

Abstract
The appearance of myofibroblasts is generally thought to be the underlying cause of the fibrotic changes that underlie idiopathic pulmonary fibrosis. However, the cellular/molecular mechanisms that account for the fibroblast-myofibroblast differentiation/activation in idiopathic pulmonary fibrosis remain poorly understood. We investigated the functional role of hyaluronan receptor CD44V6 (CD44 containing variable exon 6 (v6)) for differentiation of lung fibroblast to myofibroblast phenotype. Increased hyaluronan synthesis and CD44 expression have been detected in numerous fibrotic organs. Previously, we found that the TGFβ1/CD44V6 pathway is important in lung myofibroblast collagen-1 and α-smooth-muscle actin synthesis. Because increased EGR1 (early growth response-1) expression has been shown to appear very early and nearly coincident with the expression of CD44V6 found after TGFβ1 treatment, we investigated the mechanism(s) of regulation of CD44V6 expression in lung fibroblasts by TGFβ1. TGFβ1-mediated CD44V6 up-regulation was initiated through EGR1 via ERK-regulated transcriptional activation. We showed that TGFβ1-induced CD44V6 expression is through EGR1-mediated AP-1 (activator protein-1) activity and that the EGR1- and AP-1-binding sites in the CD44v6 promoter account for its responsiveness to TGFβ1 in lung fibroblasts. We also identified a positive-feedback loop in which ERK/EGR1 signaling promotes CD44V6 splicing and found that CD44V6 then sustains ERK signaling, which is important for AP-1 activity in lung fibroblasts. Furthermore, we identified that HAS2-produced hyaluronan is required for CD44V6 and TGFβRI co-localization and subsequent CD44V6/ERK1/EGR1 signaling. These results demonstrate a novel positive-feedback loop that links the myofibroblast phenotype to TGFβ1-stimulated CD44V6/ERK/EGR1 signaling.
Keywords: AP-1 transcription factor (AP-1), CD44, early growth response protein 1 (EGR1), extracellular-signal-regulated kinase (ERK), signaling
Introduction
Fibrosis contributes to many enduring diseases that result in end-stage organ failure and consequently is a major cause of morbidity and mortality. Lung fibrosis occurs in interstitial lung diseases, in idiopathic pulmonary fibrosis (IPF),3 in systemic scleroderma lung disease, and in response to many types of lung injury. Lung fibrosis claims more lives annually in the United States than many types of cancer; however, effective therapy is lacking (1, 2). The pathogenesis of lung fibrosis is thought to involve aberrant or overexuberant wound healing processes initiated to protect the host from injurious stimuli with subsequent myofibroblast activation (3, 4). Activation of myofibroblasts primarily occurs through increased expression of transforming growth factor β1 (TGFβ1) mRNA and protein synthesis by parenchymal cells and/or infiltrating lymphoid cells, particularly macrophages (5, 6). However, the extent to which TGFβ1 protein can contribute to the diverse pathologies in lung fibrosis and the mechanisms that regulate TGFβ1-induced myofibroblast functions in vivo remain poorly understood.
A recent study provides evidence that hyaluronan synthase 2 (Has2), transgenically overexpressed by α-smooth-muscle actin (α-SMA)-producing myofibroblasts, promotes a severe lung fibrotic phenotype in bleomycin-induced lung injury that also requires CD44, a receptor for hyaluronan (7). We have previously reported that lung myofibroblast activation by TGFβ1 is associated with the overexpression of CD44v6 and activation of MAPK/ERK1/2 (8). The EGR1 transcription factor has been implicated in mediating the fibrotic responses induced by TGFβ1 (9). EGR1 (early growth response-1)- and AP-1 (activator protein 1)-binding sites for the CD44 promoter are located at positions 235 and 110 upstream of the transcriptional start site. EGR1 mediates its effects by regulating the transcription of a wide array of downstream genes, including CD44. However, our knowledge of how TGFβ1 stimulates CD44V6 expression/activity is limited and does not explain how TGFβ1 stimulates CD44V6 while simultaneously inducing EGR1 and AP-1. Last, expression and regulation of CD44v6 are stimulated in response to the hepatocyte growth factor, in particular by the Ras/ERK pathway (10), or through activation of EGR1 (11) or AP-1 (12). However, the role of EGR1 and/or AP-1 in TGFβ1-induced CD44V6 expression/activity has not been defined in any cell type.
To determine the functions of TGFβ1-induced CD44V6 and the mechanisms that mediate CD44V6 expression and activity, we investigated activation of EGR1 in normal lung fibroblasts treated with TGFβ1. CD44V6 has been shown to be stimulated through activation of Ras/ERK signaling (10). The EGR1 gene is induced by growth factors through different signaling pathways, including the Ras/MAP kinase pathways (13). Given the crucial role of CD44V6 signaling in cellular processes, including cell survival, proliferation, and migration, it is likely that CD44V6 expression is also regulated in fibrogenic conditions.
Previous studies from our laboratory demonstrate a crucial role for TGFβ1 in controlling CD44V6 splicing in human lung fibrogenic fibroblast proliferation/activation through MAPK/ERK1/2 (8). Despite its importance, however, the mechanisms underlying the sustained activity of MAPK/ERK1/2 signaling have remained less understood.
In this study, we investigated the mechanisms underlying the TGFβ1-dependent regulation of fibroblast differentiation through EGR1 and CD44V6. Our results show that TGFβ1-induced ERK/EGR1 signal transduction is both necessary and sufficient to stimulate CD44V6. In conjunction with EGR1-induced AP-1, CD44V6 subsequently facilitates lung myofibroblast differentiation. Our results show that a positive-feedback loop couples sustained ERK/EGR1 signaling to CD44v6 in response to TGFβ1. However, we have also demonstrated that TGFβ1-stimulated HAS2 overexpression is required to initiate CD44V6/ERK/EGR1 coupling to mediate differentiation of fibroblasts to myofibroblasts. Our data indicate that HAS2 activation in TGFβ1-stimulated human normal lung fibroblasts (HNLFbs) mediates co-localization of CD44 with TGFβ receptor 1 (TGFβRI), leading to phosphorylation of ERK and, subsequently, EGR1 signaling. Therefore, HAS2-regulated HA synthesis is a major determining factor in the phenotypic activation of fibroblasts. This change in phenotype also involves the simultaneous activation of the following cooperating pathways. 1) TGFβ1/ERK/EGR1-dependent signaling stimulates CD44V6 expression/activity; 2) EGR1-dependent AP-1 activity mediates enhanced CD44V6 expression in response to TGFβ1-induced signaling; 3) a feedback loop between activated ERK/EGR1 and CD44V6 sustains CD44V6 expression in response to TGFβ1 stimulation; and 4) HA facilitates TGFβ1-dependent fibroblast differentiation through HA-CD44V6 binding and promoting interaction between the CD44V6 and TGFβRI. This then promotes specific intracellular signal transduction through the ERK pathway and subsequently through EGR1, both acting to cooperate with the TGFβ1/ERK/EGR1/CD44V6 feedback loop pathway, resulting in fibroblast-to-myofibroblast differentiation.
Results
Myofibroblasts and fibroblasts of PBS (saline)-treated lungs and bleomycin-injured lungs were enriched in lineage-negative cells
We have previously shown that expression of CD44V6 is directly related to fibrogenic function of human lung myofibroblasts (8). At the peak of lung collagen gene expression at day 14 after bleomycin lung injury in mice, the cells primarily responsible for fibrosis are activated myofibroblasts, as defined by expression of α-SMA. Several key features of fibrotic reactions in mammalian lung tissues, including TGFβ1 up-regulation, contractile filament-laden stromal cells, and myofibroblast differentiation and activation, are recapitulated in the bleomycin-injured mouse model of fibrosis (14, 15). Therefore, we used the mouse model of acute pulmonary fibrosis, initiated by tracheal installation of the bleomycin, to define the in vivo role of CD44V6 in the acute inflammatory (weeks 1–3) and reparative (weeks 3–7) phases of lung injury (15).
However, the functional similarities and differences between myofibroblasts and fibroblasts are not fully understood because they have not been separately isolated from a living tissue. To understand the biological properties of myofibroblasts and fibroblasts in injured lung tissues, we isolated them from saline control and bleomycin-injured mouse lungs at the fibrogenic phase (day 14 after injury) by using fluorescence-activated cell sorting as described previously (16). To isolate myofibroblasts and fibroblasts from the lungs, many cell types need to be eliminated when using FACS. Lungs are composed of many types of cells: epithelial cell adhesion molecule (EpCAM)-positive epithelial cells; CD31-positive vascular endothelial cells; lymphatic vessel endothelial hyaluronan receptor (Lyve-1)-positive lymphatic endothelial cells, CD45-positive leukocytes, pericytes, and mesothelial cells. Initially, lineage-negative cells, which do not express these lineage-specific cell surface markers, were isolated, and α-Sma was compared between unfractionated cells and lineage-negative cells using quantitative real-time polymerase chain reaction (real-time PCR). In saline-treated lungs, the α-SMA mRNA expression level in lineage(−) cells was 3.2-fold higher than in corresponding lineage(+) cells (Fig. 1A). In 14-day post-bleomycin-treated lungs, the α-SMA expression level in lineage(−) cells was 13-fold higher than in corresponding lineage(+) cells (Fig. 1A).
Figure 1.

Identification of myofibroblast and fibroblast populations isolated from saline-treated lungs and bleomycin-injured lungs. Flow-cytometry sorting and analysis was used to separate lineage(+) fibroblasts (positive for lineage-specific cell surface markers (CD31, CD45, CD146, EpCAM, and Lyve-1)) from lineage(−) fibroblasts (negative for these lineage-specific cell surface markers). A, mRNA expression levels of α-SMA were compared for lineage(+) and lineage(−) fibroblasts isolated from saline-treated lungs and from bleomycin-injured lungs at day 14 using real-time PCR. The α-SMA mRNA was selectively present in the lineage(+) cells with a much higher level in the 14-day bleomycin-injured fibroblasts. B–D, real-time PCR analyses for α-SMA, CD44v6, and COL1A1 are shown for lineage(−) Sca(+) cells from saline-treated lungs (L(−)/S(+)/PBS cells), from bleomycin-injured lungs at day 14 (L(−)/S(+)/14dBleo cells), and from lineage(−) Sca(−) CD49e(+) cells from bleomycin-injured lungs at day 14 (L(−)/S(−)/C(+)/14dBleo cells). Data from various groups are expressed as means ± S.E. (error bars) (n = 3). A,*, p < 0.05 compared with lineage(−); B–D, *, p < 0.05 compared with control (PBS-treated) using Student's two-tailed t test.
Because stem cell antigen-1 (Sca-1) is expressed in perivascular fibroblasts in normal lung tissue, we sorted the lineage(−) cells for Sca-1. In saline-treated lungs, lineage(−)/Sca(+) cells (L(−)/S(+)/PBS cells) had 3.5-fold higher α-Sma (Fig. 1B), 0.23-fold higher CD44v6 (Fig. 1C), and 4.7-fold higher collagen-1 (Col1a1) (Fig. 1D) mRNA expressions, respectively, compared with unfractionated fibroblasts. In 14-day post-bleomycin-treated lungs, lineage(−)/Sca(+) cells (L(−)/S(+)/14dBleo cells) had 5.4-fold higher α-Sma (Fig. 1B), 1.3-fold higher CD44v6 (Fig. 1C), and 8.7-fold higher Col1a1 (Fig. 1D) mRNA expressions compared with unfractionated fibroblasts. Because CD49e, but not Sca-1, was expressed in myofibroblasts in bleomycin-injured lungs (16), we isolated lineage(−)/Sca(−)/CD49e(+) cells from 14-day post-bleomycin-treated lungs (L(−)/S(−)/C(+)/14dBleo cells). The expressions of α-Sma (14-fold), CD44v6 (15-fold), and Col1a1 (22-fold) genes are very high compared with unfractionated fibroblasts (Fig. 1, B–D). Approximately 64,000 ± 15,000 cells (n = 3) of a lineage(−)/Sca(+) population were isolated from a lung of a PBS-treated mouse (L(−)/S(+)/PBS cells), and ∼26,000 ± 7000 cells (n = 3) of a lineage(−)/Sca(+) population were isolated from 14-day post-bleomycin-treated lungs (L(−)/S(+)/14dBleo cells). Approximately 44,000 ± 12,000 cells (n = 3) of a lineage(−)/Sca(−)/CD49e(+) population were isolated from a lung of a 14-day post-bleomycin-injured mouse (L(−)/S(−)/C(+)/14dBleo cells). The number of lineage(−)/Sca(−)/CD49e(−) cells (L(−)/S(−)/C(−)/14dBleo cells) was very low (2356 ± 785 cells (n = 3)), compared with the above fractions (L(−)/S(+)/14dBleo cells and L(−)/S(−)/C(+)/14dBleo cells) isolated from a 14-day post-bleomycin-treated mouse lung. Therefore, we did not further analyze these cells. These gene expression profiles show that fibroblasts isolated from PBS-treated murine normal lung (MNLFbs) and fibroblasts isolated from 14-day-post-bleomycin-injured lung (14dBLMFbs) are enriched in lineage(−)/Sca(+) cells (L(−)/S(+)/14dBleo cells). Consistent with a previous study (17), the myofibroblasts from the bleomycin-injured lung (14dBLMFbs) are enriched in lineage(−)/Sca(−)/CD49e(+) cells (L(−)/S(−)/C(+)/14dBleo cells).
TGFβ1 induces feedback up-regulation of CD44v6 in lung mesenchymal fibroblasts through EGR1
We previously reported that normal lung fibroblasts treated with TGFβ1 for 8 h up-regulated CD44V6 expression and that CD44V6, which is highly expressed in human lung myofibroblasts and in bleomycin-induced murine lung myofibroblasts, is responsible for TGFβ1 autoregulation (8). Expression levels of CD44 and regulation of the alternative splicing of the variants are stimulated in response to hormones, cytokines, and growth factors, particularly by the Ras/ERK pathway (18–20) through the activation of transcription factors, including EGR1 and AP-1 in other cell types (21–23). However, the specific mechanism to define a CD44v6/EGR-1/AP-1 signaling axis for activation of TGFβ1 expression and TGFβ1-mediated responses in lung fibrosis are unknown. Thus, we hypothesized that a feedback loop, in which expression of both EGR1 and CD44V6 is up-regulated in fibrogenic fibroblasts in response to exposure to TGFβ1, represents an important mechanism whereby fibrogenic fibroblasts establish and maintain constitutive stimulation of proliferative and differentiation-promoting pathways.
Up-regulation of CD44v6 expression in the fibrotic lung after bleomycin injury
First, consistent with our previous findings (8), we determined whether primary fibroblasts isolated from lungs after bleomycin-induced pulmonary fibrosis in wild type (WT) mice demonstrate a time-dependent increase of CD44v6 protein. After bleomycin instillation, CD44v6 increased ∼1.4-fold by day 3, peaked at ∼11-fold by days 14–21, and decreased to ∼3.4-fold by day 45 (Fig. 2A, top panel shows Western blot analyses for CD44v6 and β-tubulin protein, and bottom panel shows the densitometry graph of expression of these proteins from three sets of experiments). We also examined collagen-1 expression by Masson's trichrome staining and expression of CD44v6 by immunohistochemical staining of tissue sections of 0-, 21-, and 45-day post-bleomycin-treated murine lungs. Fig. 2B shows that collagen-1 expression is greatly increased at day 21 after bleomycin injury and is decreased toward normal level at day 45. Similarly, CD44v6 was barely detectable in normal lung but was significantly localized 21 days after bleomycin injury, with significant decrease to near control by day 45. These results (Fig. 2) suggest that CD44v6 induction occurs specifically in the sites of lung injury in this mouse model.
Figure 2.

