

Abstract
Idiopathic pulmonary fibrosis (IPF) is a progressive clinical syndrome of fatal outcome. The lack of information about the signaling pathways that sustain fibrosis and the myofibroblast phenotype has prevented the development of targeted therapies for IPF. Our previous study showed that isolated fibrogenic lung fibroblasts have high endogenous levels of the hyaluronan receptor, CD44V6 (CD44 variant containing exon 6), which enhances the TGFβ1 autocrine signaling and induces fibroblasts to transdifferentiate into myofibroblasts. NADPH oxidase 4 (NOX4) enzyme, which catalyzes the reduction of O2 to hydrogen peroxide (H2O2), has been implicated in the cardiac and lung myofibroblast phenotype. However, whether CD44V6 regulates NOX4 to mediate tissue repair and fibrogenesis is not well-defined. The present study assessed the mechanism of how TGF-β-1-induced CD44V6 regulates the NOX4/reactive oxygen species (ROS) signaling that mediates the myofibroblast differentiation. Specifically, we found that NOX4/ROS regulates hyaluronan synthesis and the transcription of CD44V6 via an effect upon AP-1 activity. Further, CD44V6 is part of a positive-feedback loop with TGFβ1/TGFβRI signaling that acts to increase NOX4/ROS production, which is required for myofibroblast differentiation, myofibroblast differentiation, myofibroblast extracellular matrix production, myofibroblast invasion, and myofibroblast contractility. Both NOX4 and CD44v6 are up-regulated in the lungs of mice subjected to experimental lung injury and in cases of human IPF. Genetic (CD44v6 shRNA) or a small molecule inhibitor (CD44v6 peptide) targeting of CD44v6 abrogates fibrogenesis in murine models of lung injury. These studies support a function for CD44V6 in lung fibrosis and offer proof of concept for therapeutic targeting of CD44V6 in lung fibrosis disorders.
Keywords: CD44, differentiation, hyaluronan, NOx, reactive oxygen species (ROS)
Introduction
Idiopathic pulmonary fibrosis (IPF),3 the hallmark of impaired repair of lung injury, is characterized by persistent activation and differentiation of resident fibroblasts to myofibroblasts (2, 3). However, the underlying etiology in IPF and the progressive nature of the fibrotic process in IPF are generally unknown (3).
Accumulated evidence indicates that the increased oxidative stress associated with an oxidant-antioxidant imbalance and elevated reactive oxygen species (ROS) production have a key role in the lungs of patients with IPF (4–6). NADPH oxidases (NOXs) are a family of flavoenzymes that are unique in that they are solely responsible for primary ROS (H2O2) production (7, 8). These multisubunit protein complexes consist of five subunits: a membrane-associated cytochrome b558 containing gp91phox and p22phox and a cytosolic complex of p40phox, p47phox, and p67phox. The family of human NOX proteins consists of five members (NOX1- to -5) and two related dual oxidases (DUOX1 and -2). NOX1, NOX2, and NOX3 show a high degree of homology and require p40phox, p47phox, and p67phox subunits and activation by Rac protein. In addition, the p22phox subunit is important for the stability and functioning of NOX1 to NOX4 (9–11). NOX5 (absent in mice) (12) is not regulated by p22phox (11, 13). Recent studies have reported that specific NOX (NOX2 and NOX4) and DUOX isoforms (DUOX1 and DUOX2), and subunits p22phox and p47phox are involved in the pathogenesis of obstructive lung disorders and that they correlate with an enhanced H2O2 production (14). NOX enzymes are critical determinants of the redox status of the lung tissue and have a critical role in the myofibroblast activation in IPF disease (6, 15, 16).
Fibrosis is caused by unrestrained wound-healing responses during tissue repair in pathological conditions, including IPF (17). During wound repair, both fibroblasts and myofibroblasts invade the wound and synthesize a variety of extracellular matrix (ECM) proteins and the glycosaminoglycan hyaluronan (HA) in the fibrotic tissue (18, 19). At sites of tissue damage and wound healing, fibroblasts with a contractile phenotype are essential for the synthesis of the collagen-rich scar and for providing the force for wound contraction (20). Fibrotic scars are characterized by accumulation of contractile matrix proteins produced by differentiation of fibroblasts to myofibroblasts, which can then compromise normal tissue functions. One of the major profibrotic cytokines involved in fibrogenesis is TGFβ1, as shown in the pathogenesis of IPF (21, 22). In addition to the direct effect of TGFβ1 on ECM turnover, it can drive fibroblast-to-myofibroblast differentiation and activation, which is associated with disease pathologies (2, 23–27). In fibrogenic circumstances, the myofibroblasts comprise the crucial cell population (2). In contrast to wound healing, however, a persistent accumulation of myofibroblasts is associated with the pathological reorganization and expansion of ECM components that may not normally be present or are present at only low levels in normal tissue (28).
One of the earliest fibrogenic responses is the deposition of HA in the pericellular matrix surrounding the differentiated cells (29–32). HA is synthesized by HA synthase enzymes, of which three distinct genes have been isolated and characterized, HAS1, HAS2, and HAS3 (33). HA turnover occurs under normal physiologic settings, and the increase in HA levels during pathologic conditions reveals the balance between its synthesis and catabolism. Hyaluronidases are enzymes that degrade HA, and two distinct hyaluronidase genes (HYAL1 and HYAL2) have been reported to be involved in the intracellular and extracellular catabolism of HA in a CD44-dependent manner (34). It has been shown that lung tissue from human IPF patients and from mice in which lung fibrosis was induced by bleomycin share a potentially important clue as to the cause of fibrosis, the overexpression of Has2 (hyaluronan synthase 2) by myofibroblasts, the enzyme that can synthesize HA (19). A recent study also provides evidence that deletion of Has2 in mouse mesenchymal cells increased the cellular senescence of fibroblasts in bleomycin-induced mouse lung injury (35). The authors also suggest that targeting Has2 to induce fibroblast senescence could be an attractive approach to resolve tissue fibrosis (35). On the other hand, studies also show that senescent human fibroblasts also resist apoptosis caused by growth factor deprivation and oxidative stress, indicating that resistance to apoptosis might partly explain why senescent and apoptosis-resistant myofibroblasts are highly stable in culture and induce persistent fibrosis in lungs of aged mice (6, 36, 37). Myofibroblast apoptosis is crucial to the regression of fibrotic scars and the regeneration of healthy tissue during wound repair and may be aberrant in diseases such as IPF. It is now recognized that myofibroblasts have several origins, contribute significantly to connective tissue remodeling by exerting contractile forces and producing ECM components, regress and disappear by apoptosis on wound epithelialization, and may persist in fibrotic situations and cause organ dysfunction (38).
CD44, a major receptor for HA, is involved in intracellular signaling (1, 39–56) and is an avid regulator of apoptosis in fibroblasts (1, 57). Increased synthesis of HA and expression of CD44 have been detected in numerous fibrotic conditions associated with organ dysfunction (58–61). CD44 is known to have a major role in regulating cell-cell adhesion (49, 62), migration (44, 49, 63–66), differentiation (1, 67, 68), and proliferation (69), and it therefore has an important role in wound healing (50). The human CD44 gene contains 19 exons (70). Exons 6–14 are alternatively spliced to produce CD44 variant isoforms (70). CD44 is important in maintaining the integrity of the fibroblast actin cytoskeleton (64, 72–73) and in facilitating an organized, directional migratory response to injury (74). CD44V6, in which exon 6 is expressed, is of particular interest because interaction of HA with CD44V6 alters cellular function in response to various growth factors and cytokines (75). Our recent study showed that a unique functional activity of CD44V6 includes stimulation of ERK activation, increased collagen-1 (COL1A1) synthesis, and induction of α-smooth-muscle actin (α-SMA) in isolated lung myofibroblasts through TGFβ1 autocrine signaling in fibrogenic lung disease of human subjects (1). Studies using IPF lung-derived fibroblasts (IPFFbs) demonstrated that TGFβ1-dependent profibrotic responses, including resistance to apoptosis, up-regulation of α-SMA gene expression, and the secretion of the ECM components fibronectin and COL1A1, are NOX4-dependent and contribute to persistent fibrosis in IPF disease (6, 15, 16). A recent study also indicates that the expression of CD44 is regulated in a NOX-dependent manner in atherosclerosis disease (76). In addition, ROS can degrade HA under oxidative conditions (77). Thus, understanding myofibroblast activation is crucial to understanding this aspect of injury and repair. Thus, we hypothesize that CD44V6 may be a critical determinant of the fate of fibrosis by promoting myofibroblast activation through its interaction with NOX4 in the fibroblasts from IPF patients and that this leads to the progressive fibrosis.
Proinflammatory cytokines, such as tumor necrosis factor α (TNFα), are key features of inflammatory processes and may cause tissue damage and destruction (78). TNFα acts in vasodilation, edema, leukocyte adhesion to the epithelium, macrophage activation, fever, and metalloprotease activation and contributes to the oxidative stress at inflammation sites (79, 80). Recent observations also indicate that eosinophils found in fibrotic lung express elevated levels of cytokines known to be important in lung fibrosis. These findings suggest a possible role for TNFα in eosinophil recruitment and cytokine expression in this disease. Eosinophil-derived cytokines include MCP-1 (monocyte chemotactic factor-1) and TGFβ1. This cytokine networking orchestrated by TNFα could, in turn, amplify the inflammatory response and drive the progression to fibrosis and end-stage lung disease (81). Furthermore, TNFα inhibits the anti-inflammatory cytokine interleukin-10 (IL-10) production (82), triggering chronic inflammation (83) and autoimmune diseases (84). Recent studies suggest that cells of the monocyte/macrophage lineage are also present, especially at the early stages of the fibrosis disease (85). Infiltrating leukocytes are potential sources of a number of immunomodulatory or profibrotic cytokines. It seems likely that complex cytokine-mediated networks of interaction between different cell types are involved in the development of tissue fibrosis. As a consequence of cytokine stimulation, an activated population of fibrogenic fibroblasts deposit excessive extracellular matrix, which is the pathological hallmark of the established disease (86).
The pleiotropic cytokine TGFβ1 and its signaling activation of downstream molecules has been proposed as a control mechanism for the induction of inflammatory cells and molecules. TGFβ1 binds directly to TGFβ receptor II (TGFβRII), which is a constitutively active kinase. Bound TGFβ1 is then recognized by receptor I (TGFβRI), which is recruited into the complex and becomes phosphorylated by TGFβRII. Phosphorylation allows TGFβRI to propagate the signal to downstream substrates, including SMAD2 and SMAD3. This provides a mechanism by which a cytokine can generate the first step of a canonical or a non-canonical signaling cascade (87, 88). The growing interest in the TGFβ1 pathway as a treatment option for inflammatory diseases, especially due to the possibility of controlling reactive inflammatory cells, led us to the development of novel TGFβ1-like peptides from the CD44V6 exon sites that could outperform the effects of TGFβ1.
Using fibroblasts from normal human lungs, we show that sustained production of CD44v6 involves TGFβ1/SMAD3-mediated NOX4 induction and ROS production and that NOX4 regulates CD44v6 gene expression through transcription factor AP-1, which then regulates Has2 mRNA transcription in murine lung fibroblasts. In addition, our data indicating that NOX4 regulates hyaluronidase expression in isolated mouse normal lung fibroblasts (MNLFbs) treated with TGFβ1 is significant because increased HA during patho-physiologic conditions reflects the balance between its synthesis and its catabolism. Genetic targeting of CD44v6 and an endogenous CD44v6 competing peptide (V6-PEP) tested in leukocytes significantly down-modulated fibrogenic fibroblast-mediated transendothelial migration of leukocytes induced by TNF-α. Importantly, our CD44v6 shRNA and V6-PEP peptide provide a preferred balanced immune response, with increased IL-10, which is essential for the immune response modulation, especially when associated with decreased TNF-α and abrogation of leukocyte-endothelial interactions, which may prevent excessive inflammation and tissue damage (89). Our proof-of-concept data suggest that non-toxic/anti-inflammatory CD44V6 shRNA (V6 shRNA)/transferrin (Tf)-PEG-PEI (nanoparticles) or endogenous V6-PEP/nanoparticles abrogate fibrogenesis in a murine model of lung injury by reducing NOX4/ROS activity, reducing TNF-α production, and stimulating IL-10 production, with subsequent abrogation of transendothelial migration of leukocytes.
Results
TGFβ1-induced CD44V6 synthesis regulates NOX4 and NOX4-regulated H2O2 during activation of human and mouse mesenchymal lung fibroblasts
The ability of TGFβ1 to stimulate cellular production of reactive oxygen species (H2O2) through NOX in human and mouse lung fibroblasts has been established (16), and NOX is activated through CD44 in an atherosclerosis model (90). Therefore, we investigated the role of NOX in TGFβ1-induced regulation of CD44V6. Fig. 1A shows that expression of all of the components of the NOX pathway was found in cultures of IPFFbs. Fig. 1B shows that NOX4 mRNA is highly expressed in human normal lung fibroblasts (HNLFbs) stimulated with TGFβ1, whereas other members of the NOX gene family were not affected at the mRNA level.
Figure 1.

NOX4 is induced during the fibrogenic phase of IPF, and CD44V6 regulates both NOX4 expression and NOX4-mediated H2O2. A, mRNA expressions of NOX components are shown in HNLFbs and IPFFbs. B, mRNA expressions by real-time PCR for NOX1, NOX2, and NOX4 in NLFbs treated with or without TGFβ1 and expressed relative to β-actin. C, effects of control siRNA, NOX4 siRNA, and CD44V6 siRNA on NOX4 mRNA expression in HLFbs treated with or without TGFβ1 are shown and expressed relative to β-actin. D, effects of inhibiting NOX4 by NOX4-specific siRNA or by pharmacologic inhibitor GKT137831 (1 μm) on extracellular release of H2O2 by HLFbs treated with or without 2.5 ng/ml TGFβ1 for 48 h. E, effects of blocking CD44V6 and SMAD3 using specific siRNAs on extracellular release of H2O2 by HNLFbs treated with or without 2.5 ng/ml of TGFβ1 for 48 h. The data in A are representative of three independent experiments. The experimental data in B–E are from three sets of HNLFbs with three independent experiments and are expressed as means ± S.D. (error bars). Statistical analysis was with ANOVA (B, *, p ≤ 0.005 versus TGFβ1-untreated control group; C, *, p ≤ 0.01 versus TGFβ1-treated control shRNA group; D and E, *, p ≤ 0.01 versus TGFβ1-treated control siRNA group).
To define the specific role of TGFβ1-induced CD44V6 on the regulation of NOX4, we treated HNLFb cultures with CD44V6 siRNA. Fig. 1C shows that CD44V6 siRNA efficiently blocked NOX4 induction by TGFβ1. Further, NOX4 siRNA also inhibited the induction of NOX4 by TGFβ1 (Fig. 1C). Because the TGFβ1-induced NOX4 synthesis has been largely attributed to SMAD3 signaling (16), we determined whether SMAD3 is required for H2O2 generation in HNLFbs. Fig. 1D shows that NOX4 knockdown greatly inhibits the TGFβ1-induced synthesis of H2O2, as does the siRNA knockdown of CD44V6 and SMAD3 (Fig. 1E), to levels equivalent to or better than treatment with GKT137831, a small molecule NOX4/NOX1 inhibitor from Genkyotex, Switzerland (GKT; Fig. 1D).
Our results in Fig. 2 (A and B) validated the use of these three siRNAs for the in vivo experiments used in this study. Fig. 2A shows that the elevated levels of TGFβ1-induced NOX4 mRNA are significantly reduced by both CD44V6 siRNA and NOX4 siRNA, which are increased to near-control levels by the respective cDNA treatments in IPFFbs. Fig. 2B shows that H2O2 release is inhibited by CD44V6 siRNA and SMAD3 siRNA, which are increased to control level by CD44V6 cDNA and partially increased by SMAD3 cDNA. In the experiments in Fig. 2 (C and D), we confirmed that these siRNAs (and their respective shRNAs) used alone or used with Tf-PEG-PEI (nanoparticles) (for better transfection) do not have any significant off-target effects in the in vivo settings by comparing the effects of poly(I:C) with the siRNAs and shRNAs on IFNα secretion in C57BL/6 mice (Fig. 2, C and D). The shRNAs were carefully tested for off-target effects in in vitro settings in Fig. 12 (G and H) of our companion paper (71). Synthetic siRNA duplexes or poly(I:C) were administered as i.p. injections using “low pressure” (low-pressure tail vein (LPTV); 1% (v/w)). Plasma collected at 2 h was analyzed by ELISA for IFNα secretion levels. The strong induction by poly(I:C) was absent with the treatments with siRNAs against NOX4, CD44V6, or SMAD3 and with the shRNAs against NOX4 or CD44V6. RNase-treated poly(I:C) was used as a control. Fig. 2D shows that nanoparticles for V6 shRNA, NOX4 shRNA, or V6-PEP injected (i.p. or by tail vein) with low pressure (LPTV; 1% (v/w)) or “high pressure” (HPTV; 10% (v/w)) also do not induce plasma IFNα. Further, we verified the blocking of CD44V6, or SMAD3 by specific shRNAs. The shRNA experiments were corroborated with a knockdown or with a rescue experiment. We verified the blocking of CD44V6 or SMAD3 by specific shRNAs for the coding sequences (CDS), by co-transfecting the shRNA for the target gene with or without corresponding cDNA transfection, or by replacing the knocked down gene(s) (i.e. a gene-replacement strategy, designed to circumvent the shRNA knockdown). This is accomplished by the indicated shRNA-mediated knockdown and corresponding knock-in (KI) gene transfection (see Table 1 and Fig. 12 (A–F) and the accompanying legend in our companion paper (71).
Figure 2.

Confirming the specificity of siRNA and shRNA experiments. A, IPF fibroblasts expressing control siRNA, NOX4 siRNA, or CD44V6 siRNA were further transfected with NOX4 cDNA or CD44V6 cDNA or treated with or without 2.5 ng/ml TGFβ1 for 12 h to induce NOX4 mRNA expression. Total RNAs were examined by real-time PCR analysis for NOX4 mRNA expressed relative to β-actin (A, *, p ≤ 0.005 versus control siRNA-transfected group). B, IPFFbs expressing control siRNA, CD44V6 siRNA, or SMAD3 siRNA were further transfected with CD44V6 cDNA or SMAD3 cDNA or treated with or without 2.5 ng/ml TGFβ1 for 48 h to induce extracellular release of H2O2. The averages of three replicate fibroblast cultures are presented, and error bars represent S.D.; *, p ≤ 0.005. C, plasma IFN-α induction levels in female C57BL mice are shown. 2.5 mg/kg (unless otherwise indicated) of nucleic acid was injected through the tail vein by high pressure (10% (v/w)), and plasma was collected 2 h after injection. The averages of three replicate mice are presented, and error bars represent S.D.; *, p ≤ 0.005. D, effects of administration of D5W (5% (w/v) glucose in water), control shRNA/nanoparticle, poly(I:C), CD44V6 shRNA/nanoparticle, NOX4 shRNA/nanoparticle, or V6-PEP/nanoparticle are shown. Mice received 2.5 mg/kg nucleic acid either intraperitoneally (IP) or through the tail vein via LPTV (1% (v/w)) or HPTV (10% (v/w)). The averages of three replicate mice are presented, and error bars represent S.D.; *, p ≤ 0.005. E and F, the shRNA experiments were corroborated with a knockdown or with a rescue experiment. We verified the blocking of CD44V6 or SMAD3 by specific shRNAs for the CDS, by co-transfecting the shRNA for the target gene with or without corresponding cDNA transfection, or by replacing the knocked down gene(s) (i.e. a gene-replacement strategy, designed to circumvent the shRNA knockdown); this is accomplished by indicated shRNA-mediated knockdown and corresponding KI gene transfection (see Table 1, Fig. 12 (A–F), and the legend to Fig. 12 in our companion paper (71)). Total mRNAs were examined by real-time PCR analysis and expressed relative to β-actin. The averages of three replicate mice are presented, and error bars represent S.D.; *, p ≤ 0.005.
