our data suggest that the loss of ERK stimulation caused by 17-AAG treatment is the mechanism by which 17-AAG reduces opioid anti-nociception in the paw-incision, tail-flick, and HIV peripheral neuropathy models.
Discussion
During the course of this study, we first sought to establish an in vitro model for Hsp90 regulation of MOR signaling, as Hsp90 is expressed ubiquitously. We found that changes in both protein expression and ERK signal transduction varied considerably between the four cell models used (Fig. 1). We took this to mean that the context of Hsp90 activity is important in determining which proteins Hsp90 regulates and thus presumably the resultant signal transduction and opioid-induced behaviors. This may be due to differential expression of Hsp90 co-chaperones (Hsp70, Hsp40, Hop/STIP1, Cdc37, etc.) in different cell and tissue types (14, 16, 33). These co-chaperones, particularly Hop/STIP1 and Cdc37, are very important in directing the activity and targeting of Hsp90 and should be investigated in the context of the brain, both to determine the mechanism of Hsp90 regulation of MOR signaling and opioid-induced behaviors and as potential targets for therapeutic intervention. Targeting the co-chaperone complex may be more selective by tissue type, thus reducing the potential side effects of targeting the ubiquitously expressed Hsp90. An analogous approach has been used to design Hsp70-targeted therapeutics and could be adapted for Hsp90 and pain (17). We note, however, that the neuroblastoma cell line SH-SY5Y displayed a similar ERK signaling profile after 17-AAG treatment to that we observed in the brain (Figs. 1C and 5, D and E). SH-SY5Y cells may express a similar profile of co-chaperones as found in the brain and should be investigated as a potential cell model for Hsp90 activity in the brain.
We also found that Hsp90 inhibition strongly or completely blocked morphine anti-nociception in the clinically relevant paw-incision post-surgical pain and HIV peripheral neuropathy pain models and slightly blocked DAMGO anti-nociception in the naive tail-flick model (Fig. 3). These results are striking and strongly implicate Hsp90 for a role in regulating opioid anti-nociception in both acute and chronic pain states. It's also notable that Hsp90 inhibition in the brain itself was able to block (79.3% reduction) systemic morphine anti-nociception from a peripherally induced pain state in the paw-incision model. This may indicate that Hsp90 in brain descending modulatory circuits is more critical than in the spinal circuits or primary afferents, at least in this particular model. Along these lines, Hsp90 inhibition only slightly reduced centrally administered DAMGO anti-nociception in the tail-flick model. This may also indicate a more important role for Hsp90 in descending modulatory circuits rather than centrally mediated affective anti-nociception (34), but this remains to be demonstrated. Because the two pain models differed by drug, opioid route, and pain model/type, these other variables must also be explored and may explain the differences seen. Morphine was chosen for the paw-incision and HIV neuropathy models due to clinical relevance of the drug and route, and DAMGO was chosen for the tail-flick model as a highly MOR-selective drug that would remain in the brain, helping to isolate mechanisms of action.
Hsp90 inhibition was even more effective in blocking opioid anti-nociception in the HIV neuropathy model than in either the paw-incision or tail-flick models (Fig. 3 and Table 2). Pain is induced in this model from the pro-inflammatory activity of the gp120 protein in the spinal cord (29). This may further indicate that Hsp90 is more effective in descending modulatory circuits to the spinal cord than in central circuits. These results also demonstrate that Hsp90 is involved in anti-nociception against multiple pain types (mechanical, thermal, inflammatory, and neuropathic) and in both acute and chronic pain states. Further investigation should focus on the molecular and circuit mechanisms by which Hsp90 regulates this broad array of anti-nociception.
We also found that Hsp90 inhibition worsened gastrointestinal effects, but not jumping behavior, in both acute and chronic morphine dependence and withdrawal (Fig. 4, A and B). Hsp90 thus has effects beyond anti-nociception, and Hsp90 inhibition worsens the opioid therapeutic index (decreasing anti-nociception and increasing side effects). These effects are likely specific to the opioid system and not off-target, as Hsp90 inhibition did not alter anti-nociceptive naive, paw-incision, or HIV neuropathy baselines or performance in the rotarod test (Fig. 4C). These results also imply that an Hsp90 activator could improve the opioid therapeutic index, which could be a basis for improving opioid therapy. Tamoxifen (35) and DMSO (19) are both Hsp90 activators, which we will use in future studies to test this hypothesis. Long-term, new and more selective Hsp90 activators need to be developed through drug discovery and development.
This study suggests that the mechanism for Hsp90 regulation of anti-nociception is through the signaling kinase ERK. 17-AAG treatment caused an elevation in the ERK signaling baseline in both the PAG and the caudal brain stem, which was unable to be stimulated above that baseline with DAMGO (Fig. 5). We also saw similar results with the kinase Akt. By using the ERK-selective inhibitor U0126 to model this effect, we were able to completely account for the anti-nociceptive changes caused by 17-AAG in tail-flick, post-surgical, and HIV neuropathy pain, with nearly identical curve shapes and AUC reductions (Fig. 6 and Table 2). Hsp70 was also up-regulated by 17-AAG treatment, but an Hsp70 inhibitor had no effect on tail-flick anti-nociception (Fig. 6A). Taken together, our results suggest that Hsp90 promotes anti-nociception in these pain models by maintaining the ability of ERK to be stimulated by opioid drugs. Our findings are summarized in Fig. 7. The magnitude of the ERK effect is different in the different pain models, with a small contribution to tail-flick anti-nociception and a very large or even total contribution to post-surgical and HIV neuropathy anti-nociception. These findings should be explored in greater detail. From the literature, it is clear that ERK has a substantial role in the pathophysiology of pain states and opioid-receptor function and side effects. However, that role appears to be very context-dependent, as ERK located in different circuits or regions or manipulated in different models can have different or opposing effects (36–41). We found ∼5 studies each that tested ERK specifically in paw-incision and tail-flick pain, and none of these tested these models as we did, specifically by testing ERK in the opioid anti-nociceptive response. We could find no studies that tested the role of ERK in HIV neuropathic pain. Thus our findings contribute to the growing understanding of the role of ERK in pain and opioid response, which should be further investigated for possible therapeutic use.
