

Abstract
Disruption of the Gys2 gene encoding the liver isoform of glycogen synthase generates a mouse strain (LGSKO) that almost completely lacks hepatic glycogen, has impaired glucose disposal, and is pre-disposed to entering the fasted state. This study investigated how the lack of liver glycogen increases fat accumulation and the development of liver insulin resistance. Insulin signaling in LGSKO mice was reduced in liver, but not muscle, suggesting an organ-specific defect. Phosphorylation of components of the hepatic insulin-signaling pathway, namely IRS1, Akt, and GSK3, was decreased in LGSKO mice. Moreover, insulin stimulation of their phosphorylation was significantly suppressed, both temporally and in an insulin dose response. Phosphorylation of the insulin-regulated transcription factor FoxO1 was somewhat reduced and insulin treatment did not elicit normal translocation of FoxO1 out of the nucleus. Fat overaccumulated in LGSKO livers, showing an aberrant distribution in the acinus, an increase not explained by a reduction in hepatic triglyceride export. Rather, when administered orally to fasted mice, glucose was directed toward hepatic lipogenesis as judged by the activity, protein levels, and expression of several fatty acid synthesis genes, namely, acetyl-CoA carboxylase, fatty acid synthase, SREBP1c, chREBP, glucokinase, and pyruvate kinase. Furthermore, using cultured primary hepatocytes, we found that lipogenesis was increased by 40% in LGSKO cells compared with controls. Of note, the hepatic insulin resistance was not associated with increased levels of pro-inflammatory markers. Our results suggest that loss of liver glycogen synthesis diverts glucose toward fat synthesis, correlating with impaired hepatic insulin signaling and glucose disposal.
Keywords: fatty acid metabolism, glycogen, glycogen storage disease, insulin resistance, lipogenesis, fatty liver
Introduction
After ingestion of a meal, glucose is cleared from the bloodstream primarily by conversion to glycogen in skeletal muscle and liver, the liver disposing as much as one-third of the glucose load (1). Defects in insulin-mediated regulation of glycogen synthase (GS)5 a key biosynthetic enzyme (2) are present in the muscle of most type 2 diabetes mellitus (T2DM) patients, who display post-prandial hyperglycemia and compromised glucose disposal (3, 4). Two GS genes exist in mammals, GYS1, encoding the isoform expressed in muscle and many other tissues, and GYS2, encoding an apparently liver-specific isoform (LGS). Mice lacking GYS2 (LGSKO) have a severe decrease in their ability to store glycogen in hepatocytes (5), and display most of the symptoms of glycogen storage disease in patients who have loss-of-function mutations in the GYS2 gene (6). LGSKO mice are glucose intolerant and exhibit impaired suppression of gluconeogenesis upon insulin stimulation (5). Mice with a disruption of the GYS1 gene are unable to synthesize glycogen in several tissues, but surprisingly glucose tolerance was actually improved (7, 8). One conclusion from these studies, and studies using other genetically engineered murine models (9, 10), was that, in rodents, muscle glycogen represents a much smaller fraction of the total body glycogen stores than in humans, liver therefore having a greater role in overall glucose disposal and insulin sensitivity (11, 12).
Insulin resistance is often accompanied by increased hepatic steatosis (13, 14). However, it is still unclear whether insulin resistance is responsible for excessive fat deposition in the liver or whether increased fat content is a prerequisite for the development of insulin resistance (15). Compromised glucose disposal as glycogen may further contribute to hepatic steatosis by diverting excess carbohydrates into fatty acids by the de novo lipogenesis pathway (DNL) (16, 17). In patients with non-alcoholic fatty liver disease (NAFLD), it has been estimated that as much as 26% of the liver triglyceride derives from DNL (18). The first committed step in lipid synthesis is the conversion of acetyl-CoA to malonyl-CoA, catalyzed by acetyl-CoA carboxylase (ACC). Malonyl-CoA participates in two opposing pathways, as a precursor for fatty acid synthesis and as a negative regulator of fatty acid oxidation. There are two isoforms of ACC: ACC1, associated with fatty acid synthesis (19), and ACC2, linked to the regulation of fatty acid oxidation (20). For fatty acid biosynthesis, fatty acid synthase (FAS) uses malonyl-CoA as a two-carbon donor for chain elongation. The DNL pathway is activated at the transcriptional level by the synergistic action of the transcription factors SREBP1c (sterol regulatory element binding protein-1c) and chREBP (carbohydrate response element-binding protein) acting on lipogenic and glycolytic genes (13, 21). Insulin is the most potent regulator of the SREBP1c gene, which encodes a precursor that is embedded in the endoplasmic reticulum. Under the appropriate signals, SREBP1c is cleaved in the Golgi into the mature transcriptionally active form, which translocates into the nucleus (13). Glucose regulates chREBP increasing its gene expression (21) and inducing its dephosphorylation, favoring its nuclear translocation and DNA binding capacity (22).
It is well established that insulin resistance and hepatosteatosis lead to compromised glycogen synthesis (17, 23) and that increased liver glycogen synthesis improves glucose tolerance (24) independently of insulin signaling (25). What is not well understood is the significance of impaired liver glycogen synthesis in inducing insulin resistance and liver fat overaccumulation (5). Therefore, we analyzed insulin signaling and lipid metabolism in LGSKO mice. We report that the inability to synthesize liver glycogen causes enhanced lipid synthesis and ectopic liver fat deposition, accompanied by impairment in insulin signaling restricted to liver.