Induction of CD44v6 protein expression after bleomycin injury in mice. A, Western blots for expressions of CD44v6 (using anti-CD44v6 (VFF-18 clone) from Chemicon) and β-tubulin (EMD Millipore (AA2)) in the lungs isolated at the indicated days after bleomycin injury are shown for representative results from two animals for each time point. -Fold inductions (relative abundance of CD44v6 normalized to β-tubulin from the Western blots in this figure and from two independent experiments) of CD44v6 in the lungs at different time points after bleomycin injury are shown. The data presented in the relative abundance figure are from three sets of BLMFbs with three independent experiments and are expressed as means ± S.D. (error bars). Statistical analysis was by analysis of variance. *, p ≤ 0.005. B, C57BL/6 young (2 months) mice were subjected to bleomycin lung injury as described under “Experimental procedures.” Representative micrographs illustrate the time course of bleomycin-induced fibrosis by Masson's trichrome blue staining for collagen in the lung tissues harvested at day 0 (tissue section from continuous PBS-treated control mice) and days 21 and 45 after bleomycin-induced lung injury. Representative micrographs (scale bars, 100 μm) show the localization of CD44v6 in the lung sections on day 0 (PBS control) and 21 and 45 days after bleomycin injury.
Bleomycin-induced pulmonary fibrosis is associated with Col1a1, hyaluronan, and CD44v6 synthesis
The pattern of CD44v6 expression in the fibrotic lung (Fig. 2) is similar to the changes in expression of TGFβ1 and TGFβ receptor 1 (TGFβRI), which are induced as early as 3 days after bleomycin injury (15, 24). This close time-based association suggests that increased level of TGFβ1 may up-regulate CD44v6 through its receptor and that TGFβ1 may be, at least partially, responsible for CD44v6 induction in BLMFbs. Therefore, we examined whether other fibrogenic parameters, such as Col1a1 synthesis, hyaluronan secretion, and α-Sma synthesis, are associated with the CD44v6 synthesis in ex vivo primary BLMFbs cultures (7, 14, 21, and 45 days post-bleomycin injury) and in MNLFb cultures. Fig. 3A shows that body weights of bleomycin-injured mice decreased in days 7–14 due to systemic effects of lung injury, with increases to normal during the fibrosis resolving phase, days 21–45. Collagen secretion was demonstrated by extracellular acid-soluble collagen measured using the Sircol assay (Fig. 3B), and acid-insoluble total collagen contents were determined by the hydroxyproline assays of lung tissue homogenates (Fig. 3C). Both increased from day 7 to 21 with some decrease by day 45, in contrast to the constant concentrations in the saline control lungs, consistent with the Masson's trichrome staining results in Fig. 2B. BLMFbs and MNLFbs were isolated by flow-cytometry sorting and analyzed in cultures as reported previously (17). Hyaluronan contents in the MNLFb cultures isolated from lungs at indicated days 7–45 remained constant. In contrast, hylauronan contents in BLMFb cultures isolated from the bleomycin-treated mice at day 7 increased ∼7-fold compared with the MNLFb cultures, with a modest reduction in BLMFb cultures from day 21 to 45 after bleomycin treatment (Fig. 3D). These results provide evidence that sustained hyaluronan content may contribute to the reparative phase. Because HA is synthesized on the cytoplasmic surface of the plasma membrane by three mammalian HA synthase isoenzymes (25), we measured the Has1/2/3 mRNA expressions in the isolated fibroblasts from post-bleomycin-injured lung. Results (Fig. 3E) show that Has2 expression declines in the resolution phase (45 days post-bleomycin injury in the lung), whereas Has1 and Has3 expressions remain the same as were found in the fibrogenic phase (Fig. 3E, 21 days versus 45 days). The α-Sma contents in the lungs also increased ∼6-fold by day 21 with reduction to basal level at day 45 (Fig. 3F). CD44v6 protein peaked at days 14–21 and decreased substantially at day 45 (Fig. 3F). In contrast, CD44s levels were low in the fibrogenic phase and increased substantially at day 45 (Fig. 3F), suggesting that sustained hyaluronan content (Fig. 3D) and increased CD44s (Fig. 3F) may contribute to the resolution phase after bleomycin-mediated lung injury.
Figure 3.

Increased expression of CD44v6 is associated with collagen-1 and hyaluronan synthesis in the fibrogenic fibroblasts in the bleomycin-induced pulmonary fibrosis model. A, systemic effects in response to bleomycin-induced lung fibrosis or PBS control were assessed by a time-course evaluation of body weights. Whole-lung homogenates were analyzed for acid-soluble collagen by Sircol assay (B) and for acid-insoluble collagen by a hydroxyproline assay (C) for the indicated days after bleomycin-induced lung injury. D, fibroblasts (BLMFbs) were isolated from lung tissues at days 7, 14, 21, and 45 after bleomycin-induced lung injury, and fibroblasts were isolated from PBS (MNLFBs)-treated mouse lung tissues at the same times as described under “Experimental procedures.” The effects on hyaluronan synthesis in response to bleomycin-induced lung fibrosis or to PBS control were assessed by measuring hyaluronan secreted into the medium by an ELISA-like assay. E, total RNAs were examined by real-time PCR analysis for the indicated mRNAs expressed relative to β-actin. F, lysates from BLMFbs at days 7, 14, 21, and 45 after bleomycin-induced lung injury and MNLFBs at day 0 were immunoblotted (WB) for CD44v6, CD44s, α-Sma, collagen-1, and β-actin (loading control) (using anti-CD44v6 (VFF-18 clone) from Chemicon), CD44s using anti-CD44 (ab157107) from Abcam, α-SMA (using anti-α-SMA (IH8) from Novus Biologicals, collagen-1 (using anti-collagen-1 (ab1209539) from Abcam), and β-actin (from EMD Millipore (RM112)). Densitometry analyses are shown below the Western blots after normalization with β-actin. The data in the experiments (A–F) are from three independent experiments and are expressed as means ± S.D. (error bars). Statistical analysis was by analysis of variance. B–E, *, p ≤ 0.001 versus PBS-treated control or day 0 control groups; F, *, p ≤ 0.001 versus day 0 control group.
These results suggest a close association between CD44v6 and fibrogenic properties in ex vivo lung fibroblast cultures from the bleomycin-injured mice. Such close time-based association and spatial correlation between TGFβ1 and CD44v6 induction suggest a potential mechanism of TGFβ1 and its type I receptor (TGFβRI) in CD44v6 induction, which was analyzed for their interaction/binding in fibroblast cultures from human normal and IPF lungs (as shown in Fig. 7A of our companion paper by Ghatak et al. (91)).
Figure 7.

Characterization of CD44v6 mRNA stability in 21dBLMFbs untreated or treated with TGFβ1 after the addition of ACTD. 21dBLMFbs were serum-deprived for 48 h followed by treatment with or without TGFβ1 (5 ng/ml) in the presence of ACTD (2 μg/ml) for the indicated times and analyzed for CD44v6 mRNA, CD44s mRNA, and Gapdh mRNA. As positive and negative controls, mRNA expression levels were determined from total RNAs from cells that were exposed with TGFβ1 plus ACTD with TGFβ1 antibody (5 μg/ml) or TGFβRI antibody (5 μg/ml) and with 2 μg/ml in DMSO solution plus TGFβ1 antibody or TGFβRI antibody (5 μg/ml) in serum-starved medium for the same time points (data not shown). A, real-time PCR of CD44v6 mRNA after normalization with Gapdh mRNA during the 20 h of ACTD treatment. B, representative mRNA by RT-PCR analyses. Data are representative of three independent experiments.
TGFβ1 induces both CD44v6 protein and CD44v6 mRNA expression in ex vivo mouse lung fibroblasts
We investigated the regulation of CD44v6 expression by TGFβ1 in MNLFb cultures at both mRNA and protein levels by using real-time PCR and WB analyses, respectively. Protein levels of CD44v6 were greatly increased after treatment in a dose-dependent (Fig. 4A, scatter plots) and time-dependent (Fig. 4C, scatter plots) manner. At 24 h, TGFβ1 at 2 ng/ml induced CD44v6 > 4-fold, and this level was close to the level for 5 ng/ml (Fig. 4A). At 2.5 ng/ml, CD44v6 protein was significantly increased already by 2 h after incubation and reached a maximum (∼5-fold) by 8 h (Fig. 4C). Similar to the protein levels, real-time PCR analysis revealed that TGFβ1 induced CD44v6 mRNA in a dose-dependent (Fig. 4B) and time-dependent manner (Fig. 4D). These results also support a likely role for TGFβ1 induction of CD44v6 isoform in ex vivo lung fibroblast cultures from the bleomycin-injured mice.
Figure 4.

TGFβ1 induces CD44v6 protein and CD44v6 mRNA in lung fibroblasts. A and C, Western blots demonstrate the induction of CD44v6 protein (using anti-CD44v6 (VFF-18 clone) from Chemicon) and β-actin (EMD Millipore (RM112)) after treating MNLFbs with different concentrations of TGFβ1 for 24 h (left) (A) and with 2.5 ng/ml TGFβ1 for different times (C). The same samples were probed with β-actin to ensure equal loading. Densitometry analyses are given below the Western blots after normalization with β-actin. B and D, real-time PCR analyses show the mRNA levels of CD44v6 in the MNLFb cultures with different concentrations of TGFβ1 for 24 h (B) and with 2.5 ng/ml TGFβ1 for different times (D). Data (A–D) are presented as means ± S.E. (error bars) (n = 3) after normalization with β-actin. *, p ≤ 0.005 versus control.
Trans-activation of the CD44v6 promoter by transcription factor AP-1
TGFβ1 has been implicated in the activation of Ap-1 (26), and Ap-1 has been shown to regulate CD44 expression during an inflammatory response in vascular smooth-muscle cells (27). Therefore, we investigated activation of Ap-1 in MNLFbs treated with TGFβ1. To determine whether the CD44 promoter can be activated by the AP-1 family members c-Fos and c-Jun protein, we co-transfected the WT CD44-luciferase reporter construct with a c-Fos or a c-Jun expression vector or with their combination in cultures of MNLFbs. Fig. 5A shows that transfection of c-Fos or c-Jun expression plasmids alone significantly increased luciferase activity in the WT CD44 promoter after 24 h (orange and violet) in comparison with control cells (green) treated with empty vector, and their combined transfection increased the activity additively (blue).
Figure 5.

Activation of CD44 gene transcription by c-Fos and c-Jun. A, MNLFb cultures were transiently transfected with the WT CD44 (−1262/+109) reporter construct (1 μg) with c-Fos or c-Jun expression plasmids alone (0.1 μg each), with their combination, or with empty pcDNA3 vector. All constructs were co-transfected with a β-galactosidase control plasmid to correct for differences in transfection efficiency. After 72 h, luciferase and β-galactosidase activities were measured, and luciferase activities were normalized to β-galactosidase activities. Data are presented as -fold induction of the activity from the respective control. The data are from three sets of MNLFbs with three independent experiments for each luciferase activity level and are expressed as the means ± S.E. (error bars); n = 5. B, MNLFb cultures were transiently transfected with the WT CD44 (−1262/+109) reporter construct or with a mutated AP-1 (−1262/+109/AP-1-M) construct with binding sequences described under “Experimental procedures.” Both constructs were co-transfected with c-Fos and c-Jun and with a β-galactosidase control plasmid to correct for differences in transfection efficiencies. The cultures were then treated with or without TGFβ1 (2.5 ng/ml) for 24 h. Normalized luciferase activities are plotted as the -fold induction over the activity in control cells expressing no c-Fos or c-Jun and not treated with TGFβ1. Data are expressed as means ± S.E. (n = 3; *, p < 0.05) compared with baseline for each group using Student's two-tailed t test.
To assess the effect of TGFβ1 on Ap-1-mediated trans-activation of the CD44 promoter, cultures of MNLFbs with the AP-1 subunit c-Fos and c-Jun expression plasmids were treated with 2.5 ng/ml TGFβ1 for 24 h. Fig. 5B shows a ∼5-fold increase in CD44 promoter activity in the MNLFbs treated with both c-Fos and c-Jun expression plasmids (yellow), which was significantly increased to ∼10-fold by TGFβ1 (red). This enhanced response to TGFβ1 depended upon Ap-1 promoter activity, as shown by the lack of increase in the TGFβ1-treated cultures with the mutated Ap-1-M Luc construct (Fig. 5B, green). These data provide strong evidence that enhanced trans-activation of the CD44 promoter by TGFβ1 treatment requires an intact AP-1 site at positions −110 to −104 of the CD44 5′-flanking sequence (Fig. 6, underlined AP-1).
Figure 6.

Nucleotide sequence of the upstream regulatory region of the mouse CD44 gene. A, the nucleotide sequence numbering of the mouse promoter fragment, 602 nucleotides upstream of the transcription initiation site, is on the left. Several putative transcription factor-binding sites are shown. Double-underlining indicates the ATG translation initiation site. The EGR-1 site is indicted in green. The sequences corresponding to the oligonucleotides used in ChIP primers are shown in red. B, schematic representation of deletion sites in relation to consensus DNA sequences for known transcription factors.
CD44v6, α-Sma, and Col1a1 induction by TGFβ1 involves posttranscriptional stabilization of these mRNAs in murine lung myofibroblasts
To address whether TGFβ1 induces both transcription of new mRNA and mRNA stability, 21dBLMFb cultures were serum-starved for 48 h and then pretreated with or without TGFβ1 for 16 h before actinomycin D (ACTD) was added to block gene transcription. The cultures were then continued for the indicated times (Fig. 7A). Fig. 7B shows real-time PCR results for CD44v6, CD44s, and Gapdh mRNAs at times after adding ACTD in the presence or absence of TGFβ1, and Fig. 7A shows the relative abundances of the CD44v6 mRNA normalized to Gapdh mRNA. In the absence of TGFβ1, CD44v6 mRNA decreased with a half-life of ∼2.5 h. In the presence of TGFβ1, CD44v6 mRNA increased greatly in the first 2 h and was sustained with a half-life of ∼16 h, an increase of > 6-fold (Fig. 7A). In this experiment, the cells were treated with or without TGFβ1 prior to administering ACTD. Because pretreatment of ACTD stops transcription, the much longer (16 h versus 2.5 h) half-life of CD44v6 mRNA after TGFβ1 treatment indicates that TGFβ1 increased CD44v6 mRNA stability (Fig. 7, A and B). In contrast, the half-life of CD44s mRNA after ACTD treatment was < 2 h in the absence or presence of TGFβ1 (Fig. 7B). Thus, the results in Figs. 4, 5, and 7 (A and B) indicate that induction of CD44v6 mRNA by TGFβ1 increased both CD44v6 mRNA and its stability.
The mRNA levels of α-Sma (Fig. 8A) and collagen-1 (Fig. 8B) normalized to Gapdh mRNA are shown for the time course cultures. Cultures treated with and without ACTD treatment decreased α-Sma mRNA after 4 h in the absence of TGFβ1. In contrast, cultures treated with TGFβ1 alone increased α-Sma mRNA to a higher level (∼3-fold) that was sustained through 12 h before decreasing to control level at 24 h (Fig. 8A), which was prevented by ACTD treatment. Similar results were observed for Col1a1 mRNA, in which TGFβ1-treated cultures increased ∼4-fold over non-treated control cultures (Fig. 8B). Furthermore, unlike the more rapidly reversible expression of the α-Sma mRNA, elevated Col1a1 mRNA persisted at high levels (∼4–5-fold) through 48 h after the addition of TGFβ1 with subsequent decreases to control level by 60 h. These data suggest that TGFβ1 autocrine signaling can both stabilize already transcribed mRNAs for CD44v6, Col1a1, and α-Sma and increase their half-life through various TGFβ1-induced signaling pathways, including the NADPH oxidase pathway (as shown in our companion paper (91)). Increased synthesis of CD44v6, Col1a1, and α-Sma proteins, which are key molecules, can have crucial roles for myofibroblast activation and differentiation. Knowledge of the half-lives helped us to select the treatment schedules of CD44v6 shRNA/nanoparticles, and V6-PEP/nanoparticle in bleomycin induced lung fibrosis in mice. Specifically, CD44v6 shRNA/nanoparticles or V6-PEP/nanoparticles were administered every other day from day 2 to day 30 by intratracheal delivery to the lungs of young mice during the onset of inflammation by bleomycin injury. This delivery schedule reduces CD44v6 expression, or the CD44v6-mediated fibrogenic effect, at the onset of the inflammatory phase (as shown in Fig. 13 of our companion paper (91)).
Figure 8.
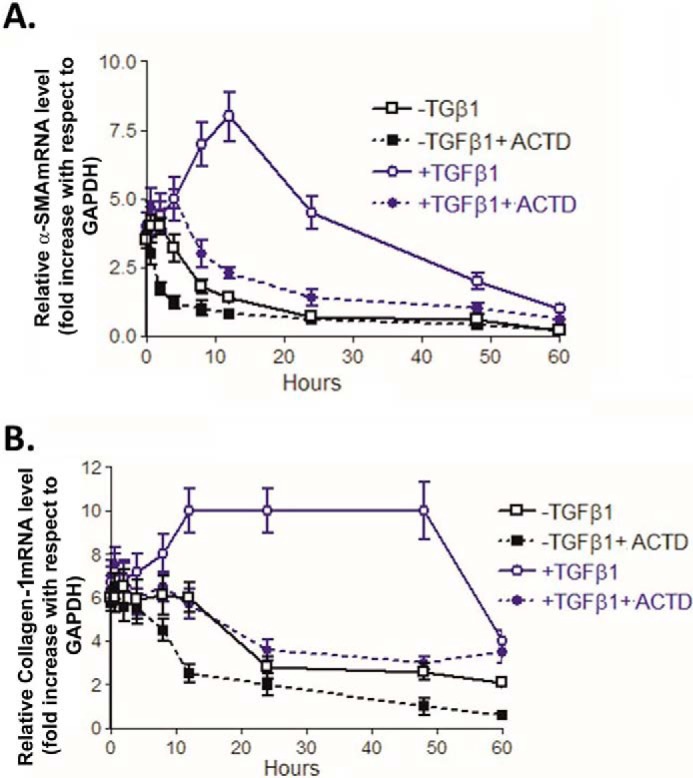
Kinetics of TGFβ1-induced α-Sma and Col1a1 mRNA expression. 21dBLMFbs were serum-deprived for 48 h followed by treatment with or without TGFβ1 (5 ng/ml) for 60 h in the presence or absence of ACTD (2 μg/ml in DMSO). Every 16 h, the serum-free medium was changed to fresh medium, and the ACTD and TGFβ1 addition was continued. RNA was isolated from the cultures at the indicated times after the addition of ACTD. A, time course for real-time PCR analyses for α-Sma mRNA. B, time course for real-time PCR analyses for Col1a1 mRNA. Positive and negative controls are used in the same manner as in Fig. 7 (data not shown). α-SMA and Col1a1 mRNA were normalized against Gapdh and expressed as relative mRNA levels compared with serum-starved MNLFbs. Data are expressed as means ± S.E. (error bars) (n = 3).
Figure 13.