Figure 12.

Co-culture with 21dBLMFbs, MNLFbs grown in the lower chamber with PBMCs (mouse leukocytes), U937 cells (human leukocytes) on confluent EC (C166), or HUVECs to determine the leukocyte migration across the endothelial cell monolayers (see “Experimental procedures.” A, leukocyte migration is expressed as mean percentage ± S.E. (error bars) of added cells migrating across control EC monolayers on inserts over 16 h. Data are from replicate wells in three independent experiments using three different 21dBLMFb cultures or three different MNLFb cultures. Migrated cells underneath the inserts were counted by hemocytometer. 21dBLMFbs significantly increased migration of PBMCs through the EC layer, which was further increased by cytokine activation of 21dBLMFbs (1 μg/ml LPS for 8 h) prior to co-culture with ECs. *, p < 0.01, statistically significantly greater than migration across resting EC layer. B, PBMCs attached to the insert 16 h after migration were measured by colorimetry (see “Experimental procedures”). Activation of the EC cell layer with cytokines reduced the number of PBMCs migrating across the EC but increased the PBMC binding to the EC monolayer. C and D, pretreatment of 21dBLMFbs with 1 μg/ml LPS for 8 h followed by treatment with 1 μg/ml V6-PEP/nanoparticle or 1 μg/ml V6 shRNA/nanoparticle reduced PBMC migration through the control EC monolayer (C) and also reduced U937 (leukocyte) cell migration on the HUVEC monolayer (D). *, p < 0.005. E, promotion of U937 (leukocyte) cell migration by CM derived from IPFFbs, by recombinant monocyte chemoattractant protein-1 (MCP-1), and by IL-8 are shown. ∼250 × 103 U937 cells were put onto confluent HUVEC monolayers that were treated with human recombinant MCP-1, with CM from IPFFb cultures alone or cultures that were treated with IL-8 (50 ng/ml), with 100 μg of each of the indicated nanoparticles, or with a 500 μg/ml concentration of either anti-MCP-1 or anti-CD44v6 for 12 h. U937 cell numbers in the lower chamber were counted in triplicate wells 16 h after the addition of the reagents and the CMs to the U937 cells and HUVECs on the inserts.
Table 1.
PCR amplification primers
| Genes | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| h-CD44v6 | AGGAACAGTGGTTTGGCAAC | CGAATGGGAGTCTTCTCTGG |
| h-p47 phox | GAGTACCGCGACAGACATCA | TCTTCCGTCTCGTCAGGACT |
| h-p67 phox | ATTACCTAGGCAAGGCGACG | TCTGGGTGGAGGCTCAGCT |
| h-p22 phox | CCAGTGGTACTTTGGTGCCT | GAGAGCAGGAGATGCAGGAC |
| h-NOX2 | CAAGATGCGTGGAAACTACCTAAGAT | TCCCTGCTCCCACTAACATCA |
| h-NOX4 | TCTGTTGTGGACCCAATTCA | AGCTGATTGATTCCGCTGAG |
| h-β-Actin | CCCAAGGCCAACCGCGAGAAG | CCTCGTAGATGGGCACAGTGT |
| h-GAPDH | GAAGGTGAAGGTCGGAGT | GAAGATGGTGATGGGATTTC |
| m-CD44v6 | CCTTGGCCACCACTCCTAATAG | CAGTTGTCCCTTCTGTCACATG |
| m-p47 phox | TATCTGGGAGCCCCTTGACAGTCCC | TCACACAGCGGACGTCAGCTTCCG |
| m-p67 phox | CAAGCCCTACGGTTGTAGCAT | CTTGGATGATAGCACTGCAC |
| m-p22 phox | GTTTCACACAGTGGTATTTCGGCG | GTTGGTAGGTGGTTGCTTGATGGT |
| m-Nox2 | TGTCATTCTGGTGTGGTTGG | CCCCTTCAGGGTTCTTGATT |
| m-Nox4 | CCTCATGGTTACAGCTTCTACCTACGC | TGACTGAGGTACAGCTGGATGTTCAC |
| m-β-Actin | CCCAGCACAATGA AGATCAA | GATCCACACGGAGTACTTG |
| m-Gapdh | AGTATGACTCCACTCACGGCAA | TCTCGCTCCTGGAAGATGGT |
| m-α-Sma | ATCGTCCACCGCAAATGC | AAGGAACTGGAGGCGCTG |
| m-Col1a1 | TGCTGCTTGCAGTAACTCG | TCAACACCATCTCTGCCTCG |
To define the in vivo role of NOX4 in the fibrogenic and reparative response to injury of the murine lung, we used a murine model of bleomycin instillation that causes epithelial injury with consequential TGFβ1 up-regulation, myofibroblast activation, and lung fibrosis (91). We identified Nox4 as one of the most highly induced genes in murine lung myofibroblasts isolated from lung tissues from 21-day bleomycin-treated mice (21dBLMFbs) compared with MNLFbs from lung tissues of PBS-treated mice (Fig. 3A). We then determined whether Nox4 protein expression was induced in fibroblasts isolated from the fibrogenic and reparative phases of the bleomycin-induced lung injury. Nox4 protein expression was induced in a time-dependent manner, increasing from day 7 up to day 21 and then returning to near-baseline levels at day 45 (Fig. 3B). Similar induction of CD44v6 expression was observed during these fibrogenic phases (see Figs. 2 (A and B) and 3F in our companion paper (71)), supporting cross-talk between Nox4 and CD44v6 expression that can lead to myofibroblast activation and fibrosis after lung injury. However, expression of Nox2, which is predominantly expressed in phagocytic cells, was increased on day 14 and returned to near-baseline levels at day 21 during the fibrogenic phase and at day 45 after bleomycin injury, when inflammatory responses had subsided (Fig. 3B).
Figure 3.

Nox4 is induced during the fibrogenic phase of bleomycin-induced lung injury in mice, and the roles of NOX4 and CD44v6 in mediating myofibroblast phenotype of fibroblasts on day 21 after bleomycin injury are shown. A, mRNAs of NADPH oxidase components were expressed in cultures of MNLFbs and BLMFbs. B, protein expressions are shown for Nox4, Nox2, and β-tubulin in BLMFbs on days 14, 21, and 45 after treatment and on PBS-treated MNLFbs (day 0) as determined by SDS-PAGE and Western blotting. The data in A are representative of three independent experiments. The data in B are from three sets of BMLFbs with three independent experiments, and are expressed as the means ± S.D. (error bars) The results of densitometry obtained for Western blots of the indicated proteins are shown in the bottom panels of the experiments shown in the top panels of B. Statistical analysis was by ANOVA; *, p ≤ 0.005 versus PBS-treated 0-day group.
The myofibroblast phenotype contributes to the healing of tissue injury by secreting ECM proteins by remodeling and contracting the ECM (16, 92, 93). Fig. 4A shows that by silencing Nox4 or CD44v6, the α-Sma, fibronectin, and pro-collagen-1 proteins, as well as Nox4 and CD44v6 proteins, are decreased in 21dBLMFbs (Fig. 4A) and IPFFbs (Fig. 4B). Interestingly, Nox4 regulates CD44v6 expression and vice versa in 21dBMLFbs and in IPFFbs (Fig. 4, A and B). In addition, we confirmed the knockdown experiments, where we always use shRNAs for CDS, or shRNAs against noncoding sequences (NCDS) (designed to target the 3′-untranslated region (UTR) of the mRNA) as ideal negative control. Total cell lysates were examined by Western blot analysis for the indicated proteins expressed relative to β-tubulin. The data in the experiments are from three sets of 21dBLMFbs with three independent experiments. The densitometry results obtained for Western blots of the indicated proteins are shown in bottom panels of the experiments in Fig. 4A. We also verified the effect of the Nox4 shRNA and CD44v6 shRNA on the α-SMA, fibronectin, and pro-collagen-1 proteins, as well as the NOX4 and CD44v6 proteins, with rescue of the observed shRNA-mediated knockdown phenotype by expression of a resistant form of the targeted mRNA. This is performed by transfecting the cells with specific shRNAs for the CDS of the target gene, by co-transfecting the shRNA for the target gene with or without corresponding cDNA transfection, or by indicated shRNA mediated knockdown and corresponding KI gene transfection in IPFFbs (Fig. 4B; as shown in Fig. 2 (E and F)). Total cell lysates of IPFFbs of the indicated treatments were examined by Western blot analysis for the indicated proteins expressed relative to β-tubulin. The densitometry results obtained for Western blots of the indicated proteins are shown in the bottom panel of Fig. 4B and validated the role of regulation of NOX4 on CD44V6 expression and vice versa.
Figure 4.

The effects of shRNA-mediated silencing of NOX4 and CD44v6 on endogenous expression of fibrogenic proteins. A and B, the effects of shRNA-mediated silencing of NOX4 and CD44v6 on endogenous expression of α-SMA, fibronectin, procollagen-1, NOX4, CD44v6, and β-actin in 21dBLMFbs (bottom panel of A). In addition, we confirmed the knockdown experiments, where we always use shRNAs for CDS, a scrambled control shRNA, or shRNAs against NCDS (designed to target the 3′-UTR of the mRNA) as ideal negative control. Total cell lysates were examined by Western blot analysis for the indicated proteins expressed relative to β-tubulin. The data in the experiments are from three sets of 21dBLMFbs with three independent experiments. The densitometry results obtained for Western blots of the indicated proteins are shown in the bottom panels of the experiments in A (bottom panel of B). In this experiment, the shRNA experiments were corroborated with a knockdown or with rescue of the observed shRNA-mediated knockdown phenotype by expression of a resistant form of the targeted mRNA. This is performed by transfecting the cells with specific shRNAs for the CDS of the target gene, by co-transfecting the shRNA for the target gene with or without corresponding cDNA transfection, or by replacing the knocked down gene(s) (i.e. a gene-replacement strategy, designed to circumvent the shRNA knockdown). This is accomplished by the indicated shRNA-mediated knockdown and corresponding KI gene transfection (see Fig. 12 (A–G) and the Fig. 12 legend in our companion paper (71)). Total cell lysates were examined by Western blot analysis for the indicated proteins expressed relative to β-tubulin. The data in the experiments are from three sets of IPFFbs with three independent experiments. The densitometry results obtained for Western blots of the indicated proteins are shown in the bottom panels of the experiments in B. A and B, densitometry graphs, statistical analysis with ANOVA; *, p ≤ 0.005 versus control shRNA (Contsh)-transfected group; NS, statistically nonsignificant. Error bars, S.D.
Redox-dependent activation of IPF mesenchymal cells by NOX4 is known to be involved in regulation of myofibroblast activation during lung fibrosis (16). To determine the role of CD44V6 in conjunction with NOX4, we examined lung tissue sections from human subjects with IPF. Both NOX4 and CD44V6 are highly expressed in cells that are presumably from inflammatory areas of the injured IPF lung (Fig. 5A). We then examined lung fibroblasts isolated from explants of IPF lung tissue (IPFFbs). Fig. 5B shows that in IPFFb TGFβ1 also induces CD44V6 and NOX4 proteins, which in turn are necessary for induction of ECM proteins fibronectin and pro-collagen-1. This result also indicates that basically IPFFbs respond the same way to TGFβ1 as do NLFbs. CD44V6 and NOX4 are also required for the induction of α-SMA, COL1A1, and fibronectin mRNAs (Fig. 5, C–G) and for their protein expression (Fig. 5B). Similar to our findings in 21dBLMFbs, both CD44V6 and NOX4 regulate each other's mRNA expression (Fig. 5, F and G) and protein expression (Fig. 5B).
Figure 5.

CD44v6 and NOX4 expression are increased in lung sections of human subjects with IPF, and they mediate profibrotic protein expression in IPFFbs. A, immunohistochemical staining shows increased expression of NOX4 and CD44v6 in inflammatory cells comparing sections from a representative IPF patient lung and a normal lung. B–G, total cell lysates were examined by Western blot analysis for the indicated protein expressed relative to β-tubulin. The data in the experiments in B are from three sets of IPFFbs with three independent experiments. The densitometry results, expressed as the means ± S.D. (error bars) obtained for representative Western blots of the indicated proteins in C–G, are shown in the top panel of B. The effects of shRNA-mediated knockdown of NOX4 in IPFFbs treated with or without TGFβ1 (2 ng/ml) on the expression of α-SMA mRNA (C) and protein (B), on fibronectin mRNA (D) and protein (B), on COL1A1 mRNA (E) and protein (B), on NOX-4 mRNA (F) and protein (B), and on CD44v6 mRNA (G) and protein (B) were determined by real-time PCR (quantified relative to β-actin at 24 h) and by Western immunoblotting (at 72 h) after TGFβ1 treatment. The densitometry results obtained for Western blotting of the proteins are shown in the bottom panel of B. The experimental data in B–G are from three sets of IPFFbs with three independent experiments and are expressed as the means ± S.D. Statistical analysis was with ANOVA (B, *, p ≤ 0.005 versus control shRNA-transfected group; C–G, *, p ≤ 0.01 versus control shRNA-transfected group).
Fig. 6A shows that shRNA-mediated knockdown of CD44V6 and NOX4 inhibited serum-stimulated proliferation of IPFFbs to the same extent as the ROS inhibitor, N-acetyl-l-cysteine (NAC). Fig. 6B shows that the CD44v6/NOX4/ROS/AKT pathway inhibits MMP2 and MMP9 expression. Consistent with this result, the suppression of NOX4, CD44v6, or ROS significantly decreased cell invasion of IPFFbs compared with the control cells (Fig. 6D). The results in Fig. 6C validated the knockdown effect of NOX4 shRNA and CD44V6 shRNA by comparing these shRNAs (CDS) with those of NCDS transfection in human IPFFbs. In summary, the data in Figs. 1–6 support the key role for the CD44V6-NOX4 pathway in myofibroblast invasion, proliferation, and secretion of ECM and expression of contractile proteins necessary for differentiation of human IPFFbs.
Figure 6.

CD44V6 and NOX4 regulate serum-stimulated proliferation and invasion in IPFFbs. A, effects of shRNA-mediated knockdown of NOX4 and CD44v6 and of 2.0 μm ROS inhibitor N-acetyl-l-cysteine (NAC) on proliferation of IPFFbs treated with or without 10% serum were determined at 24 h by the BrdU incorporation assay. B, the effects of knocking down NOX4 or CD44v6 and of inhibiting ROS on phospho-Akt, MMP2, and MMP9 expression are shown by Western blotting analysis. The densitometry results obtained for Western blotting of the indicated proteins are shown in the bottom panel of the experiment in the top panel of B. Statistical analysis was with ANOVA; B, *, p ≤ 0.005 versus control shRNA-transfected group. C, NOX4 mRNA analyses are shown for IPFFbs expressing control shRNA, NOX4 shRNA for CDS, CD44v6 shRNA (CDS), NOX4 shRNA for NCDS, or CD44v6 shRNA (NCDS). Total RNAs were examined by real-time PCR analysis for NOX4 mRNAs expressed relative to β-actin. C, *, p ≤ 0.005 versus control shRNA-transfected group. D, effects of knocking down NOX4 and CD44v6 and of inhibiting ROS on cell invasion by IPFFbs. The experimental data in A–D are from three sets of IPFFbs with three independent experiments and are expressed as the means ± S.D. (error bars). Statistical analysis was with ANOVA; D, *, p ≤ 0.005 versus control shRNA-transfected group.
CD44v6 regulates TGFβRI kinase activity to induce downstream TGFβ1 signaling in IPF lung disease
HA has been reported to induce ROS in a CD44-dependent manner (94, 95), and NOX mediates increases in ROS production (16). Therefore, signaling events that mediate CD44v6 and TGFβ1 are important in lung fibrosis of human subjects (1). In this study, we demonstrated the association of TGFβRI and CD44v6 in the Duolink assay of IPFFbs and its near absence in HNLFbs (Fig. 7A). Therefore, we determined whether the interaction between CD44V6 and the TGFβ1 receptor(s) has a significant role in regulating fibrogenic fibroblast cell-specific behaviors, such as p-SMAD activation and NOX4 production. The immunoblots in Fig. 7B from HNLFb cultures show that overexpression of HAS2 significantly up-regulates phosphoserine and phosphothreonine on TGFβ1RI (lane 2, HAS2 + Cont shRNA), which is blocked by CD44V6 shRNA (Fig. 7B, lane 3). Further, TGFβ1-induced up-regulation of phosphoserine and phosphothreonine (lane 4, TGFβ1 + Cont shRNA) was also blocked by CD44V6 shRNA (Fig. 7B, lane 5). These results show that serine/threonine kinase(s) actively phosphorylates TGFβRI when it is associated with CD44V6 and when HAS2 is overexpressed. These results also provide strong evidence that HA-mediated TGFβRI kinase activity is CD44V6-dependent and that TGFβ1-stimulated TGFβRI kinase activity involves CD44V6. Although the TGFβRI is known to activate SMAD2 and SMAD3, the profibrotic effects of TGFβ1 signaling have been largely attributed to SMAD3 signaling (96). Fig. 7C shows that serine/threonine phosphorylation of SMAD3 is also up-regulated by HAS2 overexpression and TGFβ1 treatments in HNLFb cultures, which is blocked by CD44V6 shRNA. Therefore, these results provide strong evidence that HA, the physiological ligand for CD44V6, has an important role in activating CD44V6-associated TGFβRI kinase activity that is required for the onset of the SMAD3-mediated TGFβ1 response that promotes the progressive fibrosis in IPF.
Figure 7.