Results
Insulin signaling in the liver and muscle of LGSKO mice
LGSKO mice are insulin intolerant based on impaired glucose tolerance tests and insulin-dependent suppression of gluconeogenesis (5), although interpretation of the insulin tolerance tests was complicated by a lower starting blood glucose in LGSKO mice. However, the percentage reduction in blood glucose was comparable between the two genotypes. Here, we analyzed insulin signaling in the two main repositories of glycogen, liver and muscle. Thus, CN and LGSKO mice were injected IP with 5 units/kg of insulin, the consequent percentage lowering of blood glucose was similar in CN and LGSKO mice (Fig. 1A), consistent with previous results (5). The basal levels of phosphorylation of downstream insulin signaling components (Akt(Thr-308), GSK3α/β (Ser-21/9), and FoxO1 (Thr-24)) were significantly lower in the livers of LGSKO mice (Fig. 1, B and D-F), consistent with lower basal circulating insulin in fed LGSKO mice (5). After 5 units/kg of insulin administration, phosphorylation of these proteins reached a maximum after 5 min and was maintained up to 15 min in the livers of control mice (Fig. 1, C-G). However, in LGSKO mouse liver, there was a clear delay in the phosphorylation of Akt, and downstream targets GSK3α and -β and FoxO1, reaching the same maximum value as the controls after only 10 min. In addition, the response was less sustained, with some signals already decreasing by 15 min. Moreover, we observed that the ability of insulin to exclude FoxO1 from the nuclear fraction (26) was impaired in the LGSKO mice (Fig. 1H), correlating with a less sustained insulin-dependent FoxO1 phosphorylation. Upstream of Akt, we observed that both basal- and insulin-stimulated tyrosine phosphorylation of IRS1 was decreased in LGSKO livers (Fig. 1I). In an insulin dose response, at 15 min, the depression in blood glucose, as percentage of the starting value, was similar in control and LGSKO mice (Fig. 2A). The amplitude of the insulin-dependent increase in the phosphorylation of Akt and Gsk3α and -β was significantly suppressed in the livers from LGSKO mice (Fig. 2, B–D and F). FoxO1 phosphorylation likewise tended to be less in the LGSKO mice (Fig. 2E). We conclude that the absence of liver glycogen correlates with disturbances in the insulin signaling pathway.
Figure 1.

In vivo insulin response during a time course is reduced in liver of LGSKO mice. 5 units/kg (body weight) of insulin was injected i.p. and the liver was harvested after 5, 10, or 15 min. As control, saline was injected for 15 min (n = 6–7). A, % of blood glucose difference before and after injection of insulin/vehicle in CN (black bars) and LGSKO mice (white bars). B–F, fold-change compared with CN saline of the phosphorylated form, normalized by total corresponding protein content, of Akt (Thr-308) (B and C), GSK3α (Ser-21) (D), GSK3β (Ser-9) (E), and FoxO1 (Thr-24) (F). Panel G shows representative blots. The marks on the right represent the molecular weight markers in kDa. Samples in B are the saline-injected group to highlight the difference between genotypes under the basal state for Akt (Thr-308) phosphorylation, using long exposure blots for quantification. In panel C these samples are shown with the remaining insulin stimulation time course, using the short exposure blot for quantification purposes. H, FoxO1 was found in the cytoplasm (crude extract) and the nuclear extract of CN and LGSKO mice liver stimulated with insulin or saline for 15 min (n = 4–5). I, immunoprecipitated IRS1 from liver was immunoblotted for total IRS1 and phosphotyrosine (n = 3). Groups with the same letter are not significantly different from each other (p < 0.05). In panel C, *, p < 0.05 versus CN by Student's t test.
Figure 2.

In vivo response at different insulin amounts is blunted in liver of LGSKO mice. 0.05, 0.5, and 5 units/kg (body weight) of insulin was injected i.p. and then the liver was harvested after 15 min. As control, saline was injected for 15 min (n = 6). A, % of blood glucose difference before and after injection of insulin/vehicle in CN (black bars) and LGSKO mice (white bars). B–E, fold-change compared with CN saline of the phosphorylated form, normalized by total corresponding protein content, of Akt (Thr-308) (B), GSK3α (Ser-21) (C), GSK3β (Ser-9) (D), and FoxO1 (Thr-24) (E). Panel F shows representative blots. The marks on the right represent the molecular weight markers in kDa. Groups with the same letter are not significantly different from each other (p < 0.05).
We performed a similar analysis of skeletal muscle from LGSKO mice treated with insulin. There was a trend toward lower basal phosphorylation, significant only in the case of GSK3α (Figs. 3, A–C, and 4, A–C), possibly explained by lower basal insulin levels (5). We observed no difference in the time courses of insulin-induced phosphorylation of Akt and GSK3 (Fig. 3, A–D). The insulin dose response of Akt phosphorylation was indistinguishable between control and LGSKO mice (Fig. 4, A–D). Phosphorylation of GSK3, especially GSK3α, appeared slightly less sensitive to insulin in the LGSKO animals but the amplitude of the response was unchanged (Fig. 4, C and D). Overall, we conclude that insulin signaling in skeletal muscle is only minimally affected by the lack of liver glycogen.
Figure 3.

In vivo response during insulin time course is unchanged in skeletal muscle of LGSKO mice. 5 units/kg (body weight) of insulin was injected i.p. and then skeletal muscle was collected after 5, 10, or 15 min. As control, saline was injected for 15 min (n = 6–7). Fold-change compared with CN saline of the phosphorylated form, normalized by the corresponding total protein, of Akt (Thr-308) (A), GSK3α (Ser-21) (B), and GSK3β (Ser-9) (C) in CN (black bars) and LGSKO mice (white bars). Panel D shows representative blots. The marks on the right represent the molecular weight markers in kDa. Groups with the same letter are not significantly different from each other (p < 0.05).
Figure 4.

In vivo response at different insulin amounts is unchanged in skeletal muscle of LGSKO mice. 0.05, 0.5, and 5 units/kg (body weight) of insulin was injected i.p. and then the skeletal muscle was harvested after 15 min. As control, saline was injected for 15 min (n = 6–7). Fold-change compared with CN saline of the phosphorylated form, normalized by the corresponding total protein, of Akt (Thr-308) (A), GSK3α (Ser-21) (B), and GSK3β (Ser-9) (C) in CN (black bars) and LGSKO mice (white bars). Panel D show representative blots. The marks on the right represent the molecular weight markers in kDa. Groups with the same letter are not significantly different from each other (p < 0.05).