TGFβ1-induced DNA-binding activity of AP-1. A, MNLFbs were transiently co-transfected with the 4 × Ap-1-pGL-2-luciferase reporter construct and an Egr-1-overexpressing plasmid and then cultured with 0.5 or 10% FBS for 48 h. Cells cultured with 10% FBS were then treated with either 20 μm Egr1-S (5′-ATGGCAGCGGCCAAGGCGG-3′) for 16 h or first transfected with c-Fos shRNA, c-Jun shRNA, or control shRNA for 24 h and then treated with Egr1-S nucleotides for another 16 h. Then the cultures were treated with TGFβ1 (5 ng/ml) for 24 h. Data are presented as -fold increase of luciferase activity for cultures in 20% FBS with respect to control cultures in 0.5% FBS without any treatment. B, nuclear run-on analysis was used in conjunction with real-time PCR to assay CD44v6 transcription in MNLFbs. MNLFb cultures were pretreated with 10 μm U0126 for 2 h or transfected with vector or with dominant-negative (DN) c-Fos for 48 h before treatment with 20 μm Egr1-S nucleotide for 16 h with or without the addition of TGFβ1 (5 ng/ml) for another 24 h. CD44v6 mRNA expression, represented as -fold change, was normalized to Gapdh and then expressed relative to untreated MNLFb control cultures. C, AP-1-binding activities were assayed in MNLFb cultures treated with or without TGFβ1 (5 ng/ml) for the indicated times. D, MNLFb cultures were first treated with 10 μm U0126 for 2 h or transfected with control shRNA (scrambled shRNA) or with c-Fos shRNA, followed by treatment with 20 μm Egr1-S oligonucleotide for 16 h with or without the addition of TGFβ1 (5 ng/ml) for another 24 h. Ap-1-binding activities were assayed. E, ChIP assays with AP-1 (c-Jun) antibody show the amplification of the 402-bp fragment containing the Ap-1-binding site within the CD44 promoter after treating MNLFbs that were previously transfected with Egr1 shRNA or c-Fos shRNA with 2.5 ng/ml of TGFβ1 for 24 h. Representative results of at least two independent immunoprecipitation experiments and multiple independent real-time PCR analyses are shown. F, Ap-1-dependent CD44 promoter activity was assayed in MNLFb cultures treated with TGFβ1 (2.5 ng/ml for 24 h). Cells were transiently co-transfected with a WT CD44 (−1262/+109) promoter construct or with a mutant CD44 (−1262/+109/AP-1-M) construct and with a β-galactosidase control plasmid. After 72 h, cells were treated with vehicle or TGFβ1 (2.5 ng/ml for 24 h), followed by luciferase and β-galactosidase activity measurements. Values are expressed as relative luciferase units/min/mg of protein, normalized to β-galactosidase activity in TGFβ1-treated cells compared with their respective controls. G, MNLFb cultures were treated with 20 μm Egr1-S nucleotide for 16 h before TGFβ1 (2.5 ng/ml for 24 h) treatment and luciferase assays. Data are expressed from various groups (A–D, F, and G) as means ± S.E. (error bars) (n = 3).*, p < 0.05 compared with baseline for each group using Student's two-tailed t test.
A positive-feedback loop couples EGR1 and CD44v6 for sustained activation of CD44v6 in response to TGFβ1
TGFβ1-induced CD44v6 is independent of Smad signaling in lung fibroblasts
Studies indicate that TGFβ1, upon binding to its specific TGFβ1 receptor (TGFβRI), stimulates diverse cellular activities by initiating multiple signal transduction pathways, including Smad, p38 MAPK, and AKT kinase in lung fibroblasts (28–30). To address whether Smad signaling is involved in mediating CD44v6 induction, we examined the consequences of overexpressing inhibitory Smad7 for CD44v6 expression. Fig. 9A (inset) shows that Smad7 overexpression by Smad7 cDNA transfection increased the content of Smad7 mRNA in MNLFbs in a concentration-dependent manner within 24 h of transfection. To investigate whether Smad7 modulates the responsiveness to TGFβ, we transfected TGFβ-inducible p3TPLux luciferase reporter construct, which contains the PAI-1 promoter (31, 32), into the MNLFbs cells in the absence or presence of Smad7 cDNA. Fig. 9A shows that TGFβ-induced luciferase activity is inhibited by expression of Smad7 but not by expression of Smad2. Fig. 9B (Western blot and the densitometric analyses) shows that TGFβ1 markedly induced CD44v6 expression in vector-transfected MNLFbs. However, SMAD7 overexpression failed to block CD44v6 induction in Smad7-overexpressing MNLFbs. Thus, the results in Fig. 9 provide evidence that TGFβ1-induced CD44v6 expression in lung fibroblasts is independent of intact Smad signaling.
Figure 9.

MAPK-ERK signaling, but not TGFβ1/TGFβRI signaling, is required for the induction of CD44v6 expression by TGFβ1. A, effects of mouse Smad7 transfection on TGFβ1-induced p3TPLux expression in MNLFbs. For the transcriptional response assay, MNLFb cells were transfected with p3TPLux as described under “Experimental procedures.” In each experiment, total equal amounts of DNA were transfected as described under “Experimental procedures.” Luciferase activity was measured and normalized for transfection efficiency. Results shown are representative of at least three independent experiments. Validation of transfection of Smad7 with different concentrations of Smad7 cDNA (pSmad7) for 24 h is shown in RT-PCR analyses of Smad7 and Gapdh in the inset. B, MNLFbs were transfected with empty vector or Smad7 expression vector. After 48 h of transfection, the cultures were treated with 2.5 ng/ml TGFβ1 for the indicated times. Western blots show the expression of CD44v6 and β-actin (using anti-CD44v6 (VFF-18 clone) from Chemicon) and β-actin (EMD Millipore (RM112)). C, MNLFbs were pretreated with the kinase inhibitors, 10 μm U0126, 100 nm wortmannin, and 10 μm SC-68376 for 2 h before the addition of 2.5 ng/ml TGFβ1 for 8 h. The mRNA levels for CD44v6 were determined by real-time PCR. Data are presented as means ± S.E. (error bars) (A, n = 3; B, n = 3) after normalization with β-actin. *, p ≤ 0.005 versus controls (without TGFβ1); *, p ≤ 0.005 versus TGFβ1-treated control.
In addition to TGFβ1/Smad signaling, TGFβ1 is also capable of stimulating parallel downstream signal pathways that lead to activation of p38 MAPK and AKT kinase in lung fibroblasts (28–30). To examine the potential implication of these pathways in CD44v6 mRNA induction, we used specific pharmacological inhibitors to block particular signaling pathways. Fig. 9C shows that specific inhibitions of AKT by wortmannin and of p38 MAPK by SC-68376 did not inhibit CD44v6 induction by TGFβ1 (Fig. 9C, violet and orange compared with yellow). However, specific inhibition of MAPK/ERK activation by U0126 did inhibit the CD44v6 induction by TGFβ1. Therefore, it is unlikely that Smad, PI3K/AKT, or p38 MAPK signaling pathways have key roles in mediating CD44v6 induction by TGFβ1 in lung fibroblasts, whereas the MAPK/ERK pathway probably does (Fig. 9, A–C, green compared with yellow).
TGFβ1 up-regulates EGR1 and CD44v6 mRNAs and proteins via the ERK1/2 pathway
EGR1 is an inducible zinc finger transcription factor, capable of binding to specific GC-rich DNA sequences (33, 34). The EGR1-binding site for the CD44v6 promoter is located at position 235 upstream of the transcriptional start site. EGR1 mediates its effects by regulating the transcription of a wide array of downstream signaling molecules, including CD44 (33, 35–38). Fig. 3E shows that Has2 is an important mediator of differentiation in bleomycin-induced lung fibroblasts. Thus, we focused on transcriptional mechanisms mediated by TGFβ1 and their contribution to earlier regulatory processes by EGR1, which may be a mediator for TGFβ1-stimulated CD44v6 and Has2 expressions. Western blots in Fig. 10A using MNLFbs show that Egr1 protein is induced by TGFβ1 ∼1.8-fold by 2 h and increases to ∼4-fold by 4 h. CD44v6 protein levels were also elevated by TGFβ1 ∼4-fold by 4 h and ∼6-fold by 8 h (Fig. 10A) and remained increased through 24 h (data not shown). Fig. 10B shows WB analysis of nuclear protein extracts from primary human lung mesenchymal cells (HNLFbs; IMR-90 cells) after a 4-h treatment with TGFβ1 in the presence and absence of specific inhibitors for ERK1/2 (U0126) and PI3K/AKT (LY94002). The up-regulation of both nuclear EGR1 and CD44V6 was mediated through the ERK1/2 pathway but not by the PI3K/AKT pathway. To determine the involvement of the EGR1/ERK pathway in lung fibroblast function, we measured proliferation of HNLFbs in the presence and absence of EGR1 or EGR1 plus ERK inhibitor. Fig. 10C shows that proliferation of HNLFbs induced by up-regulation of EGR1 is also inhibited by the ERK inhibitor U0126.
Figure 10.

TGFβ1 up-regulates EGR1 and CD44v6 protein expression and cell proliferation via the EGR-1 pathway, and blocking TGFβ1 by TGFβ1 shRNA down-regulates EGR1 and CD44v6 mRNA expressions via down-regulation of the EGR1/ERK1/2 pathway. A, a time course is shown for CD44v6 and β-actin up-regulation by TGFβ1 (5 ng/ml) in MNLFbs. B, WB analyses are shown for 10 μg of nuclear extracts from cultures of human lung mesenchymal cells (HNLFbs; IMR-90 cells) treated 2 h with specific inhibitors for ERK1/2 (10 μm U0126) and PI3K (5 μm LY294002) followed by TGFβ1 (5 ng/ml) for 24 h. C, effects of ERK suppression on EGR1-mediated proliferation of HNLFbs treated with or without serum were determined at 24 h by a BrdU incorporation assay. D, translocation of EGR-1 to the nucleus was measured in response to treatment with or without TGFβ1 (5 ng/ml) for 24 h using immunofluorescence. The cells were probed with CD44V6 primary Ab followed by Alexa 488 (secondary) Ab (green), and counterstained with EGR-1 (primary) Ab and labeled with Alexa 594 (secondary) Ab (red). Images were taken by confocal microscopy (scale bars, 100 μm). Data are expressed as means ± S.E. (error bars) (n = 3). *, p < 0.05 compared with baseline for each group using Student's two-tailed t test. E, TGFβ1/TGFβ1RI interaction induces CD44v6 mRNA expression. 14dBLMFb and 21dBLMFb cultures were transfected with control shRNA or TGFβ1RI shRNA for 24 h and then treated with TGFβ1 (5 ng/ml) for 24 h. RNA isolates were analyzed for CD44v6 mRNA. F, EGR1 and TGFβ1-induced EGR1 promote CD44v6 mRNA expression. HNLFb and MNLFb cultures were treated with 20 μm EGR1-S, as described under “Experimental Procedures,” or with TGFβ1R1 shRNA for 24 h and then treated with TFGβ1 (5 ng/ml) for 24 h. RNA isolates were analyzed for CD44V6 mRNA by real-time PCR, normalized against GAPDH, and expressed as -fold increase compared with serum-starved IRM-90 cells. Data are expressed as means ± S.E. (n = 3). *, p < 0.01 compared with baseline for each group using Student's two-tailed t test.
It has been shown that EGR1 activation involves nuclear translocations depending on stimuli (33, 35, 36). Fig. 10D shows the effects of the exogenous addition of TGFβ1 on EGR1 translocation in HNLFbs. Localizations of EGR1 and CD44v6 were determined by indirect immunofluorescence microscopy with monoclonal anti-CD44v6 and polyclonal anti-EGR1 antibody, followed by appropriate fluorescence-labeled (rabbit anti-mouse and goat anti-rabbit) secondary antibodies. The majority of the induced EGR1 (red) after overexpression of TGFβ1 in HNLFbs was detected on the nucleus (blue), whereas the majority of CD44v6 stays in the cytoplasmic compartment (green) of HNLFbs. Moreover, we found (Fig. 10E) that blocking the TGFβRI by TGFβRI shRNA inhibited CD44v6 mRNA expression in both 14dBLMFbs and 21dBLMFbs, and this suppression was not overcome by adding TGFβ1, indicating that TGFβ1 interacting with TGFβRI induces CD44v6 mRNA. In addition, CD44V6 mRNA is significantly increased 4–5-fold in both HNLFbs and MNLFbs treated with an EGR1 sense nucleotide (EGR1-S), which was increased further by treatment with TGFβ1 (Fig. 10F). In contrast, treatment with EGR1 antisense nucleotide (EGR1-AS) decreased the response to TGFβ1 (Fig. 10F).
HAS2-regulated HA synthesis is a key mediator of TGFβ1-dependent myofibroblast differentiation and proliferation
Fig. 3E shows that HAS2 is an important mediator of differentiation in bleomycin-induced lung fibroblasts. This finding is supported by the studies indicating that HAS2 is primarily responsible for TGFβ1-induced differentiation of fibroblasts to myofibroblasts (25, 39, 40). Here we confirm that transfection with shRNA targeting HAS2 (HAS2 shRNA) was sufficient to knock down HAS2 mRNA expression (Fig. 11A) and inhibit α-SMA up-regulation (Fig. 11B). CD44V6 co-immunoprecipitation with TGFβRI was also prevented following silencing HAS2 by HAS2 shRNA transfection (Fig. 11C). We therefore determined whether HA production by HAS2 was a direct regulator of the ERK and EGR1 intracellular signaling pathways leading to phenotypic change. Primary lung fibroblasts from the lung of a healthy subject (HNLFbs) were transfected with HAS2 shRNA or control (scrambled) shRNA, and EGR1 expression and ERK phosphorylation were assessed by Western blotting. When stimulated with TGFβ1, the fibroblasts transfected with HAS2 shRNA inhibited activation of EGR1 (Fig. 11D, immunoblot (top) and corresponding densitometry graph (bottom)) and also of pERK (Fig. 11E, immunoblot (top) and corresponding densitometry graph (bottom)). In addition, both the early and late peaks of the biphasic signaling pattern were lost. The loss of early ERK phosphorylation peaks supports previous studies, which have shown that HAS2 impairment inhibits fibroblast differentiation (41). In addition, the attenuation of the late phosphorylation peaks supports previous findings highlighting HAS2 and HA as necessary mediators of fibroblast proliferation (42, 43). These data suggest that HAS2 production of the lung fibroblast in response to TGFβ1 was partly responsible for the co-localization of CD44v6 with TGFβRI and enables the resulting differentiation signaling response through the ERK/EGR1 pathway.
Figure 11.

HAS2-derived HA is required for interaction of CD44V6 with TGFβRI and consequent ERK/EGR1 intracellular signaling. Serum-starved confluent HNLFbs were transfected with control shRNA or HAS2 shRNA for 48 h before treatment with 2.5 ng/ml TGFβ1 for up to 48 h (every 12 h, fresh TGFβ1 was replaced in the medium). A, real-time PCR analysis validates knockdown of HAS2 mRNA. B, real-time PCR analysis demonstrates that HAS2 knockdown inhibits α-SMA mRNA expression. A and B, data are expressed as means ± S.E. (error bars) (n = 3). *, p < 0.05 compared with baseline for each group using Student's two-tailed t test. C, cell lysates were immunoprecipitated (IP) with TGFβRI antibody, followed by WB analysis of CD44v6 and TGFβRI expressions. The blot shown here is representative of three separate experiments. D and E, immunoblotting for EGR1, β-tubulin, pERK, and ERK at the indicated times; the corresponding densitometry graphs are shown in the bottom panels. The blot shown here is representative of three separate experiments, and densitometry graphs show mean ± S.E. of three separate experiments. *, p < 0.01.
Confirmation of specificity of shRNAs used in this study by rescue experiments using shRNA-immune cDNA
To facilitate the use of our protocol, an overview of the entire procedure is shown in Table 1. After evaluating the knockdown effects of each shRNA, the most effective shRNAs from each gene can be accepted for targeting a specific gene. Finally, after generating a stable cell line with effective shRNA and confirming the knockdown efficiency of each gene, the cell line can be transfected with the gene replacement vector, containing the modified target gene (KNOCK-IN (KI; shRNA-immune cDNA)) that no longer contains target sites for the shRNAs but still encodes a functional protein. This construct restores full function and rescues any loss-of-function phenotype. This can often be achieved by utilizing one or more silent third-codon point mutations within the targeted region. In the experiments of Fig. 12 (A–F), CD44v6, TgfβRI, Egr1, Erk1, c-Fos, and c-Jun were targeted for removal by transfecting 21dBLMFbs with corresponding shRNA knockdown constructs. The cells were then transfected with the KI construct that circumvented the targeting vector, resulting in re-expression of knocked down cells, and the resulting proteins were analyzed following immunoblotting of cell lysates and real-time PCR and total RNA analyses. Introduction of shRNAs for coding sequences (CDS) effectively targeted the genes. Reintroduction of the CD44v6 cDNA, TgfβRI cDNA, Egr1 cDNA, Erk1 cDNA, c-Fos cDNA, or c-Jun cDNA in 21dBLMFbs that were pretransfected with their specific shRNAs (for CDS) for 24 h did not result in expression of these mRNAs due to the presence of the corresponding shRNAs generated from the transfected shRNA vectors. However, the corresponding proteins and mRNAs were expressed in these shRNA transfectants after co-transfection with the vectors carrying KI genes with mutations in the regions corresponding to the shRNAs to circumvent the targeting region (KI) genes in Fig. 12 (A–F). The results in Fig. 12 (A–F) validated the use of these six shRNAs for the experiments in the proposed studies in this paper and in our companion paper (91).
Table 1.
Steps to validating the specificity of an shRNA experiment

Figure 12.