Figure 7.
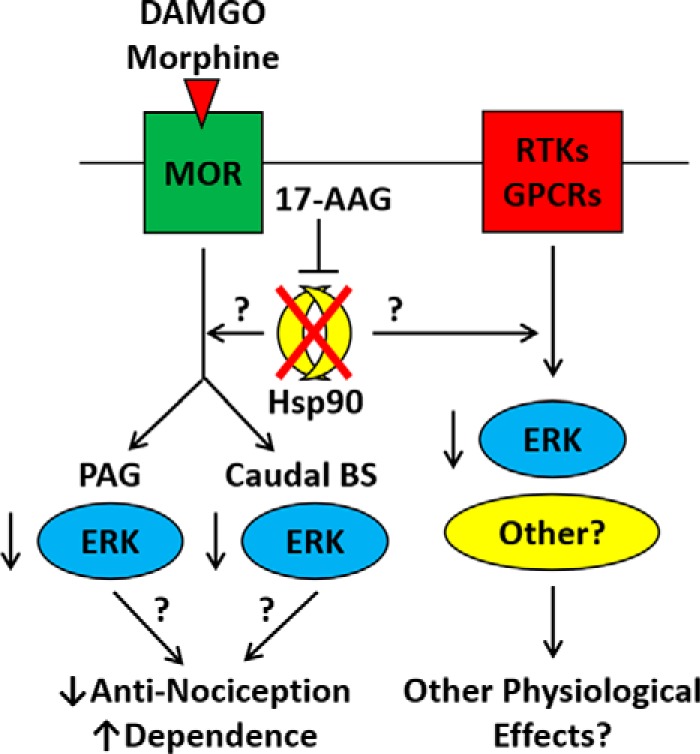
Summary model of the role of Hsp90 in opioid signaling and behavior. This model summarizes our findings that Hsp90 inhibition causes ERK MAPK to lose the ability to be stimulated through opioid-receptor activation in the brain. This loss of stimulation may account for the loss of opioid anti-nociception seen in three pain models. The molecular mechanisms linking Hsp90 to ERK and ERK to anti-nociception and potentially to dependence/withdrawal are currently unknown. We also note in this figure that Hsp90 effects on signaling kinases are likely to impact other receptor systems, including GPCRs and receptor tyrosine kinases. Stimulating these systems while Hsp90 is inhibited could cause a variety of other physiological effects.
From known roles in the literature and our data showing changes in ERK and Akt generally in the brain, it is very likely that numerous other receptor systems are affected by Hsp90 inhibition. These could include receptor tyrosine kinases, other GPCRs, and other receptor systems generally. Specificity to the opioid system was achieved in this study by using selective opioid drugs and assessing opioid-mediated behaviors. This represents an opportunity, in that our model will be useful for exploring the role of Hsp90 in many brain systems and responses. This also represents a challenge to developing Hsp90 as a therapeutic target, as there are likely to be off-target effects. As discussed above, targeting selective co-chaperones could be a means of decreasing off-target interactions (17). In addition, biased ligands or similar approaches could be developed to manipulate the Hsp90-regulated signaling cascade downstream of a specific receptor. This would be analogous to the method used to target another universal regulator, βarr2 (8). The likelihood that Hsp90 affects other receptor systems is also included in the summary presented in Fig. 7.
As discussed above, Hsp90 has been investigated in 4 studies for its role in pain and opioid response. The Devi group (21) found that Hsp90 inhibition immediately prior to precipitation of morphine withdrawal reduced jumping behavior; and a separate group found that Hsp90 inhibition blocked adenylyl cyclase superactivation, a marker of dependence, in a cell model (20). In contrast, we found that Hsp90 inhibition promoted the gastrointestinal effects of dependence and withdrawal (Fig. 4, A and B). For the Devi study (21), the differences observed may be because they applied the inhibitor immediately prior to withdrawal via the i.p route, whereas we applied inhibitor chronically using the i.c.v. route. We suggest that chronic treatment may more closely model chronic pain treatment, but these differences are still notable. We will investigate the chronic application of 17-AAG systemically in future studies to explore these differences. For the adenylyl cyclase superactivation study, Koshimizu et al. (20) applied the inhibitor chronically as did we. However, as we discovered in our in vitro studies (Fig. 1), the effects of Hsp90 inhibition can be highly heterogeneous between lines. The SK-N-SH cells used in that study may or may not accurately model Hsp90 in the brain. Further investigation into the role of Hsp90 in dependence and withdrawal is needed.