Fat metabolism in LGSKO mice
From DEXA analysis of body composition, we observed an increase in whole body fat in young (4 months old) and old (14 months old) LGSKO mice (Fig. 5A). In young animals, this correlated with an elevation in epididymal fat mass that was lost in the old mice (Fig. 5B). We previously showed (5) that liver triglyceride content trended to be higher in 4-month-old LGSKO mice with statistically significant elevations at 7 and 15 months of age. Indeed, staining of liver sections with Oil Red O indicated an age-dependent increase in fat in both CN and LGSKO mice (Fig. 5F). Fat accumulation was more intense in the pericentral area of the liver from CN mice and this zonation was maintained up to 14 months, although not as clearly as at 4 or 7 months. In the LGSKO livers, we observed defective zonation in the acinus, with more lipid deposits in the periportal area. This ectopic fat accumulation was intensified at 7 months and by 14 months the gradation in fat deposits between periportal and pericentral hepatocytes was completely absent, which also correlated with a more pronounced impairment of insulin signaling at 14 months of age in LGSKO liver than in younger animals (Fig. 5E), leading us to consider that the glucose intolerance of LGSKO mice is linked to increased hepatic fat accumulation. Triglyceride export from the liver, which contributes to hepatic fat levels, was also measured (Fig. 5C). In both young and old mice, there were no differences in hepatic triglyceride export by CN or LGSKO mice, which correlated with no changes in the mRNA levels of lipoprotein assembly markers apolipoprotein B (Apob) and microsomal triglyceride transport protein (Mttp) under any feeding condition (Fig. 5D).
Figure 5.

LGSKO mice have more body fat without change in liver triglyceride export. The percentage in body fat measured by DEXA scan (A) and the epididymal fat weight by body weight (mg/g) (B) were measured in 4- and 14-month-old CN (black bars) and LGSKO mice (white bars). n = 6–12 per group. Hepatic triglyceride accumulation in plasma samples after the intravenous injection tyloxapol (C) in CN (full symbols and solid lines) and LGSKO (open symbols and dashed lines) 4-month (circles) and 14-month (triangles)-old mice. n = 5–7 per group. Fold-change of Apob and Mttp (D) mRNA content normalized by 18S rRNA measured by quantitative real-time PCR in liver of CN fed (black), 6 h fasted (heavy hatched) or fasted overnight (light hatched), and LGSKO fed (white), 6-h fasted (horizontally crossed) or fasted overnight (diagonally crossed) mice (n = 5–8). E, Western blotting of pAkt (Thr-308), Akt, and GAPDH in livers of 4- or 14-month-old CN and LGSKO mice injected i.p. with 5 units/kg (body weight) of insulin for 15 min. As control, saline was injected for 15 min (n = 3). The marks on the right of the blots represent the molecular weight markers in kDa. F, neutral lipid staining by Oil Red-O in frozen liver sections of 4- (left), 7- (central), or 14- (right) month-old CN (top) and LGSKO (bottom) mice. The bar represent 200 μm. Notice that the gradation of the fat deposits between periportal and pericentral areas (arrows) of the liver are more noticeable in CN mice at 4 and 7 months. Groups with the same letter are not significantly different from each other (p < 0.05).
We observed an enhanced capacity for hepatic lipogenesis and glycolysis in LGSKO mice as judged by increased ACC1, SREBP1c, and chREBP protein levels (Fig. 6, A and B) as well as increased glucokinase activity (Fig. 6C). Moreover, chREBP was more dephosphorylated, and thus activated, in the LGSKO liver, as judged by the disappearance of the chREBP upper band in Western blots (Fig. 6D). To test whether the electrophoretic band shift corresponded to dephosphorylation of chREBP, lysates were treated with calf intestine phosphatase, which resulted in loss of the upper band and an increase in the lower band of CN samples (Fig. 6D). We confirmed the efficacy of the phosphatase treatment by the reduction of the signal from phospho-GSK3β (Fig. 6D). In other experiments, we investigated whether exposure of fasted mice to glucose pushes the liver of LGSKO mice into a more lipogenic state. Monitoring liver glycogen stores gave the expected results (Fig. 6E). Following glucose gavage, expression of several genes relevant to fatty acid synthesis, ACC, fatty acid synthase, SREBP1c, and chREBP, was significantly increased in the liver of LGSKO mice but not in CN mice (Fig. 6F). The expression of pyruvate kinase was similarly increased, consistent with a need for increased glycolytic capacity to fuel enhanced lipogenesis. SREBP1c and chREBP protein levels were also increased (Fig. 6, G and H). To test whether increased lipogenic factors contributed to enhanced de novo lipogenesis, we cultured primary hepatocytes from 3-month-old mice and monitored lipid formation from [U-14C]acetate (Fig. 7A). Only hepatocytes from CN mice accumulated glycogen, as expected (Fig. 7B). Insulin signaling, as judged by monitoring phospho-Akt, was intact in the isolated hepatocytes (Fig. 7, D and E), but in cells derived from LGSKO mice the response was more transient under these conditions, similar to insulin stimulation of the animals (Fig. 1C). Moreover, we observed a 40% increase in de novo lipogenesis in LGSKO hepatocytes (Fig. 7C). SREBP1c protein was elevated in LGSKO hepatocytes (Fig. 7, D and F), but not chREBP (Fig. 7, D and G).
Figure 6.

LGSKO mice have enhanced lipogenesis in liver. Fold-change (A) and representative blots (B) of ACC1, SREBP1c, and chREBP normalized by GAPDH in liver of LGSKO (white bars) compared with CN (black bars) 4-month-old fed mice (n = 8). The marks on the right represent the molecular markers in kDa. C, liver glucokinase activity (n = 5). D, blot for chREBP, pGSK3β(S9), and GAPDH after treatment with (+) or without (−) calf intestine phosphatase of CN and LGSKO liver homogenates. The chREBP upper band (only evident in CN samples) and pGSK3β(S9) band disappear with phosphatase treatment. E, liver glycogen content in animals fed, fasted for 24 h, and fasted for 24 h then administered an oral glucose bolus (3.6 g/kg body weight) and tissues were harvested 2 h post-gavage (fast+2h glc) (n = 4). Groups with the same letter are not significantly different from each other (p < 0.05). F, fold-change of pyruvate kinase (pklr), acetyl-CoA carboxylase 1 (Acc1), fatty acid synthase (Fasn), SREBP1C (Srebf1), and chREBP (mlxipl) mRNA content normalized by 18S rRNA in liver measured by quantitative real-time PCR (n = 4). Fold-change quantification of SREBP1c and chREBP normalized by GAPDH (G) and representative blots (H) from liver of 24-h fasted CN (black bars) and LGSKO (white bars) and fasted for 24 h and glucose bolus for 2 h (fast + 2 h glc) CN (checked bars) and LGSKO (hatched bars) mice (n = 4). The marks on the right of the blots are molecular weight markers in kDa. Groups with the same letter are not significantly different from each other (p < 0.05). *, p < 0.05 versus CN.