Confirming the specificity of shRNA experiments. Schematic overview of the procedure used to confirm the shRNA experiments (Table 1). A–F, in 21dBLMFFbs, we verified the blocking of CD44v6, TGFβRI (TR1), EGR1, ERK1, c-Fos, or c-Jun by specific shRNAs for the CDS by replacing the knocked down gene(s) (i.e. a gene replacement strategy, designed to circumvent the shRNA knockdown). This was accomplished by the indicated shRNA-mediated knockdown and corresponding KI gene transfections. Total RNAs were examined by real-time PCR analysis for the indicated mRNAs expressed relative to β-actin. Cell lysates were processed for WB analysis for the indicated proteins. The blot shown is representative of three separate experiments, and the real-time PCR graphs show the mean ± S.E. (error bars) of three separate experiments. **, p < 0.01. G, lack of off-target effects of shRNAs (interferon response) is shown in cultured mouse alveolar macrophage MH-S cells. Poly(I:C), but not 1 μg/ml shRNAs, induces IFN-α secretion by MH-S cells. IFN-α levels in cell culture supernatants were measured after 24-h exposure to the indicated nucleic acids and doses. The average of three replicate treatments is presented, and error bars represent S.D. An shRNA dose of 100 μg/20 kg of mouse weight was chosen (56). H, plasma IFN-α induction levels in female C57BL mice treated with the indicated shRNAs are shown. 100 μg/mouse of shRNAs was injected by high pressure (10% (v/w)) in the tail, and plasma was collected 2 h after injection. The averages of three replicate mice are presented, and error bars represent S.D.; *, p ≤ 0.005.
Off-target effects are the gene perturbations caused by unintentional interactions between the siRNA and shRNA molecules and cellular components (44), which can complicate the interpretation of siRNA data (45). The off-target effects of shRNA and siRNA are different due to the fundamental differences between these two RNAi approaches. For example, because shRNA is expressed in the nucleus and processed by the endogenous machinery, it is less likely to trigger an immune response in vivo. Also, the 5′ ends of endogenously spliced shRNA oligomers are less inflammatory than the 5′ ends of exogenous siRNA molecules (46–48). Further, synthetic siRNA delivered to primary hematopoietic cells induced up-regulation of type I IFN genes and increased IFN synthesis (49, 50). Therefore, we compared the effects of poly(I:C), siRNA, and shRNA on cultured mouse alveolar macrophage MH-S cells that can be induced to secrete IFN-α by stimulation with poly(I:C). MH-S cells were treated with various doses (2, 5, and 10 μg) of poly(I:C) used as a positive control or with shRNA duplex against TgfβRI, CD44v6, or EGR1 for 24 h, and the levels of secreted IFN-α in MH-S cell supernatants were measured by enzyme-linked immunosorbent assay (ELISA). Poly(I:C) induced a clear dose-dependent IFN-α response, approaching the response to lipopolysaccharide (LPS), which is known to induce a strong IFN-α response (Fig. 12G) through interaction with macrophages (51). In contrast, two sets of synthetic TgfβRI shRNAs failed to induce a measurable IFN-α response at any dose (Fig. 12G). We also examined the response of mice to naked shRNAs (Fig. 12H, TGFβRI shRNA-1, CD44v6 shRNA-1, EGR1 shRNA-1, ERK shRNA-1, c-Fos shRNA-1, c-Jun shRNA-1) and found that serum IFN-α levels were induced in mice injected with poly(I:C) (used as a positive control) but were not induced with any of our shRNAs. We conclude that our shRNAs are well tolerated in MH-S cells and by mice without showing measurable IFN-α responses at any dose (Fig. 12, G and H).
TGFβ1 induces CD44v6 expression in lung fibroblasts through Egr1 and activation of Ap-1
The AP-1 transcriptional activating complex is made up of Jun and Fos proteins. To address the effect of overexpression of EGR1 on the activity of other genes, such as Ap-1, we examined whether overexpressed EGR1 cDNA alone or in combination with c-Fos shRNA or c-Jun shRNA resulted in modified transcriptional activity of Ap-1 with an in vitro luciferase assay. Egr1 cDNA increased Ap-1 luciferase activity ∼6-fold (Fig. 13A, green compared with yellow). This increase of luciferase activity by Egr1 cDNA-overexpressing cells was decreased significantly by transfection with c-Fos shRNA (Fig. 13A, blue) and with c-Jun shRNA (Fig. 13A, dark pink) compared with control shRNA transfection (Fig. 13A, light pink).
We determined whether the increases in CD44v6 mRNA levels in Egr1-S-treated cells (Fig. 10F) were due to the effect of the Erk → Egr1 pathway (Fig. 13B) or due to Ap-1 protein component (c-Jun), which is downstream of Egr1 (52). TGFβ1 treatment resulted in a ∼4-fold increase in transcription of CD44v6 mRNA in MNLFbs (Fig. 13B, green compared with yellow). The TGFβ1-induced increase in CD44v6 mRNA expression was further increased by Egr1-S treatment to the level of the TGFβ1 +Egr1-S + Vect control (Fig. 13B, light pink and dark pink), which was effectively blocked by pretreatment with the ERK inhibitor U0126 (Fig. 13B, blue) and by transfection with dominant-negative c-Fos expression plasmid (Fig. 13B, gray).
Next, we determined whether TGFβ1 is an effective Ap-1 activator. MNLFbs were pretreated with 2.5 ng/ml TGFβ1 for 0, 1, 2, 4, 8, and 12 h. Fig. 13C shows a time-dependent increase in Ap-1 DNA binding after 2 h that increased ∼4–5-fold by 6–8 h and remained elevated through 12 h. Strong TGFβ1 induction of Ap-1 binding was also observed by 24 h (data not shown). We investigated whether activation of Ap-1 is through the AP-1 subunit (c-Fos). MNLFbs that were pretreated with U0126 or Egr1-S or pretransfected with c-Jun shRNA were treated with TGFβ1 for 24 h. MNLFBs pretreated with Egr1-S oligonucleotide increased Ap-1-binding activity in response to TGFβ1 (∼40%) (Fig. 13D, blue and dark pink compared with green) and ∼4-fold over control (Fig. 13D, blue compared with yellow). The pretreatment with c-Fos shRNA inhibited the TGFβ1 increase to near the level inhibited by the Erk inhibitor U0126 (Fig. 13D, gray compared with orange) in contrast with the control shRNA (Fig. 13D, dark pink).
Next, we analyzed the direct binding of Ap-1 to the CD44v6 promoter using a ChIP assay and determined whether this activity is through an Egr1/Ap-1 pathway. The MNLFbs were pretreated with Erk inhibitor U0126 for 2 h or pretransfected with control shRNA, Egr1 shRNA, or c-Fos shRNA for 24 h and then treated with TGFβ1 for 4 h (Fig. 13E). Specific chromatin·protein complexes were then immunoprecipitated using an anti-AP-1 (c-Jun) antibody. The region homologous to the Ap-1-binding site was then subjected to PCR amplification using as a template the DNA from the AP-1-specific immunoprecipitated complexes (Fig. 13E). The appropriate band (402 bp) corresponding to the fragment containing the predicted Ap-1-binding site was detected only in the TGFβ1-treated cells (Fig. 13E, lanes 3–6). Both Egr1 shRNA (Fig. 13E, lane 5) and c-Fos shRNA (Fig. 13E, lane 6) greatly inhibited this band compared with the control shRNA (Fig. 13E, lane 4). Controls without exposure to Ap-1 antibody and untreated MNLFbs cells showed no band (Fig. 13E, lanes 1 and 2).
To determine whether TGFβ1-dependent increase of Ap-1 DNA-binding activity is the mechanism for increased CD44 expression, MNLFbs were transfected with WT CD44 promoter constructs containing either an AP-1-binding sequence (from −1262 to + 109 bp) or with a mutated AP-1 site (−110 to −104, TTAGTCA to CTAGGCA) (Fig. 6) that disrupts the function of this site (53). Three days post-transfection, the cultures were treated with 2.5 ng/ml TGFβ1 for 24 h. Fig. 13F shows that TGFβ1 increased luciferase activity greatly with the WT CD44 (−1262/+109) promoter construct (dark pink) but only modestly with the mutant CD44 (−1262/+109)/AP-1 M construct in MNLFbs (green). Fig. 13G shows that treatment of MNLFbs with Egr1-S increased luciferase activity for the CD44v6 promoter with the AP-1-binding sequence ∼3-fold (dark pink), which increased further to ∼6-fold when combined with TGFβ1 (green). In contrast, the combined treatment of MNLFbs with the mutant Ap-1 promoter showed luciferase activity nearly down to control level (gray). The Egr1-S and Ap-1-binding sites for the mouse promoter upstream of the transcriptional start sites are shown (Fig. 6, A and B). The results in Fig. 13 indicate that 1) TGFβ1 regulates CD44v6 expression by Egr1-activated Ap-1, and 2) this Ap-1 activity requires Egr1 activity.
Down-regulation of TGFβ1-induced ERK and of AP-1 activation inhibits fibroblast-myofibroblast differentiation
Our previous study showed that TGFβ1-induced CD44v6 stimulates α-SMA expression (8). To further confirm the role of ERK and AP-1 in TGFβ1-induced α-SMA expression and fibroblast-myofibroblast differentiation, MNLFbs were treated with ERK inhibitor U0126 for 2 h or were transfected with c-Jun shRNA or CD44V6 shRNA (as positive control) followed by TGFβ1, as described above. The results show that U0126, c-Jun shRNA, and CD44V6 shRNA inhibit the TGFβ1-induced fibroblast-to-myofibroblast differentiation measured by suppression of collagen gel contraction (Fig. 14, A and B).
Figure 14.

ERK/AP-1/CD44v6 pathway has a crucial role in α-SMA synthesis and TGFβ1-induced myofibroblast contractility. A, areas of the gels calculated from the gel diameters are shown as a measure of contraction from the experiment of B. *, p < 0.05 compared with untreated control cells. B, a photograph shows the contraction obtained in each gel. The stressed fibroblast-populated collagen lattices were prepared and stressed as described under “Experimental procedures.” The stressed HNLFbs were then mechanically released from the culture plate and allowed to contract rapidly within 2 h. The pictures were taken at the end of contraction. Data are expressed as means ± S.E. (error bars) (n = 3).
TGFβ1-induced activation of ERK/EGR1 signaling depends on CD44V6 and stimulates CD44V6 expression
To investigate whether CD44v6 is important for activation of ERK/EGR1 signaling in response to specific cytokines, we examined the effects of TGFβ1 on HNLFbs. Previous studies have shown that TGFβ1 stimulation of ERK activation depends on hyaluronan interaction with CD44 (54).
We previously showed that CD44V6 overexpression leading to ERK activation in human fibrogenic lung fibroblasts depends on autocrine TGFβ1 signaling (8). A recent study demonstrated that expression of the CD44V6 variant is up-regulated by hepatocyte growth factor/scatter factor (HGF/SF) through a feedback loop requiring the presence of EGR1 in melanoma cells (11). However, the mechanism of TGFβ1 in sustained CD44V6 induction remains largely unknown. We propose that in human lung fibroblasts, CD44V6 and MAPK/ERK signaling in response to TGFβ1 are components of a positive-feedback loop. If CD44V6 isoforms and ERK do form a feedback loop, then expression of CD44V6 isoforms should depend on activated ERK, because ERK has been shown to be important for TGFβ1-dependent ERK/EGR1 signal transduction (Figs. 10B and 13B). Thus, down-regulation of the CD44V6-containing isoforms would be expected to result in disruption of the positive-feedback loop between pERK/EGR1 and CD44V6.
To investigate whether CD44V6 is important for activation of ERK/EGR1 signaling in response to TGFβ1, we examined the effects of TGFβ1 on HNLFbs. Our CD44V6 shRNA, which only knocks down V6-containing isoforms, as was shown in our in vivo studies of colon cancer (55, 56), was used to knock down CD44V6 expression and activity in HNLFbs. CD44V6 shRNA transfection resulted in an ∼8-fold knockdown of expression of CD44V6 mRNAs containing this exon compared with a control shRNA, and no change of CD44s mRNA was observed (Fig. 15A, left). WB analysis using a CD44V6-specific antibody also showed 3-fold or greater knockdown in its protein level (data not shown).
Figure 15.
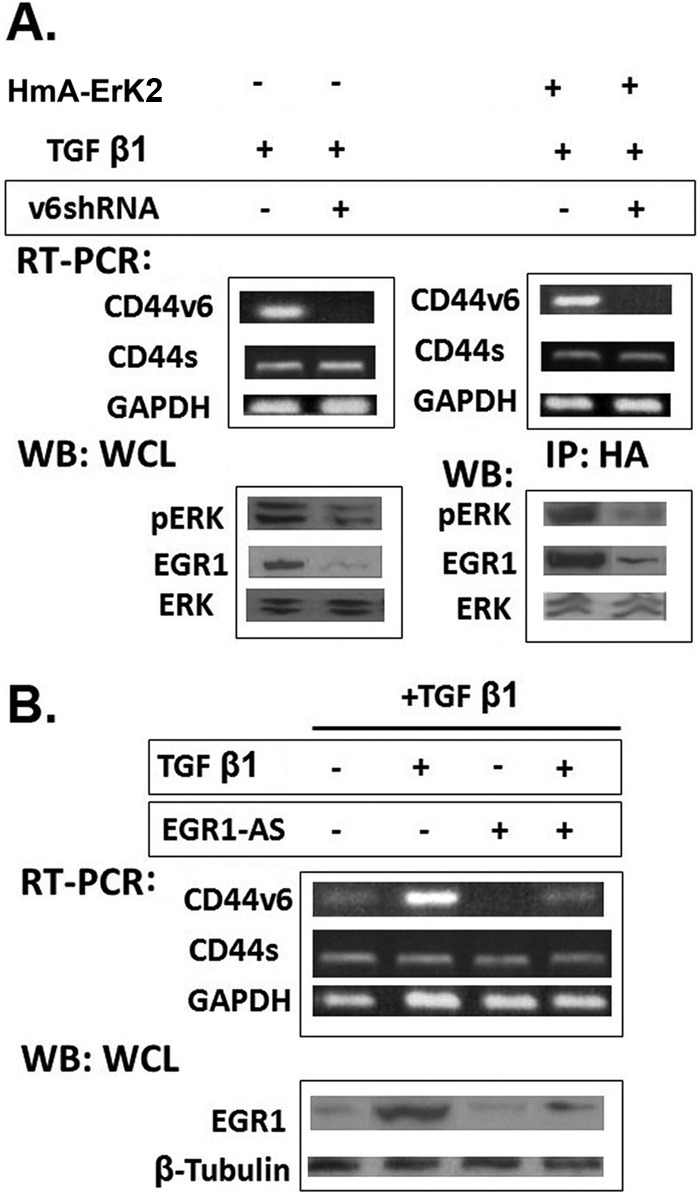
shRNA-mediated knockdown of CD44V6 down-regulates ERK induced EGR-1 activity. A (left), RT-PCR analyses of CD44V6 in HNLFbs transfected with control shRNA or CD44V6 shRNA followed by TGFβ1 (5 ng/ml) stimulation for 24 h. GAPDH was used as a loading control. WB analyses of ERK phosphorylation following TGFβ1 (2.5 ng/ml) stimulation for 16 h in cells transfected with control or CD44V6 shRNA were done with antibodies recognizing pERK, EGR1, or total ERK. Right, RT-PCR analyses are shown for CD44v6 in HNLFbs transfected with control shRNA or CD44v6 shRNA or treated with 10 μm U0126 for 2 h or with an additional hemagglutinin (HmA-ERK2) construct, followed by TGFβ1 (5 ng/ml) stimulation for 24 h. Immunoprecipitation with HmA antibody was followed by WB analyses for pERK, ERK, and EGR1. B, RT-PCR analyses of CD44V6 and Western blot analyses of EGR1 expression are shown for HNLFbs that were stimulated with TGFβ1 (5 ng/ml) for 24 h in the absence or presence of EGR-AS nucleotides. Data are representative of three (n = 3) independent experiments.
If CD44V6 splicing and ERK activation are part of a feed-forward loop, then conversely, constitutive activation of ERK should increase CD44V6 in the absence of CD44V6 shRNA treatment, and accordingly, CD44V6 shRNA treatment should no longer increase the CD44v6 splicing after TGFβ1 treatment if the ERK expression vector (hemagglutinin-tagged (HmA-ERK)) is co-transfected with CD44V6 shRNA. Fig. 15A (right) shows that overexpression of the HmA-ERK plasmid with CD44V6 shRNA decreased both ERK phosphorylation (pERK) and EGR1 expression greatly compared with control shRNA-treated cells after 24-h transfection followed by TGFβ1 stimulation for 24 h. The amount of ERK protein, however, was not altered. The involvement of EGR1 in the CD44V6 and ERK feedback loop was further supported by measuring the levels of CD44V6 mRNA in the presence and absence of EGR1-AS treatment followed by TGFβ1 stimulation. Fig. 15B shows that this treatment inhibited the up-regulation of CD44V6 expression, indicating that ERK/EGR1 signaling is required for TGFβ1-induced CD44V6 expression. The results in Fig. 15 provide further support for a positive-feedback loop between CD44V6 and ERK/EGR1 activation that sustains CD44V6 stimulation by TGFβ1 treatment.
To investigate whether TGFβ1 stimulation up-regulates CD44 variants, serum-depleted lung fibroblasts from IPF patients (IPFFbs; from our companion paper (91)) were stimulated by the addition of TGFβ1 to medium. We first examined the expression profile of CD44 variants in IPFFbs and also in HNLFbs after stimulation with TGFβ1 by exon-specific reverse transcription-PCR (RT-PCR) analysis (Fig. 16A). Exon V3 is covered by two primers (V3I and V3II) due to the existence of an alternative splice acceptor site in the human V3 exon (57). Several variant isoforms are indeed expressed. Interestingly, exon V6 seems to be expressed together with exons V4–V6 (shown both in IPFFbs and TGFβ1-stimulated HNLFbs by the ladder) and probably also alone (indicated by the lower band in the V6 lane). Note that V6 seems not to be coexpressed with exon V3, which appears as an independent isoform. In addition, on the protein level, CD44V6-containing isoforms were detected in these cells (Fig. 16B). The levels of CD44 variants were monitored by RT-PCR using different sets of primers, as shown in Fig. 16C. The variants were detected using a 3′ primer (red arrows) from a constitutive exon 6 and two different 5′ primers (green) complementary to either V4 or V6 exon, respectively. In addition, the CD44S standard form was detected using primers that base-pair to the constitutive exons. The V6 primer and standard primer each predominantly amplified a single product (Fig. 16, C–F). The V4 primer gave rise to three spliced CD44 variants containing 1) variant exons V4, V5, and V6 (depicted as V4–V6); 2) variant exons V4 and V7 (depicted as V4V7); and 3) variant exon V4 (V4), all joined to the 3′-constitutive exon 6 (Fig. 16, C–F). All products were confirmed by DNA sequencing. The expression levels of CD44 variants were also examined using 3′ primers complementary to individual variable exons and a primer to the 5′-constitutive exon. Similar results were obtained with these primer sets (data not shown). Fig. 16 shows that, after stimulation with TGFβ1, spliced variants containing V6 (V4–V6) mRNAs as well as protein expressions were strongly inhibited by down-regulation of V6-containing CD44 isoforms by CD44V6 shRNA treatment as compared with control shRNA-transfected cells. However, the levels of the other two variants without V6 (V4V7 and V4) remained unaltered after treatment with either CD44V6 shRNA or control shRNA in IPFFbs treated with TGFβ1 (Fig. 16D, right). Western blotting data (Fig. 16D, right) shows that CD44V6 shRNA down-regulated both pERK and EGR1 protein expression, keeping ERK level unchanged. Importantly, CD44V6 shRNA down-regulated only CD44V6-containing isoforms and did not affect CD44S expression (Fig. 16D, right). To further support that ERK is involved in a positive-feedback loop between CD44V6 and ERK/EGR1 signaling, we used the ERK shRNA in IPFFbs (Fig 16E). Fig. 12H shows that the transfection of MHS-1 cells with ERK shRNA and CD44V6 shRNA reduces the production of IFN-α in plasma samples from mice, which nullifies the off-target effects of these shRNAs. Treatment with ERK shRNA to TGFβ1-stimulated cells inhibited the up-regulation of all of the examined CD44 variants, indicating that ERK signaling is required for TGFβ1-stimulated alternative splicing (Fig. 16E). Moreover, ERK shRNA down-regulated both pERK and EGR1 protein expression, keeping ERK level unchanged. Importantly, ERK shRNA treatment in the absence of TGFβ1 also inhibited CD44V6 mRNA and CD44V6 protein expression without affecting CD44S mRNA and CD44S protein expression (Fig. 16E), indicating that another signaling cascade other than TGFβ1 involving ERK may also regulate alternative splicing of CD44. Use of EGR1-AS treatment only depleted CD44V6-containing isoforms (both mRNA and protein expression) (Fig. 16F), whereas the mRNA expression of the other two variants (V4V7 and V4) remained unaltered after treatment with EGR1-AS compared with control cells. Again, we demonstrated that EGR1-AS treatment only depleted CD44V6-containing isoforms without affecting CD44S mRNA and protein expression levels (Fig. 16F). These findings provide strong support for a positive-feedback loop for CD44 V6-dependent activation of ERK/EGR1 signaling in response to TGFβ1. Overall, these results indicate that CD44 V6 isoforms are up-regulated by TGFβ1.
Figure 16.