For the pain studies, it was found that Hsp90 inhibition reversed diabetes-induced sensory hypoalgesia in mice (18) and that Hsp90 inhibition reversed CCI-induced mechanical allodynia and potentiated morphine anti-nociception (19). The hypoalgesia results are too orthogonal to our model for a direct comparison, but the reduction in pain and enhancement of morphine anti-nociception in the CCI model is opposed to our results, particularly our findings with HIV neuropathy, as CCI is also a chronic neuropathic pain model. Important differences between our studies include that the Hsp90 inhibitor was given systemically over a 3-h or less time scale, whereas we administered inhibitor i.c.v. for 24 h. CCI also has a different mechanism of pain induction than HIV neuropathy. Either of these differences could explain the discrepancy, and future studies will explore CCI neuropathic pain and different routes of administration in our pain models. Given the importance of context to Hsp90 function, future therapy may need to be closely tailored to the specific pain state of the patient.
In summary, we have demonstrated that Hsp90 promotes opioid anti-nociception in three different pain models by an ERK-dependent mechanism. Based on our signaling findings in the brain, this is likely accomplished by Hsp90 maintaining ERK in a competent state for opioid signal transduction activation. Despite the role of Hsp90 as a ubiquitous and crucial signaling regulator, very little work has been done on the role of this protein in opioid signal transduction and pain. Our results thus make a significant contribution to this field and provide the rationale for testing Hsp90 activators as therapies to enhance the opioid therapeutic index. Future studies will expand on the molecular mechanisms downstream of Hsp90 and ERK responsible for these effects and the specific cell types, regions, and circuits in the brain responsible for the effects of Hsp90 on opioid signal transduction and anti-nociception.
Experimental procedures
Materials
17-AAG, DAMGO, LPA, naloxone, and U0126 were all purchased from Tocris (subsidiary of R&D Systems, Minneapolis, MN). Pifithrin-μ was purchased from Sigma-Aldrich. Morphine sulfate pentahydrate was obtained through the National Institute on Drug Abuse Drug Supply Program and distributed through the Research Triangle Institute. Gp120 IIIb protein was obtained from SPEED BioSystems, LLC (Gaithersburg, MD, catalog no. YCP1549). 17-AAG, U0126, and pifithrin-μ were prepared as stock solutions in DMSO, and DAMGO and LPA were prepared as stock solutions in water. Naloxone and morphine were prepared fresh for each experiment in USP saline. Appropriate vehicle controls were used for each experiment: 0.1% DMSO in serum-free culture media for 17-AAG and DAMGO in the culture experiments; 10% DMSO in water for 17-AAG i.c.v. injections; water for DAMGO i.c.v. injections; USP saline for systemic morphine and naloxone injections; and 10% DMSO, 10% Tween-80, and 80% USP saline for the 17-AAG/pifithrin-μ and U0126 i.c.v. injections.
Cell lines, culture, and in vitro experiments
Parental CHO-K1, HEK, and SH-SY5Y cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). Human MOR-β-arrestin2EGFP-U2OS cells were a kind gift from Dr. Larry Barak of the Duke University Addiction Research GPCR Assay Bank (42). The SH-SY5Y and U2OS cells were used without further modification. The CHO and HEK cells were developed into stable cell lines by electroporation of a 3X-HA N-terminally tagged human MOR expression construct (Genecopoeia, Rockville, MD) and by selection with 500 μg/ml G418. The populations were enriched for MOR expression by two cycles of flow cytometry, selecting the top 2–3% of the population in each round. The resulting stable, high-expressing cell lines were characterized by immunocytochemistry and Western blot analysis of ERK activation in response to DAMGO, and the CHO line was further characterized by saturation radioligand binding with [3H]diprenorphine (KD = 2.81 ± 0.72 nm; Bmax = 435 ± 76 fmol/mg; n = 3 independent experiments). Both lines were also characterized in multiple signal transduction assays for MOR function for other projects (data not shown).
The cells were all maintained in growth medium (DMEM/F12 for CHO and SH-SY5Y cells and MEM for HEK and U2OS cells) with 10% heat-inactivated FBS (Gibco brand, ThermoFisher Scientific) and 1× penicillin/streptomycin. The CHO and HEK cells were further maintained with 500 μg/ml G418, and the U20S cells with 200 μg/ml G418 and 100 μg/ml Zeocin. All lines were incubated at 37 °C in a 5% CO2 humidified incubator. All lines were monitored for mycoplasma contamination by DAPI staining and microscopy, and all cells were mycoplasma-negative for these experiments.
For experiments, the cells were plated in 6-well plates in a minimum of two replicate wells/condition (averaged for a technical replicate of n = 1), recovered overnight, and then treated with 1 μm 17-AAG or vehicle for a further 24 h. Then the cells were serum-starved with the appropriate serum-free culture medium: 30 min for HEK, 60 min for CHO, and 24 h for U2OS and SH-SY5Y cells (serum starvation concurrent with 17-AAG treatment for U2OS and SH-SY5Y cells). The cells were then treated with vehicle, 10 μm DAMGO, or 10 μm LPA for 10 min. The cells were washed in ice-cold PBS and then collected with a plastic cell scraper in the presence of lysis buffer (20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 2 mm EDTA, 0.1% SDS, 1% Nonidet P-40 or equivalent, 0.25% deoxycholate, 1 mm sodium orthovanadate, 1 mm PMSF, 1 mm sodium fluoride, and a protease inhibitor mixture from EMD Millipore, Billerica, MA). The lysates were sheared twice with a 27-gauge needle and centrifuged at 13,000 rpm at 4 °C for 10 min, and the supernatants were stored at −80 °C.