Figure 7.

De novo lipogenesis is increased in cultured primary hepatocytes of LGSKO mice. A, experimental protocol of the hepatocyte culture and DNL experiment. First, hepatocytes from 3-month-old mouse livers were isolated, plated, and cultured overnight with medium supplemented with 10% FBS, high glucose (25 mm), and insulin (100 nm) (HG/HI). Afterward, cells were starved for 5 h with a medium with low glucose (5 mm) and insulin (1 nm) (LG/LI) and no FBS. This was referred to as time 0 (starvation). Finally, hepatocytes were challenged by adding medium with 10% FBS, high glucose, and insulin and supplemented with 1 mCi/ml of [14C]acetate for 15, 60, or 180 min. B, glycogen accumulation and [14C]acetate incorporation in the lipid fraction (after 180 min), as a measure of de novo lipogenesis; C, during the challenge in hepatocytes from CN (black bars) and LGSKO mice (open bars). We did not detect glycogen in LGSKO hepatocytes. Lipogenesis results are calculated from three independent experiments. D, blots, from a representative experiment, of phospho-Akt (Thr-308), total Akt, SREBP1c, chREBP, and GAPDH as loading control. The marks on the right of the blots are molecular weight markers in kDa. Fold-change compared with CN basal (0 min) of Akt (Thr-308) normalized by total Akt (E), SREBP1 (F), and chREBP (G) normalized by GAPDH. Groups with the same letter are not significantly different from each other (p < 0.05). In panel C, * p < 0.05 versus CN by Student's t test.
No liver inflammation in LGSKO mice
There is growing evidence connecting obesity, NAFLD, and insulin resistance to a chronic low-grade inflammatory state in adipose tissue and liver (27). In liver, lipid accumulation promotes inflammation by activation of a pro-inflammatory program from the Kupffer cells that in turn increases the production of TNFα and other cytokines, enhancing hepatic macrophage infiltration, and the activation of c-Jun N-terminal kinase (JNK) in hepatocytes, all contributing to insulin resistance. To monitor chronic inflammation in livers of LGSKO mice, we examined several common markers. We did not observe any increase in JNK phosphorylation, but rather a decrease (Fig. 8, A and B). In parallel, the expression of other inflammation markers, tumor necrosis factor α (TNFα) and the integrin α-M/CD11b, characteristic of pro-inflammatory macrophages, was not increased in LGSKO livers (Fig. 8C), suggesting that inflammation was not inducing insulin resistance.
Figure 8.
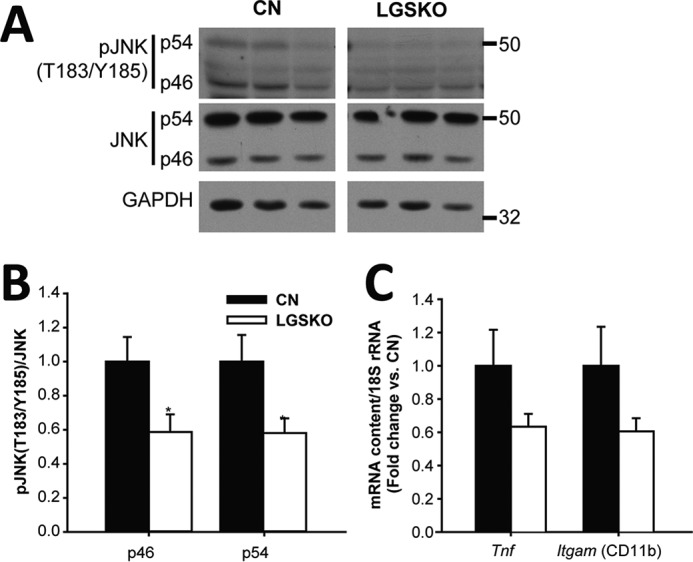
Inflammatory response in liver of LGSKO mice. Representative blots (A) and fold-change (B) of CN (black bars) and LGSKO (white bars) of pJNK (Thr-183/Tyr-185) p46 and p54 normalized with total JNK in liver (n = 6). C, fold-change of tumor necrosis factor (Tnf) and integrin αM/CD11b (Itgam) mRNA content normalized by 18S rRNA in liver of CN and LGSKO mice measured by quantitative real-time PCR (n = 5). Integrin αM/CD11b amplicon recognizes transcript variants 1 and 2. Marks in the blots are the molecular markers of the noted weight. *, p < 0.05 versus CN by Student's t test.
Discussion
In the present study, we report that the inability to synthesize glycogen in liver impairs local insulin action by causing an early decrease in the response of several components of the transduction cascade, namely IRS, Akt, GSK3, and FoxO1. From the dose responses and in agreement with previous reports (28), downstream targets such as GSK3 and FoxO1 were more sensitive to insulin than Akt itself, consistent with the amplification inherent in the protein kinase cascade. In LGSKO mice, the basal activation states of all the signaling components were significantly diminished and, although insulin still elicited activation, in general the degree and extent of stimulation was decreased. Temporally, the onset of activation by insulin was delayed and the duration reduced.
In contrast to liver, the impact of glycogen on insulin signaling has been thoroughly studied in skeletal muscle (see Ref. 29 for a review). In healthy skeletal muscle, there is an inverse relationship between glycogen content and insulin response, where decreased glycogen enhances Akt phosphorylation and glucose uptake (30, 31). Furthermore, lack of glycogen stores improves whole body glucose tolerance (8). However, the muscle of T2DM patients has defects in muscle GS regulation and non-oxidative glucose disposal by insulin, but has normal insulin-stimulated Akt and GSK3α phosphorylation responses (3). These results suggest that the mechanisms by which glycogen content influences insulin signaling are different in liver as compared with skeletal muscle.