CD44V6/TGFβRI kinase and signaling events in lung fibrosis. A, Duolink assays were performed in IPFFbs and HNLFbs to analyze the association between TGFβRI and CD44v6. Data in A (bottom) are expressed in relative fluorescence units adjusted for protein concentration (mean ± S.E. (error bars); n = 3; *, p ≤ 0.005). B, HNLFbs were transfected with vector control, HAS2 cDNA, or TGFβ1 cDNA or pretransfected with control shRNA or CD44V6 shRNA either alone or followed by HAS2 or TGFβ1 overexpression. After 72 h, lysates were prepared and immunoprecipitated (IP) with anti-TGFβRI antibody. The components in the immunoprecipitate were separated by SDS-PAGE and analyzed by Western blotting with anti-phosphoserine, anti-phosphothreonine, and anti-TGFβRI antibodies. C, the lysates were immunoprecipitated with anti-Smad3 antibody, separated by SDS-PAGE, and analyzed by Western blotting with anti-phosphoserine, anti-phosphothreonine, and anti-Smad3 antibodies. D, real-time PCR analyses are shown for CD44v6 in HNLFbs transfected with control shRNA or CD44V6 shRNA, followed by incubation without (left) or with TGFβ1 for 24 h (right). β-Actin was used as a loading control. E, Western blots are shown for Smad3 phosphorylation, NOX4, and β-actin in lysates from cells harvested from the experiment in C. F, IPFFbs expressing control shRNA or two different V6 shRNAs (V6 shRNA (lane 1) (CDS) or V6 shRNA (lane 2) (CDS)) or V6 shRNA (lane 1) (NCDS). Total RNAs were examined by real-time PCR analysis for CD44V6 mRNA expressed relative to GAPDH. G, HNLFbs were treated without or with control siRNA, SMAD3 siRNA, NOX4 siRNA, or CD44V6 siRNA (v6) for 24 h and then treated with TGFβ1 for 12 h. For comparison, HNLFbs were pretreated with 1 μm GKT137831 (GKT; a NOX1/NOX4 inhibitor) alone for 8 h and then treated with TGFβ1 for 12 h. Intracellular ROS generation was detected by measuring H2DCFDA fluorescence. C–F show data representative of three experiments. The experimental data in B and G are from three sets of three independent experiments. Statistical analysis was with ANOVA; B and G, *, p ≤ 0.005 versus the respective control group.
CD44v6 enhances hepatocyte growth factor (HGF), VEGF, and EGF-dependent tyrosine kinase receptor (c-MET, VEGF receptor, and EGF receptor) activation of the downstream MAPK-signaling cascade through a positive-feedback loop (97–99). Therefore, we examined whether down-regulation of CD44V6 inhibits TGFβRI/SMAD3 signaling and whether constitutive activation of TGFβ1 can recover production of CD44V6 in HNLFb cultures in which CD44V6 expression is depleted. RT-PCR results in Fig. 7D show that CD44V6 siRNA effectively inhibits up-regulation of CD44V6 by TGFβ1, and Western blots in Fig. 7E show that CD44V6 siRNA also effectively inhibits TGFβRI phosphorylation and activation of p-SMAD3. Western blots showed that the amount of SMAD3 protein in this experiment was not altered (Fig. 7E). The results in Fig. 7F validated the effect of CD44V6 shRNA (CDS) on CD44V6 mRNA expression with the relevant shRNA (NCDS; proper negative control).
CD44V6 regulates TGFβ1/SMAD3-mediated NOX4 induction and ROS production
The findings for Fig. 7 (A–E) provide support for a positive-feedback loop in which CD44V6-dependent activation of TGFβRI/SMAD3 can result in synthesis of the NOX4 expression. Therefore, we determined the role of the SMAD3/NOX4 pathway in ROS generation and whether CD44V6 is required for initiating the TGFβRI/SMAD3/NOX4-induced fibrogenic phenotype. Fig. 7G shows the results of analyses of dichlorofluorescein diacetate (H2DCFDA) fluorescence (a measurement of intracellular ROS) in TGFβ1-treated HNLFb cultures. A marked increase in fluorescence showed that intracellular ROS increased in HNLFbs treated with TGFβ1, which was significantly decreased by transfecting the cells with either SMAD3 shRNA, NOX4 shRNA, or CD44V6 shRNA before TGFβ1 treatment. Further, an optimal concentration of GKT137831 (1 μm; the dose obtained from the dose-response curve of GKT137831 on NOX4 expression (data not shown)), a pharmacologic inhibitor of NOX1/NOX4, also inhibited ROS ∼50% in the TGFβ1-treated HNLFb cultures. These data support a role for CD44V6 signaling via TGFβRI and SMAD3 in the induction and activation of the NOX4 response in TGFβ1-treated HNLFbs. In addition, the positive-feedback loop coupling CD44V6 and TGFβRI activation shown in Fig. 7E can produce sustained SMAD3/NOX4 signaling, which can promote progressive fibrosis. This is supported by evidence that SMAD3/NOX4 signaling is required for lung myofibroblast activation and differentiation (16).
The CD44V6-NOX4 pathway induces TGFβ1-induced ECM contractility
Tissue repair is mediated by myofibroblasts through secretion of ECM proteins and by remodeling and contracting the extracellular matrix (92, 100). Therefore, we investigated the role of the CD44V6-NOX4 pathway in contractile function of myofibroblasts by measuring the contractility of three-dimensional collagen matrices and by analyzing the expression of α-SMA, the cytoskeletal protein responsible for contractile actin stress fibers (51). Fig. 8 shows that TGFβ1-induced collagen gel contractility (A) and expression of α-SMA (B) were significantly inhibited by the GKT137831 (NOX4/NOX1 inhibitor), by knockdown of NOX4, by blocking CD44V6 with an inhibitory CD44V6 peptide (V6-PEP; see Fig. 10 for the peptide function), and by inhibiting intracellular ROS with catalase expression vector. Overall, these results indicate a crucial role for CD44V6-NOX4-dependent ROS activation in ECM contractile properties that occurs when myofibroblasts differentiate in response to TGFβ1.
Figure 8.
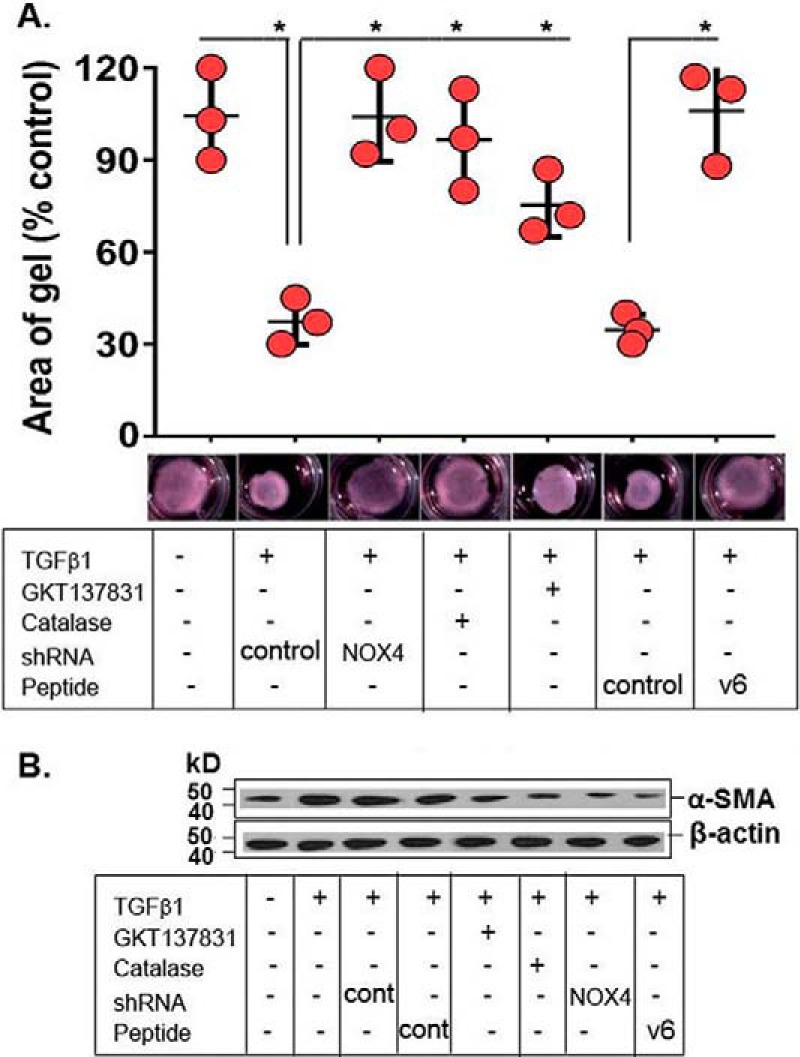
Effects of NOX4, CD44V6, and ROS on TGFβ1-induced HNLFb collagen matrix contractility. A, HNLFbs embedded in three-dimensional collagen matrices were treated with NOX4 shRNA, a NOX inhibitor, GKT137831 (1 μm (obtained from a dose-response experiment for NOX4 inhibition)), an endogenous CD44V6 competing peptide (V6-PEP), or a catalase expression vector (a ROS inhibitor) before treatment with TGFβ1 (2.5 ng/ml for 72 h) followed by measurements of matrix contractility (area of gel). Data represent mean ± S.E. (error bars); n = 3; *, p < 0.005 compared with untreated controls. B, representative Western blots are shown for expression of α-SMA and β-actin in lysates from cells transfected with control shRNA (cont) or NOX4 shRNA, or treated with 1 μm GKT137831, or transfected with catalase, or treated with control peptide (cont), or treated with CD44v6-blocking peptide (V6-PEP) for 24 h and then incubated with TGFβ1 (2.5 ng/ml for 72 h). The experimental data in A and B are from three sets of three independent experiments. Statistical analysis was with ANOVA; A, *, p ≤ 0.01 versus the respective control group.
Figure 10.

A linear peptide comprising three V6-specific amino acids abrogates TGFβ1-mediated p-SMAD3 activation, and anti-TGFβ1 antibody recognizes V6-PEP. A, schematic diagram of murine and human CD44v6 peptide sequences. CD44v6 peptides in the boxed region of murine (red) and human (blue) CD44V6 were used in competitive experiments in which IPFFbs (B–D) or BLMFbs (E–F) were treated with or without a 1 μg/ml concentration of the peptides (based on dose-response curves (data not shown)) and with 2.5 ng/ml TGFβ1 for 24 h, followed by analysis for SMAD3 and phosphorylated p-SMAD. B, TGFβ1-mediated p-SMAD3 activation was abrogated by human V6-PEP but not by human V6-PEP-1, which is outside the box in A. C, M mutations were made for each amino acid (shown in green in A) in the Lys, Glu, and Arg locations in human V6-PEP and tested for their effects on the p-SMAD3 activation assay. These mutations prevented the V6-PEP inhibition of TGFβ1-induced phosphorylation of p-SMAD3. D, the commercial anti-TGFβ1 antibody recognized the V6-PEP, bound on the surfaces of PBMCs (leukocytes), confirming that the TGFβRII has equally recognized both TGFβ1 and the V6-PEP peptide. (± S.D.; n = 3; *, p ≤ 0.005.) E and F, TGFβ1-mediated p-SMAD3 activation is abrogated by CD44V6-specific peptides in 21dBLMFbs stimulated with 2.5 ng/ml TGFβ1 for 24 h. Peptides corresponding to the V6 exon sequences (human V6-PEP and murine v6-PEP-2) inhibited TGFβ1-induced phosphorylation of p-SMAD3 in contrast to a control peptide (Cont-PEP) and to the human V6-PEP-1 (E). The mutated (M) human V6-PEP peptides (C) and the mutated (M) mouse v6-PEP-2 peptides (F) were unable to inhibit TGFβ1-induced p-SMAD3. B, C, E, and F are representative of three sets of three independent experiments.
Nox4/Ap-1-dependent mechanism of CD44v6 and Has2 expressions in primary lung fibroblasts
A previous report (76) provides evidence that NOX-dependent ROS regulates CD44 expression in atherogenesis. Therefore, we determined whether NOX4 has a critical role in the regulation of CD44v6 and HA expression in MNLFbs treated with TGFβ1. Fig. 9A shows that TGFβ1 treatment for 4 h resulted in a 2.5-fold increase in transcription of CD44v6 mRNA in wild-type MNLFbs, which increased to 7-fold by 8 h, whereas TGFβ1 had no significant effect in Nox4-null lung fibroblasts (Nox4-null MNLFbs). Further support for redox-dependent regulation of CD44v6 mRNA expression was shown by its significant inhibition in MNLFbs that had been pretransfected with NOX4 shRNA or pretreated with the Nox1/Nox4 inhibitor GKT137831 followed by TGFβ1 treatment (Fig. 9A).
Figure 9.

NOX4-Ap-1-dependent regulation of CD44V6 and of hyaluronan synthesis and degradation. A, real-time PCR analysis of CD44v6 expression in MNLFbs and Nox4 null MNLFbs treated with 2.5 ng/ml TGFβ1 are shown for the indicated times. Data are expressed as -fold change in gene expression relative to untreated controls. B, DNA-binding activity of Ap-1 was assayed in MNLFbs and Nox4-null MNLFbs after TGFβ1 treatment for the indicated times. Data are expressed as absorbance at 450 nm. C, Ap-1-dependent CD44 promoter activity was assayed in NLFbs and Nox4-null MNLFbs and in IPFFbs pretransfected with either control siRNA or NOX4 siRNA. Cells were transiently co-transfected with WT CD44-Luc or CD44-AP-1 mutant (M) plasmids and with a β-galactosidase control plasmid. After 48 h, cells were treated with vehicle or with 2.5 ng/ml TGFβ1 for 24 h, followed by luciferase and β-galactosidase activity measurements. Data represent -fold change in luciferase activity, normalized to β-galactosidase activity in TGFβ1-treated cells compared with their respective controls (mean ± S.E. (error bars); n = 3; *, p < 0.005). D, real-time PCR analysis of Has2 mRNA expression in MNLFbs and Nox4-null MNLFbs treated with 2.5 ng/ml TGFβ1 for times up to 12 h. Data represent -fold change in gene expression, normalized to β-actin and relative to untreated MNLFb control. E, MNLFbs and Nox4-null MNLFbs were treated with 2.5 ng/ml TGFβ1 or 50 μg/ml Healon (1000-kDa HA), HA500, or HA150 for 12 h. Nox activity (ROS generation) was detected by measuring H2DCFDA fluorescence. F, MNLFbs, NOX4-null MNLFbs, and CD44-null MNLFbs were treated with 2.5 ng/ml TGFβ1 or HA150 alone for 12 h or pretreated with 2.5 ng/ml TGFβ1 for 12 h and then treated with HA150 for another 12 h. ROS generation was detected by measuring H2DCFDA fluorescence. G, MNLFbs and Nox4-null MNLFbs were treated with 2.5 ng/ml TGFβ1 or 50 μg/ml Healon (1000 kDa), HA500, HA150 for 12 h or first treated with or without 12.5 units of heat-inactivated (95 °C for 1 h) Streptomyces hyaluronidase for 1 h. ROS generation was detected by measuring H2DCFDA fluorescence. H, real-time PCR analysis of Hyal1, Hyal2, and Hyal3 mRNA expression in MNLFbs and Nox4-null MNLFbs treated with 2.5 ng/ml TGFβ1 for times up to 24 h. Data represent -fold change in gene expression, normalized to β-actin and relative to untreated MNLFbs control (mean ± S.E.; n = 3; *, p < 0.005). The data in the experiments (A–D and H) are representative of three sets of three independent experiments. Data in E–G are expressed in relative fluorescence units adjusted for protein concentration. (mean ± S.E.; n = 3; *, p < 0.005).
Because recent studies from our laboratory and other show that (i) proinflammatory TGFβ1 up-regulates CD44V6 (1), (ii) TGFβ1 up-regulates NADPH oxidase (16), and (iii) NOX-derived ROS regulates Ap-1 (101), we investigated whether Nox4-derived ROS, CD44v6, and Ap-1 activation are interrelated during lung fibrosis. Fig. 9B shows that Ap-1 DNA-binding activity increased significantly (∼60%) at 2 h and remained elevated through 12 h (∼300%) after TGFβ1 treatment in MNLFbs. In contrast, Nox4-null cells treated with TGFβ1 showed only a minimal response (Fig. 9B). To determine whether increased Ap-1 DNA-binding activity induced by NADPH oxidase is the mechanism for increased CD44v6 expression, MNLFbs, IPFFbs, and Nox4-null MNLFbs were co-transfected with CD44 promoter-luciferase reporter gene constructs containing either an Ap-1-binding sequence (WT CD44 Luc) or an Ap-1 mutant site CD44 Luc (AP-1-M; see Fig. 6B in our companion paper (71)). Fig. 9C shows that the luciferase activities in TGFβ1-treated MNLFbs and IPFFbs transfected with the WT CD44 Luc construct had significantly more luciferase activity than the same cells transfected with the AP-1-M construct. In contrast, transfection of Nox4-null MNLFbs, or IPFFbs that were transfected with NOX4 siRNA, with either WT CD44 Luc or the CD44 Luc (AP-1-M) construct showed no differences in luciferase activities (Fig. 9C; the inset validates the NOX4 shRNA). The CD44 promoter regulates transcription of CD44s and variants, including the v6 exon. Because NOX4 shRNA down-regulated CD44v6 expression in both fibrogenic mouse lung fibroblasts (21dBLMFbs) and IPFFbs (Fig. 4, A and B), one of the interpretations of Fig. 9C is that lowering of Nox4 activity in Nox4-null MNLFbs down-regulates CD44v6 transcription. Because blocking CD44V6 inhibits CD44V6-containing isoforms (see Fig. 16 in our companion paper (71)) and because NOX4 regulates CD44V6 (Fig. 4, A and B), the experiment in Fig. 9C cannot rule out the down-regulations of transcriptions of all other CD44V6-containing isoforms.
ROS production in inflammatory atherosclerosis disease is largely mediated through the interaction of CD44 with HA (76). To address this, HAS2 mRNA in MNLFb and NOX4-null MNLFb cultures was measured after TGFβ1 treatment for various times by real-time PCR in quiescent cells. MNLFbs show very little expression of Has1 and Has3 (Fig. 9D). Therefore, Has2 is the major hyaluronan synthase in MNLFbs. Fig. 9D shows that Has2 mRNA expression increased continuously over 12 h with TGFβ1 in the MNLFbs but showed minimal response in the Nox4-null MNLFbs. Consistent with the increased CD44v6 expression by Nox4, this result indicates that Nox4 has a putative role in regulating Has2 expression and also provides evidence of involvement of HA interaction with CD44s and/or its variants. TGFβ1 also had a smaller effect on Has1 and Has3 expression in either cell type (Fig. 9D).
Low-molecular-weight HA (LMWHA) (presumably through hyaluronidase cleavage of high-molecular-weight HA (HMWHA)) has been implicated in augmenting ROS (76, 95). Therefore, we analyzed H2DCFDA fluorescence as a measure of Nox4 activity (intracellular ROS production) in normal lung fibroblast cultures prepared from lungs of CD44-null mice (CD44-null MNLFbs) and NOX4-null mice (NOX4-null MNLFbs) that were treated with TGFβ1, LMWHA (150- or 500-kDa HA), HMWHA (Healon; 1000 kDa), or both TGFβ1 and LMWHA (HA150). Fig. 9E shows that a 12-h treatment with TGFβ1 significantly induced ROS synthesis (H2DCFDA fluorescence) in wild-type MNLFbs compared with Nox4-null MNLFbs. Further, HA150 and HA500 both increased ROS synthesis. In contrast, the 1000-kDa HA (HMWHA) inhibited the TGFβ1-induced ROS synthesis in MNLFbs. Fig. 9F shows that TGFβ1 increased H2DCFDA fluorescence ∼3-fold in MNLFbs, similar to the increase by HA150, and their combination was additive. However, HA150 alone or pretreatment with TGFβ1 followed by HA150 treatment had no significant effect on H2DCFDA fluorescence in CD44-null cells and in Nox4-null cells (Fig. 9F). These results suggest that interaction of 150-kDa HA with TGFβ1-induced CD44v6 and/or Nox4 induces ROS generation in MNLFbs.