Animals
Male CD-1 mice in age-matched controlled cohorts from 4–8 weeks of age were used for all experiments and were obtained from Charles River Laboratories (Wilmington, MA). CD-1 (a.k.a. ICR) mice are commonly used in opioid research as a line with a strong response to opioid drugs (e.g. see Refs. 43 and 44). Mice were allowed to recover for a minimum of 5 days after shipment before being used in experiments. The mice were kept in AAALAC-accredited vivaria at the University of New England and the University of Arizona under temperature control and 12-h light/dark cycles with food (standard lab chow) and water available ad libitum. No more than five mice were kept in a cage. The animals were monitored daily, including after surgical procedures, by trained veterinary staff. All experiments performed were in accordance with IACUC-approved protocols at both the University of New England and the University of Arizona.
Dose-response and brain-signaling experiments
For the dose-response experiments, the mice were injected i.c.v. with 0.05, 0.5, or 5 nmol of 17-AAG or vehicle. For the brain signaling experiments, the mice were injected with 0.5 nmol of 17-AAG i.c.v. This was performed by anesthetizing the mice with ∼2% isoflurane in standard air and incising the skin by ∼5 mm with a no. 10 scalpel blade. The i.c.v. injection was made using a jacketed 10-μl Hamilton syringe in a 5-μl maximum volume, 1 mm posterior and 1 mm lateral to bregma and 2.5 mm deep. The skin was closed with tissue glue, and the mice were allowed to recover for 24 h in their home cage. The next day, the signaling experiment mice had the incision reopened, and an i.c.v. injection of 0.1 nmol of DAMGO was done as above using the same site. After 10 min, the mice were sacrificed by rapid cervical dislocation. The dose-response mice proceeded immediately to sacrifice after 24 h. For both groups of mice, the brains were removed and dissected on an ice-cooled block. PAG and caudal brainstems (pons and medulla) were removed and rapidly frozen on liquid nitrogen. Protein was extracted using the same lysis buffer and general protocol as described above, except that a Polytron homogenizer was used to disrupt the tissue (maximum speed, 3 × 10 s, on ice) for PAG, and a 1-ml glass/glass Dounce homogenizer was used for the caudal brain stem. For a subset of the brain signaling experiments, MOR and βarr2 were immunoprecipitated from PAG using the protocol reported by Schmid and Bohn (45).
Western blotting and blot analysis
Cell and brain protein lysates were quantified with a BCA protein quantitation assay using the manufacturer's protocol (Bio-Rad). The protein was run on precast Tris-glycine gels using a mini-PROTEAN Tetra Western blotting apparatus (Bio-Rad) following the manufacturer's instructions. The gels were transferred to nitrocellulose membrane (Bio-Rad) using a wet transfer system (30 V, minimum of 1 h on ice). The blots were blocked with 5% nonfat dry milk in TBS and incubated with primary antibody in 5% BSA in TBS + 0.1% Tween-20 (TBST) overnight rocking at 4 °C. The blots were then washed three times for 5 min in TBST, incubated with secondary antibody (see below) in 5% milk in TBST for 1 h of rocking at room temperature, washed again, and imaged with a LiCor Odyssey infrared imaging system (LiCor, Lincoln, NE). Some blots were then stripped with 25 mm glycine-HCl and 1% SDS, pH 2.0, for 30–60 min of rocking at room temperature prior to being washed and re-exposed to primary antibody.
The resulting image bands were quantified using Scion Image (based on NIH Image). All images were quantified in the linear signal range, which is easier to ensure because the Odyssey imager is a dynamic imager that allows for fine control of exposure. Scion Image also indicated any saturated pixels. The intensity levels of all proteins except ERK and Akt were normalized to the housekeeping protein GAPDH, the pERK signal was normalized to the tERK signal, and pAkt was normalized to tAkt, with both measured from the same blot as the primary target. Separate quantitation of GAPDH, tERK, and tAkt indicated that the drug treatments used did not change the expression levels of these proteins (Fig. 5, A–C, data not shown). The normalized intensities were further normalized to a vehicle control present on the same blot.
Antibodies
All antibodies were validated prior to use by in vitro experiments using a cell line expressing the target in question with a negative control cell line that did not express the target in question or, in the case of Hsp70 and ERK, by treatment with a drug as a positive control. Hsp70 and ERK were validated by proper response to the drug (17-AAG and DAMGO/10% FBS/LPA); the other targets were validated by comparison with the negative control.
The antibodies used were: Hsp70 (Cell Signaling 4872S, lot 4, rabbit, 1:1000), GAPDH (ThermoFisher MA5-15738, lot PI209504, mouse, 1:1000), STAT3 (Cell Signaling 9139S, lot 7, mouse, 1:1000), pERK (Cell Signaling 4370S, lot 12, rabbit, 1:1000), tERK (Cell Signaling 4696S, lot 16, mouse, 1:1000), pAkt (Cell Signaling 9018P, lot 3, rabbit, 1:1000), tAkt (Cell Signaling 2920S, lot 3, mouse, 1:1000), MOR (Abcam ab10275, lot GR81301-2, 1:500 for immunoprecipitation), β-arrestin2 (Abcam ab54790, rabbit, 1:500 for immunoprecipitation), β-arrestin2 (Cell Signaling 3857S, 2, rabbit, 1:1000 for Western blotting), HA tag for in vitro MOR (Cell Signaling 3724S, lot 3, rabbit, 1:1000), secondary GαM680 (LiCor 926-68020, lot C50721-02, goat, 1:10,000–1:20,000), and secondary GαR800 (LiCor 926-32211, lot C50602–05, goat, 1:10,000–1:20,000).