Compromised glucose disposal as glycogen in muscle (3, 4, 17), excess dietary carbohydrate (16), or defects in glucose production by the liver (32) have all been associated with increased hepatic lipid deposition via DNL. LGSKO mice have impaired glucose disposal, but at 4 months of age we did not observe increased lipid deposition in the liver. Only upon aging was fat overaccumulated in the LGSKO liver (5). There was an age-dependent progressive disruption of the hepatic zonation as judged by histological analysis starting at 4 months. Steatosis in liver could also be caused by impaired β-oxidation (33) or increased dietary fat consumption (18). However, we do not favor these explanations because gluconeogenesis, fueled by oxidation of fat, is indeed enhanced in LGSKO mice (5). Thus, we focused on studying DNL as a potential cause of lipid accumulation. The liver of LGSKO mice had enhanced lipogenic capacity upon refeeding, which can be explained by increased expression and transcription factor levels of SREBP1c and chREBP, which in turn regulate glucokinase, pyruvate kinase, ACC1, and FAS gene expression. Therefore, this program enhances DNL synergistically by increasing lipogenic enzyme content and substrate availability through glycolysis, as previously reported (21). The LGSKO mouse parallels some aspects of NAFLD and T2DM, displaying a mixed pattern of insulin resistance (defective suppression of glucose production) and sensitivity (enhanced lipid synthesis) in liver, known as the “insulin signaling paradox” (reviewed in Refs. 14 and 34). Although fasting fails to suppress liver lipogenesis in NAFLD patients (13), it is effective in 4-month-old LGSKO mice. This consideration is relevant to interpreting the results of the present model. Hepatic fat accumulation could also be caused by decreased lipid secretion as VLDL. Fat export from the liver of LGSKO mice was no different from CN mice, suggesting that increased fat deposition is most likely caused by enhanced lipogenesis. Also, the results indicate that initially fat from excess glucose accumulated in white adipose tissue and with age also in the liver in LGSKO mice. Another aspect of hepatic lipid accumulation is its distribution along the portocentral axis of the liver. In NAFLD patients and/or as a result of high carbohydrate diets, there is a disturbance of the acinus zonation resulting in progressive steatosis from the pericentral to the periportal hepatocytes (35). In the absence of glycogen synthesis, the same age-dependent progression occurred along with total triglyceride accumulation (5) and insulin resistance without macrophage infiltration or inflammation. Therefore, as suggested by Hijmans et al. (35), we can speculate that ectopic fat accumulation in the periportal area of LGSKO liver is linked to the defective insulin response. Furthermore, impairment of hepatic zonation could explain how gluconeogenesis and lipogenesis are both enhanced in liver lacking glycogen, despite the fact that the two pathways are mutually exclusive (15), possibly resolving the insulin paradox (34). Ectopic fat accumulation in periportal hepatocytes causes insulin resistance, leading to impaired suppression of gluconeogenesis and enhanced glucose production. This would result in the exposure of pericentral hepatocytes to an excess of glucose (from the diet, due to the lack of glycogen synthesis and increased gluconeogenesis) converted into fat by enhanced lipogenesis, which requires intact insulin signaling (36). Another factor contributing to hepatosteatosis could be excessive uptake of circulating fatty acids derived from white adipose tissue lipolysis (18). Short term fasting (6 h) increases the blood non-esterified fatty acid levels in LGSKO mice above those of CN mice (5). Moreover, the newly synthesized pericentral lipids in the LGSKO liver could also be exported and eventually taken up by periportal hepatocytes, probably due to the “first-pass” effect (35), altogether explaining the ectopic steatosis. Exploring the insulin paradox in liver, Titchenell et al. (37) studied a series of genetically modified mice and proposed that gluconeogenesis is increased because elevated circulating fatty acids become a dominant inducer of gluconeogenesis under insulin resistant conditions, a hypothesis consistent with the metabolic conditions of the present model. Alternatively, Accili and colleagues (38) proposed that in hepatocytes there is a selective response to insulin, DNL being activated by ∼4-fold lower insulin levels than the suppression of gluconeogenesis. This hypothesis could also provide a satisfactory mechanism to reconcile the results reported in this LGSKO mouse.
The exact mechanism linking the absence of liver glycogen with disruptions of hepatic insulin signaling and enhanced lipogenesis remains to be established. The traditional view of the glycogen molecule simply as an energy store has perhaps evolved in recent years with the recognition that a growing number of proteins, besides the basic metabolic enzymes, are associated with the glycogen granule (39–41). Could the glycogen particle additionally function as a regulatory complex? Could the lack of glycogen particles cause a disturbance in the localization of some proteins? Mislocalization of proteins that are important for the signaling or metabolic properties of the cell can cause disorders, including aberrant cell signaling (42). Much more work will be needed to explore this hypothesis.
Experimental procedures
Mouse background and husbandry
Generation of Gys2lox/lox control (CN) and Gys2lox/lox/Alb-CRE (LGSKO) mice was described previously (5). We used male mice for all experiments. All mice were maintained in temperature- and humidity-controlled conditions with a 12-h light, 12-h dark cycle and were allowed food (Harlan Teckland global diet number 2019S) and water ad libitum. Animals were maintained in the Association for Assessment of Accreditation of Laboratory Animal Care approved animal facility at Indiana University. All procedures were approved by the Indiana University Animal Care and Use Committee.
In vivo insulin stimulation, fasting and glucose refeeding, and liver triglyceride secretion experiments
Randomly fed, 3–4-month-old mice were injected between 10–11 a.m. intraperitoneally (i.p.) with insulin (HumulinR, Lilly) at the doses and times specified before sacrifice by cervical dislocation. Tissues were immediately harvested, frozen in liquid nitrogen, and stored at −80 °C until further analyses. For refeeding studies, 7–9-month-old mice were either fasted for 24 h, or fasted for 24 h and then given an oral bolus of glucose (3.6 g/kg body weight) for 2 h. To analyze liver triglyceride secretion, 300 mg/kg of body weight of Tyloxapol (Cole-Parmer) was injected via tail vein to 4-h fasted mice to prevent peripheral lipolysis. Plasma samples from the tail vein were obtained at 0, 30, 60, and 90 min after Tyloxapol injection for analysis of triglycerides.