shRNA-mediated knockdown of CD44V6 affects alternative splicing of CD44 and down-regulates EGR-1 expression and ERK phosphorylation. A and B, CD44 variant exon-specific RT-PCR analysis in IPFFbs and TGFβ1-stimulated HNLFbs. The primers are described in Table 2. The PCR primers used to amplify CD44 variable and standard (S) isoforms are shown. S refers to the use of two primers in the CD44 constant exons 5 and 6 (black boxes in the schematic drawing of the relevant parts of the CD44 gene in A; the other lanes refer to PCRs performed with the forward primers in variant exons and the reverse primer in constant exon 6. The conditions for amplifications used were described previously (90). M, a DNA ladder. The Western blot (B) shows a staining of IPFFbs and TGFβ1-stimulated HNLFbs cell lysates with the CD44V6-specific and CD44S antibodies. C, the expression levels of CD44 variants were also examined using 5′ primers complementary to individual variable exons and a primer to the 3′-constitutive exon as indicated by arrowheads. A scheme of human CD44 gene exon structure is shown. Red boxes, constitutive exons; green box, the variable exons that can be alternately or entirely spliced out. Green arrows, forward primer positions; red arrows, the common reverse primer located at the 3′ gene constant region. D–F, real-time PCR analyses of CD44V6 in cultures of fibroblasts isolated from human IPFFbs, as described in our companion paper (91). IPFFbs were transfected with control shRNA or CD44v6 shRNA (B) or ERK shRNA (E) or treated with Egr-1-AS (F) followed by stimulation with TGFβ1 (2.5 ng/ml for 24 h). RNA was extracted from the IPFFb cultures, and real-time PCR analyses done on agarose gels for the PCR products are shown. The numbers of PCR cycles and exposure length of the images for each set of PCR primers vary and cannot be directly compared. Normalization was done by using GAPDH as a housekeeping control gene (bottom panels). The results show real-time PCR for an IPF patient representative of analyses of IPFFbs from seven patients. The primers for both the CD44V6 and CD44s predominantly generate one PCR product, whereas the primers for the CD44V4 variants amplify three splice variants. Structures of the PCR products are depicted to the right of the individual example gels. WB analyses from whole cell lysates from the treated or transfected cells were done with antibodies recognizing pERK, EGR1, or total ERK (D and E) or EGR1 and β-tubulin (F). Data are representative of three (n = 3) independent experiments.
Discussion
During the last few years, great progress has been made in understanding the molecular aspects of intracellular signaling downstream of the TGFβ1 receptors. In particular, when tissues are challenged by injury, wound healing is initiated and is associated with increased expression of TGFβ1 by infiltrating cells, including lymphocytes, monocytes/macrophages, and platelets (58). The extracellular matrix can store TGFβ isoforms (TGFβ1, -2, and -3) that are sequestered as latent precursor TGFβ molecules, often complexed with latent TGFβ-binding proteins. After injury, intracellular signal transduction is initiated with sequential activation of latent TGFβ and downstream Smad signaling pathways (59, 60). The canonical Smad pathway is uniquely associated with TGFβ signaling and is deregulated in fibrosis (61), and profibrotic effects of TGFβ1 signaling in lung fibrosis have been largely attributed to Smad3 signaling (62). Although it is clear that TGFβ downstream effectors of Smad signaling are critical, discrete cellular phenotypes result although the same Smad signaling pathways (Smad2/Smad3) are inactivated (62). A possible explanation for the complexity of this variability in the cellular responses to TGFβ is the existence of cell type-specific signaling pathways, the presence of other stimuli in the local microenvironment, and the ability of TGFβ1 to exert its responses via different effector pathways (63, 64). Consistent with the ability of TGFβ1 to induce CD44V6 expression and function for lung fibroblast activation, we have identified that CD44V6 is a potential target that is activated by TGFβ1 in a subset of fibrogenic human lung fibroblasts (8). In agreement with this, CD44v6 induction is considerably induced at time points (Fig. 2) when a sustained activation of TGFβ1 was found in the bleomycin-induced lung injury model (14, 15).
EGR1 is induced at sites of injury and repair by a variety of stimuli, including cytokines, oxidized lipids, angiotensin II, H2O2, and injury-related diseases, through regulation of downstream genes involved in tissue injury and remodeling (33, 65). Crucial EGR1 targets include PDGF, fibroblast growth factor 2, vascular endothelial growth factor, CD44, fibronectin, and matrix metalloproteinases. Studies have shown that EGR1 can stimulate TGFβ1 production and can also be stimulated by TGFβ1 (33, 35–38). Studies demonstrate that EGR1 is a central mediator of TGFβ1-induced apoptosis, fibrosis, and alveolar remodeling in vivo (9). In melanoma cancer cells, HGF/SF promotes the autocrine stimulation of CD44V6 through the transcriptional activation of EGR1 (11). However, the role of TGFβ1 in EGR1-mediated CD44V6 induction was unknown in that study. This also suggests that in lung fibrosis, TGFβ1 may simultaneously induce EGR1 and stimulate CD44V6 and that CD44V6 is required for the fibrosis to occur.
When activated EGR1 enters into the nucleus, it recognizes and binds to the EGR1 site (GCGCACGG) found in the EGR1 target genes, including CD44. In addition to EGR1 signaling, studies indicate that TGFβ1 may also elicit its activity by activating several MAPKs, including p38, MAPK, ERK1/2, and JNK, in different cell systems (66). Our study shows that CD44v6 expression is markedly induced in the lung after bleomycin injury (Figs. 1 and 2). Our previous studies showed that TGFβ1-induced CD44V6 regulates COL1A1 and α-SMA synthesis in fibrogenic human lung fibroblasts (8), and these findings are supported by our present studies in which CD44v6, Col1a1, and α-SmA are cooperatively stimulated in isolated lung fibroblasts during the fibrogenic phase in the bleomycin-induced mouse model (Fig. 3). TGFβ1 induces CD44v6 mRNA and CD44v6 protein expressions in isolated MNLFbs and in IPFFbs (Fig. 4, A and B), and this induction by TGFβ1 is independent of intact Smad signaling, because Smad7 overexpression failed to block CD44v6 induction in Smad7-overexpressing MNLFbs (Fig. 9). Post-transcriptional regulation and stability of mRNAs for CD44v6 (Fig. 7) and Col1a1 and α-Sma (Fig. 8) in lung fibroblasts depended on culturing conditions. Because we also see an increase in promoter activity (Fig. 5, A and B), the increase in CD44v6 mRNA by TGFβ1 after ACTD treatment primarily may be due to both an increase in mRNA stability and an increase in transcription Figs. 4 (A and B) and 7 (A and B). Thus, post-transcriptional regulation may have a significant role in CD44v6 expression. Interestingly, the magnitude of CD44v6 mRNA induction by TGFβ1 is parallel to its increase in protein (Fig. 4, A and B). Similarly, α-Sma mRNA and Col1a1 mRNA have estimated half-lives of 4 and 12 h, respectively, in quiescent 21dBLMFbs but are increased to 20 and 54 h, respectively, in TGFβ1-stimulated 21dBLMFbs (Fig. 8). Thus, the present findings in Figs. 5 and 6 provide strong evidence that TGFβ1 autocrine signaling can stabilize already transcribed mRNAs of CD44v6, Col1a1, and α-Sma and that increased synthesis of these proteins provides key molecules for fibroblast-to-myofibroblast differentiation. These results indicate that the myofibroblast phenotype with increased synthesis of CD44v6 and α-Sma is induced before up-regulation of Col1a1 induction, and CD44v6 and α-Sma induction do not persist as does Col1a1 in response to TGFβ1. Knowing their half-lives, then, can help in selecting treatment duration for genetic and pharmacologic inhibitors to inhibit profibrogenic effects in lung after belomycin injury (as shown in Fig. 13 of our companion paper (91)).
However, CD44v6 induction by TGFβ1 in lung fibroblasts appears primarily dependent on MAPK/ERK pathways (Figs. 10B and 13 (B and D)), and the induction of CD44v6 expression in fibrogenic lungs is probably mediated by a TGFβ1 effector, ERK1/2, that mediates EGR1 protein expression (Fig. 10, A and B). Moreover, the results in Fig. 10 indicate that 1) EGR1 appears before CD44V6 in response to TGFβ1, 2) EGR1 is required for TGFβ1-mediated CD44V6 up-regulation, 3) ERK1/2 regulates TGFβ1-increased EGR1 and CD44v6, and 4) PI3K has no effect on the nuclear CD44v6- and EGR1-stimulatory function of TGFβ1. EGR1 then translocates into the cell nucleus for stimulation of the CD44V6 promoter, a mechanism that is supported by several observations. First, a putative Egr1 is found in the CD44 promoter (Fig. 13G) that is functionally responsive to Egr1-S overexpression (Figs. 10F and 13G). Second, TGFβ1 stimulates translocation of EGR1 to the nucleus of HNLFbs (Fig. 10D). Finally, preincubation of quiescent lung fibroblasts (MNLFbs and HNLFbs) with antisense oligonucleotide of EGR1 before TGFβ1 stimulation reduces CD44V6 expression, or preincubation with sense oligonucleotides of EGR1 before TGFβ1 stimulation increases CD44V6 expression (Fig. 10F).
Fig. 11 showed that silencing HAS2 attenuated co-localization of CD44v6 with TGFβRI and suppressed EGR1 expression and phosphorylation of ERK. This indicates that increased hyaluronan synthesis through up-regulation of HAS2 is a necessary step leading to receptor (CD44V6) co-localization with TGFβRI and subsequent ERK-EGR1 intracellular signaling; this interaction of increased HA with CD44v6 is probably necessary to initiate transdifferentiation of fibroblast to myofibroblast (Fig. 14).
Most of the genes that encode AP-1, JUN proteins (c-JUN, JUNB, and JUND), and FOS proteins (c-FOS, FOSb, FRA-1, and FRA-2) behave as “immediate-early” genes (i.e. genes whose transcription is rapidly induced, independently of de novo protein synthesis, following stress responses, including cell proliferation, apoptosis, inflammation, wound healing, and cancer) (67). Three different types of MAPKs (ERKs, p38 MAPK, and JNK) contribute to Ap-1 activity in response to a diverse array of extracellular stimuli (68). In addition to the differences in substrate specificities, the three types of MAPKs that affect Ap-1 activities differ in their responses to extracellular stimuli (68, 69). We hypothesized that the TGFβ1/MAPK (Erk)/Egr1/Ap-1 signaling pathways may regulate CD44v6 expression in TGFβ1-stimulated MNLFbs. Interestingly, CD44v6 induction by TGFβ1 not only depends on Egr1 signaling, but also needs the abundant Ap-1 transcription factor for participation and cooperation (Fig. 13F). This mechanism is supported by the following. 1) Egr1 regulates Ap-1 promoter activity through AP-1 components c-Fos and c-Jun (Fig. 13, A, D, and E). 2) The TGFβ1-induced increase in CD44v6 mRNA expression was also effectively stimulated by Egr1-S oligonucleotide-treated MNLFbs with a significant effect in ERK-inhibited or in c-Fos-suppressed cells followed by treatment with Egr1-S (Fig. 13B). 3) Ap-1 DNA-binding activity increased significantly by 4 h and remained elevated for up to 12 h after TGFβ1 treatment (Fig. 13C). 4) Egr1-S treatment further increased AP-1 DNA-binding activity in TGFβ1-treated but not in c-Fos-suppressed MNLFbs (Fig. 13D). 5) Previous studies demonstrated the functionality of the Egr1- and Ap-1-binding sites in the human and mouse CD44 promoter (11, 21, 22, 27). Although the mouse Egr1- and Ap-1-binding site sequence is not identical to the human sequence, the three nucleotides essential for the binding of Egr1 and Ap-1 to the homologous human site are conserved (21, 22). However, the TGFβ1-stimulated ERK-mediated EGR1 function in mediating AP-1 activity in lung fibroblasts has not been reported previously. 6) The Egr1 and c-Fos suppression experiment in TGFβ1-stimulated MNLFbs. The ChIP assay demonstrated in vivo interaction of Ap-1 factors with the Ap-1-binding region of CD44v6 promoter in MNLFbs. In addition, treating MNLFbs with Egr1 shRNA or c-fos shRNA followed by 2.5 ng/ml TGFβ1 treatment for 24 h abolished the ability of Ap-1 factors to interact with the Ap-1-binding region of CD44v6 promoter in MNLFbs (Fig. 13E). 7) Our study showed that a conserved Ap-1 site 110 base pairs from the transcription start site of the CD44 promoter-luciferase reporter gene is important for basal activity (Fig. 6B), and its activity depends on Egr1. Mutation of the AP-1 site significantly reduced induction of promoter activity by the Egr1-S oligonucleotide (Fig. 13, F and G). 8) c-Fos and c-Jun were present in the CD44·AP-1 DNA-binding complex after TGFβ1 stimulation (Figs. 6 and 13E), and this response depends on Egr1 activity (Fig. 13G).
An AP-1 signaling complex was shown to be involved in the phenotypic differentiation of pancreatic and hepatic stellate cells induced by sustained culture (70). We found that during the course of lung fibroblast-to-myofibroblast differentiation induced by TGFβ1, α-SMA protein synthesis (Fig. 8) and myofibroblast contractility function were stimulated (Fig. 14), and Ap-1 DNA-binding activity and Ap-1 promoter activity were increased significantly by TGFβ1 in MNLFbs (Fig. 13, A–E). We also found that TGFβ1-induced Erk/Egr1 regulates Ap-1 activity (Figs. 10 (B and F) and 13 (E and G)), and TGFβ1-induced CD44v6 transcription level depends on Erk, Egr1, and the Ap-1 component c-Fos (Fig. 13D). Further, our previous study showed that CD44v6 regulates α-SMA protein in human fibrogenic lung fibroblasts (8). Thus, we used the U0126 (ERK inhibitor) and specific shRNA for c-Jun as AP-1 inhibitors to block the AP-1-mediated fibroblast-to-myofibroblast differentiation in TGFβ1-stimulated MNLFbs. Our results showed that they effectively inhibited TGFβ1-induced α-Sma protein expression (Fig. 8) and collagen gel contractility (myofibroblast differentiation), as did CD44V6 shRNA (Fig. 14, A and B). The antifibrotic effect of CD44V6 shRNA may be explained by its prevention of myofibroblast differentiation through inhibition of AP-1 activity. This provides direct evidence that TGFβ1-induced ERK/EGR1-mediated AP-1 may regulate α-SMA expression in human/murine lung fibroblasts to mediate myofibroblast differentiation.
Our previous study indicated that fibrotic lung fibroblasts, which already possess a strong TGFβ1/TGFβRI autocrine loop, also express CD44V6 (8). The presence of the V6 regions promotes CD44 oligomerization, which in turn helps binding to ligands, including hyaluronan and growth factors, such as HGF (10, 56, 71). Furthermore, TGFβ1 promotes the feedback up-regulation of CD44V6-containing isoforms through a new mechanistic model for the activation of the ERK/EGR1 pathway (Fig. 17). Upon growth factor/cytokine stimulation, receptor kinases generate an early burst of ERK activation independent of CD44V6 variants. Although this signal is rapidly down-regulated, it initiates a positive-feedback loop between TGFβ1-induced ERK/EGR1 and CD44V6 by stimulating V6 isoform comprising CD44 isoform expression through alternative splicing (Figs. 15 and 16). The up-regulated CD44V6, in conjunction with TGFβ1/TGFβRI, then facilitates ERK/EGR1 signaling for a sufficient time to drive fibroblast-myofibroblast differentiation. The positive-feedback loop described above may be important for the transition from normal to fibrogenic phenotypes. Maintenance of this positive-feedback loop could constitute a mechanism for persistent ERK/EGR1 signaling in fibroblasts to maintain a fibrogenic phenotype.
Figure 17.