Behavioral experiments
Prior to any behavioral experiment or testing, the animals were brought to the testing room in their home cages for at least 1 h for acclimation. Testing always occurred within the same approximate time of day between experiments, and environmental factors (noise, personnel, and scents) were minimized. All testing apparatus (cylinders, grid boxes, etc.) were cleaned between uses. The experimenter was blinded to treatment group by another laboratory member delivering coded drug vials, which were then decoded after collection of all data.
Paw incision and mechanical allodynia
Mechanical thresholds were determined prior to surgery using calibrated Von Frey filaments (Ugo Basile, Varese, Italy) with the up-down method and four measurements after the first response per mouse (28). The mice were housed in a homemade apparatus with Plexiglas walls and ceiling and a wire-mesh floor (3-inch wide × 4-inch long × 3-inch high with 0.25-inch wire mesh). The surgery was then performed by anesthesia with ∼2% isoflurane in standard air, preparation of the left plantar hind paw with iodine and 70% ethanol, and a 5-mm incision made through the skin and fascia with a no. 11 scalpel. The muscle was elevated with curved forceps leaving the origin and insertion intact, and the muscle was split lengthwise using the scalpel. The wound was then closed with 5-0 polyglycolic acid sutures. For the 17-AAG experiments, the mice were then injected i.c.v. and left to recover for 24 h. For the U0126 experiments, the mice went directly to recovery. The next day, the mechanical threshold was again determined as described above, and i.c.v. injections took place for the U0126 experiments with a 15-min treatment time. Both the 17-AAG and the U0126 mice were then injected with 1, 3.2, or 10 mg/kg morphine s.c., and mechanical thresholds were determined over a time course. No animals were excluded from these studies.
Tail-flick assay
Pre-injection tail-flick baselines were determined in a 52 °C tail-flick assay with a 10-s cutoff time. The mice were then injected i.c.v. with 17-AAG/pifithrin-μ or U0126 with a 24-h (17-AAG/pifithrin-μ) or 15-min (U0126) treatment time. 24 h post-injection baselines were determined for the 17-AAG experiments. The mice were then injected i.c.v. with 0.01, 0.032, or 0.1 nmol of DAMGO, and tail-flick latencies were determined over a time course. After completing the study, the mice that did not respond to DAMGO were excluded from the analysis, suggesting a missed i.c.v. injection, as it was clear that the 17-AAG and other treatments did not cause a large decrease in anti-nociception.
HIV peripheral neuropathy
HIV peripheral neuropathy was induced by intrathecal injection of gp120 protein (15 ng/μl in 0.1 m PBS and 0.1% BSA, 7-μl volume) using an established protocol (29) on days 1, 3, and 5. Mechanical thresholds were monitored every 2 days on both hind paws using Von Frey filaments as described above. For the 17-AAG experiments, 17-AAG was injected i.c.v. as described above on day 20 with a 24-h treatment time. For the U0126 experiments, U0126 was injected i.c.v. on day 21 with a 15-min treatment time. For both experiments, morphine (10 mg/kg s.c.) was then injected, and mechanical thresholds were measured over a time course on both hind paws. Both hind paws gave highly similar results, so only the morphine results from the left paw are shown (data not shown). No animals were excluded from these studies.
Dependence and withdrawal
For the acute dependence and withdrawal paradigm, mice were injected i.c.v. with 17-AAG or vehicle and left to recover for 20 h. They were then injected with 100 mg/kg morphine s.c. and left to recover for 4 h, and then withdrawal was precipitated with 10 mg/kg naloxone i.p. For the chronic dependence and withdrawal paradigm, a 4-day escalating-dosage protocol was used. Morphine was injected twice daily, with 1× A90 on day 1, 2× A90 on day 2, 3.5× A90 on day 3, and finally one injection of 3.5× A90 on day 4 followed 4 h later by 30 mg/kg naloxone i.p. 17-AAG or vehicle was injected i.c.v. daily from days 0–3. For both paradigms, behavior was observed in 16-inch-high × 6-inch-deep Plexiglas cylinders for 20 min. The number of jumps was recorded, and the weight of the urine and feces was recorded at the end of the observation period. One whole technical replicate of the chronic dependence and withdrawal was excluded (both treatment groups) because the majority of the vehicle-treated animals did not display any withdrawal behavior. No animals were excluded from the acute dependence and withdrawal studies.
Rotarod test
Mice were subjected to three training trials of 3 min each on a Rotarod device, with the machine off for trial 1, the machine on but not rotating for trial 2, and the machine rotating at 4 rpm for trial 3 (30). An automatic timer in the unit was used to record fall latencies with a 3-min maximum time. The mice were then injected i.c.v. with 17-AAG or vehicle and allowed to recover for 24 h, and another 3-min Rotarod trial was performed without additional treatments or interventions. This trial was done with an accelerating 4–16 rpm over the 3-min trial time. No mice were excluded from these studies.