Primary hepatocyte isolation and de novo lipogenesis assay
Primary hepatocytes were isolated from 3-month-old mice as described previously (43). The day after isolation, the medium was changed to serum-free DMEM with low glucose (4.5 mm), 1 nm dexamethasone and insulin, and 100 units/ml of penicillin and 0.1 mg/ml of streptomycin for 5 h (Fig. 6A). Then the medium was changed to high glucose (25 mm), 10 nm dexamethasone, 100 nm insulin, 1 microcurie/ml of [U-14C]acetate, and 10% fetal bovine serum to promote de novo lipogenesis. After 0, 15, 60, or 180 min of incubation, cells were rinsed with ice-cold PBS, lysis buffer was added (50 mm Tris-HCl, pH 7.48, 100 mm NaCl, 100 mm NaF, 4 mm EDTA, 2 mm EGTA and protease and phosphatase inhibitors, as described below), and cells were flash-frozen in liquid N2. Hepatocytes were then lysed by sonication for 15 s. Lipids were isolated following a previously described method (44) and lipogenesis was monitored as the incorporation of radiolabeled acetate into the lipid pool. Glycogen was measured in hepatocytes following a method described previously (10).
Sample preparation, biochemical and RNA analyses, and Western blotting
Glycogen content was measured as described by Suzuki et al. (10). Plasma triglyceride concentration was measured using an enzymatic method (triglyceride determination kit, Sigma). Blood glucose was assessed utilizing a Breeze2 glucometer (Bayer). Mouse body composition was determined by DEXA scan (45). Ten-micrometer thick frozen liver sections obtained in embedding media (OCT, Tissue-Tek) were stained for neutral lipids with Oil-Red O and counterstained with hematoxylin. RNA isolation and quantitative real-time PCR was performed as described by Irimia et al. (5). The fold-change in target mRNA content normalized by 18S rRNA was calculated as described previously (46). Glucokinase activity was measured as previously described (47). For Western blotting, tissue samples were homogenized with a tissue-tearor in 30 volumes of ice-cold buffer (100 mm HEPES, pH 7.5, 4 mm EDTA, 2 mm EGTA, 20 mm KF, 300 mm NaCl, 20% glycerol, 0.1% Igepal, 0.35% β-mercaptoethanol, 20 mm β-glycerophosphate, 2 mm sodium pyrophosphate, 1 mm Na3VO4, 10 μg/ml of aprotinin, 10 μg/ml of leupeptin, 2 mm benzamidine, 0.5 mm PMSF), then were sonicated for 10 s on ice, rotated for 1 h at 4 °C, centrifuged at 18,000 × g for 20 min at 4 °C, and the supernatant was collected. Laemmli buffer was added and the samples boiled for 10 min and stored at −80 °C. Total liver extracts obtained in NEB3 buffer with protease inhibitors were treated with 10 units/μl of calf intestine phosphatase (New England Biolabs) at 37 °C for 1 h. To immunoprecipitate IRS1, 750 μg of protein obtained as described above (without Igepal) were incubated with the antibody overnight at 4 °C under rotation and then precipitated with Dynabeads-Protein A (Invitrogen) following the manufacturer's instructions. Fractionation of liver tissue samples was done using the NE-PER nuclear and cytoplasmic extraction kit (Thermo Scientific). Protein was measured in the lysates using the Bradford reagent kit (Sigma number 6916). Immunoblots were performed as described previously (8) using the following antibodies: Akt pan, pAkt(Thr-308), pGSK3α/β(Ser-21/9), FoxO1, pFoxO1(Thr-24), SAPK/JNK, pSAPK/pJNK(Thr-183/Tyr-185), (Cell Signaling numbers 4685, 4056, 9331, 9454, 9464, 9252, and 9251, respectively), IRS-1 (Millipore#06–248), p-Tyrosine (PY20 BD number 610000), GSK3α/β and GAPDH (Invitrogen numbers 44-610 and 39-8600, respectively), SREBP1c (H-160 Santa Cruz number 8984), and chREBP (Novus Biologicals number NB400-135B). All samples were transferred to the same membrane to ensure similar blotting conditions. In the figures showing representative blots, a break between sample lanes corresponds to non-adjacent parts of the same autoradiography film.
Statistical analyses
The results were analyzed using Statgraphics plus 5.0 (Statistical Graphics Corporation, Herndon, VA). For each parameter, the kurtosis and skewness were calculated to test normal distribution. When normality was reached, multivariate analysis of variance, followed by LSD post hoc test, was performed to compare the data. When comparing two samples, Student's t test was used. All data are presented as mean ± S.E.
Author contributions
J. M. I., N. M., and P. J. R. conceived and coordinated the study. J. M. I. designed, performed, and analyzed all experiments. C. M. M. and D. M. S. maintained the animal colonies and provided technical assistance. S. S. helped with hepatocyte preparation for the experiments of Fig. 6. A. D. P. R., J. M. I., S. S., N. M., and P. J. R. contributed to discussion of results and conclusions and manuscript preparation. All authors reviewed the results and approved the final version of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants DK 27221 and NS 056454. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- GS
- glycogen synthase
- DNL
- de novo lipogenesis
- NAFLD
- non-alcoholic fatty liver disease
- SREBP
- sterol regulatory element-binding protein
- chREBP
- carbohydrate response element-binding protein
- GSK3
- glycogen synthase kinase 3
- ACC
- acetyl-CoA carboxylase.