Model of a positive-feedback loop between ERK/EGR-1 activation and CD44V6. EGR1-induced AP-1 activity stimulates CD44V6 splicing.
These findings suggest a novel regulatory mechanism dependent on CD44v6 expression, in which EGR1 and AP-1 can interact in a TGFβ1-dependent manner and work cooperatively to enhance CD44V6 transcription in lung myofibroblasts. Together with our previous finding (8), this study provides a unique example of how ubiquitous transcription factors, such as EGR1 and AP-1, have a crucial role in regulating expression of a particular gene(s) in the lung during fibrogenic processes.
Conclusion
Our findings reveal for the first time that HA/CD44V6 controls TGFβ1-dependent differentiation of fibroblasts to myofibroblasts through the interaction of HA/CD44V6 with activated ERK and EGR1, providing mechanistic insights into the role of HA/CD44V6 signaling in IPF pathogenesis. On the basis of several findings of induced activity of TGFβ1 on CD44V6 expression/activity, we investigated 1) the agonistic activity of TGFβ1 on CD44V6-mediated induction of α-SMA expression; 2) the role of the ERK/EGR1 signaling in TGFβ1-induced AP-1 activity for CD44v6 promoter activity expression; 3) whether TGFβ1 agonistic activity on CD44v6 is through a feedback loop between ERK/EGR1 signaling and CD44V6 expression; and 4) whether HAS2-derived HA regulates fibroblast differentiation through CD44V6/ERK/EGR1-regulated intracellular signaling pathways. Our data provide strong evidence for a new mechanistic model (Fig. 17) for a novel positive-feedback loop that operates between EGR1 and CD44v6 mediated by TGFβ1 to sustain increased collagen-1 synthesis that defines the progressive nature of lung fibrotic diseases. Thus, targeting cell surface receptor CD44V6 with CD44V6 shRNA, which specifically down-regulates CD44V6 isoform-containing mRNA and CD44V6 protein expression without affecting CD44S mRNA and CD44S protein expression (Figs. 15 and 16), may provide a novel therapeutic avenue in IPF disease by reversing myofibroblast activation by inhibiting HA-CD44V6-mediated downstream signaling by blocking CD44V6 using CD44v6 shRNA or competing endogenous CD44v6 by CD44v6 peptides (See Figs. 10 and 13 from our companion paper (91)).
Experimental procedures
Materials
Dulbecco's modified Eagle's medium (DMEM) (normal 5.5 mm (low glucose)), glutamine, and pyruvate were from Life Technologies, Inc. Fetal bovine serum (FBS) was from Atlanta Biologicals, and l-glutamine, gentamicin sulfate, and amphotericin B were from Hyclone. Actinomycin D, cycloheximide, Nonidet P-40, EGTA, sodium orthovanadate, glycerol, phenylmethylsulfonyl fluoride, leupeptin, pepstatin A, aprotinin, and HEPES were purchased from Sigma. Recombinant human TGFβ1 was purchased from R&D Systems (Minneapolis, MN). The antibodies against EGR1, CD44v6, α-SMA, pERK, ERK, GAPDH, β-actin, β-tubulin, horseradish peroxidase-linked anti-rabbit and anti-mouse antibodies, and Luminol reagent were purchased from commercial sources (Santa Cruz Biotechnology, Inc., Abcam, Ebioscience, Thermo Fisher, Cell Signaling Technology, and Southern Biotechnology Associates Inc.).
Management of animals, human lung samples, and lung fibroblasts
HNLFbs were from ATCC. Six-week-old mice (C57BL/6 strain) were obtained from the Jackson Laboratories. Bleomycin (0.05 units/20 g of animal) was instilled intratracheally. All animal care and experimentation were done in accordance with the institutional animal care and use committee protocol (AR 3220) approved by the Medical University of South Carolina according to the rules of the National Institutes of Health. Lung tissues at 21 days after vehicle or bleomycin (Sigma) instillations were perfused with z-fix (Anatech Ltd., Battle Creek, MI) and processed for paraffin sections. We used flow-cytometry sorting and analysis and demonstrated that α-SMA-positive fibroblasts were negative for lineage-specific cell surface markers (CD31, CD45, CD146, EpCAM, and Lyve-1), and these fibroblasts were regarded as lineage-negative cells. MNLFbs isolated from saline-treated lungs were shown to be enriched in lineage(−)/Sca-1(+). In contrast, myofibroblasts at day 14 or 21 isolated from bleomycin-injured mouse lungs (14dBLMFbs and 21dBLMFbs) were enriched in lineage(−)/Sca-1(−)/CD49e(+) following the method described (17).
Cell culture
Fibroblasts were cultured as reported previously (72). Briefly, lung tissues were diced (∼0.5 × 0.5-mm pieces) and cultured in DMEM with normal 5.5 mm glucose, glutamine, and pyruvate (Life Technologies) supplemented with 10% fetal bovine serum, 2 mm l-glutamine, gentamicin sulfate (50 μg/ml), and amphotericin B (5 μg/ml) at 37 °C in 10% CO2. The medium was changed every 3 days to remove dead and non-attached cells until fibroblasts reached confluence. Monolayer cultures were maintained in the same medium. Lung fibroblasts were used between the second and fourth passages in all experiments. The purity of isolated lung fibroblasts was determined by crystal violet staining and by immunofluorescence staining using monoclonal antibody 3C4 against human fibroblasts, as described previously (73). All of the treatments and transfection experiments were done with cells that were serum-starved for 24 h.
Cell lysis and immunoblotting
Fibroblasts were cultured until they were confluent. Cells were washed twice at 4 °C with 1× phosphate-buffered saline (PBS), harvested with 0.05% Versene and then washed in cold PBS again. The cells were pelleted by centrifugation at 5000 × g for 2 min at 4 °C. The pellets were treated with the lysis buffer containing 1% Nonidet P-40, 0.5 mm EGTA, 5 mm sodium orthovanadate, 10% (v/v) glycerol, 100 μg/ml phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 1 μg/ml aprotinin, and 50 mm HEPES, pH 7.5. The lysates were clarified by centrifugation at 12,000 × g for 10 min at 4 °C and then stored at −80 °C as described previously (71, 74–80). Cell lysates (normalized for protein concentration) were analyzed by immunoblotting as described previously (56, 71, 76, 77). The proteins on the blots were analyzed with antibodies for EGR1, CD44v6, CD44s, ERK, and pERK (β-tubulin and β-actin were used as internal standards) and detected by luminol reagent (Santa Cruz Biotechnology) following treatment with horseradish peroxidase-linked anti-rabbit or anti-mouse antibodies as secondary antibodies. Each protein was analyzed in samples from at least three independent experiments from each set of fibroblasts.
RNA silencing
For determining shRNA sequences used in this study, 1) coding nucleotide sequences of the genes were obtained from the NCBI, National Institutes of Health, website (www.ncbi.nlm.nih.gov); 2) we designed hairpin shRNAs to target a transcript sequence using the Broad Institute GPP Web Portal (http://portals.broadinstitute.org/gpp/public/)4; and 3) sequences for cloning in pSico/pSicoR vectors were designed following the MIT Jacks Lab website (http://web.mit.edu/jacks-lab/protocols/pSico.html).4 For example, sense sequence for CD44v6 for cloning in pSico/pSicoR vector (56) was 5′-TTAGTAGTACAACGGAAGAAACTTCAAGAGAGTTTCTTCCGTTGTACTACTA, and sense sequence for EGR1 for cloning in pSico/pSicoR vector was 5′-TCCACGCCGAACACTGACATTTTTCAAGAGAAAATGTCAGTGTTCGGCGTGG.
Similarly, scrambled shRNA sequence, c-Fos shRNA, TGFβRI shRNA, and c-Jun shRNA obtained from the website were cloned in pSico/pSicoR vector. The resulting pSicoR-CD44v6 shRNA (CD44v6 shRNA), pSicoR-EGR1 shRNA (EGR1 shRNA), pSicoR-TGFβRI shRNA (TGFβRI shRNA), pSicoR c-Fos shRNA (c-Fos shRNA), and pSicoR c-Jun shRNA (c-Jun shRNA) transfectants constitutively silence respective CD44v6, EGR1, TGFβRI, c-Fos, and c-Jun genes in the cells. PSicoR-scrambled shRNA (control shRNA) transfectants were used as control to the above shRNA transfectants.
Confirming the specificity of shRNA experiments
Synthetic shRNAs that are 21–23 nucleotides in length have been shown to effectively silence specific target genes by promoting mRNA degradation in cultured mammalian cells and mice (56, 81, 82). However, in cultured cells, one potential source of off-target effects by either transfected shRNA duplexes or endogenously expressed shRNAs is the unintentional activation of the interferon response (44, 49, 83). Previous work has shown that off-target gene suppression and up-regulation of type I interferon (IFN) response are induced by dsRNAs of ≥30 bp in length (84). There are also recent reports of shRNA-induced stimulation of Toll-like receptors (TLR3 (dsRNA) and TLR9 (unmethylated CpG)) and downstream IFN-α and IFN-β responses (85). Despite the highly publicized in vivo miRNA-related toxicity of shRNA, it has been shown that shRNA has several advantages over siRNA: fewer off-target effects, multiple-target-silencing capacity without a corresponding increase in dose, durability of effect, and inducible application (86, 87). Because we use siRNAs and shRNAs extensively to study CD44v6-induced fibrogenic function, we follow several steps that can avoid this off-target problem, as shown in Table 1.
Measurement of IFN-α induction by shRNAs; in vitro studies for IFN-α measurement
Mouse alveolar macrophage MH-S was purchased from ATCC (Manassas, VA). Cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated low LPS fetal bovine serum, 1% penicillin-streptomycin, and 1% glutamine at 37 °C under 5% CO2. We plated 5 × 106 cells/well of a 24-well plate in medium containing 1.2% DMSO. After 24 h, LPS (1 μg/well; Sigma), poly(I:C) (from Invivogen) or CD44v6 shRNA, and Egr1 shRNA with indicated doses (as shown in Fig. 12G) were added directly to the appropriate wells for 24 h before harvesting the supernatant. IFN-α levels were measured by ELISA (BD Biosciences) according to the manufacturer's instructions.
Transient transfection of HNLFbs and of idiopathic fibrogenic lung fibroblasts
All transfections were done using Lipofectamine (Invitrogen) in cultures at ∼75% confluence. After transfection, the cultures were grown for another 72–96 h for analyses.
CD44v6 mRNA expression analysis by real-time PCR
Preparation of mRNA from tissue samples was done using a commercially available mRNA purification kit. cDNA was synthesized with mRNA as template for an oligo(dT)-primed reaction catalyzed by reverse transcriptase. The quality of mRNA preparations and of cDNA syntheses was checked by including GAPDH-specific primers (Table 2) as an internal RT-PCR control. The primers used in the experiments were described in our recent paper (56) and in Table 2.
Table 2.
RT-PCR primers
| Genes | Forward sequence (5′–3′) | Reverse sequence (5′–3′) |
|---|---|---|
| C5′ | GATGGAGAAAGCTCTGGAGCTAC | |
| C6′ | TTTGCTCCACCTTCTTGACTCC | |
| h-CD44V2 | GATGAGCACTAGTGCTACAG | |
| h-CD44V3I | ACGTCTTCAAATACCATCTC | |
| h-CD44V3II | TGGGAGCCAAATGAAGAAAA | |
| h-CD44V4 | TCAACCACACCACGGGCTTT | |
| h-CD44V5 | GTAGACAGAAATGGCACCAC | |
| h-CD44V6 | GAGGCAACTCCTAGTAGTAC | |
| h-CD44V7 | CAGCCTCAGCTCATACCAGC | |
| h-CD44V8 | TCCAGTCATAGTACAACGCT | |
| h-CD44V9 | CAGAGCTTCTCTACATCACA | |
| h-CD44V10 | GGTGGAAGAAGAGACCCAAA | |
| h-CD44V6 | AGGAACAGTGGTTTGGCAAC | CGAATGGGAGTCTTCTCTGG |
| h-CD44s | AAGACATCTACCCCAGCA | GGTAGCAGGGATTCTGTC |
| h-Gapdh | GAAGGTGAAGGTCGGAGT | GAAGATGGTGATGGGATTTC |
| m-CD44v6 | CCTTGGCCACCACTCCTAATAG | CAGTTGTCCCTTCTGTCACATG |
| m-CD44s | AGCAGCGGCTCCACCATCGAGA | TCGGATCCATGAGTCACAGTG |
| m-GAPDH | AGTATGACTCCACTCACGGCAA | TCTCGCTCCTGGAAGATGGT |
| h-HAS1 | TCAAGGCGCTCGGAGATTC | CTACCCAGTATCGCAGGCT |
| h-HAS2 | AAGAACAACTTCCACGAAAAGGG | GGCTGGGTCAAGCATAGTGT |
| h-HAS3 | CGCAGCAACTTCCATGAGG | AGTCGCACACCTGGATGTAGT |
| m-Has1 | CACCATCTCAGCCTACCAAGA | ATCGGCGAAGACTTCTCGGA |
| m-Has2 | TGAACAAAACGCTAGCACTCTG | ACTTTAATCCCAGGGTAGGTCAG |
| m-Has3 | CAATCGCCAGGAAGATACCTAC | GGAAATTGCTACGCCACACAA |
| m-α-Sma | ATCGTCCACCGCAAATGC | AAGGAACTGGAGGCGCTG |
| m-collagen-1 | TGCTGCTTGCAGTAACTCG | TCAACACCATCTCTGCCTCG |
| m-Smad7 | TCAAGAGGCTGTGTTGCTGT | CAGGCTCCAGAAGAAGTTGG |
| m-Egr1 | TCCTCTCCATCACATGCCTG | CACTCTGACACATGCTCCAG |
| m-c-Jun | GTT GCG GCC GCG AAA CTT | CAT TGC CCT CGA GCC CTG |
| m-c-Fos | CTGTCCGTCTCTAGTGCCAACTT | ATCTGTCTCCGCTTGGAGCGTAT |
| m-TgfβRI | TGCCATAACCGCACTGTCA | AATGAAAGGGCGATCTAGTGATGGGACCAGATCCAAAAGGACA |
Sense EGR1 experiments
20-mer sense human EGR1 oligonucleotide AGTGTGCCCCTGCACCCCGC as well as control scrambled oligonucleotides were obtained via an NCBI Blast search for human EGR1 mRNA and cloned into pSicoR vector as described previously (56). Cells were transfected with the pSicoR vectors containing sense oligonucleotides for 24 h in serum-free medium. Total RNA and protein were then extracted for further analyses.
Antisense EGR1 experiments
For antisense experiments, phosphorothioate antisense oligonucleotides were synthesized. The antisense oligonucleotide contains a stretch of a DNA mimic (phosphorothioate DNA), which can recruit RNase H to degrade the target RNA. In 20 μl of FuGENE 6, the EGR1 sense oligonucleotide AgtGTTCCCCGCGCCCCGca and the antisense EGR1 oligonucleotide tgCGGGGCGCGGGGAACacT (where the bases in lowercase letters were phosphothiolated) were mixed with 0.5 ml of serum-free medium and then added dropwise to 10 μg of the control scrambled oligonucleotide and the antisense oligonucleotide. The sample was then added to 10 ml of cells (500,000 cells/ml) for 24 h in serum-free medium. Total RNA and protein were then extracted, and mRNA expression and protein expression were analyzed by real-time PCR and immunoblotting as described (8).
Real-time PCR of CD44v6 in MNLFbs and HNLFbs
Total RNA was isolated from MNLFbs and HNLFbs after various treatments and transfections as described in the figure legends for each specified experiment using the RNeasy minikit (Qiagen) according to the standard protocol provided by the manufacturer, with on-column DNA digestion. RNA integrity and concentration were analyzed using Bioanalyzer, and 1 μg of RNA was retrotranscribed into cDNA using the First Strand cDNA synthesis kit from Roche Applied Science (Qiagen). SYBR Green technology (Bio-Rad) was used for all real-time PCR experiments. Amplification was done with the real-time PCR analyzer (Bio-Rad). The PCR mixture (25 μl) contained 12.5 μl of 2× SYBR Green PCR Master Mix (Bio-Rad), 5 μl of diluted RT product (1:20), and 0.5 μm sense and antisense primer sets. The primers used are shown in Table 2.
The real-time PCR assays were done in three individual experiments with triplicate samples using standard conditions. After sequential incubations at 50 °C for 2 min and 95 °C for 10 min, respectively, the amplification protocol consisted of 50 cycles of denaturing at 95 °C for 15 s, annealing, and extension at 60 °C for 60 s. The standard curve was made from a series dilution of template cDNA. Expression levels of α-Sma, collagen-1 (Col1a1), and CD44v6 mRNA were calculated after normalization with the housekeeping gene Gapdh.