Statistical analysis
All data were reported as the mean ± S.D. and normalized where appropriate as described above. The behavioral data were reported raw without maximum possible effect (MPE) or other normalization. Biological and technical replicates are described in the figure legends. Comparisons between two groups (protein expression and withdrawal) were performed by unpaired 2-tailed t tests. Comparisons of more than two groups (ERK and Akt signaling, paw incision, tail flick, HIV neuropathy, and rotarod) were performed by two-way ANOVA with Fisher's least significant difference post-hoc test. In all cases, significance was defined as p < 0.05. For the dose-response experiments, the A50 was calculated by performing linear regression analysis of the data points and solving for A50 by the equation y = α + β(logA50), where y is the effect at the desired percentage (i.e. 1.6 at the A50 in the 17-AAG dose-response experiment), α is the y intercept of the line, and β is the slope. The stimulation caused by 5 nmol of 17-AAG was assumed to be the maximum stimulation for the 17-AAG dose-response analysis. AUC analysis was performed in the tail-flick, paw-incision, and HIV neuropathy experiments by setting the lowest point from both summary curves as the baseline in each analysis. The dose-response curves for the paw-incision and tail-flick experiments were constructed using the AUC from the individual curve of each mouse. All graphing, AUC, and statistical analyses were performed using GraphPad Prism 7.0 (San Diego, CA).
Author contributions
W. L. performed most of the in vivo and some of the in vitro work, analyzed the data, and participated in the experimental design and project direction. N. M. and C. B. performed both in vitro and in vivo Western blot experiments, analyzed the data, and participated in the experimental design. S. M. and P. R. performed significant numbers of in vitro experiments, analyzed the data, and participated in the experimental design. J. C. performed the dependence and withdrawal experiments and part of the initial tail-flick experiments, analyzed the data, and participated in the experimental design. K. E. performed a few of the in vitro experiments, analyzed the data, and supervised N. M., S. M., C. B., and P. R. in the performance and analysis of their work. E. J. B. supervised J. C. in the design and performance of the dependence and initial tail-flick experiments. J. M. S. conceived and directed the project at all stages, including the supervision of personnel and analysis of data, and wrote the manuscript. All authors had editorial input to the manuscript.
Acknowledgments
We thank Kayla Lindros of the University of New England for technical assistance with the dependence and withdrawal experiments and Dr. Larry Barak of the Duke University Addiction Research GPCR Assay Bank for the kind gift of the MOR-β-arrestin2EGFP-U2OS cell line.
This work was supported by the University of New England and the University of Arizona and by National Institutes of Health Pilot Project Grant P20GM103643 (Ian Meng PI) under an institutional COBRE award. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- MOR
- μ-opioid receptor
- βarr2
- β-arrestin2
- Hsp
- heat-shock protein
- CCI
- chronic constriction injury
- 17-AAG
- 17-N-allylamino-17-demethoxygeldanamycin
- pERK
- phosphorylated ERK
- tERK
- total ERK
- LPA
- lysophosphatidic acid
- i.c.v.
- intracerebroventricular
- PAG
- periaqueductal gray
- s.c.
- subcutaneous
- GPCR
- G-protein-coupled receptor
- ANOVA
- analysis of variance
- AUC
- area under the curve
- DAMGO
- [d-Ala2, N-MePhe4, Gly2-ol]-enkephalin.
References
- 1. Gaskin D. J., and Richard P. (2012) The economic costs of pain in the United States. J. Pain 13, 715–724 [DOI] [PubMed] [Google Scholar]
- 2. Breivik H., Cherny N., Collett B., de Conno F., Filbet M., Foubert A. J., Cohen R., and Dow L. (2009) Cancer-related pain: a pan-European survey of prevalence, treatment, and patient attitudes. Ann. Oncol. 20, 1420–1433 [DOI] [PubMed] [Google Scholar]
- 3. National Clinical Guideline Centre for Acute and Chronic Conditions (Great Britain) (2012) in Opioids in Palliative Care: Safe and Effective Prescribing of Strong Opioids for Pain in Palliative Care of Adults, National Collaborating Centre for Cancer, Cardiff, UK: [PubMed] [Google Scholar]
- 4. Raehal K. M., Walker J. K., and Bohn L. M. (2005) Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther. 314, 1195–1201 [DOI] [PubMed] [Google Scholar]
- 5. Woodcock J., Witter J., and Dionne R. A. (2007) Stimulating the development of mechanism-based, individualized pain therapies. Nat. Rev. Drug Discov. 6, 703–710 [DOI] [PubMed] [Google Scholar]
- 6. Bohn L. M., Lefkowitz R. J., Gainetdinov R. R., Peppel K., Caron M. G., and Lin F. T. (1999) Enhanced morphine analgesia in mice lacking β-arrestin 2. Science 286, 2495–2498 [DOI] [PubMed] [Google Scholar]
- 7. Raehal K. M., and Bohn L. M. (2011) The role of β-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology 60, 58–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. DeWire S. M., Yamashita D. S., Rominger D. H., Liu G., Cowan C. L., Graczyk T. M., Chen X. T., Pitis P. M., Gotchev D., Yuan C., Koblish M., Lark M. W., and Violin J. D. (2013) A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared to morphine. J. Pharmacol. Exp. Ther. 344, 708–717 [DOI] [PubMed] [Google Scholar]