To determine the specificity of HA for ROS production, the MNLFbs and CD44-null MNLFbs were treated with Streptomyces hyaluronidase (control; heat-inactivated hyaluronidase) prior to HA treatment. Hyaluronidase removes extracellular HA and thus demonstrates the specificity of HA treatment. Fig. 9G shows that the hyaluronidase treatment abrogated the HA150- and HA500-increased ROS production in MNLFbs, whereas, in Nox4-null MNLFbs, these treatments had no effect. Hyaluronidases have been reported to be involved in the intracellular and extracellular catabolism of HA in a CD44-dependent manner (34). The hyaluronidase-treated cultures may have produced LMWHA species; thus, we cannot rule out the effect of LMWHA produced by hyaluronidase on the exogenous addition of LMWHA contributing to NOX activity, as seen in Fig. 9G. Further, TGFβ1 treatment also caused a time-dependent increase in the mRNA expression of hyaluronidases Hyal3 and Hyal1 in MNLFbs, but not in Nox4-null cells (Fig. 9H). Hyal3 mRNA expression was 3.5-fold higher, and Hyal1 mRNA expression was 2.7-fold higher in MNLFbs than in Nox4-null cells within a short time period (at 8 h) after TGFβ1 treatment. TGFβ1 had no significant effect on Hyal2 expression in either cell type (Fig. 9H). Thus, these data suggest that Nox4 activation mediates TGFβ1-induced Has2 and hence HA levels in MNLFbs by regulating both Has2 (synthetic enzyme) and Hyal3/1 (degradation enzymes). Furthermore, ROS-regulated expression of Hyal3 and Hyal1 may lead to enhanced degradation of HA and generation of ROS, stimulating LMWHA. Thus, NOX4 activation increases HA synthesis, and LMWHA (HA150 and HA500) enhances Nox4-mediated intracellular ROS levels, suggesting a positive-feedback loop between HA and Nox4 activation, and this function of LMWHA requires CD44v6. Conversely, when HA150 and HA500 were treated with heat-inactivated hyaluronidase, this effect was reversed, indicating the specificity of LMWHA for regulating ROS production in a Nox4-dependent way (Fig. 9G).
Therapeutic targeting of CD44v6 is effective in fibrosis resolution
The co-receptor function of CD44v6 requires the molecule to be membrane-bound and to express the ectodomain of CD44v6 (see Figs. 15 and 16 in our companion paper (71)). Moreover, the CD44V6 shRNA/nanoparticle was well described in our previous paper (45) and was found to down-regulate the TGFβ1-stimulated α-SMA and collagen-1 production in fibrogenic lung fibroblasts (1). In this study, we determined whether CD44v6 peptides block co-receptor function of the CD44v6 isoform.
CD44V6 peptides block the co-receptor function of the CD44v6 isoform
We prepared three peptides from murine and human CD44V6 sequences (human V6-PEP and mouse v6-PEP-2) (box in Fig. 10A) and human V6-PEP-1 (outside the box). Bioinformatic analyses indicate that the Lys and Glu (Fig. 10A, green KE) of the V6-PEP peptide can interact with Glu119 and Ser49 of the TGFβRII activation site, and similar results have been reported for interaction of a TGFβ1 binding site in the TGFβRII activation site (102). Moreover, V6-PEP has an Arg residue (Fig. 10A, green R), which interacts with the TGFβRII by two hydrogen bonds, and this finding parallels a recent published study (102). The V6-PEP-1 peptide did not present any residue interacting in the same region, and for this reason, it was used as a control.
We tested whether these peptides would compete with endogenous CD44v6 and inhibit the activation of TGFβ1 with its receptor TGFβR. The human V6-PEP effectively inhibited TGFβ1-dependent activation of p-SMAD in IPFFbs (Fig. 10B) and BLMFbs (Fig. 10E), whereas the human V6-PEP-1 did not. Each of the 3 amino acids (Lys, Glu, and Arg, indicated in Fig. 10A) within the V6-PEP sequence were mutated, and the mutated (M) peptides were unable to inhibit the TGFβ1-induced activation of p-SMAD3 in the IPFFb cultures (Fig. 10C). Fig. 10 (E and F) shows that the mouse v6-PEP-2 inhibited the TGFβ1 activation of p-SMAD3 in 21dBLMFbs cultures, and mutations of the Glu and Lys (indicated in Fig. 10A) did not (Fig. 10F).
Interestingly, the peptides are not species-specific, and the addition of the murine peptide (v6-PEP-2) as well as the human V6-PEP to murine 21dBLMFbs cultures inhibited the activation of p-SMAD3 (Fig. 10, E and F). Similarly, the murine v6-PEP-2 also inhibited p-SMAD3 activation significantly in IPFFbs (data not shown). Therefore, it is likely that both the human and murine homologous sequences confer the co-receptor function of CD44V6-containing isoforms for TGFβ1/TGFβRI/p-SMAD3 activation, and we used the human V6-PEP in subsequent experiments.
Fig. 10D shows that a monoclonal antibody to TGFβ1 interacts with the V6-PEP peptide bound to peripheral blood mononuclear cell (PBMC) cultures equivalent to its binding to TGFβ1 bound to the isolated PBMC cultures. This indicates that the v6-PEP peptide mimics TGFβ1, confirming that the TGFβ1/TGFβR interaction on the isolated PBMCs has equally recognized both TGFβ1 and the V6-PEP peptide.
The V6-PEP/nanoparticle and the CD44V6 shRNA (V6 shRNA)/nanoparticle are non-cytotoxic and mediate an anti-inflammatory response
To demonstrate that the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle mimic TGFβ1 without cytotoxic action, we performed the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays using PBMC cultures. Fig. 11A shows that the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle tested at 1, 10, and 100 μg/ml concentrations did not affect PBMC viability and presented no significant differences from controls. To confirm whether the V6-PEP/nanoparticle and V6 shRNA/nanoparticle have the same ability as TGFβ1 to modulate an immune response, we stimulated PBMCs with either LPS, our V6 shRNA/nanoparticles, or V6-PEP/nanoparticles and measured TNFα and IL-10 production by an ELISA. The V6-PEP/nanoparticle and the V6 shRNA/nanoparticle were not able to induce TNFα (Fig. 11B) or IL-10 (Fig. 11C) production in the absence of inflammatory stimulus. PBMCs pretreated with the different concentrations of the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle followed by LPS stimulation for 24 h significantly decreased TNFα production when compared with LPS-treated cells (Fig. 11D). Interestingly, all concentrations of both the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle significantly increased anti-inflammatory IL-10 production when compared with controls (Fig. 11E). Furthermore, the efficiency of both nanoparticles in down-modulating TNFα and up-regulating IL-10 was sustained after a 48-h stimulus of PBMCs before LPS treatment for 24 h (data not shown). Overall, the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle are non-toxic, and they significantly reduced inflammatory responses. Thus, the peptide and shRNA against CD44V6 could down-regulate pro-inflammatory TNFα and up-regulate anti-inflammatory IL-10 production during inflammatory stimuli, a profile that is required in the treatment of inflammatory diseases (103).
Figure 11.

The CD44V6 shRNA (V6 shRNA) and the V6-PEP/nanoparticles are non-cytotoxic and mediate anti-inflammatory responses. A, cytotoxicity analysis (by MTT assay) results are shown for V6 shRNA/nanoparticles and V6-PEP/nanoparticles in PBMCs (leukocytes). The V6 shRNA/nanoparticle (pSico-CD44V6 shRNA/Tf-PEG-PEI plus FSP-1-Cre/Tf-PEG-PEI) and v6-PEP/nanoparticle (1–100 μg/ml) showed no statistical difference when compared with control (PBMCs with control peptide treatment; the data of other control (PBMCs with control shRNA/nanoparticle (pSico-scrambled shRNA/Tf-PEG-PEI plus FSP-1-Cre/Tf-PEG-PEI) treatment) show the same cytotoxicity as PBMCs with control peptide treatment (data not shown)), indicating that the tested concentrations did not have a cytotoxic effect. B–E, analyses of cytokine production by PBMCs after stimulation for 24 h. Analyses of pro-inflammatory TNF-α (B) and regulatory IL-10 (C) cytokines released in the absence of inflammatory stimulus are shown. Analysis of TNF-α (D) and IL-10 (E) cytokines released after PBMC stimulation with 1 μg/ml LPS are shown. PBMC cultures pretreated with the V6-PEP/nanoparticle or with V6 shRNA/nanoparticle at 1 μg/ml were able to decrease TNF-α production (*, p < 0.001). PBMC cultures pretreated with the V6-PEP/nanoparticle or with V6 shRNA/nanoparticle followed by stimulation with 1 μg/ml LPS resulted in a significant increase in IL-10 production when compared with control (± S.D.; n = 3; *, p ≤ 0.001).
The V6-PEP/nanoparticle and the V6 shRNA/nanoparticle inhibit the fibrogenic lung fibroblasts' ability to promote migration of mononuclear leukocytes across endothelial cell monolayers
Perivascular infiltrates of inflammatory cells are a hallmark of injured tissue in tissue fibrosis. Thus, we determined whether the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle can inhibit fibrogenic lung fibroblast-mediated mononuclear leukocyte migration across endothelial cell (EC) monolayer cultures. A three-cell co-culture system, together with complementary conditioned medium transfer experiments, was used to determine whether fibrogenic lung fibroblast cultures promote migration of mononuclear leukocytes across EC (C166 cells) monolayers. Fig. 12A shows that co-culture of ECs with 21dBLMFbs in the lower Boyden chamber increased leukocyte (PBMC) migration through the EC layer by ∼2.3-fold, which increased to ∼3.5-fold when the 21dBLMFbs were treated with the inflammatory stimulant LPS, compared with migration through the EC layer in the absence of the 21dBMLFbs in the lower chamber. In contrast, EC co-culture with normal MNLFbs showed only a minimal increase in leukocyte migration. Further, co-culture of ECs treated with LPS with untreated 21dBMLFbs did not increase leukocyte migration (Fig. 12A), possibly, at least in part, due to the greatly increased adhesion of the leukocytes to the LPS-treated ECs (Fig. 12B). No increase of leukocyte adhesion was observed with the other co-culture experiments (Fig. 12B), where leukocytes migrate substantially through the EC layer (Fig. 12A). Next, we showed (Fig. 12C) that treatment of the 21dBLMFbs with LPS followed by treatment with the V6-PEP/nanoparticle or with the V6 shRNA/nanoparticle effectively prevents the ability of the LPS treatment to stimulate PBMC migration through the EC layer. Similarly, treatment of human IPFFbs with LPS and then with the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle also prevents the subsequent human U937 (leukocytes) migration response on an HUVEC monolayer (Fig. 12D). These results indicate that the fibrogenic lung fibroblasts promote transendothelial leukocyte migration by altering the properties of the EC layer.
To investigate this further, a series of conditioned medium (CM) transfer experiments were done using IPFFb CM to measure U937 monocyte migration through the HUVEC layer. Fig. 12E shows that IPFFb CM substantially increased U937 leukocyte migration into the lower chamber. Further experiments investigated whether transmigration of leukocytes might involve chemokine mediators. IL-8, a C-X-C chemokine, has been implicated as a promoter of leukocyte extravasation in fibrosis (104), whereas the C-C class of chemokines predominantly affects the migration of mononuclear leukocytes (105) and includes MCP-1 (monocyte chemoattractant protein-1). Fig. 12E shows that IL-8 consistently induced a small increase in U937 cell migration compared with control wells, whereas MCP-1 induced transendothelial migration of U937 cells into the lower chamber equivalent to IPFFb CM alone. Further, preincubation of IPFFb CM with anti-MCP-1 antibody, with anti-CD44v6 antibody, with V6-PEP/nanoparticles, or with V6 shRNA/nanoparticles reduced U937 migration back to the level of IL-8 and near the level seen in control wells (Fig. 12E). These results indicate that release or activation of the MCP-1 C-C chemokine by fibrogenic fibroblasts provides another likely mechanism by which fibrogenic lung fibroblasts can promote transendothelial leukocyte migration and that this chemokine is at least partly regulated by CD44v6. This is further supported by evidence that hyaluronan binding with CD44 regulates MCP-1 in renal tubular epithelial cells (106). These data suggest that fibrogenic lung fibroblasts promote leukocyte migration across endothelial cell monolayers in tissue culture via a CD44v6-dependent mechanism that possibly includes MCP-1, which is likely to be relevant to the perivascular mononuclear leukocyte infiltrates characteristic of early fibrotic lesions (107).
Role of the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle in bleomycin-induced lung fibrosis
Previous studies have shown that intratracheal instillation of bleomycin induces epithelium injury that leads to fibrosis, which peaks 14–28 days post-injury followed by gradual resolution (108) (see timeline in Fig. 13A). Although CD44v6 was induced in mice starting at 3–7 days after bleomycin injury, its expression remained elevated in the fibrogenic phase (Fig. 2 in our companion paper (71)). Using our non-toxic and non-inflammatory shRNA/nanoparticle delivery method (45, 109), we evaluated the efficacy of targeting CD44v6 in the bleomycin fibrotic lung model by intratracheal delivery of v6 shRNA/nanoparticles and v6-PEP/nanoparticles starting on day 2 and every 2 days for 30 days after bleomycin injury (details of nanoparticle delivery are discussed under “Experimental procedures”). The preparation of nanoparticles and the delivery was done following our published method (45, 109). The nanoparticles contained 1) V6 shRNA/nanoparticle (genetic approach) and 2) the V6-PEP/nanoparticle (a small-molecule approach). Fig. 13B shows that both the CD44v6 knockdown by the V6 shRNA/nanoparticle and blocking by the V6-PEP/nanoparticle restored the capacity for resolving fibrosis by day 30 as determined by less Masson's trichrome blue staining for collagen (Fig. 9B), by significantly reducing CD44v6 and Nox4 mRNA and protein expression (Fig. 13, C and D), and by suppressing hydroxyproline (collagen) content (Fig. 13E). Fig. 13F shows that lung weights for the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle treatments were significantly lower compared with control shRNA/nanoparticle treatment. Fig. 13G shows that the mice given the V6-PEP/nanoparticle or the V6 shRNA/nanoparticle had less body weight loss and returned body weights to control levels by 30 days, in contrast to the mice receiving the control shRNA/nanoparticles. Fig. 13H shows that the survival of mice treated with the control shRNA/nanoparticle or control peptide/nanoparticle decreased during 21–45 days after treatment, whereas the mice treated with the V6-PEP/nanoparticle or the V6 shRNA/nanoparticle survived.
Figure 13.

In vivo targeting of CD44v6 by genetic modification, and with the CD44v6-blocking v6-PEP. A, the CD44v6 shRNA/nanoparticle (pSico-CD44v6 shRNA/Tf-PEG-PEI plus FSP-1-Cre/Tf-PEG-PEI), control (Con) shRNA/nanoparticle (pSico-scrambled shRNA/Tf-PEG-PEI plus FSP-1-Cre/Tf-PEG-PEI), or V6-PEP/nanoparticle were administered every other day from day 2 to day 30 by intratracheal delivery to the lungs of young mice during the onset of inflammation by bleomycin injury and then left untreated for another 15 days (see “Experimental procedures” for a detailed description of the shRNA/nanoparticle preparation and delivery method). Lung tissue was harvested at the indicated times up to 45 days after injury. The time course of fibrosis induction, treatment schedule, and resolution of fibrosis are shown. Nanoparticle preparation and delivery method was validated by our group previously in an intestinal/colon cancer murine model (45, 153). B, fibrosis was assessed by Masson's trichrome blue staining for collagen in sections of lungs isolated at day 24. Fibroblasts were isolated from lungs at 0, 7, 14, 21, and 45 days after bleomycin treatment in mice treated intratracheally with or without CD44V6 shRNA/nanoparticle, control shRNA/nanoparticle, or V6-PEP/nanoparticle and cultured ex vivo. C, total RNA was isolated from these fibroblasts, and real-time PCR analyses were done for CD44v6, Nox4, and Gapdh mRNA expression. D, whole-cell lysates were prepared from the isolated fibroblasts from the indicated days after bleomycin injury. Immunoblotting analyses were done for CD44v6, Nox4, and GAPDH protein expressions. E, quantitative hydroxyproline collagen assays were done for tissue samples from lungs collected at 0, 7, 14, 21, and 45 days after bleomycin injury in mice treated with or without CD44V6 shRNA/nanoparticle or control shRNA/nanoparticle. F–H, lung weight (F), body weight (G), and survival of mice over 45 days (H) were measured for the indicated days after treatment. The data in the experiments (A–H) are representative of three sets of three independent experiments with 10 mice in each experiment. Statistical analysis was with ANOVA (± S.D.; n = 3; *, p ≤ 0.001).
Discussion
IPF is a chronic progressive fibrosis interstitial lung disease. The underlying etiology in IPF is generally unknown. Currently, IPF affects about 100,000 people in the United States, with a high 5-year mortality among those with severe progressive disease (110, 111). Pirfenidone and nintedanib were each shown to reduce the average decline in lung function in randomized clinical trials (112). Both drugs have demonstrated efficacy in phase III clinical trials by retarding the rate of progression of IPF, but neither drug appears to be able to completely arrest disease progression (112). Thus, lung transplantation remains the only viable intervention in end-stage disease (111). Patients with IPF are recognized as a high unmet medical need with increasing prevalence within the aging population. Understanding the key cellular processes, including proliferation, acquisition of a contractile phenotype, extracellular matrix protein synthesis and secretion, and cell fate decisions, including cell survival and apoptosis, provides an opportunity to explore potential novel therapeutic approaches. One of the major profibrotic cytokines promoting fibrogenesis is TGFβ1. The pathologic changes in IPF (excessive accumulation of ECM and remodeling of the lung architecture) result when the balance between fibroblast proliferation and apoptosis is shifted toward accelerated proliferation and/or slowed apoptosis of fibroblasts, the primary ECM producers (113). A number of studies, including our own, have demonstrated enhanced autocrine TGFβ1 signaling in myofibroblasts, the principal cell type associated with the pathogenesis of disease (1, 26, 114–117). Our study (7) demonstrated that the HA receptor CD44V6 induces autocrine TGFβ1 signaling. However, the signal transduction cascades by which CD44V6 induces TGFβ1 responses were not elucidated.