Nuclear run-on assay
We used a nonradioactive-based nuclear run-on method in conjunction with real-time PCR to assay for CD44v6 transcription rate in the MNLFbs treated with Egr1-S oligonucleotide (20 μm for 16 h), or TGFβ1 (2.5 ng/ml for 24 h) or their combination in the presence or absence of U0126 (10 μm) treatment, or after dominant-negative c-Jun transfection in these cells. Briefly, 50 × 106 cells were trypsinized, washed twice in Ca2+ and Mg2+-free PBS, and centrifuged (2500 × g for 10 min). The cell pellets were resuspended in cell lysis buffer containing 10 mm Tris-HCl, pH 7.4, 3 mm MgCl2, 10 mm NaCl, 150 mm sucrose, and 0.5% Nonidet P-40. The nuclei were collected by centrifugation and stored frozen in 40% glycerol at −70 °C until used. The run-on reaction was done in a 2× transcription buffer containing 200 mm KCl, 20 mm Tris-HCl, pH 8.0, 5 mm MgCl2, 5 mm dithiothreitol, 4 mm each of ATP, GTP, and CTP, 200 mm sucrose, and 20% glycerol. The reaction was initiated with the addition of 4 mm biotin-16-UTP (Roche Applied Science) and incubated for 1 h at 29 °C. The RNA was isolated with TRIzol reagent (Invitrogen) according to the manufacturer's protocol. Dynabeads M-280 (Dynal, Thermo Fisher), magnetic beads with covalently linked streptavidin, were used to isolate the biotin-labeled run-on RNA according to the manufacturer's protocol. The beads were resuspended in RNase-free water and then stored at −20 °C until used. Reverse transcription and real-time PCR were done with the primers for mouse CD44v6 and GAPDH as described above.
Ap-1 transcription factor-binding activity
Nuclear extracts after various treatments and transfections as described in the figure legends for each specified experiment were prepared with the NucBuster protein extraction kit (Novagen) according to the manufacturer's protocol. AP-1 DNA-binding activity was measured using the Trans Binding AP-1 assay kit (Panomics). Briefly, nuclear extracts were incubated with biotinylated Ap-1-consensus-binding sequence oligonucleotides, and the complexes bound to the oligonucleotides were detected using a primary AP-1 antibody and a secondary antibody conjugated to horseradish peroxidase. AP-1 activity was assayed by measuring the absorbance at 450 nm.
ChIP assay
The ChIP assay was done using the ChIP assay kit (Upstate Biotechnology) following the manufacturer's directions and as described (11). MNLFbs were transfected with or without Egr1 shRNA, c-Jun shRNA, or control shRNA for 48 h and then treated with or without 2.5 ng/ml TGFβ1 for 24 h. Nuclear Ap-1-associated chromatins were immunoprecipitated with an anti-AP-1/c-Jun mouse monoclonal antibody. Sequences of the primers for amplifying the CD44 promoter were chosen from the 542 nucleotides upstream of the transcription initiation site shown in Fig. 6. Control IgGs were used as negative controls for immunoprecipitation. Chromatin inputs were used as loading controls for PCR.
Transient transfection and luciferase activity assays
MNLFbs were transfected with CD44 promoter luciferase reporter or pCMV-β-galactosidase constructs using the in vitro DNA transfection kit as recommended by the manufacturer. Cell suspensions were transfected with 1 μg of CD44 promoter constructs and 0.5 μg of pCMV-β-galactosidase constructs mixed in serum-free DMEM. After 24 h, cells were quiesced in 0.1% FBS/DMEM for 72 h. Cells were treated with various reagents as described in the figure legends for the specified experiment. Cell lysates were mixed with either luciferin substrate or β-Glo galactosidase substrate (Promega) in equal amounts. After a 10-min incubation, luminescence was measured using a luminometer as described (71).
Three-dimensional collagen gel contraction assay
HNLFbs were treated and transfected with various reagents as described in the figure legends for the specified experiment. Collagen gels were reconstituted by mixing one part 3 mg/ml neutralized rat tail collagen type 1 and two parts cell suspension in serum-free medium. Cell suspensions were seeded at a density of 200,000 cells/ml into 4-well tissue culture plates, and the gels were allowed to polymerize at 37 °C for 1 h before adding 1 ml of medium. A volume of 600 μl/gel was fabricated in a 4-well plastic culture dish, which ensured that the gel would remain attached throughout the culture period. The stressed HNLFbs were cultured in 10% FBS for 20 h after polymerization. After 20 h of recovery in 10% FBS, the stressed HNLFbs were incubated in 0.5% FBS with or without TGFβ1 (5 ng/ml) for another 48 h in serum-deprived medium. The edges of the gels were gently detached from the walls of the well using a sterile spatula. Images were photographed, and gel areas were measured using ImageJ software (National Institutes of Health).
Sircol assay for collagen
Acid-soluble collagen in whole-lung homogenates was analyzed by the Sircol assay as described previously (88).
Hydroxyproline content of whole lung
Mouse whole lungs were homogenized in PBS and then acidified (by adding an equal volume of 12 n HCl), hydrolyzed (by heating at 120 °C for 24 h), and processed for hydroxylproline measurements as described previously (89).
Lung histology and immunohistochemical staining
Paraffin-embedded tissue sections were processed for Masson's trichrome staining for collagen as described previously (8).
Statistical analysis
Data from various groups are expressed as means ± S.E. (n = 3–5). Statistical comparisons were done using Student's t test for samples.
Author contributions
The experiments of this work were designed and carried out by S. M. and S. G. The paper was written by S. M. and S. G. V. C. H. reviewed and edited the draft and final versions of the text, figures, and figure legends and gave advice regarding draft corrections wherever necessary. R. R. M. and V. J. T. commented on the final version of the paper. W. D. performed RT-PCR experiments and data acquisition, R. G. L. was responsible for tissue culture experiments and data acquisition, J. E. B., G. B., and C. C. B. supplied Tissue culture reagents, and M. A. P. provided CD44-Luciferase (both WT and Ap-1 mutant constructs).
Acknowledgments
We thank Dr. B. Ogretmen (Department of Biochemistry and Molecular Biology, Medical University of South Carolina) for advice regarding the rescue experiments of siRNA and shRNA.
This work was supported by National Institutes of Health (NIH) Grant 1R03CA167722-01A1 (to S. M. and S. G.); NIH Grants 1RO1 (HL33756), P30 GM103342-03, 5P20GM103499-16, AHA16GRNT31330032, and 1P20 GM103444 (to S. M., S. G., and R. R. M.); NIH Grants 1 R01HL113325 and 1 P01HL107147 (to V. C. H.); and a pilot project award (to S. M.) from the COBRE in Lipidomics and Pathobiology (NIH Grant 5P30GM103339-03) at the Medical University of South Carolina. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- IPF
- idiopathic pulmonary fibrosis
- HA
- hyaluronan
- MNLFb
- primary mouse normal lung fibroblast
- BLMFb
- primary murine lung myofibroblast isolated from lung tissues from bleomycin-treated mice
- IPFFb
- serum-depleted lung fibroblast from IPF patients
- HNLFb
- primary human normal lung fibroblast
- ACTD
- actinomycin
- control shRNA
- pSicoR-scrambled shRNA
- CD44v6 shRNA
- pSicoR-CD44v6 shRNA
- WB
- Western blot
- EpCAM
- epithelial cell adhesion molecule
- HGF
- hepatocyte growth factor
- SF
- scatter factor
- pERK
- phosphorylated ERK
- CDS
- coding sequences
- 14d and 21d
- 14- and 21-day, respectively.
References
- 1. Wynn T. A. (2004) Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 4, 583–594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duffield J. S., Lupher M., Thannickal V. J., and Wynn T. A. (2013) Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 8, 241–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tomasek J. J., Gabbiani G., Hinz B., Chaponnier C., and Brown R. A. (2002) Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363 [DOI] [PubMed] [Google Scholar]
- 4. Hinz B., Phan S. H., Thannickal V. J., Galli A., Bochaton-Piallat M. L., and Gabbiani G. (2007) The myofibroblast: one function, multiple origins. Am. J. Pathol. 170, 1807–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Desmoulière A., Geinoz A., Gabbiani F., and Gabbiani G. (1993) Transforming growth factor-β1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Branton M. H., and Kopp J. B. (1999) TGF-β and fibrosis. Microbes Infect. 1, 1349–1365 [DOI] [PubMed] [Google Scholar]
- 7. Li Y., Jiang D., Liang J., Meltzer E. B., Gray A., Miura R., Wogensen L., Yamaguchi Y., and Noble P. W. (2011) Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 208, 1459–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ghatak S., Bogatkevich G. S., Atnelishvili I., Akter T., Feghali-Bostwick C., Hoffman S., Fresco V. M., Fuchs J. C., Visconti R. P., Markwald R. R., Padhye S. B., Silver R. M., Hascall V. C., and Misra S. (2014) Overexpression of c-Met and CD44v6 receptors contributes to autocrine TGF-β1 signaling in interstitial lung disease. J. Biol. Chem. 289, 7856–7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee C. G., Cho S. J., Kang M. J., Chapoval S. P., Lee P. J., Noble P. W., Yehualaeshet T., Lu B., Flavell R. A., Milbrandt J., Homer R. J., and Elias J. A. (2004) Early growth response gene 1-mediated apoptosis is essential for transforming growth factor β1-induced pulmonary fibrosis. J. Exp. Med. 200, 377–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Orian-Rousseau V., Chen L., Sleeman J. P., Herrlich P., and Ponta H. (2002) CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 16, 3074–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Recio J. A., and Merlino G. (2003) Hepatocyte growth factor/scatter factor induces feedback up-regulation of CD44v6 in melanoma cells through Egr-1. Cancer Res. 63, 1576–1582 [PubMed] [Google Scholar]
- 12. Foster L. C., Arkonac B. M., Sibinga N. E., Shi C., Perrella M. A., and Haber E. (1998) Regulation of CD44 gene expression by the proinflammatory cytokine interleukin-1β in vascular smooth muscle cells. J. Biol. Chem. 273, 20341–20346 [DOI] [PubMed] [Google Scholar]
- 13. Lo L. W., Cheng J. J., Chiu J. J., Wung B. S., Liu Y. C., and Wang D. L. (2001) Endothelial exposure to hypoxia induces Egr-1 expression involving PKCα-mediated Ras/Raf-1/ERK1/2 pathway. J. Cell Physiol. 188, 304–312 [DOI] [PubMed] [Google Scholar]
- 14. Zhang K., Rekhter M. D., Gordon D., and Phan S. H. (1994) Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis: a combined immunohistochemical and in situ hybridization study. Am. J. Pathol. 145, 114–125 [PMC free article] [PubMed] [Google Scholar]
- 15. Santana A., Saxena B., Noble N. A., Gold L. I., and Marshall B. C. (1995) Increased expression of transforming growth factor β isoforms (β1, β2, β3) in bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 13, 34–44 [DOI] [PubMed] [Google Scholar]
- 16. McQualter J. L., Brouard N., Williams B., Baird B. N., Sims-Lucas S., Yuen K., Nilsson S. K., Simmons P. J., and Bertoncello I. (2009) Endogenous fibroblastic progenitor cells in the adult mouse lung are highly enriched in the sca-1 positive cell fraction. Stem Cells 27, 623–633 [DOI] [PubMed] [Google Scholar]
- 17. Akamatsu T., Arai Y., Kosugi I., Kawasaki H., Meguro S., Sakao M., Shibata K., Suda T., Chida K., and Iwashita T. (2013) Direct isolation of myofibroblasts and fibroblasts from bleomycin-injured lungs reveals their functional similarities and differences. Fibrogenesis Tissue Repair 6, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Monaghan M., Mulligan K. A., Gillespie H., Trimble A., Winter P., Johnston P. G., and McCormick D. (2000) Epidermal growth factor up-regulates CD44-dependent astrocytoma invasion in vitro. J. Pathol. 192, 519–525 [DOI] [PubMed] [Google Scholar]
- 19. Richards J. D., Davé S. H., Chou C. H., Mamchak A. A., and DeFranco A. L. (2001) Inhibition of the MEK/ERK signaling pathway blocks a subset of B cell responses to antigen. J. Immunol. 166, 3855–3864 [DOI] [PubMed] [Google Scholar]
- 20. Weg-Remers S., Ponta H., Herrlich P., and König H. (2001) Regulation of alternative pre-mRNA splicing by the ERK MAP-kinase pathway. EMBO J. 20, 4194–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maltzman J. S., Carmen J. A., and Monroe J. G. (1996) Transcriptional regulation of the Icam-1 gene in antigen receptor- and phorbol ester-stimulated B lymphocytes: role for transcription factor EGR1. J. Exp. Med. 183, 1747–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fitzgerald K. A., and O'Neill L. A. (1999) Characterization of CD44 induction by IL-1: a critical role for Egr-1. J. Immunol. 162, 4920–4927 [PubMed] [Google Scholar]
- 23. Hebbard L., Steffen A., Zawadzki V., Fieber C., Howells N., Moll J., Ponta H., Hofmann M., and Sleeman J. (2000) CD44 expression and regulation during mammary gland development and function. J. Cell Sci. 113, 2619–2630 [DOI] [PubMed] [Google Scholar]
- 24. Zhao Y., and Shah D. U. (2000) Expression of transforming growth factor-β type I and type II receptors is altered in rat lungs undergoing bleomycin-induced pulmonary fibrosis. Exp. Mol. Pathol. 69, 67–78 [DOI] [PubMed] [Google Scholar]
- 25. Spicer A. P., and McDonald J. A. (1998) Characterization and molecular evolution of a vertebrate hyaluronan synthase gene family. J. Biol. Chem. 273, 1923–1932 [DOI] [PubMed] [Google Scholar]
- 26. Shih S. C., and Claffey K. P. (2001) Role of AP-1 and HIF-1 transcription factors in TGF-β activation of VEGF expression. Growth Factors 19, 19–34 [DOI] [PubMed] [Google Scholar]
- 27. Foster L. C., Wiesel P., Huggins G. S., Pañares R., Chin M. T., Pellacani A., and Perrella M. A. (2000) Role of activating protein-1 and high mobility group-I(Y) protein in the induction of CD44 gene expression by interleukin-1β in vascular smooth muscle cells. FASEB J. 14, 368–378 [DOI] [PubMed] [Google Scholar]
- 28. Horowitz J. C., Lee D. Y., Waghray M., Keshamouni V. G., Thomas P. E., Zhang H., Cui Z., and Thannickal V. J. (2004) Activation of the pro-survival phosphatidylinositol 3-kinase/AKT pathway by transforming growth factor-β1 in mesenchymal cells is mediated by p38 MAPK-dependent induction of an autocrine growth factor. J. Biol. Chem. 279, 1359–1367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hecker L., Logsdon N. J., Kurundkar D., Kurundkar A., Bernard K., Hock T., Meldrum E., Sanders Y. Y., and Thannickal V. J. (2014) Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 6, 231ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T. R., Horowitz J. C., Pennathur S., Martinez F. J., and Thannickal V. J. (2009) NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 15, 1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nakao A., Afrakhte M., Morén A., Nakayama T., Christian J. L., Heuchel R., Itoh S., Kawabata M., Heldin N. E., Heldin C. H., and ten Dijke P. (1997) Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 389, 631–635 [DOI] [PubMed] [Google Scholar]