- 9. Chen X. T., Pitis P., Liu G., Yuan C., Gotchev D., Cowan C. L., Rominger D. H., Koblish M., Dewire S. M., Crombie A. L., Violin J. D., and Yamashita D. S. (2013) Structure-activity relationships and discovery of a G protein-biased μ opioid receptor ligand, [(3-methoxythiophen-2-yl)methyl]({2-[(9R)-9-(pyridin-2-yl)-6-oxaspiro-[4.5]decan- 9-yl]ethyl})amine (TRV130), for the treatment of acute severe pain. J. Med. Chem. 56, 8019–8031 [DOI] [PubMed] [Google Scholar]
- 10. Soergel D. G., Subach R. A., Burnham N., Lark M. W., James I. E., Sadler B. M., Skobieranda F., Violin J. D., and Webster L. R. (2014) Biased agonism of the μ-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: a randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 155, 1829–1835 [DOI] [PubMed] [Google Scholar]
- 11. Viscusi E. R., Webster L., Kuss M., Daniels S., Bolognese J. A., Zuckerman S., Soergel D. G., Subach R. A., Cook E., and Skobieranda F. (2016) A randomized, phase 2 study investigating TRV130, a biased ligand of the μ-opioid receptor, for the intravenous treatment of acute pain. Pain 157, 264–272 [DOI] [PubMed] [Google Scholar]
- 12. Yue X., Varga E. V., Stropova D., Vanderah T. W., Yamamura H. I., and Roeske W. R. (2006) Chronic morphine-mediated adenylyl cyclase superactivation is attenuated by the Raf-1 inhibitor, GW5074. Eur. J. Pharmacol. 540, 57–59 [DOI] [PubMed] [Google Scholar]
- 13. Traynor J. (2012) μ-Opioid receptors and regulators of G protein signaling (RGS) proteins: from a symposium on new concepts in μ-opioid pharmacology. Drug Alcohol Depend. 121, 173–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li J., and Buchner J. (2013) Structure, function and regulation of the hsp90 machinery. Biomed. J. 36, 106–117 [DOI] [PubMed] [Google Scholar]
- 15. Ota A., Zhang J., Ping P., Han J., and Wang Y. (2010) Specific regulation of noncanonical p38α activation by Hsp90-Cdc37 chaperone complex in cardiomyocyte. Circ. Res. 106, 1404–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Odunuga O. O., Longshaw V. M., and Blatch G. L. (2004) Hop: more than an Hsp70/Hsp90 adaptor protein. Bioessays 26, 1058–1068 [DOI] [PubMed] [Google Scholar]
- 17. Assimon V. A., Gillies A. T., Rauch J. N., and Gestwicki J. E. (2013) Hsp70 protein complexes as drug targets. Curr. Pharm. Des. 19, 404–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Urban M. J., Li C., Yu C., Lu Y., Krise J. M., McIntosh M. P., Rajewski R. A., Blagg B. S., and Dobrowsky R. T. (2010) Inhibiting heat-shock protein 90 reverses sensory hypoalgesia in diabetic mice. ASN Neuro. 2, e00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hutchinson M. R., Ramos K. M., Loram L. C., Wieseler J., Sholar P. W., Kearney J. J., Lewis M. T., Crysdale N. Y., Zhang Y., Harrison J. A., Maier S. F., Rice K. C., and Watkins L. R. (2009) Evidence for a role of heat shock protein-90 in toll like receptor 4-mediated pain enhancement in rats. Neuroscience 164, 1821–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Koshimizu T. A., Tsuchiya H., Tsuda H., Fujiwara Y., Shibata K., Hirasawa A., Tsujimoto G., and Fujimura A. (2010) Inhibition of heat shock protein 90 attenuates adenylate cyclase sensitization after chronic morphine treatment. Biochem. Biophys. Res. Commun. 392, 603–607 [DOI] [PubMed] [Google Scholar]
- 21. Abul-Husn N. S., Annangudi S. P., Ma'ayan A., Ramos-Ortolaza D. L., Stockton S. D. Jr., Gomes I., Sweedler J. V., and Devi L. A. (2011) Chronic morphine alters the presynaptic protein profile: identification of novel molecular targets using proteomics and network analysis. PloS One 6, e25535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jhaveri K., Ochiana S. O., Dunphy M. P., Gerecitano J. F., Corben A. D., Peter R. I., Janjigian Y. Y., Gomes-DaGama E. M., Koren J. 3rd, Modi S., and Chiosis G. (2014) Heat shock protein 90 inhibitors in the treatment of cancer: current status and future directions. Expert Opin. Investig. Drugs 23, 611–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang Q., and Traynor J. R. (2013) Modulation of μ-opioid receptor signaling by RGS19 in SH-SY5Y cells. Mol. Pharmacol. 83, 512–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang W., Liu Y., Zhao Z., Xie C., Xu Y., Hu Y., Quan H., and Lou L. (2016) Y-632 inhibits Hsp90 function through disrupting the interaction of Hsp90-Hop and exerts antitumor activity in vitro and in vivo. Cancer Sci. 107, 782–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim N., Kim J. Y., and Yenari M. A. (2015) Pharmacological induction of the 70-kDa heat shock protein protects against brain injury. Neuroscience 284, 912–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kovelowski C. J., Ossipov M. H., Sun H., Lai J., Malan T. P., and Porreca F. (2000) Supraspinal cholecystokinin may drive tonic descending facilitation mechanisms to maintain neuropathic pain in the rat. Pain 87, 265–273 [DOI] [PubMed] [Google Scholar]