NOX4 is a component of the TGFβ1 signaling pathway, with NOX4-derived H2O2 acting as a signaling molecule responsible for activating downstream cell survival signaling pathways, including the AKT and MAPK pathways (118, 119). Studies have demonstrated that TGFβ1 is responsible for the induction in NOX4 expression by pulmonary fibroblasts surrounding pulmonary vessels, suggesting that NOX4 is a component of the TGFβ1 signaling to generate extracellular H2O2 (16, 120, 121). In addition, NOX4 has been implicated in the profibrotic responses in lung myofibroblasts (16, 122). Despite its critical role in modulating profibrotic responses stimulated by TGFβ1 and NOX4, the regulation of CD44V6 expression and activity, as well as the mechanisms underlying their mutual regulation in fibrotic diseases, has not been reported.
The present results reveal the following. 1) Levels of CD44V6 and NOX4 are significantly elevated in IPF lung biopsies and in lung biopsies from bleomycin-induced lung injury (Figs. 1A, 3A, and 5A). Moreover, the expressions of CD44V6 and NOX4 in explanted normal lung fibroblasts were markedly up-regulated by TGFβ1 (Figs. 1B, 3B, 4 (A and B), and 5 (B, D, and F)). 2) CD44V6 regulates TGFβ1-induced NOX4 expression in ex vivo normal lung fibroblasts (Figs. 6C and 7E), and stimulation of NOX4 by CD44V6 involved the activation of TGFβRI kinase and canonical TGFβRI-independent p-SMAD3 serine threonine kinase activation, which then directly stimulated NOX4 expression (Fig. 7, B, C, and E). 3) Nox4 also enhanced TGFβ1-induced CD44v6 expression via Ap-1 activation (Fig. 9, B and C). 4) Nox4 is directly involved in regulating the increased synthesis of Has2 in TGFβ1-treated murine lung fibroblasts (Fig. 9D). 5) TGFβ1 up-regulates Hyal3 and Hyal1 hyaluronidases predominantly, suggesting that degradation of HA in TGFβ1-treated lung fibroblasts is also regulated by Nox4 (Fig. 9H). 6) Interaction of LMWHA with the increased CD44v6 expression induced in TGFβ1-treated lung fibroblasts that depends on Nox4 has an important role in cell proliferation, differentiation, and ECM protein and contractile protein expressions in the ex vivo murine and human lung fibroblasts (Figs. 4 (A and B), 5B, 6A, and 8). 8) NOX4-increased ROS production activated AKT, MMP2, and MMP9 expressions, which mediated the cell invasion of lung fibroblasts (Fig. 6, B and D). 9) CD44v6-and HA-dependent Nox activity and intracellular ROS expressions are significantly up-regulated in the ex vivo wild-type MNLFbs compared with Nox4-null MNLFbs and CD44v6-null MNLFbs (Fig. 9, E and F). 10) Irrespective of the mechanism of co-receptor function, V6-PEP mimicked TGFβ1 by binding to a TGFβ1 monoclonal antibody, confirming that the PBMC cell surface TGFβR is also equally recognized by V6-PEP and TGFβ1 (Fig. 10D), and inhibited co-receptor function of CD44v6 through inhibition of p-SMAD3 activation (Fig. 10, B, C, E, and F). 11) Both V6-PEP and co-receptor are not toxic (Fig. 11A) and are anti-inflammatory by inhibiting TNFα production (Fig. 11, B and D), by increasing LPS-induced IL-10 production (Fig. 11E), and by abrogating transendothelial migration of leukocytes via fibrogenic lung myofibroblasts (Fig. 12, A, C, and E). 12) Inflammatory mediators of fibrogenic fibroblasts induce leukocyte migration through endothelial cell layers, and this function is mediated by CD44v6 and MCP-1, indicating that in addition to their role in ECM homeostasis, fibroblasts may influence inflammatory disease processes through an effect on CD44v6-mediated leukocyte trafficking, which may involve MCP-1. 13) GKT137831 (1 μm), a pharmacological inhibitor of NOX1/NOX4-based NADPH oxidases, not only inhibits TGFβ1-induced H2O2 but also attenuates TGFβ1-induced myofibroblast differentiation measured by collagen gel contraction (Fig. 8). 14) In the bleomycin-challenged mouse model of pulmonary fibrosis, the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle strategies targeted CD44V6 induction and TGFβ1 activity, respectively, and suppressed fibrosis by inhibiting gene transcription of profibrotic and proinflammatory mediators and of ECM proteins in the lungs.
HA is synthesized by three HA synthases (33); however, HAS2 is the major HA-synthesizing enzyme in IPF, based on two lines of evidence. First, targeted overexpression of Has2 by myofibroblasts produced an aggressive phenotype, leading to severe lung fibrosis and death after bleomycin-induced injury (19). Second, conditional deletion of Has2 in mesenchymal cells abrogated the invasive fibroblast phenotype, impeded myofibroblast accumulation, and inhibited the development of lung fibrosis (19). Our results indicate that increased Has2 expression and activity in TGFβ1-treated lung fibroblasts is dependent on NOX4 activation (Fig. 9D). Furthermore, our data indicating that Nox4 regulates Hyal3 and Hyal1 expression in MNLFbs treated with TGFβ1 are significant because increased HA production during pathological conditions depends on the turnover of HA (i.e. the sum of its synthesis and catabolism). This observation is substantiated by the published studies indicating that Hyal3-null mice do not show HA accumulation (123) and Hyal3 contributes to HA metabolism by augmenting the activity of Hyal1 (124). Hyal3 is also enhanced by cytokines in chondrocytes (125) and also in vascular smooth-muscle cells in atherosclerotic disease (90). In a recently proposed model, the breakdown of HA is initiated extracellularly by HYAL3 and then continued intracellularly by HYAL1 (126). In addition, HA can also be degraded into smaller fragments in a non-enzymatic way by exposure to ROS intermediates (127), which is believed to be an important mechanism for generating HA fragments at sites of inflammation (128). It is generally accepted that free radicals, especially the highly reactive hydroxyl radical, have an important role in the degradation process of HA (127–132), but the direct action of ROS on HA turnover and degradation has received scant attention to date.
Our data show that the increase in TGFβ1-induced NOX4 activity by HA500 and HA150 (Fig. 9, E and G) is due to enhanced CD44v6 expression (Fig. 7D and Figs. 15 and 16 in our companion paper (71)) caused by TGFβ1 pretreatment. We cannot rule out the possibility that HA regulates TGFβ1-induced NOX4 activity through binding to CD44v6, because HA regulates Rac1 GTPase (95), which is a regulatory subunit of NADPH oxidase. We found that CD44-null MNLFbs and Nox4-null MNLFbs suppressed ROS production (Fig. 9F), and this finding is supported by the studies in which HA-CD44 interaction has been reported to induce ROS production (95), and down-regulation of p47phox or p67phox decreased ROS levels (95). Interestingly, our studies indicate that Nox4 regulates CD44v6 gene expression (Fig. 9C); Nox4 increases Has2 expression (Fig. 9D), which is known to produce HA; and LMWHA enhances Nox4/ROS activity (Fig. 9E), suggesting the presence of a positive-feedback loop between HA and NOX4 activation. These data support a key role for NOX4 and HA in the pathogenesis of lung fibroblasts in response to TGFβ1. Our results thus suggest that the NOX4-dependent expression of CD44v6 and HA in the fibrogenic lung fibroblasts and ROS-dependent HA degradation and release of LMWHA lead to increased interaction of CD44v6 and LMWHA and induction of pro-inflammatory cytokine expression in the fibroblasts.
Increased myofibroblast differentiation, invasion, proliferation, and synthesis of pro-fibrogenic molecules, including NOX4, CD44v6, and ECM protein (collagen-1 and fibronectin), as seen in Figs. 4A, 5 (B–G), and 6 (A–D) are important events in the development of fibrogenic processes. Serum-stimulated wild-type IPFFbs proliferate more rapidly than NOX4- or CD44V6-depleted IPFFbs (Fig. 6A). Our data also indicate that the importance of NOX4-dependent CD44V6 expression in pulmonary fibrosis is supported by our findings that CD44V6 shRNA significantly decreased TGFβ1-induced α-SMA, fibronectin, collagen-1, and NOX4 expressions, endothelial cell layer invasion, and cell proliferation and differentiation of IPFFbs and TGFβ1-stimulated HNLFb (Figs. 5 (B–G), 6 (A–D), and 8). We also found that soluble CD44V6 levels in the plasma of 21-day bleomycin-treated mice were increased compared with PBS-treated mice (data not shown), which further indicates that activation of NOX4 in fibrogenic settings releases soluble CD44V6 by proteolytic cleavage of membrane-anchored CD44V6. This may well be from the increased high levels of matrix metalloproteinases in IPFFbs (Fig. 6B).
Similar to a feedback loop between NOX4 and HA (Fig. 9, D and E), our studies further indicate that CD44V6 regulates NOX4 expression and function through TGFβ1-induced p-SMAD3 activation (Fig. 7, B and C). NOX4 also enhanced TGFβ1-induced CD44v6 expression via Ap-1 activation (Fig. 9, B and C), and CD44V6 regulates NOX4 expression and activity (Figs. 6C and 9 (E and G)), suggesting another positive-feedback loop between CD44V6 and NOX4 activation. Taken together with the previous demonstration (1) that augmented CD44V6 expression in interstitial lung disease fibroblasts sensitized them to the profibrotic effects of TGFβ1, the present results provide previously unreported insight into the mechanism of fibrosis in IPF and indicate a fundamental role of HA-CD44V6-mediated NOX4/ROS in mediating this process. In this context, it is noteworthy that pharmacologic inhibition of NOX1/NOX4 in TGFβ1-treated lung fibroblasts not only attenuated ROS generation but also decreased fibroblast differentiation to myofibroblasts (Figs. 7G and 8).
To explore the role of CD44V6 in fibrotic responses to injury of the mammalian lung, we used a murine model of acute lung injury. It has been reported that histological hallmarks, such as intra-alveolar buds, mural incorporation of collagen, obliteration of the alveolar space, and epithelial injury with subsequent mesenchymal cell activation and fibrosis, are present in bleomycin-treated animals similar to IPF patients (133). This observation has led to the assumption that bleomycin reproduces typical features of the human disease, including TGFβ1 up-regulation and activation, and myofibroblast differentiation and activation are recapitulated in this animal model. Therefore, the use of this model has become very popular. Further, the bleomycin model has the advantage that it is quite easy to perform and is widely accessible and reproducible, therefore fulfilling important criteria expected from a good animal model (16, 133). We first showed that CD44v6 expression is induced during the fibrogenic phase of bleomycin-induced lung injury in a time-dependent manner, increasing from day 3 up to day 28 (Fig. 2 in our companion paper (71) and Fig. 13 (C and D)), whereas NOX4 expression was induced from day 7 up to day 28 (Fig. 13, C and D), supporting a functional relationship between Nox4 and CD44v6 expression, myofibroblast activation, and fibrosis after lung injury. In contrast, expression of the Nox2 isoform, which is predominantly expressed in phagocytic cells, was increased on day 14 and returned to baseline levels at day 45, when inflammatory responses through synthesis of Nox4 had subsided (Fig. 3B).
We also examined the effects of targeted suppression of CD44v6 induction with in vivo CD44v6 shRNA/nanoparticle and v6-PEP/nanoparticle treatments compared with control shRNA/nanoparticles in the bleomycin animal model of lung injury and fibrosis. We found 1) that these nanoparticles have no off-target effects (i.e. they produce IFNα neither in in vitro (see Fig. 12H in our companion paper (71)) nor in in vivo settings (Fig. 2, C and D)); 2) that they are non-toxic (Fig. 11A), promote anti-inflammatory responses (Fig. 11, B and D), and abrogate fibrogenic lung fibroblast-mediated leukocyte migration across an endothelial cell layer (Fig. 12, C and D); and 3) that these antagonists targeting CD44V6 up-regulate IL-10 production (Fig. 11E) during inflammatory stimuli, a profile that is required in the treatment of inflammatory diseases for immune suppression (103, 134).
These findings in Figs. 10–12 explain the reasons that led us to select these novel lung fibroblast-specific CD44V6 shRNA/nanoparticles through FSP-1-Cre/nanoparticle-mediated transactivation of the V6-PEP/nanoparticle and the V6 shRNA/nanoparticle for intratracheal delivery in the bleomycin-treated mouse model. CD44V6 knockdown by lung fibroblast-specific V6 shRNA/nanoparticle and by competing for endogenous CD44v6 co-receptor function by v6-PEP/nanoparticle mediated a marked antifibrotic effect, as determined by Masson's trichrome staining for collagen (Fig. 13B), by biochemical analyses of hydroxyproline content (Fig. 13E) in whole lung homogenates, by inhibiting increased lung weight (Fig. 13F), by restoring body weight (Fig. 13G), and by mouse survival (Fig. 13H).
Our data indicating that lung fibroblast-specific genetic inhibition of CD44V6 markedly decreases NOX4 expression in the lung fibroblasts (Fig. 13, C and D) validate our hypothesis that HA-CD44V6 activation promotes fibrosis via NOX4-dependent processes. To our knowledge, this is the first study to show that a specific peptide inhibitor of CD44V6 activity significantly decreases profibrogenic processes in bleomycin-induced lung fibrosis in mice. This inhibitor may have therapeutic potential because it inhibited Nox4, Has2 expression, and oxidative stress in lung mesenchymal cells in pulmonary fibrosis.
In summary, our data provide insight into the molecular mechanisms by which CD44V6-mediated NOX4 activation promotes fibrogenic lesion formation. NOX4 mediates a TGFβ1-induced HA synthesis by regulating both HA synthetic and degradation enzymes, and a positive-feedback loop between HA and NOX4 increases oxidative stress and enhances fibrosis (model in Fig. 14). On the other hand, CD44v6 regulates NOX4 expression and function via TGFβRI/p-SMAD3 activation, and NOX4 regulates CD44v6 gene transcription via Ap-1 activation. Further, another positive-feedback loop between CD44v6 and NOX4 increases oxidative stress and enhances fibrosis. Interaction between increased CD44v6 and HA resulting from NOX4 activation in profibrogenic background induces cytokine and ROS production, which promote pulmonary fibrosis. Taken together, these results demonstrate that the CD44V6 is itself a target of TGFβ1 signaling in lung fibroblasts. Enhanced CD44V6 expression probably contributes to NOX4-mediated persistence and progression of the fibrotic response.
Figure 14.
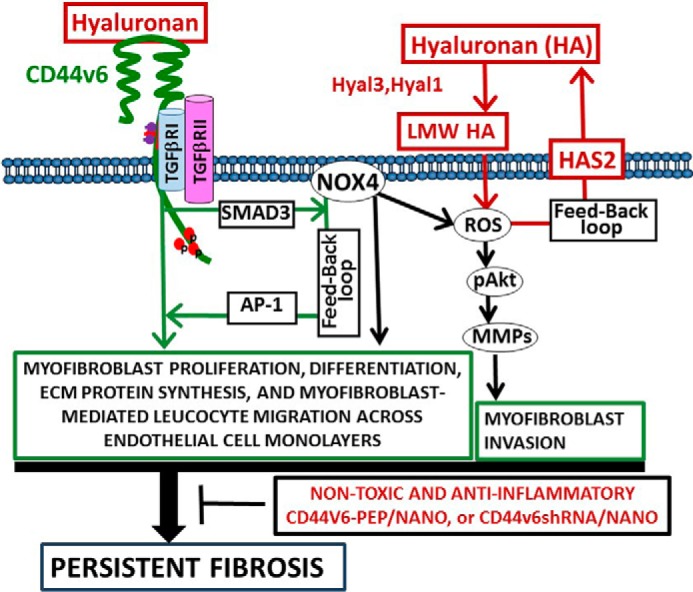
Proposed model for persistent fibrosis in mice after lung injury. NOX4-derived ROS facilitates TGFβ-mediated fibrosis by inducing differentiation of fibroblasts into myofibroblasts and by synthesis of extracellular matrix proteins. Our studies indicate that NOX4 increases HAS2 expression, and LMWHA enhances NOX4/ROS activity, which provides evidence for a positive-feedback loop between HA and NOX4 activation. ROS then induces cell invasion through the Akt → MMP pathway. Our study also provides evidence that in response to lung injury, CD44V6 induces NOX4 through regulation of TGFβ1/SMAD3 signaling in myofibroblasts, and NOX4 also regulates HAS2 production as well as AP-1-mediated CD44V6 expression. This suggests that the CD44v6 variant and HA are up-regulated by TGFβ1/SMAD3 signaling through a feedback loop requiring the presence of NOX4. These studies thus show the involvement of CD44V6 in TGFβ-induced NOX4 expression, myofibroblast activation, and profibrotic responses.
As discussed above, we evaluated the role of CD44v6 in mediating the fibrogenic response in several ways. However, CD44-null mice did not show significant protection from the development of pulmonary fibrosis (19). Compensation of proteins by members of the same family has been widely proposed to explain the lack of phenotype of several knock-out mice. However, the effect on protection from the development of pulmonary fibrosis is more pronounced when the α-Sma-Has2 transgenic mice are bred with the CD44-null mice (19). However, our results indicate that progressive fibrosis requires the generation of an invasive myofibroblast phenotype that requires TGFβ1 where NOX4, HA, and CD44V6 are critical downstream components of the TGFβ1-induced fibrogenic response (Figs. 7 (B–G) and 9 (A, B, D, F, and G)). Moreover, NOX4 and CD44V6 regulate MMP2 and MMP9 for the emergence of the invasive phenotype (Fig. 6, B and D). Our studies also indicate that fibrogenic lung fibroblasts modulate leukocyte-endothelial cell interactions in ways that facilitate migration of leukocytes across EC layers, at least partly via a CD44V6-dependent mechanism involving MCP-1 activation. These results provide further support for the view that, in addition to their role in extracellular matrix homeostasis, fibroblasts may influence inflammatory disease processes through an effect on leukocyte trafficking. The findings that coordinated gene expression with up-regulation of matrix-degrading enzymes (MMPs), inflammatory soluble mediators like MCP-1, as well as fibroblast activation through CD44V6-NOX4 signaling occur in both mouse and human pulmonary fibrosis, suggesting that the approach to abrogate CD44V6 could be used to identify therapeutic targets. The proof-of-principle experiments suggest that the non-toxic and anti-inflammatory V6-PEP/nanoparticle or CD44V6 shRNA/nanoparticle have no off-target effects and attenuated lung fibrosis in mice in vivo at the time of bleomycin injury. Therefore these proof-of-principle studies (Figs. 11–13) represent an innovative therapeutic strategy to control tissue fibrosis.