- 32. Nakao A., Imamura T., Souchelnytskyi S., Kawabata M., Ishisaki A., Oeda E., Tamaki K., Hanai J., Heldin C. H., Miyazono K., and ten Dijke P. (1997) TGF-β receptor-mediated signalling through Smad2, Smad3 and Smad4. EMBO J. 16, 5353–5362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Khachigian L. M., Lindner V., Williams A. J., and Collins T. (1996) Egr-1-induced endothelial gene expression: a common theme in vascular injury. Science 271, 1427–1431 [DOI] [PubMed] [Google Scholar]
- 34. Sukhatme V. P., Kartha S., Toback F. G., Taub R., Hoover R. G., and Tsai-Morris C. H. (1987) A novel early growth response gene rapidly induced by fibroblast, epithelial cell and lymphocyte mitogens. Oncogene Res. 1, 343–355 [PubMed] [Google Scholar]
- 35. Gashler A., and Sukhatme V. P. (1995) Early growth response protein 1 (Egr-1): prototype of a zinc-finger family of transcription factors. Prog. Nucleic Acids Res. Mol. Biol. 50, 191–224 [DOI] [PubMed] [Google Scholar]
- 36. Silverman E. S., and Collins T. (1999) Pathways of Egr-1-mediated gene transcription in vascular biology. Am. J. Pathol. 154, 665–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu C., Yao J., Mercola D., and Adamson E. (2000) The transcription factor EGR-1 directly transactivates the fibronectin gene and enhances attachment of human glioblastoma cell line U251. J. Biol. Chem. 275, 20315–20323 [DOI] [PubMed] [Google Scholar]
- 38. Kane S., Prentice M. A., Mariano J. M., Cuttitta F., and Jakowlew S. B. (2002) Differential induction of early response genes by adrenomedullin and transforming growth factor-β1 in human lung cancer cells. Anticancer Res. 22, 1433–1444 [PubMed] [Google Scholar]
- 39. Meran S., Thomas D., Stephens P., Martin J., Bowen T., Phillips A., and Steadman R. (2007) Involvement of hyaluronan in regulation of fibroblast phenotype. J. Biol. Chem. 282, 25687–25697 [DOI] [PubMed] [Google Scholar]
- 40. Spicer A. P., and Nguyen T. K. (1999) Mammalian hyaluronan synthases: investigation of functional relationships in vivo. Biochem. Soc. Trans. 27, 109–115 [DOI] [PubMed] [Google Scholar]
- 41. Simpson R. M., Wells A., Thomas D., Stephens P., Steadman R., and Phillips A. (2010) Aging fibroblasts resist phenotypic maturation because of impaired hyaluronan-dependent CD44/epidermal growth factor receptor signaling. Am. J. Pathol. 176, 1215–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Meran S., Luo D. D., Simpson R., Martin J., Wells A., Steadman R., and Phillips A. O. (2011) Hyaluronan facilitates transforming growth factor-β1-dependent proliferation via CD44 and epidermal growth factor receptor interaction. J. Biol. Chem. 286, 17618–17630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Meran S., Thomas D. W., Stephens P., Enoch S., Martin J., Steadman R., and Phillips A. O. (2008) Hyaluronan facilitates transforming growth factor-β1-mediated fibroblast proliferation. J. Biol. Chem. 283, 6530–6545 [DOI] [PubMed] [Google Scholar]
- 44. Jackson A. L., Bartz S. R., Schelter J., Kobayashi S. V., Burchard J., Mao M., Li B., Cavet G., and Linsley P. S. (2003) Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 21, 635–637 [DOI] [PubMed] [Google Scholar]
- 45. Echeverri C. J., Beachy P. A., Baum B., Boutros M., Buchholz F., Chanda S. K., Downward J., Ellenberg J., Fraser A. G., Hacohen N., Hahn W. C., Jackson A. L., Kiger A., Linsley P. S., Lum L., et al. (2006) Minimizing the risk of reporting false positives in large-scale RNAi screens. Nat. Methods 3, 777–779 [DOI] [PubMed] [Google Scholar]
- 46. Marques J. T., Devosse T., Wang D., Zamanian-Daryoush M., Serbinowski P., Hartmann R., Fujita T., Behlke M. A., and Williams B. R. (2006) A structural basis for discriminating between self and nonself double-stranded RNAs in mammalian cells. Nat. Biotechnol. 24, 559–565 [DOI] [PubMed] [Google Scholar]
- 47. Ge Q., Dallas A., Ilves H., Shorenstein J., Behlke M. A., and Johnston B. H. (2010) Effects of chemical modification on the potency, serum stability, and immunostimulatory properties of short shRNAs. RNA 16, 118–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Goodchild A., Nopper N., King A., Doan T., Tanudji M., Arndt G. M., Poidinger M., Rivory L. P., and Passioura T. (2009) Sequence determinants of innate immune activation by short interfering RNAs. BMC Immunol. 10, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sledz C. A., Holko M., de Veer M. J., Silverman R. H., and Williams B. R. (2003) Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 5, 834–839 [DOI] [PubMed] [Google Scholar]
- 50. Kim D. H., Longo M., Han Y., Lundberg P., Cantin E., and Rossi J. J. (2004) Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat. Biotechnol. 22, 321–325 [DOI] [PubMed] [Google Scholar]
- 51. Heidel J. D., Hu S., Liu X. F., Triche T. J., and Davis M. E. (2004) Lack of interferon response in animals to naked siRNAs. Nat. Biotechnol. 22, 1579–1582 [DOI] [PubMed] [Google Scholar]
- 52. Parra E., Ferreira J., and Ortega A. (2011) Overexpression of EGR-1 modulates the activity of NF-κB and AP-1 in prostate carcinoma PC-3 and LNCaP cell lines. Int. J. Oncol. 39, 345–352 [DOI] [PubMed] [Google Scholar]
- 53. Hofmann M., Rudy W., Günthert U., Zimmer S. G., Zawadzki V., Zöller M., Lichtner R. B., Herrlich P., and Ponta H. (1993) A link between ras and metastatic behavior of tumor cells: ras induces CD44 promoter activity and leads to low-level expression of metastasis-specific variants of CD44 in CREF cells. Cancer Res. 53, 1516–1521 [PubMed] [Google Scholar]
- 54. Midgley A. C., Rogers M., Hallett M. B., Clayton A., Bowen T., Phillips A. O., and Steadman R. (2013) Transforming growth factor-β1 (TGF-β1)-stimulated fibroblast to myofibroblast differentiation is mediated by hyaluronan (HA)-facilitated epidermal growth factor receptor (EGFR) and CD44 co-localization in lipid rafts. J. Biol. Chem. 288, 14824–14838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ghatak S., Hascall V. C., Berger F. G., Penas M. M., Davis C., Jabari E., He X., Norris J. S., Dang Y., Markwald R. R., and Misra S. (2008) Tissue-specific shRNA delivery: a novel approach for gene therapy in cancer. Connect. Tissue Res. 49, 265–269 [DOI] [PubMed] [Google Scholar]
- 56. Misra S., Hascall V. C., De Giovanni C., Markwald R. R., and Ghatak S. (2009) Delivery of CD44 shRNA/nanoparticles within cancer cells: perturbation of hyaluronan/CD44v6 interactions and reduction in adenoma growth in Apc Min/+ MICE. J. Biol. Chem. 284, 12432–12446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. van Weering D. H., Baas P. D., and Bos J. L. (1993) A PCR-based method for the analysis of human CD44 splice products. PCR Methods Appl. 3, 100–106 [DOI] [PubMed] [Google Scholar]
- 58. Ghatak S., Maytin E. V., Mack J. A., Hascall V. C., Atanelishvili I., Moreno Rodriguez R., Markwald R. R., and Misra S. (2015) Roles of proteoglycans and glycosaminoglycans in wound healing and fibrosis. Int. J. Cell Biol. 2015, 834893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roberts A. B. (1998) Molecular and cell biology of TGF-β. Miner. Electrolyte Metab. 24, 111–119 [DOI] [PubMed] [Google Scholar]
- 60. Varga J., and Whitfield M. L. (2009) Transforming growth factor-β in systemic sclerosis (scleroderma). Front. Biosci. 1, 226–235 [DOI] [PubMed] [Google Scholar]
- 61. Varga J. (2002) Scleroderma and Smads: dysfunctional Smad family dynamics culminating in fibrosis. Arthritis Rheum. 46, 1703–1713 [DOI] [PubMed] [Google Scholar]
- 62. Bonniaud P., Kolb M., Galt T., Robertson J., Robbins C., Stampfli M., Lavery C., Margetts P. J., Roberts A. B., and Gauldie J. (2004) Smad3 null mice develop airspace enlargement and are resistant to TGF-β-mediated pulmonary fibrosis. J. Immunol. 173, 2099–2108 [DOI] [PubMed] [Google Scholar]
- 63. Roberts A. B., Piek E., Böttinger E. P., Ashcroft G., Mitchell J. B., and Flanders K. C. (2001) Is Smad3 a major player in signal transduction pathways leading to fibrogenesis? Chest 120, 43S–47S [DOI] [PubMed] [Google Scholar]
- 64. Flanders K. C., Sullivan C. D., Fujii M., Sowers A., Anzano M. A., Arabshahi A., Major C., Deng C., Russo A., Mitchell J. B., and Roberts A. B. (2002) Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am. J. Pathol. 160, 1057–1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. McCaffrey T. A., Fu C., Du B., Eksinar S., Kent K. C., Bush H. Jr., Kreiger K., Rosengart T., Cybulsky M. I., Silverman E. S., and Collins T. (2000) High-level expression of Egr-1 and Egr-1-inducible genes in mouse and human atherosclerosis. J. Clin. Invest. 105, 653–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Javelaud D., and Mauviel A. (2005) Crosstalk mechanisms between the mitogen-activated protein kinase pathways and Smad signaling downstream of TGF-β: implications for carcinogenesis. Oncogene 24, 5742–5750 [DOI] [PubMed] [Google Scholar]
- 67. Eferl R., and Wagner E. F. (2003) AP-1: a double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868 [DOI] [PubMed] [Google Scholar]
- 68. Karin M. (1995) The regulation of AP-1 activity by mitogen-activated protein kinases. J. Biol. Chem. 270, 16483–16486 [DOI] [PubMed] [Google Scholar]
- 69. Jung Y. D., Fan F., McConkey D. J., Jean M. E., Liu W., Reinmuth N., Stoeltzing O., Ahmad S. A., Parikh A. A., Mukaida N., and Ellis L. M. (2002) Role of P38 MAPK, AP-1, and NF-κB in interleukin-1β-induced IL-8 expression in human vascular smooth muscle cells. Cytokine 18, 206–213 [DOI] [PubMed] [Google Scholar]
- 70. Fitzner B., Sparmann G., Emmrich J., Liebe S., and Jaster R. (2004) Involvement of AP-1 proteins in pancreatic stellate cell activation in vitro. Int. J. Colorectal. Dis. 19, 414–420 [DOI] [PubMed] [Google Scholar]
- 71. Ghatak S., Hascall V. C., Markwald R. R., and Misra S. (2010) Stromal hyaluronan interaction with epithelial CD44 variants promotes prostate cancer invasiveness by augmenting expression and function of hepatocyte growth factor and androgen receptor. J. Biol. Chem. 285, 19821–19832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hsu E., Shi H., Jordan R. M., Lyons-Weiler J., Pilewski J. M., and Feghali-Bostwick C. A. (2011) Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 63, 783–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bogatkevich G. S., Ludwicka-Bradley A., Highland K. B., Hant F., Nietert P. J., Singleton C. B., and Silver R. M. (2007) Down-regulation of collagen and connective tissue growth factor expression with hepatocyte growth factor in lung fibroblasts from white scleroderma patients via two signaling pathways. Arthritis Rheum. 56, 3468–3477 [DOI] [PubMed] [Google Scholar]
- 74. Misra S., Ujházy P., Varticovski L., and Arias I. M. (1999) Phosphoinositide 3-kinase lipid products regulate ATP-dependent transport by sister of P-glycoprotein and multidrug resistance associated protein 2 in bile canalicular membrane vesicles. Proc. Natl. Acad. Sci. U.S.A. 96, 5814–5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Misra S., Ujházy P., Gatmaitan Z., Varticovski L., and Arias I. M. (1998) The role of phosphoinositide 3-kinase in taurocholate-induced trafficking of ATP-dependent canalicular transporters in rat liver. J. Biol. Chem. 273, 26638–26644 [DOI] [PubMed] [Google Scholar]
- 76. Misra S., Toole B. P., and Ghatak S. (2006) Hyaluronan constitutively regulates activation of multiple receptor tyrosine kinases in epithelial and carcinoma cells. J. Biol. Chem. 281, 34936–34941 [DOI] [PubMed] [Google Scholar]
- 77. Misra S., Obeid L. M., Hannun Y. A., Minamisawa S., Berger F. G., Markwald R. R., Toole B. P., and Ghatak S. (2008) Hyaluronan constitutively regulates activation of COX-2-mediated cell survival activity in intestinal epithelial and colon carcinoma cells. J. Biol. Chem. 283, 14335–14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Misra S., Hascall V. C., Berger F. G., Markwald R. R., and Ghatak S. (2008) Hyaluronan, CD44, and cyclooxygenase-2 in colon cancer. Connect. Tissue Res. 49, 219–224 [DOI] [PubMed] [Google Scholar]
- 79. Misra S., Ghatak S., Zoltan-Jones A., and Toole B. P. (2003) Regulation of multidrug resistance in cancer cells by hyaluronan. J. Biol. Chem. 278, 25285–25288 [DOI] [PubMed] [Google Scholar]
- 80. Misra S., Ghatak S., and Toole B. P. (2005) Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J. Biol. Chem. 280, 20310–20315 [DOI] [PubMed] [Google Scholar]
- 81. Elbashir S. M., Harborth J., Lendeckel W., Yalcin A., Weber K., and Tuschl T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 [DOI] [PubMed] [Google Scholar]
- 82. Song E., Lee S. K., Wang J., Ince N., Ouyang N., Min J., Chen J., Shankar P., and Lieberman J. (2003) RNA interference targeting Fas protects mice from fulminant hepatitis. Nat. Med. 9, 347–351 [DOI] [PubMed] [Google Scholar]
- 83. Bridge A. J., Pebernard S., Ducraux A., Nicoulaz A. L., and Iggo R. (2003) Induction of an interferon response by RNAi vectors in mammalian cells. Nat. Genet. 34, 263–264 [DOI] [PubMed] [Google Scholar]
- 84. Manche L., Green S. R., Schmedt C., and Mathews M. B. (1992) Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell Biol. 12, 5238–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Yew N. S., and Scheule R. K. (2005) Toxicity of cationic lipid-DNA complexes. Adv. Genet. 53, 189–214 [PubMed] [Google Scholar]
- 86. Rao D. D., Senzer N., Cleary M. A., and Nemunaitis J. (2009) Comparative assessment of siRNA and shRNA off target effects: what is slowing clinical development. Cancer Gene Ther. 16, 807–809 [DOI] [PubMed] [Google Scholar]
- 87. Rao D. D., Vorhies J. S., Senzer N., and Nemunaitis J. (2009) siRNA vs. shRNA: similarities and differences. Adv. Drug Deliv. Rev. 61, 746–759 [DOI] [PubMed] [Google Scholar]
- 88. Vittal R., Horowitz J. C., Moore B. B., Zhang H., Martinez F. J., Toews G. B., Standiford T. J., and Thannickal V. J. (2005) Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am. J. Pathol. 166, 367–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hattori N., Degen J. L., Sisson T. H., Liu H., Moore B. B., Pandrangi R. G., Simon R. H., and Drew A. F. (2000) Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J. Clin. Invest. 106, 1341–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Konig H., Moll J., Ponta H., and Herrlich P. (1996) Trans-acting factors regulate the expression of CD44 splice variants. EMBO J. 15, 4030–4039 [PMC free article] [PubMed] [Google Scholar]
- 91. Ghatak S., Hascall V. C., Markwald R. R., Feghali-Bostwick C., Artlett C. M., Gooz M., Bogatkevich G. S., Atanelishvili I., Silver R. M., Wood J., Thannickal V. J., and Misra S. (2017) Transforming growth factor β1 (TGFβ1)-induced CD44V6-NOX4 signaling in pathogenesis of idiopathic pulmonary fibrosis. J. Biol. Chem. 292, 10490–10519 [DOI] [PMC free article] [PubMed] [Google Scholar]