- 27. Pogatzki E. M., and Raja S. N. (2003) A mouse model of incisional pain. Anesthesiology 99, 1023–1027 [DOI] [PubMed] [Google Scholar]
- 28. Chaplan S. R., Bach F. W., Pogrel J. W., Chung J. M., and Yaksh T. L. (1994) Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 [DOI] [PubMed] [Google Scholar]
- 29. Yuan S. B., Shi Y., Chen J., Zhou X., Li G., Gelman B. B., Lisinicchia J. G., Carlton S. M., Ferguson M. R., Tan A., Sarna S. K., and Tang S. J. (2014) Gp120 in the pathogenesis of human immunodeficiency virus-associated pain. Ann. Neurol. 75, 837–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vanderah T. W., Largent-Milnes T., Lai J., Porreca F., Houghten R. A., Menzaghi F., Wisniewski K., Stalewski J., Sueiras-Diaz J., Galyean R., Schteingart C., Junien J. L., Trojnar J., and Rivière P. J. (2008) Novel d-amino acid tetrapeptides produce potent antinociception by selectively acting at peripheral κ-opioid receptors. Eur. J. Pharmacol. 583, 62–72 [DOI] [PubMed] [Google Scholar]
- 31. Qin W. J., Wang Y. T., Zhang M., Wen R. T., Liu Q., Li Y. L., Chen F., Lawrence A. J., and Liang J. H. (2013) Molecular chaperone heat shock protein 70 participates in the labile phase of the development of behavioural sensitization induced by a single morphine exposure in mice. Int. J. Neuropsychopharmacol. 16, 647–659 [DOI] [PubMed] [Google Scholar]
- 32. Zeni A. L., Zomkowski A. D., Maraschin M., Rodrigues A. L., and Tasca C. I. (2012) Involvement of PKA, CaMKII, PKC, MAPK/ERK, and PI3K in the acute antidepressant-like effect of ferulic acid in the tail suspension test. Pharmacol. Biochem. Behav. 103, 181–186 [DOI] [PubMed] [Google Scholar]
- 33. McConnell J. R., and McAlpine S. R. (2013) Heat shock proteins 27, 40, and 70 as combinational and dual therapeutic cancer targets. Bioorg. Med. Chem. Lett. 23, 1923–1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ossipov M. H., Lai J., Malan T. P. Jr, and Porreca F. (2000) Spinal and supraspinal mechanisms of neuropathic pain. Ann. N.Y. Acad. Sci. 909, 12–24 [DOI] [PubMed] [Google Scholar]
- 35. Zhao R., Leung E., Grüner S., Schapira M., and Houry W. A. (2010) Tamoxifen enhances the Hsp90 molecular chaperone ATPase activity. PloS One 5, e9934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Moulédous L., Díaz M. F., and Gutstein H. B. (2007) Extracellular signal-regulated kinase (ERK) inhibition does not prevent the development or expression of tolerance to and dependence on morphine in the mouse. Pharmacol. Biochem. Behav. 88, 39–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen Y., Geis C., and Sommer C. (2008) Activation of TRPV1 contributes to morphine tolerance: involvement of the mitogen-activated protein kinase signaling pathway. J. Neurosci. 28, 5836–5845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Macey T. A., Bobeck E. N., Hegarty D. M., Aicher S. A., Ingram S. L., and Morgan M. M. (2009) Extracellular signal-regulated kinase 1/2 activation counteracts morphine tolerance in the periaqueductal gray of the rat. J. Pharmacol. Exp. Ther. 331, 412–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gudasheva T. A., Povarnina P. Y., Antipova T. A., Firsova Y. N., Konstantinopolsky M. A., and Seredenin S. B. (2015) Dimeric dipeptide mimetics of the nerve growth factor loop 4 and loop 1 activate TRKA with different patterns of intracellular signal transduction. J. Biomed. Sci. 22, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dai R. P., Li C. Q., Zhang J. W., Li F., Shi X. D., Zhang J. Y., and Zhou X. F. (2011) Biphasic activation of extracellular signal-regulated kinase in anterior cingulate cortex distinctly regulates the development of pain-related anxiety and mechanical hypersensitivity in rats after incision. Anesthesiology 115, 604–613 [DOI] [PubMed] [Google Scholar]
- 41. Tillu D. V., Melemedjian O. K., Asiedu M. N., Qu N., De Felice M., Dussor G., and Price T. J. (2012) Resveratrol engages AMPK to attenuate ERK and mTOR signaling in sensory neurons and inhibits incision-induced acute and chronic pain. Mol. Pain 8, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Barak L. S., Ferguson S. S., Zhang J., and Caron M. G. (1997) A β-arrestin/green fluorescent protein biosensor for detecting G protein-coupled receptor activation. J. Biol. Chem. 272, 27497–27500 [DOI] [PubMed] [Google Scholar]
- 43. Lowery J. J., Raymond T. J., Giuvelis D., Bidlack J. M., Polt R., and Bilsky E. J. (2011) In vivo characterization of MMP-2200, a mixed δ/μ opioid agonist, in mice. J. Pharmacol. Exp. Ther. 336, 767–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ananthan S., Saini S. K., Dersch C. M., Xu H., McGlinchey N., Giuvelis D., Bilsky E. J., and Rothman R. B. (2012) 14-Alkoxy- and 14-acyloxypyridomorphinans: μ agonist/δ antagonist opioid analgesics with diminished tolerance and dependence side effects. J. Med. Chem. 55, 8350–8363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schmid C. L., and Bohn L. M. (2010) Serotonin, but not N-methyltryptamines, activates the serotonin 2A receptor via a ss-arrestin2/Src/Akt signaling complex in vivo. J. Neurosci. 30, 13513–13524 [DOI] [PMC free article] [PubMed] [Google Scholar]