Conclusion (Fig. 14)
A network consisting of cytokines, oxygen radicals derived from inflammatory cells, closely controls activation of several ECM components to induce the progression of fibrosis. The activated inflammatory cells that accumulate in the lower airways release harmful amounts of ROS, which results in lung injury and proliferation of fibroblasts in alveolar walls (135). In this study, we show that bleomycin-induced mouse lung fibroblasts express the fibrosis-associated protein CD44V6 (1), which can facilitate presentation of TGFβ1/p-SMAD3 to its receptor TGFβRI for induction of NOX4 and associated impaired oxidant-antioxidant balance during the fibrogenic phase. NOX4 also enhanced TGFβ1-induced CD44V6 expression via AP-1 activation and HAS2 production. Furthermore, we demonstrate that CD44v6 promotes the feedback up-regulation of NOX4 through TGFβ1-TGFβRI signaling. Similarly, HA and NOX4 form a feedback loop to up-regulate ROS, which induces cell invasion through a ROS/AKT/MMP2/9 pathway. Because bleomycin is widely used in experimental models of human diseases resembling pulmonary fibrosis, in the present study, we investigated the possible protective effect of inhibiting CD44V6 against bleomycin-induced oxidative injury in the mouse lung. Our results together with biochemical and signaling data agree with this hypothesis, because the bleomycin-induced increase in fibrotic activity, as assessed by the lung collagen content, is also reduced by the lung fibroblast-specific release of CD44V6 shRNA/nanoparticle and the V6-peptide/nanoparticle treatments. In conclusion, the findings of the present study demonstrated for the first time that blocking CD44V6 has an additional protective effect on the source(s) of inflammation that induce production of NOX4 with subsequent ROS generation and deposition of pathological extracellular matrix components that result in pulmonary fibrosis .
Experimental procedures
Materials
Dulbecco's modified Eagle's medium (DMEM) low glucose, glutamine, and pyruvate were from Life Technologies, Inc. Fetal bovine serum was from Atlanta Biologicals, and l-glutamine, gentamicin sulfate, and amphotericin B were from GE Healthcare. Actinomycin D, cycloheximide, Nonidet P-40, EGTA, sodium orthovanadate, glycerol, phenylmethylsulfonyl fluoride, leupeptin, pepstatin A, aprotinin, and HEPES were purchased from Sigma. Recombinant human TGFβ1 was purchased from R&D Systems (Minneapolis, MN). The antibodies against NOX4, NrF2, CD44v6, α-SMA, SMAD2/3, phospho-ERK, ERK, GAPDH, β-actin, β-tubulin, horseradish peroxidase-linked anti-rabbit and anti-mouse antibodies, and luminol reagent were purchased from commercial sources (Santa Cruz Biotechnology, Abcam, Ebioscience, Thermo Fischer, Cell Signaling Technology, and Southern Biotechnology Associates Inc.). Silencing siRNA and shRNA oligonucleotides were synthesized by IDT Technology. GKT137831 was provided by Genkyotex SA (Geneva, Switzerland). GKT137831 is an efficient inhibitor of both NOX1 and NOX4 isoforms (Ki ∼ 100–150 nm, Emax > 90%) with substantial selectivity toward NOX2 in cell-free assays (Ki = 1500 nm, Emax = 70%). GKT137831 was solubilized in DMSO and was used at 1 and 5 μm.
Hyaluronidase isolated from Streptomyces hyaluronlyticus, which contains endotoxin, was from EMD Millipore (catalog no. 389561) and was used to digest hyaluronan. The hyaluronidase was removed by digesting with Proteinase K followed by denaturation at 95 °C for 5 min. Endotoxin was removed by extracting twice with 1% Triton X-114. The aqueous phase contains the degraded hyaluronan (136). The endotoxin level was measured by an end-point chromogenic limulus amebocyte lysate assay.
Hyaluronan from Hyalose (Oklahoma City, OK) was prepared by enzymatic synthesis of specific molecular mass and was free from endotoxin. Pharmaceutical-grade hyaluronan from Life Core Biomedical (Chaska, MN) was also used.
Management of animals, human lung samples, and lung fibroblasts
Six-week-old mice (C57BL/6 strain) were obtained from the Jackson Laboratories. Mice (0.05 units/20 g of animal) were anesthetized with isoflurane. Intratracheal administration of bleomycin (0.025 units in PBS) was used to induce lung injury as described previously (22). All animal care and experimentation were done in accordance with the institutional animal care and use committee protocol (AR 3220) approved by the Medical University of South Carolina according to the rules of the National Institutes of Health. Lung tissues at 21 days after PBS or bleomycin instillations were perfused with z-fix (Anatech Ltd., Battle Creek, MI) and processed for paraffin sections. Fibroblasts from control mouse lungs (NMLFbs) and from 14- and 21-day bleomycin-injured mouse lungs (14dBLMFbs and 21dBLMFbs) were isolated as described in our companion paper (Fig. 1 in Ref. 71).
Cell culture
HNLFbs and lung fibroblasts from IPF patients were obtained from Dr. Carol Feghali-Bostwick (Medical University of South Carolina). Briefly, lung tissues were diced (∼0.5 × 0.5-mm pieces) and cultured in DMEM with normal glucose, glutamine, and pyruvate (Life Technologies) supplemented with 10% fetal bovine serum, 2 mm l-glutamine, gentamicin sulfate (50 μg/ml), and amphotericin B (5 μg/ml) at 37 °C in 10% CO2. The medium was changed every 3 days to remove dead and non-attached cells until fibroblasts reached confluence. Monolayer cultures were maintained in the same medium. Lung fibroblasts were used between the second and fourth passages in all experiments. The purity of isolated lung fibroblasts was determined by crystal violet staining and by immunofluorescence staining using monoclonal antibody 3C4 against human fibroblasts as described previously (137). All of the treatments and transfection experiments were done with cells that were serum-starved for 24 h.
Cell lysis and immunoblotting
Isolated primary fibroblasts were cultured until they were confluent. Cells were washed twice at 4 °C with PBS, harvested with 0.05% Versene, and then washed in cold PBS again as described previously (1, 42, 43, 45, 48, 49, 51–54, 138). The cells were pelleted by centrifugation at 5000 × g for 2 min at 4 °C. The pellets were treated with the lysis buffer containing 1% Nonidet P-40, 0.5 mm EGTA, 5 mm sodium orthovanadate, 10% (v/v) glycerol, 100 μg/ml phenylmethylsulfonyl fluoride, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 1 μg/ml aprotinin, and 50 mm HEPES, pH 7.5. The lysates were clarified by centrifugation at 12,000 × g for 10 min at 4 °C and then stored at −80 °C as described previously.
For SDS-PAGE, the denatured cell lysates were loaded onto a 4–12% gradient polyacrylamide gel at 15–30 μg of protein/lane in an Invitrogen minigel apparatus. Proteins were transferred to nitrocellulose membranes and blocked for 1 h with 5% nonfat dry milk in Tris-buffered saline containing 0.1% Tween 20 followed by washing in the same Tris-Tween buffer. The membranes were probed with the appropriate antibody diluted in Tris-buffered saline containing 5% bovine serum albumin (for polyclonal antibodies) or 5% nonfat dry milk (for monoclonal antibodies) followed by treatment with peroxidase-linked secondary antibodies and luminol reagents. The proteins on the blots were detected with antibodies for NOX4, NOX2, fibronectin, collagen-1, CD44V6, α-SMA phospho-Akt, and Akt (42, 43, 45, 48, 54, 138). β-Tubulin and β-actin were used as internal standards. Sizes of proteins were estimated from prestained molecular weight standards electrophoresed in the same gel as the samples. Immunoreactive bands were quantified by densitometry. Each protein sample was analyzed from at least three independent experiments from each set of fibroblasts.
Immunohistochemical staining in lung sections
Lung sections from bleomycin- and saline-treated mice and from IPF patients and normal subjects were deparaffinized using standard procedures and permeabilized with 0.1% Triton X-100 in PBS. NOX4 and CD44V6 were localized in sections by immunohistochemical staining using NOX4 and CD44V6 antibodies from Abcam following the manufacturer's protocols. As a negative control, the primary antibody was replaced with non-immune rabbit IgG, and no staining occurred.
CD44V6 shRNA and V6-PEP blocking peptide nanoparticle delivery in the bleomycin injury lung model of mice
The sequences of the two V6 human peptides are KEQWFGNRWHEGYR (V6-PEP) and QATPSSTTEETATQ (V6-PEP-1). The sequence of the v6-murine (rat) peptide is KEKWFENEWQGKNP (v6-PEP-2). These peptides and the control peptide were prepared by Dharmacon Inc. The pSicoR-V6 shRNA/nanoparticles, the control non-targeting pSicoR-Scrambled shRNA/nanoparticles (control shRNA/nanoparticle), the V6-PEP/nanoparticles, and the control peptide/nanoparticles were prepared as described in our published paper (45). For in vivo studies, the peptide/nanoparticle and shRNA/nanoparticle were used at doses of ∼100 μg/mouse by intratracheal delivery with bleomycin in a total volume of 50 μl. The dose for shRNA/nanoparticles and for peptide/nanoparticles (∼100 μg/mouse) was determined from a pilot dose-response experiment (data not shown and as described in our previous study (45)).
RNA silencing
The shRNA-coding nucleotide sequences of the genes were obtained from the NCBI, National Institutes of Health, website (www.ncbi.nlm.nih.gov),4 and we designed hairpin shRNAs to target the transcript sequences using the Broad Institute GPP Web Portal (http://portals.broadinstitute.org/gpp/public/).4 Sequences for cloning in pSico/pSicoR vectors were designed following the MIT Jacks Laboratory website (http://web.mit.edu/jacks-lab/protocols/pSico.html).4 For example, the sense sequence for CD44v6 for cloning in pSico/pSicoR vector was 5′-TTAGTAGTACAACGGAAGAAACTTCAAGAGAGTTTCTTCCGTTGTACTACTA-3′.
Similarly, the scrambled shRNA sequence obtained from the website was cloned in pSico/pSicoR vector. c-Fos shRNA and c-Fun shRNA were purchased from Dharmacon, as were the appropriate scrambled controls. Double-stranded oligonucleotide cassettes for control shRNA, CD44V6 shRNA, and NOX4 shRNA were prepared. The linearized pSicoR vectors were ligated to the double-stranded oligonucleotide cassettes (45). The resulting pSicoR-CD44v6 shRNA (v6 shRNA) and pSicoR-NOX4 shRNA transfectants constitutively silence the respective CD44V6 and NOX4 genes in the cells. pSicoR-scrambled shRNA (control shRNA) transfectants were used as control to the above shRNA transfectants. In the in vivo experiments, we used these shRNAs in Tf-PEG-PEI (nanoparticles) following our published paper (45).
Confirming the specificity of shRNA experiments
To confirm the shRNA knockdown efficiencies, in specific experiments, we used more than one shRNA. We also confirmed the knockdown experiments, comparing the knockdown effects of shRNAs for CDS either with those of NCDS (as proper negative controls) or with rescue of the observed shRNA-mediated knockdown phenotype by expression of a resistant form of the targeted mRNA. This is performed by 1) transfecting the cells with specific shRNAs for the CDS of the target gene or 2) co-transfecting the shRNA (CDS) for the target gene with or without corresponding cDNA transfection or by the indicated shRNA-mediated knockdown and corresponding KI gene transfection (see Table 1 and Fig. 12 (A–F) in our companion paper) (71). Total cell lysates were examined by Western blot analysis for the indicated proteins and β-tubulin. Total mRNA were analyzed for the indicated mRNAs by RT-PCR and real-time PCR. Synthetic shRNAs that are 21–23 nucleotides in length have been shown to effectively silence specific target genes by promoting mRNA degradation in cultured mammalian cells and mice (45, 139, 140). However, in cultured cells, one potential source of off-target effects by either transfected shRNA duplexes or endogenously expressed shRNAs is the unintentional activation of the interferon response (141–143). Previous work has shown that nonspecific gene target effects that include off-target gene suppression and up-regulation of type I IFN response are induced by dsRNAs ≥ 30 bp in length (144). There are also recent reports of shRNA-induced stimulation of Toll-like receptors (TLR3 (dsRNA) and TLR9 (unmethylated CpG)) and downstream IFNα and IFNβ responses (145). Despite the highly publicized in vivo miRNA-related toxicity of shRNA, it has been shown that shRNA has several advantages over siRNA: fewer off-target effects, multiple-target-silencing capacity without a corresponding increase in dose, durability of effect, and inducible application (146, 147). Because we use siRNAs and shRNAs extensively to study CD44V6-induced fibrogenic functions, we follow several steps (as shown in our companion paper (71) and in Fig. 2 (A–F)) that can avoid this off-target problems as well as confirm the specificity of the shRNAs used in this study. Specifically, the confirmation of knockdown by siRNA and shRNAs was done by using more than one shRNA by demonstrating that the expression level of the target mRNA was substantially reduced, whereas the level of expression of a control shRNA (scrambled shRNA)-transfected mRNA and/or protein was unaffected (Figs. 2 (A, B, E, and F), 4 (A and B), 5 (B–G), 6 (B–D), and 7 (D–G)) and in our companion paper (71).
In vivo mouse studies for measuring IFN
C57BL/6 mice (6–8 weeks of age; Jackson Laboratories) were used for the shRNA injections. The shRNAs were formulated in D5W (5% (w/v) glucose in water). To examine the effect of various types of administration, mice received 2.5 mg/kg shRNA either i.p. or through the tail vein via LPTV (1% (v/w)) or HPTV (10% (v/w)). The HPTV method of administrating shRNA provides for cellular uptake and gene down-regulation in the livers of mice (140, 148). Mouse tails were warmed with a heating pad before injection of a 0.2-ml (for LPTV) or a 2-ml (for HPTV) volume (per 20 g of mouse weight) over ∼5 s. To measure plasma cytokine levels, blood was harvested from mice 2 h after injection by cardiac puncture, and plasma was isolated using Microtainer tubes (BD Biosciences). Preliminary experiments compared the IFN-α response with poly(I:C) at 1, 3, 6, 12, and 24 h after injection, and the maximum response was consistently observed at 3 h. Therefore, data from the 3-h time point after shRNA treatment were used. IFN-α levels were measured by ELISA according to the manufacturer's instructions.
Transient transfection using HNLFbs and IPFFbs
All transfections were done using Lipofectamine (Invitrogen) in cultures at ∼75% confluence. After transfection, the cultures were grown for another 72–96 h for analyses.
mRNA expression analysis by RT-PCR
Preparation of mRNA from tissue samples used a commercially available mRNA purification kit. cDNA was synthesized with mRNA as template for an oligo(dT)-primed reaction catalyzed by reverse transcriptase. The quality of mRNA preparations and cDNA syntheses was checked by including GAPDH- or β-actin-specific primers as an internal RT-PCR control. All cDNA sequences were obtained from the GenBankTM database. RT-PCR used human gene-specific sense and antisense primers based on sequences published in GenBankTM. PCR was done for 30 cycles for GAPDH, 32 cycles for p22phox, and 36 cycles for all other primers, with amplification at 95 °C for 1 min, 58 °C for 1 min, and 72 °C for 2 min, followed by an extension step at 72 °C for 10 min. For semiquantitative RT-PCR, PCR-amplified DNA was separated on 1.2% agarose gels, stained with ethidium bromide, and visualized and photographed under UV light.
Real-time PCR of CD44v6 in MNLFbs and HNLFbs
Total RNA was isolated from MNLFbs and HNLFbs after various treatments, and transfections were done as mentioned in the figure legends for each specified experiment using the RNeasy minikit (Qiagen) according to the standard protocol provided by the manufacturer, with on-column DNA digestion. RNA integrity and concentration were analyzed using Bioanalyzer (City State), and 1 μg of RNA was reverse-transcribed into cDNA using the First Strand cDNA synthesis kit from Roche Applied Science. SYBR Green (Bio-Rad) was used for all real-time PCR assays. Amplification was done with the real-time PCR analyzer (Bio-Rad). The PCR mixture (25 μl) contained 12.5 μl of 2× SYBR Green PCR Master Mix (Bio-Rad), 5 μl of diluted RT product (1:20), and 0.5 μm sense and antisense primer sets. The primers used for human and mouse genes are shown in Table 1. The real-time PCR assays were done in three individual experiments with triplicate samples using standard conditions. After sequential incubations at 50 °C for 2 min and 95 °C for 10 min, respectively, the amplification protocol consisted of 50 cycles of denaturing at 95 °C for 15 s, annealing, and extension at 60 °C for 60 s. The standard curve was made from a series dilution of template cDNA. Expression levels of α-SMA, NOX4, collagen-1 (COL1A1), and CD44V6 mRNAs were calculated after normalization with the housekeeping gene GAPDH.
AP-1 transcription factor-binding activity
Nuclear extracts were prepared with the NucBuster protein extraction kit (Novagen) according to the manufacturer's protocol. AP-1 DNA-binding activity was measured using the TransBinding AP-1 assay kit (Panomics). Briefly, nuclear extracts were incubated with biotinylated AP-1 consensus-binding sequence oligonucleotides, and the complexes bound to the oligonucleotides were detected using a primary AP-1 antibody and a secondary HRP-conjugated antibody. AP-1 activity was assayed by measuring the absorbance at 450 nm.
Transient transfection
All transfections were done using Lipofectamine (Invitrogen) in cultures that were ∼75% confluent. After transfection, the cells were cultured for 72–96 h prior to analyses.
Cell proliferation assays
Cell proliferation was measured by a BrdU cellular ELISA kit based on the incorporation of the pyrimidine analogue BrdU into the DNA of proliferating cells that are cultured in microtiter plates. After its incorporation into DNA, BrdU in the cells was detected by anti-BrdU monoclonal antibody. Fibroblasts (20,000) were cultured in 96-cell plates at 37 °C. BrdU (20 μm) was added to wells during the final 2–18 h of culture. Cells were fixed and permeabilized, and the DNA was denatured to enable antibody binding to the incorporated BrdU. Detector mouse anti-BrdU monoclonal antibody was pipetted into the wells and allowed to incubate for 1 h. unbound antibody was removed, HRP-conjugated goat anti-mouse IgG was added, and then tetramethylbenzidine was added. The absorbance was detected at 450 nm and was quantified in a microplate reader (BioTek, Winooski, VT).
Migration of leukocytes through endothelial cell cultures
Leukocytes patrol the vascular system, and their rolling, firm attachment, and subsequent migration across endothelial walls are necessary for their recruitment to sites of inflammation. This process involves a multistep cascade consisting of leukocyte rolling, adhesion, and transmigration. A quantitative assay for leukocyte transendothelial migration has been described using a modified Boyden chamber system (149). This is a two-chamber system with a porous membrane providing an interface between the two chambers. Mouse endothelial cells (C166 from ATCC) were cultured on top of the porous membrane that was coated with an ECM protein. Isolation of PBMCs (leukocytes) from mice was done by density gradient centrifugation of buffy coats using Lympholyte-M (Atlanta Biologicals) gradient medium, according to the manufacturer's instructions. Isolated lung fibroblasts from mice on days 0 (MNLFbs) and 21 after bleomycin treatment (21dBLMFbs) were used in these experiments as described (see Fig. 1 in our companion paper (71)). Migration of the leukocytes across the endothelial monolayer was determined by adding confluent leukocytes 10 × 103 cells/6.5-mm-diameter transwell culture insert (8-μm pore size) on the top of a confluent monolayer (10 × 103 endothelial cells). The effect of fibroblasts in the bottom chamber on leukocyte migration was examined by co-culturing lung fibroblast monolayers (MNLFbs and 21dBLMFbs; 10 × 103) in the tissue culture wells below the transwell insert. Leukocyte migration across the endothelial cell layer in the upper chamber was determined by measuring the number of cells that migrate between the endothelial cells, through the porous membrane, and into the lower chamber. The cell numbers present on the undersurface of the transwell insert were determined by direct counting under a dissecting microscope. Medium from the lower chamber with the loosely adherent leukocytes from the floor of the lower chamber was collected with fresh DMEM. The resulting suspension of cells was centrifuged at 3000 × g for 5 min and resuspended in counting fluid. Cell numbers were determined by direct counting using a hemocytometer. Based on the results of the initial experiments, a 16-h migration period was found to give consistent results, and this time point was selected for subsequent studies.