References
- 1. Ludvik B., Nolan J. J., Roberts A., Baloga J., Joyce M., Bell J. M., and Olefsky J. M. (1995) A noninvasive method to measure splanchnic glucose uptake after oral glucose administration. J. Clin. Invest. 95, 2232–2238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Roach P. J., Depaoli-Roach A. A., Hurley T. D., and Tagliabracci V. S. (2012) Glycogen and its metabolism: some new developments and old themes. Biochem. J. 441, 763–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Højlund K., Staehr P., Hansen B. F., Green K. A., Hardie D. G., Richter E. A., Beck-Nielsen H., and Wojtaszewski J. F. (2003) Increased phosphorylation of skeletal muscle glycogen synthase at NH2-terminal sites during physiological hyperinsulinemia in type 2 diabetes. Diabetes 52, 1393–1402 [DOI] [PubMed] [Google Scholar]
- 4. Højlund K., Birk J. B., Klein D. K., Levin K., Rose A. J., Hansen B. F., Nielsen J. N., Beck-Nielsen H., and Wojtaszewski J. F. (2009) Dysregulation of glycogen synthase COOH- and NH2-terminal phosphorylation by insulin in obesity and type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 94, 4547–4556 [DOI] [PubMed] [Google Scholar]
- 5. Irimia J. M., Meyer C. M., Peper C. L., Zhai L., Bock C. B., Previs S. F., McGuinness O. P., DePaoli-Roach A. A., and Roach P. J. (2010) Impaired glucose tolerance and predisposition to the fasted state in liver glycogen synthase knock-out mice. J. Biol. Chem. 285, 12851–12861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Weinstein D. A., Correia C. E., Saunders A. C., and Wolfsdorf J. I. (2006) Hepatic glycogen synthase deficiency: an infrequently recognized cause of ketotic hypoglycemia. Mol. Genet. Metab. 87, 284–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pederson B. A., Cope C. R., Schroeder J. M., Smith M. W., Irimia J. M., Thurberg B. L., DePaoli-Roach A. A., and Roach P. J. (2005) Exercise capacity of mice genetically lacking muscle glycogen synthase: in mice, muscle glycogen is not essential for exercise. J. Biol. Chem. 280, 17260–17265 [DOI] [PubMed] [Google Scholar]
- 8. Pederson B. A., Schroeder J. M., Parker G. E., Smith M. W., DePaoli-Roach A. A., and Roach P. J. (2005) Glucose metabolism in mice lacking muscle glycogen synthase. Diabetes 54, 3466–3473 [DOI] [PubMed] [Google Scholar]
- 9. Brüning J. C., Michael M. D., Winnay J. N., Hayashi T., Hörsch D., Accili D., Goodyear L. J., and Kahn C. R. (1998) A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell. 2, 559–569 [DOI] [PubMed] [Google Scholar]
- 10. Suzuki Y., Lanner C., Kim J. H., Vilardo P. G., Zhang H., Yang J., Cooper L. D., Steele M., Kennedy A., Bock C. B., Scrimgeour A., Lawrence J. C. Jr., and DePaoli-Roach A. A. (2001) Insulin control of glycogen metabolism in knockout mice lacking the muscle-specifict protein phosphatase PP1G/RGL. Mol. Cell. Biol. 21, 2683–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Michael M. D., Kulkarni R. N., Postic C., Previs S. F., Shulman G. I., Magnuson M. A., and Kahn C. R. (2000) Loss of insulin signaling in hepatocytes leads to severe insulin resistance and progressive hepatic dysfunction. Mol. Cell. 6, 87–97 [PubMed] [Google Scholar]
- 12. Accili D. (2004) Lilly lecture 2003: the struggle for mastery in insulin action: from triumvirate to republic. Diabetes 53, 1633–1642 [DOI] [PubMed] [Google Scholar]
- 13. Ferré P., and Foufelle F. (2010) Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP-1c. Diabetes Obes. Metab. 12, 83–92 [DOI] [PubMed] [Google Scholar]
- 14. Williams K. H., Shackel N. A., Gorrell M. D., McLennan S. V., and Twigg S. M. (2013) Diabetes and nonalcoholic fatty liver disease: a pathogenic duo. Endocr. Rev. 34, 84–129 [DOI] [PubMed] [Google Scholar]
- 15. Sun Z., and Lazar M. A. (2013) Dissociating fatty liver and diabetes. Trends Endocrinol. Metab. 24, 4–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schwarz J. M., Linfoot P., Dare D., and Aghajanian K. (2003) Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 77, 43–50 [DOI] [PubMed] [Google Scholar]
- 17. Petersen K. F., Dufour S., Savage D. B., Bilz S., Solomon G., Yonemitsu S., Cline G. W., Befroy D., Zemany L., Kahn B. B., Papademetris X., Rothman D. L., and Shulman G. I. (2007) The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc. Natl. Acad. Sci. U.S.A. 104, 12587–12594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Donnelly K. L., Smith C. I., Schwarzenberg S. J., Jessurun J., Boldt M. D., and Parks E. J. (2005) Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Invest. 115, 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mao J., DeMayo F. J., Li H., Abu-Elheiga L., Gu Z., Shaikenov T. E., Kordari P., Chirala S. S., Heird W. C., and Wakil S. J. (2006) Liver-specific deletion of acetyl-CoA carboxylase 1 reduces hepatic triglyceride accumulation without affecting glucose homeostasis. Proc. Natl. Acad. Sci. U.S.A. 103, 8552–8557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Choi C. S., Savage D. B., Abu-Elheiga L., Liu Z. X., Kim S., Kulkarni A., Distefano A., Hwang Y. J., Reznick R. M., Codella R., Zhang D., Cline G. W., Wakil S. J., and Shulman G. I. (2007) Continuous fat oxidation in acetyl-CoA carboxylase 2 knockout mice increases total energy expenditure, reduces fat mass, and improves insulin sensitivity. Proc. Natl. Acad. Sci. U.S.A. 104, 16480–16485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dentin R., Pégorier J. P., Benhamed F., Foufelle F., Ferré P., Fauveau V., Magnuson M. A., Girard J., and Postic C. (2004) Hepatic glucokinase is required for the synergistic action of ChREBP and SREBP-1c on glycolytic and lipogenic gene expression. J. Biol. Chem. 279, 20314–20326 [DOI] [PubMed] [Google Scholar]