Cell adhesion assay
Cells from the cell layer that adhered onto the inside part of the insert were isolated, and 100 μl of the cell suspension/well was added onto 96-well plates in DMEM with 0.1% fatty acid-free bovine serum albumin followed by incubation for 30 min at 37 °C under 5% CO2 with the lid off. After incubation, unattached cells were removed by rinsing with PBS. Attached cells were fixed in 5% glutaraldehyde for 20 min and stained with 0.1% crystal violet. After washing, stains were dissolved in 200 μl of 10% acetic acid, and color was read at 575 nm in a BioTek plate reader.
Invasion assay
As described in detail previously (49, 54, 150), the invasive potential of the transfected cells was evaluated by measuring the number of cells invading the Matrigel-coated transwell chambers (BD Biosciences).
Cytotoxicity assay
2 × 105 PBMCs were treated with the synthetic peptide/nanoparticles or the shRNA/nanoparticles at 1, 10, and 100 μm for 24 h. Then 10 μl of MTT (Calbiochem) solution (5 mg/ml) was added to each well, and the culture was further incubated for 4 h at 37 °C. A total of 50 μl of N-dimethylmethanamide solution was added to each well, followed by overnight incubation. The absorbance of each well was determined on a microplate reader at 592 nm. The relative cell viability (percentage) was calculated using the formula, % viability = (A592(treated cells)/(A592(untreated cells)) × 100. Negative control cells were treated with medium.
Preparation of conditioned media
Monolayers of fibroblasts in 75-cm2 tissue culture flasks were rinsed twice with serum-supplemented (10% FBS) DMEM, and 10 ml of fresh medium was added. After a 24-h conditioning period, media were harvested, and cellular debris was removed by centrifugation at 3000 × g for 10 min. Media were either used immediately or stored frozen at −80 °C for later experiments.
Measurement of cytokine levels after cellular stimulations
2 × 106 PBMCs were treated with the V6-PEP/nanoparticles or the CD44V6 shRNA/nanoparticles at 1, 10, and 100 μm for 1 h, followed by incubation with 1 μg/ml LPS (Sigma-Aldrich) or the recombinant 2.5 ng/ml TGFβ1 (Sigma-Aldrich) for 24 h. Pro-inflammatory (TNF-α) and regulatory anti-inflammatory (IL-10) cytokines were subsequently quantified in triplicates by commercial ELISA kits, according to the manufacturer's instructions.
V6-PEP detection by ELISA
2 × 106 PBMCs diluted (1:1 and 1:2 dilution) in bicarbonate buffer (0.1 m, pH 9.4) were coated in a 96-well Maxisorp microtiter plate (Nunc, ThermoFisher Scientific) and incubated overnight at 4 °C. After blocking with 5% BSA in PBS (Sigma-Aldrich) at 37 °C for 1 h, each well was washed once with PBS. V6-PEP (1 μg/ml) or TGFβ1 (1 ng/ml) was added, and the plate was incubated at 37 °C for 1 h. Each well was washed five times with PBS, and anti-TGFβ1 antibody labeled with HRP (BD Bioscience) diluted at 1:250 was added into each well, followed by incubation for 1 h at 4 °C. Each well was washed five times and treated with o-phenylenediamine dihydrochloride 1 mg/ml (Sigma-Aldrich), a soluble substrate for the detection of peroxidase activity in ELISA. The reaction was stopped by adding 20 μl of 4 n H2SO4 and read at 492 nm in a microplate reader.
Intracellular ROS detection
Fibroblasts grown in glass-bottom plates were washed with Hanks' buffered salt solution and loaded with H2DCFDA (Invitrogen) dissolved in Hanks' buffered saline at 10 μm. Fluorescence was read at 535 nm using a plate reader.
Three-dimensional collagen gel contraction assay
HNLFbs were treated or transfected with various reagents as mentioned in the figure legends for the specified experiment. Collagen gels were reconstituted by mixing one part 3 mg/ml neutralized rat tail collagen-1 and two parts cell suspension in serum-free medium. Cell suspensions were seeded at a density of 200,000 cells/ml into 4-well tissue culture plates, and the gels were allowed to polymerize at 37 °C for 1 h before adding 1 ml of medium. A volume of 600 μl/gel was fabricated in a 4-well plastic culture dish, which ensured that the gel would remain attached throughout the culture period. The stressed HNLFbs were cultured in 10% FBS for 20 h after polymerization. The stressed HNLFbs were then incubated in 0.5% FBS with or without TGFβ1 (5 ng/ml) for another 48 h in serum-deprived medium, and the edges of the gels were then gently detached from the walls of the well using a sterile spatula. Gels were then photographed after 16 h, and the areas were measured using ImageJ software (National Institutes of Health).
In vivo targeting of CD44V6 by genetic modification and with the CD44V6-blocking V6-PEP
For genetic targeting of CD44V6, we planned to test whether intratracheal treatment with engineered nanoparticle delivery systems to convey CD44V6 shRNA in the fibrogenic lung cells. The idea was to transactivate a conditionally silenced plasmid with CD44V6 oligonucleotide by Cre recombinase produced in response to a tissue-specific promoter. The principle is as follows. The recombinase, produced under the influence of a tissue-specific promoter in the cells will eliminate the (CMV-EGFP)-cassette from U6-(CMV-EGFP)f/f-CD44V6 shRNA from the pSico-CD44V6 shRNA, and the U6 promoter will induce synthesis of CD44V6 shRNA. Normal cells in different organs will not be affected because they rely mostly on the standard CD44s expression, which does not have any variant exons. Even if the cells express CD44V6, shRNA will not be produced due lack of response to the tissue-specific promoter. The plasmids will be delivered through nanoparticle (Tf-PEG-PEI) carrier. Unused plasmids in other organs will be progressively destroyed by cytoplasmic nucleases, and the nanoparticles will be cleared because PEG and PEI are biodegradable, thus avoiding any toxicity problem. It has been shown that FSP-1 increases in the lung fibroblast cells after bleomycin treatment (151, 152). A recent study also indicates that conditional deletion of Has2 by use of the FSP-1-Cre mouse line in mesenchymal lung fibroblast cells abrogated the invasive fibroblast phenotype, impeded myofibroblast accumulation, and inhibited the development of lung fibrosis (19). Thus, to assess the contribution of fibroblast expression of HAS2/CD44v6, we used a FSP-1 promoter-driven Cre plasmid. Further, using our non-toxic and non-inflammatory shRNA/nanoparticle delivery method (45, 109), V6-PEP/nanoparticles, CD44V6 shRNA/nanoparticles (pSico-CD44v6 shRNA/Tf-PEG-PEI plus FSP-1-Cre/Tf-PEG-PEI), control peptide/nanoparticles, or control shRNA/nanoparticle (pSico-scrambled shRNA/Tf-PEG-PEI plus FSP-1-Cre/Tf-PEG-PEI) were administered every other day from day 2 to day 30 by intratracheal delivery to the lungs of young mice during the onset of inflammation by bleomycin injury, and then the mice were left untreated for another 15 days. Lung tissue was harvested at the indicated times up to 45 days after injury. The time course of fibrosis induction, treatment schedule, and resolution of fibrosis was measured.
Preparation of pSico-CD44v6 shRNA and pFSP-1-Cre
Preparation of Tf-coated PEG-PEI nanoparticles and nanoparticle size determination were carried out as validated by our group previously in an intestinal/colon cancer murine model (45, 109, 153).
Sircol assay for collagen
Acid-soluble collagen in whole-lung homogenates was analyzed by the Sircol assay as described previously (154).
Hydroxyproline content of whole lung
A lobe of each mouse lung was homogenized in PBS and then acidified by adding an equal volume of 12 n HCl, hydrolyzed by heating at 120 °C for 24 h, and processed for hydroxyproline measurements as described previously (155).
Lung histology staining
Paraffin-embedded tissue sections were processed for Masson's trichrome staining for antibodies against CD44V6 and NOX4 as described previously (1).
Measurement of hydrogen peroxide production
Extracellular H2O2 released from cultured cells was assayed by Amplex® Red. The Amplex® Red H2O2 assay kit uses the Amplex® Red reagent (10-acetyl-3,7-dihydroxyphenoxazine) to detect H2O2 released from cells. In the presence of HRP, the Amplex® Red reagent reacts with H2O2 in a 1:1 stoichiometry to produce the red-fluorescent oxidation product, resorufin. Fluorescence was read at 585 nm using a plate reader.
Proximity ligation Duolink assay
We used the Duolink in situ PLA reagent from Olink Biosciences (Uppsala, Sweden) to characterize interaction between endogenous CD44v6 and TGFβ1RI. Cells were grown on glass-bottom culture dishes (MatTek) according to the protocol supplied by Duolink reagents from Sigma-Aldrich. After incubating overnight with the CD44V6 and TGFβRI antibodies, the cells were further incubated with the oligonucleotide labeled with anti-goat plus and anti-mouse minus PLA probes, as recommended by the manufacturer. Negative control slides were incubated with secondary antibodies only before incubation with PLA probes. Samples were mounted with the Duolink mounting medium containing DAPI to counterstain nuclear DNA. PLA images (fluorescence dots) were acquired using a Zeiss LSM 880 NLO confocal microscope (Cell and Molecular Imaging Shared Resource, Hollings Cancer Center, Medical University of South Carolina) with a 63×/1.4 numeric aperture objective and analyzed by the Duolink Image Tool (Sigma). Counted dots are expressed as mean ± S.E.
Statistical analysis
Each experiment was done in triplicate and was repeated three times (n = 9). Data were pooled for statistical analyses. Animal studies, Western blot analyses, mRNA analyses, and proliferation experiments for each separate experiment were repeated three or four times. Data from various groups are expressed as means ± S.E. (n = 3–4). Statistical comparisons were done using Student's t test for unpaired samples or using two-way analysis of variance (ANOVA) with Bonferroni post-test correction on grouped data as mentioned in the figure legends.
Author contributions
The experiments of this work were designed and carried out by S. M. and S. G. The paper was written by S. M. and S. G. V. C. H. reviewed and edited the draft and final versions of the text, figures, and figure legends and gave advice regarding draft corrections wherever necessary. R. R. M., V. J. T., C. M. A., and C. F.-B. commented on the final version of the paper. J. W. supplied the NOX4 inhibitor and gave advice on its pharmacology and how to use it for the studies to ensure effective blockade of NOX4-dependent ROS formation. M. G. took the microscope picture of the Duolink experiment. G. S. B. and I. A. helped in designing bleomycin treatments in mice, and R. M. S. supplied tissue culture reagents.
This work was supported by National Institutes of Health (NIH) Grant 1R03CA167722-01A1 (to S. M. and S. G.); NIH Grants 1RO1 (HL33756), P30 GM103342-03, 5P20GM103499-16, AHA16GRNT31330032, and 1P20 GM103444 (to S. M., S. G., and R. R. M.); NIH Grants 1 R01HL113325 and 1 P01HL107147 (to V. C. H.); a pilot project award from the COBRE in Lipidomics and Pathobiology (NIH Grant 5P30GM103339-03) at the Medical University of South Carolina (to S. M.); and the Cell and Molecular Imaging Shared Resource, Hollings Cancer Center, Medical University of South Carolina (NIH Grant P30 CA138313) and Shared Instrumentation Grant S10 OD018113. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- IPF
- idiopathic pulmonary fibrosis
- NOX
- NADPH oxidase
- ROS or H2O2
- reactive oxygen species
- HA
- hyaluronan
- ECM
- extracellular matrix
- IPFFb
- primary fibroblast derived from IPF patient lungs
- HNLFb
- primary humn normal lung fibroblast
- BLMFb
- primary murine lung myofibroblast isolated from lung tissues from bleomycin-treated mice
- MNLFbs
- primary mouse normal lung fibroblasts
- NOX4-null MLFbs
- NOX4-null lung fibroblasts
- CD44-null MNLFbs
- CD44-null lung fibroblasts
- LMWHA
- low-molecular weight HA (150-kDa HA, 500-kDa HA)
- HMWHA
- high-molecular weight HA H2DCFDA, dichlorofluorescein diacetate
- TGFβRI and TGFβRII
- TGFβ1 receptor I and II, respectively
- control shRNA
- pSicoR-scrambled shRNA
- Tf
- transferrin
- CD44v6 shRNA
- pSicoR-CD44v6 shRNA
- NOX4 shRNA
- pSicoR-NOX4 shRNA
- PBMC
- peripheral blood mononuclear cell
- LPTV
- low-pressure tail vein
- HPTV
- high-pressure tail vein
- v6-PEP
- CD44v6-blocking peptide
- CDS
- coding sequences
- NCDS
- noncoding sequences
- KI
- knock-in
- 14d and 21d
- 14- and 21-day, respectively
- p-SMAD
- phospho-SMAD
- HA150 and HA500
- 150- and 500-kDa HA, respectively
- ANOVA
- analysis of variance
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- EC
- endothelial cell
- Tf
- transferrin
- HUVEC
- human umbilical vein endothelial cell
- CM
- conditioned medium
- MMP
- matrix metalloproteinase
- NLFb
- normal lung fibroblast.