- 22. Sakiyama H., Wynn R. M., Lee W. R., Fukasawa M., Mizuguchi H., Gardner K. H., Repa J. J., and Uyeda K. (2008) Regulation of nuclear import/export of carbohydrate response element-binding protein (ChREBP): interaction of an α-helix of ChREBP with the 14-3-3 proteins and regulation by phosphorylation. J. Biol. Chem. 283, 24899–24908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Samuel V. T., Liu Z. X., Qu X., Elder B. D., Bilz S., Befroy D., Romanelli A. J., and Shulman G. I. (2004) Mechanism of hepatic insulin resistance in non-alcoholic fatty liver disease. J. Biol. Chem. 279, 32345–32353 [DOI] [PubMed] [Google Scholar]
- 24. Ros S., Zafra D., Valles-Ortega J., García-Rocha M., Forrow S., Domínguez J., Calbó J., and Guinovart J. J. (2010) Hepatic overexpression of a constitutively active form of liver glycogen synthase improves glucose homeostasis. J. Biol. Chem. 285, 37170–37177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ros S., García-Rocha M., Calbó J., and Guinovart J. J. (2011) Restoration of hepatic glycogen deposition reduces hyperglycaemia, hyperphagia and gluconeogenic enzymes in a streptozotocin-induced model of diabetes in rats. Diabetologia 54, 2639–2648 [DOI] [PubMed] [Google Scholar]
- 26. Guo S. (2014) Insulin signaling, resistance, and the metabolic syndrome: insights from mouse models into disease mechanisms. J. Endocrinol. 220, T1–T23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tilg H., and Moschen A. R. (2008) Insulin resistance, inflammation, and non-alcoholic fatty liver disease. Trends Endocrinol. Metab. 19, 371–379 [DOI] [PubMed] [Google Scholar]
- 28. Hoehn K. L., Hohnen-Behrens C., Cederberg A., Wu L. E., Turner N., Yuasa T., Ebina Y., and James D. E. (2008) IRS1-independent defects define major nodes of insulin resistance. Cell Metab. 7, 421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jensen J., Rustad P. I., Kolnes A. J., and Lai Y. C. (2011) The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front. Physiol. 2, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Derave W., Lund S., Holman G. D., Wojtaszewski J., Pedersen O., and Richter E. A. (1999) Contraction-stimulated muscle glucose transport and GLUT-4 surface content are dependent on glycogen content. Am. J. Physiol. 277, E1103–E1110 [DOI] [PubMed] [Google Scholar]
- 31. Derave W., Hansen B. F., Lund S., Kristiansen S., and Richter E. A. (2000) Muscle glycogen content affects insulin-stimulated glucose transport and protein kinase B activity. Am. J. Physiol. Endocrinol. Metab. 279, E947–E955 [DOI] [PubMed] [Google Scholar]
- 32. Bandsma R. H., Prinsen B. H., van Der Velden Mde S., Rake J. P., Boer T., Smit G. P., Reijngoud D. J., and Kuipers F. (2008) Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a. Pediatr. Res. 63, 702–707 [DOI] [PubMed] [Google Scholar]
- 33. Grefhorst A., Hoekstra J., Derks T. G., Ouwens D. M., Baller J. F., Havinga R., Havekes L. M., Romijn J. A., and Kuipers F. (2005) Acute hepatic steatosis in mice by blocking β-oxidation does not reduce insulin sensitivity of very-low-density lipoprotein production. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G592–G598 [DOI] [PubMed] [Google Scholar]
- 34. Brown M. S., and Goldstein J. L. (2008) Selective versus total insulin resistance: a pathogenic paradox. Cell Metab. 7, 95–96 [DOI] [PubMed] [Google Scholar]
- 35. Hijmans B. S., Grefhorst A., Oosterveer M. H., and Groen A. K. (2014) Zonation of glucose and fatty acid metabolism in the liver: mechanism and metabolic consequences. Biochimie 96, 121–129 [DOI] [PubMed] [Google Scholar]
- 36. Biddinger S. B., Hernandez-Ono A., Rask-Madsen C., Haas J. T., Alemán J. O., Suzuki R., Scapa E. F., Agarwal C., Carey M. C., Stephanopoulos G., Cohen D. E., King G. L., Ginsberg H. N., and Kahn C. R. (2008) Hepatic insulin resistance is sufficient to produce dyslipidemia and susceptibility to atherosclerosis. Cell Metab. 7, 125–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Titchenell P. M., Quinn W. J., Lu M., Chu Q., Lu W., Li C., Chen H., Monks B. R., Chen J., Rabinowitz J. D., and Birnbaum M. J. (2016) Direct hepatocyte insulin signaling is required for lipogenesis but is dispensable for the suppression of glucose production. Cell Metab. 23, 1154–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cook J. R., Langlet F., Kido Y., and Accili D. (2015) Pathogenesis of selective insulin resistance in isolated hepatocytes. J. Biol. Chem. 290, 13972–13980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roach P. J., Cheng C., Huang D., Lin A., Mu J., Skurat A. V., Wilson W., and Zhai L. (1998) Novel aspects of the regulation of glycogen storage. J. Basic Clin. Physiol. Pharmacol. 9, 139–151 [DOI] [PubMed] [Google Scholar]
- 40. Graham T. E., Yuan Z., Hill A. K., and Wilson R. J. (2010) The regulation of muscle glycogen: the granule and its proteins. Acta Physiol. (Oxf.) 199, 489–498 [DOI] [PubMed] [Google Scholar]
- 41. Stapleton D., Nelson C., Parsawar K., McClain D., Gilbert-Wilson R., Barker E., Rudd B., Brown K., Hendrix W., O'Donnell P., and Parker G. (2010) Analysis of hepatic glycogen-associated proteins. Proteomics 10, 2320–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hung M. C., and Link W. (2011) Protein localization in disease and therapy. J. Cell Sci. 124, 3381–3392 [DOI] [PubMed] [Google Scholar]
- 43. Park J. S., Surendran S., Kamendulis L. M., and Morral N. (2011) Comparative nucleic acid transfection efficacy in primary hepatocytes for gene silencing and functional studies. BMC Res. Notes 4, 8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bligh E. G., and Dyer W. J. (1959) A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 [DOI] [PubMed] [Google Scholar]
- 45. Nagy T. R., and Clair A. L. (2000) Precision and accuracy of dual-energy X-ray absorptiometry for determining in vivo body composition of mice. Obes. Res. 8, 392–398 [DOI] [PubMed] [Google Scholar]
- 46. Pfaffl M. W. (2001) A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Davidson A. L., and Arion W. J. (1987) Factors underlying significant underestimations of glucokinase activity in crude liver extracts: physiological implications of higher cellular activity. Arch. Biochem. Biophys. 253, 156–167 [DOI] [PubMed] [Google Scholar]