References
- 1. Ghatak S., Bogatkevich G. S., Atnelishvili I., Akter T., Feghali-Bostwick C., Hoffman S., Fresco V. M., Fuchs J. C., Visconti R. P., Markwald R. R., Padhye S. B., Silver R. M., Hascall V. C., and Misra S. (2014) Overexpression of c-Met and CD44v6 receptors contributes to autocrine TGF-β1 signaling in interstitial lung disease. J. Biol. Chem. 289, 7856–7872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thannickal V. J. (2012) Mechanisms of pulmonary fibrosis: role of activated myofibroblasts and NADPH oxidase. Fibrogenesis Tissue Repair 5, S23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thannickal V. J., Toews G. B., White E. S., Lynch J. P. 3rd, Martinez F. J. (2004) Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 55, 395–417 [DOI] [PubMed] [Google Scholar]
- 4. Kinnula V. L. (2008) Redox imbalance and lung fibrosis. Antioxid. Redox Signal. 10, 249–252 [DOI] [PubMed] [Google Scholar]
- 5. Kinnula V. L., and Myllärniemi M. (2008) Oxidant-antioxidant imbalance as a potential contributor to the progression of human pulmonary fibrosis. Antioxid. Redox Signal. 10, 727–738 [DOI] [PubMed] [Google Scholar]
- 6. Hecker L., Logsdon N. J., Kurundkar D., Kurundkar A., Bernard K., Hock T., Meldrum E., Sanders Y. Y., and Thannickal V. J. (2014) Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 6, 231ra47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Thannickal V. J., and Fanburg B. L. (1995) Activation of an H2O2-generating NADH oxidase in human lung fibroblasts by transforming growth factor β1. J. Biol. Chem. 270, 30334–30338 [DOI] [PubMed] [Google Scholar]
- 8. Waghray M., Cui Z., Horowitz J. C., Subramanian I. M., Martinez F. J., Toews G. B., and Thannickal V. J. (2005) Hydrogen peroxide is a diffusible paracrine signal for the induction of epithelial cell death by activated myofibroblasts. FASEB J. 19, 854–856 [DOI] [PubMed] [Google Scholar]
- 9. Lambeth J. D. (2004) NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189 [DOI] [PubMed] [Google Scholar]
- 10. Ambasta R. K., Kumar P., Griendling K. K., Schmidt H. H., Busse R., and Brandes R. P. (2004) Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase. J. Biol. Chem. 279, 45935–45941 [DOI] [PubMed] [Google Scholar]
- 11. Kawahara T., Ritsick D., Cheng G., and Lambeth J. D. (2005) Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J. Biol. Chem. 280, 31859–31869 [DOI] [PubMed] [Google Scholar]
- 12. Fulton D. J. (2009) Nox5 and the regulation of cellular function. Antioxid. Redox Signal. 11, 2443–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. BelAiba R. S., Djordjevic T., Petry A., Diemer K., Bonello S., Banfi B., Hess J., Pogrebniak A., Bickel C., and Görlach A. (2007) NOX5 variants are functionally active in endothelial cells. Free Radic. Biol. Med. 42, 446–459 [DOI] [PubMed] [Google Scholar]
- 14. Griffith B., Pendyala S., Hecker L., Lee P. J., Natarajan V., and Thannickal V. J. (2009) NOX enzymes and pulmonary disease. Antioxid. Redox Signal. 11, 2505–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hecker L., Cheng J., and Thannickal V. J. (2012) Targeting NOX enzymes in pulmonary fibrosis. Cell Mol. Life Sci. 69, 2365–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hecker L., Vittal R., Jones T., Jagirdar R., Luckhardt T. R., Horowitz J. C., Pennathur S., Martinez F. J., and Thannickal V. J. (2009) NADPH oxidase-4 mediates myofibroblast activation and fibrogenic responses to lung injury. Nat. Med. 15, 1077–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ghosh A. K., Quaggin S. E., and Vaughan D. E. (2013) Molecular basis of organ fibrosis: potential therapeutic approaches. Exp. Biol. Med. 238, 461–481 [DOI] [PubMed] [Google Scholar]
- 18. Hunt T. K., Knighton D. R., Thakral K. K., Goodson W. H. 3rd, Andrews W. S. (1984) Studies on inflammation and wound healing: angiogenesis and collagen synthesis stimulated in vivo by resident and activated wound macrophages. Surgery 96, 48–54 [PubMed] [Google Scholar]
- 19. Li Y., Jiang D., Liang J., Meltzer E. B., Gray A., Miura R., Wogensen L., Yamaguchi Y., and Noble P. W. (2011) Severe lung fibrosis requires an invasive fibroblast phenotype regulated by hyaluronan and CD44. J. Exp. Med. 208, 1459–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Grinnell F. (1994) Fibroblasts, myofibroblasts, and wound contraction. J. Cell Biol. 124, 401–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Munger J. S., Huang X., Kawakatsu H., Griffiths M. J., Dalton S. L., Wu J., Pittet J. F., Kaminski N., Garat C., Matthay M. A., Rifkin D. B., and Sheppard D. (1999) The integrin αvβ6 binds and activates latent TGFβ1: a mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328 [DOI] [PubMed] [Google Scholar]
- 22. Fichtner-Feigl S., Strober W., Kawakami K., Puri R. K., and Kitani A. (2006) IL-13 signaling through the IL-13α2 receptor is involved in induction of TGF-β1 production and fibrosis. Nat. Med. 12, 99–106 [DOI] [PubMed] [Google Scholar]
- 23. Desmoulière A., Geinoz A., Gabbiani F., and Gabbiani G. (1993) Transforming growth factor-β1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell Biol. 122, 103–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vaughan M. B., Howard E. W., and Tomasek J. J. (2000) Transforming growth factor-β1 promotes the morphological and functional differentiation of the myofibroblast. Exp. Cell Res. 257, 180–189 [DOI] [PubMed] [Google Scholar]
- 25. Evans R. A., Tian Y. C., Steadman R., and Phillips A. O. (2003) TGF-β1-mediated fibroblast-myofibroblast terminal differentiation: the role of Smad proteins. Exp. Cell Res. 282, 90–100 [DOI] [PubMed] [Google Scholar]
- 26. Serini G., Bochaton-Piallat M. L., Ropraz P., Geinoz A., Borsi L., Zardi L., and Gabbiani G. (1998) The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-β1. J. Cell Biol. 142, 873–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hinz B., Mastrangelo D., Iselin C. E., Chaponnier C., and Gabbiani G. (2001) Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 159, 1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Desmoulière A. (1995) Factors influencing myofibroblast differentiation during wound healing and fibrosis. Cell Biol. Int. 19, 471–476 [DOI] [PubMed] [Google Scholar]
- 29. Li Y., Rahmanian M., Widström C., Lepperdinger G., Frost G. I., and Heldin P. (2000) Irradiation-induced expression of hyaluronan (HA) synthase 2 and hyaluronidase 2 genes in rat lung tissue accompanies active turnover of HA and induction of types I and III collagen gene expression. Am. J. Respir. Cell Mol. Biol. 23, 411–418 [DOI] [PubMed] [Google Scholar]
- 30. Jones S., and Phillips A. O. (2001) Regulation of renal proximal tubular epithelial cell hyaluronan generation: implications for diabetic nephropathy. Kidney Int. 59, 1739–1749 [DOI] [PubMed] [Google Scholar]
- 31. Göransson V., Hansell P., Moss S., Alcorn D., Johnsson C., Hallgren R., and Maric C. (2001) Renomedullary interstitial cells in culture: the osmolality and oxygen tension influence the extracellular amounts of hyaluronan and cellular expression of CD44. Matrix Biol. 20, 129–136 [DOI] [PubMed] [Google Scholar]
- 32. Jenkins R. H., Thomas G. J., Williams J. D., and Steadman R. (2004) Myofibroblastic differentiation leads to hyaluronan accumulation through reduced hyaluronan turnover. J. Biol. Chem. 279, 41453–41460 [DOI] [PubMed] [Google Scholar]
- 33. Spicer A. P., and McDonald J. A. (1998) Characterization and molecular evolution of a vertebrate hyaluronan synthase gene family. J. Biol. Chem. 273, 1923–1932 [DOI] [PubMed] [Google Scholar]
- 34. Harada H., and Takahashi M. (2007) CD44-dependent intracellular and extracellular catabolism of hyaluronic acid by hyaluronidase-1 and -2. J. Biol. Chem. 282, 5597–5607 [DOI] [PubMed] [Google Scholar]
- 35. Li Y., Liang J., Yang T., Monterrosa Mena J., Huan C., Xie T., Kurkciyan A., Liu N., Jiang D., and Noble P. W. (2016) Hyaluronan synthase 2 regulates fibroblast senescence in pulmonary fibrosis. Matrix Biol. 10.1016/j.matbio.2016.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Campisi J., and d'Adda di Fagagna F. (2007) Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 8, 729–740 [DOI] [PubMed] [Google Scholar]
- 37. Chen Q. M., Liu J., and Merrett J. B. (2000) Apoptosis or senescence-like growth arrest: influence of cell-cycle position, p53, p21, and bax in H2O2 response of normal human fibroblasts. Biochem. J. 347, 543–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hinz B., Phan S. H., Thannickal V. J., Prunotto M., Desmouliére A., Varga J., De Wever O., Mareel M., and Gabbiani G. (2012) Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am. J. Pathol. 180, 1340–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lesley J., Hascall V. C., Tammi M., and Hyman R. (2000) Hyaluronan binding by cell surface CD44. J. Biol. Chem. 275, 26967–26975 [DOI] [PubMed] [Google Scholar]
- 40. Lesley J., and Hyman R. (1992) CD44 can be activated to function as an hyaluronic acid receptor in normal murine T cells. Eur. J. Immunol. 22, 2719–2723 [DOI] [PubMed] [Google Scholar]
- 41. Lesley J., English N., Charles C., and Hyman R. (2000) The role of the CD44 cytoplasmic and transmembrane domains in constitutive and inducible hyaluronan binding. Eur. J. Immunol. 30, 245–253 [DOI] [PubMed] [Google Scholar]
- 42. Misra S., Ghatak S., and Toole B. P. (2005) Regulation of MDR1 expression and drug resistance by a positive feedback loop involving hyaluronan, phosphoinositide 3-kinase, and ErbB2. J. Biol. Chem. 280, 20310–20315 [DOI] [PubMed] [Google Scholar]
- 43. Misra S., Ghatak S., Zoltan-Jones A., and Toole B. P. (2003) Regulation of multidrug resistance in cancer cells by hyaluronan. J. Biol. Chem. 278, 25285–25288 [DOI] [PubMed] [Google Scholar]
- 44. Misra S., Hascall V. C., Berger F. G., Markwald R. R., and Ghatak S. (2008) Hyaluronan, CD44, and cyclooxygenase-2 in colon cancer. Connect. Tissue Res. 49, 219–224 [DOI] [PubMed] [Google Scholar]
- 45. Misra S., Hascall V. C., De Giovanni C., Markwald R. R., and Ghatak S. (2009) Delivery of CD44 shRNA/nanoparticles within cancer cells: perturbation of hyaluronan/CD44v6 interactions and reduction in adenoma growth in Apc Min/+ MICE. J. Biol. Chem. 284, 12432–12446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Misra S., Hascall V. C., Markwald R. R., and Ghatak S. (2015) Interactions between hyaluronan and its receptors (CD44, RHAMM) regulate the activities of inflammation and cancer. Front. Immunol. 6, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Misra S., Heldin P., Hascall V. C., Karamanos N. K., Skandalis S. S., Markwald R. R., and Ghatak S. (2011) Hyaluronan-CD44 interactions as potential targets for cancer therapy. FEBS J. 278, 1429–1443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Misra S., Toole B. P., and Ghatak S. (2006) Hyaluronan constitutively regulates activation of multiple receptor tyrosine kinases in epithelial and carcinoma cells. J. Biol. Chem. 281, 34936–34941 [DOI] [PubMed] [Google Scholar]
- 49. Ghatak S., Hascall V. C., Markwald R. R., and Misra S. (2010) Stromal hyaluronan interaction with epithelial CD44 variants promotes prostate cancer invasiveness by augmenting expression and function of hepatocyte growth factor and androgen receptor. J. Biol. Chem. 285, 19821–19832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ghatak S., Maytin E. V., Mack J. A., Hascall V. C., Atanelishvili I., Moreno Rodriguez R., Markwald R. R., and Misra S. (2015) Roles of proteoglycans and glycosaminoglycans in wound healing and fibrosis. Int. J. Cell Biol. 2015, 834893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ghatak S., Misra S., Norris R. A., Moreno-Rodriguez R. A., Hoffman S., Levine R. A., Hascall V. C., and Markwald R. R. (2014) Periostin induces intracellular cross-talk between kinases and hyaluronan in atrioventricular valvulogenesis. J. Biol. Chem. 289, 8545–8561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ghatak S., Misra S., and Toole B. P. (2002) Hyaluronan oligosaccharides inhibit anchorage-independent growth of tumor cells by suppressing the phosphoinositide 3-kinase/Akt cell survival pathway. J. Biol. Chem. 277, 38013–38020 [DOI] [PubMed] [Google Scholar]
- 53. Ghatak S., Misra S., and Toole B. P. (2005) Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J. Biol. Chem. 280, 8875–8883 [DOI] [PubMed] [Google Scholar]
- 54. Misra S., Obeid L. M., Hannun Y. A., Minamisawa S., Berger F. G., Markwald R. R., Toole B. P., and Ghatak S. (2008) Hyaluronan constitutively regulates activation of COX-2-mediated cell survival activity in intestinal epithelial and colon carcinoma cells. J. Biol. Chem. 283, 14335–14344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brecht M., Mayer U., Schlosser E., and Prehm P. (1986) Increased hyaluronate synthesis is required for fibroblast detachment and mitosis. Biochem. J. 239, 445–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tammi R., and Tammi M. (1991) Correlations between hyaluronan and epidermal proliferation as studied by [3H]glucosamine and [3H]thymidine incorporations and staining of hyaluronan on mitotic keratinocytes. Exp. Cell Res. 195, 524–527 [DOI] [PubMed] [Google Scholar]
- 57. Henke C., Bitterman P., Roongta U., Ingbar D., and Polunovsky V. (1996) Induction of fibroblast apoptosis by anti-CD44 antibody: implications for the treatment of fibroproliferative lung disease. Am. J. Pathol. 149, 1639–1650 [PMC free article] [PubMed] [Google Scholar]
- 58. Svee K., White J., Vaillant P., Jessurun J., Roongta U., Krumwiede M., Johnson D., and Henke C. (1996) Acute lung injury fibroblast migration and invasion of a fibrin matrix is mediated by CD44. J. Clin. Invest. 98, 1713–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Laurent T. C., Laurent U. B., and Fraser J. R. (1996) Serum hyaluronan as a disease marker. Ann. Med. 28, 241–253 [DOI] [PubMed] [Google Scholar]
- 60. Laurent U. B., Laurent T. C., Hellsing L. K., Persson L., Hartman M., and Lilja K. (1996) Hyaluronan in human cerebrospinal fluid. Acta Neurol. Scand. 94, 194–206 [DOI] [PubMed] [Google Scholar]
- 61. Lewis A., Steadman R., Manley P., Craig K., de la Motte C., Hascall V., and Phillips A. O. (2008) Diabetic nephropathy, inflammation, hyaluronan and interstitial fibrosis. Histol. Histopathol. 23, 731–739 [DOI] [PubMed] [Google Scholar]
- 62. Kosaki R., Watanabe K., and Yamaguchi Y. (1999) Overproduction of hyaluronan by expression of the hyaluronan synthase Has2 enhances anchorage-independent growth and tumorigenicity. Cancer Res. 59, 1141–1145 [PubMed] [Google Scholar]
- 63. Ghatak S., Hascall V. C., Karamanos N. K., Markwald R. R., and Misra S. (2012) Targeting the tumor microenvironment in cancer progression. In Extracellular Matrix: Pathobiology and Signaling (Karamanos N., ed), pp. 723–740, DeGruyter, Berlin, Germany [Google Scholar]
- 64. Legg J. W., Lewis C. A., Parsons M., Ng T., and Isacke C. M. (2002) A novel PKC-regulated mechanism controls CD44 ezrin association and directional cell motility. Nat. Cell Biol. 4, 399–407 [DOI] [PubMed] [Google Scholar]
- 65. Itano N., Atsumi F., Sawai T., Yamada Y., Miyaishi O., Senga T., Hamaguchi M., and Kimata K. (2002) Abnormal accumulation of hyaluronan matrix diminishes contact inhibition of cell growth and promotes cell migration. Proc. Natl. Acad. Sci. U.S.A. 99, 3609–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ito T., Williams J. D., Al-Assaf S., Phillips G. O., and Phillips A. O. (2004) Hyaluronan and proximal tubular cell migration. Kidney Int. 65, 823–833 [DOI] [PubMed] [Google Scholar]
- 67. Zoltan-Jones A., Huang L., Ghatak S., and Toole B. P. (2003) Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J. Biol. Chem. 278, 45801–45810 [DOI] [PubMed] [Google Scholar]
- 68. Camenisch T. D., Schroeder J. A., Bradley J., Klewer S. E., and McDonald J. A. (2002) Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat. Med. 8, 850–855 [DOI] [PubMed] [Google Scholar]
- 69. Evanko S. P., Angello J. C., and Wight T. N. (1999) Formation of hyaluronan- and versican-rich pericellular matrix is required for proliferation and migration of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 19, 1004–1013 [DOI] [PubMed] [Google Scholar]
- 70. Screaton G. R., Bell M. V., Jackson D. G., Cornelis F. B., Gerth U., and Bell J. I. (1992) Genomic structure of DNA encoding the lymphocyte homing receptor CD44 reveals at least 12 alternatively spliced exons. Proc. Natl. Acad. Sci. U.S.A. 89, 12160–12164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ghatak S., Markwald R. R., Hascall V. C., Dowling W., Lottes R. G., Baatz J. E., Beeson G., Beeson C. C. Perrella M. A., Thannickal V. J., and Misra S. (2017) Transforming growth factor β1 (TGFβ1) regulates CD44V6 expression and activity through extracellular signal-regulated kinase (ERK)-induced EGR1 in pulmonary fibrogenic fibroblasts. J. Biol. Chem. 292, 10465–10489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morrison H., Sherman L. S., Legg J., Banine F., Isacke C., Haipek C. A., Gutmann D. H., Ponta H., and Herrlich P. (2001) The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev. 15, 968–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tsukita S., Oishi K., Sato N., Sagara J., Kawai A., and Tsukita S. (1994) ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J. Cell Biol. 126, 391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Acharya P. S., Majumdar S., Jacob M., Hayden J., Mrass P., Weninger W., Assoian R. K., and Puré E. (2008) Fibroblast migration is mediated by CD44-dependent TGFβ activation. J. Cell Sci. 121, 1393–1402 [DOI] [PubMed] [Google Scholar]
- 75. Cheng C., and Sharp P. A. (2006) Regulation of CD44 alternative splicing by SRm160 and its potential role in tumor cell invasion. Mol. Cell Biol. 26, 362–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vendrov A. E., Madamanchi N. R., Hakim Z. S., Rojas M., and Runge M. S. (2006) Thrombin and NAD(P)H oxidase-mediated regulation of CD44 and BMP4-Id pathway in VSMC, restenosis, and atherosclerosis. Circ. Res. 98, 1254–1263 [DOI] [PubMed] [Google Scholar]
- 77. McNeil J. D., Wiebkin O. W., Betts W. H., and Cleland L. G. (1985) Depolymerisation products of hyaluronic acid after exposure to oxygen-derived free radicals. Ann. Rheum Dis. 44, 780–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Dinarello C. A. (1992) Interleukin-1 and tumor necrosis factor: effector cytokines in autoimmune diseases. Semin. Immunol. 4, 133–145 [PubMed] [Google Scholar]
- 79. Zelová H., and Hošek J. (2013) TNF-α signalling and inflammation: interactions between old acquaintances. Inflamm. Res. 62, 641–651 [DOI] [PubMed] [Google Scholar]
- 80. Kodama S., Davis M., and Faustman D. L. (2005) The therapeutic potential of tumor necrosis factor for autoimmune disease: a mechanistically based hypothesis. Cell Mol. Life Sci. 62, 1850–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Zhang K., Gharaee-Kermani M., McGarry B., Remick D., and Phan S. H. (1997) TNF-α-mediated lung cytokine networking and eosinophil recruitment in pulmonary fibrosis. J. Immunol. 158, 954–959 [PubMed] [Google Scholar]
- 82. Evans H. G., Roostalu U., Walter G. J., Gullick N. J., Frederiksen K. S., Roberts C. A., Sumner J., Baeten D. L., Gerwien J. G., Cope A. P., Geissmann F., Kirkham B. W., and Taams L. S. (2014) TNF-α blockade induces IL-10 expression in human CD4+ T cells. Nat. Commun. 5, 3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Murakami M., and Hirano T. (2012) The molecular mechanisms of chronic inflammation development. Front. Immunol. 3, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sabat R., Grütz G., Warszawska K., Kirsch S., Witte E., Wolk K., and Geginat J. (2010) Biology of interleukin-10. Cytokine Growth Factor Rev. 21, 331–344 [DOI] [PubMed] [Google Scholar]
- 85. Kräling B. M., Maul G. G., and Jimenez S. A. (1995) Mononuclear cellular infiltrates in clinically involved skin from patients with systemic sclerosis of recent onset predominantly consist of monocytes/macrophages. Pathobiology 63, 48–56 [DOI] [PubMed] [Google Scholar]
- 86. Denton C. P., Shi-Wen X., Sutton A., Abraham D. J., Black C. M., and Pearson J. D. (1998) Scleroderma fibroblasts promote migration of mononuclear leucocytes across endothelial cell monolayers. Clin. Exp. Immunol. 114, 293–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wrana J. L., Attisano L., Wieser R., Ventura F., and Massagué J. (1994) Mechanism of activation of the TGF-beta receptor. Nature 370, 341–347 [DOI] [PubMed] [Google Scholar]
- 88. Wrana J. L., Attisano L., Cárcamo J., Zentella A., Doody J., Laiho M., Wang X. F., and Massagué J. (1992) TGFβ signals through a heteromeric protein kinase receptor complex. Cell 71, 1003–1014 [DOI] [PubMed] [Google Scholar]
- 89. Maher K., Završnik J., Jerič-Kokelj B., Vasiljeva O., Turk B., and Kopitar-Jerala N. (2014) Decreased IL-10 expression in stefin B-deficient macrophages is regulated by the MAP kinase and STAT-3 signaling pathways. FEBS Lett. 588, 720–726 [DOI] [PubMed] [Google Scholar]
- 90. Vendrov A. E., Madamanchi N. R., Niu X. L., Molnar K. C., Runge M., Szyndralewiez C., Page P., and Runge M. S. (2010) NADPH oxidases regulate CD44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. J. Biol. Chem. 285, 26545–26557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zhang K., Rekhter M. D., Gordon D., and Phan S. H. (1994) Myofibroblasts and their role in lung collagen gene expression during pulmonary fibrosis. A combined immunohistochemical and in situ hybridization study. Am. J. Pathol. 145, 114–125 [PMC free article] [PubMed] [Google Scholar]
- 92. Tomasek J. J., Gabbiani G., Hinz B., Chaponnier C., and Brown R. A. (2002) Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 3, 349–363 [DOI] [PubMed] [Google Scholar]
- 93. Hinz B. (2007) Formation and function of the myofibroblast during tissue repair. J. Invest. Dermatol. 127, 526–537 [DOI] [PubMed] [Google Scholar]
- 94. Basoni C., Reuzeau E., Croft D., Génot E., and Kramer I. M. (2006) CD44 and TGFβ1 synergise to induce expression of a functional NADPH oxidase in promyelocytic cells. Biochem. Biophys. Res. Commun. 343, 609–616 [DOI] [PubMed] [Google Scholar]
- 95. Kim Y., Lee Y. S., Choe J., Lee H., Kim Y. M., and Jeoung D. (2008) CD44-epidermal growth factor receptor interaction mediates hyaluronic acid-promoted cell motility by activating protein kinase C signaling involving Akt, Rac1, Phox, reactive oxygen species, focal adhesion kinase, and MMP-2. J. Biol. Chem. 283, 22513–22528 [DOI] [PubMed] [Google Scholar]
- 96. Bonniaud P., Kolb M., Galt T., Robertson J., Robbins C., Stampfli M., Lavery C., Margetts P. J., Roberts A. B., and Gauldie J. (2004) Smad3 null mice develop airspace enlargement and are resistant to TGF-β-mediated pulmonary fibrosis. J. Immunol. 173, 2099–2108 [DOI] [PubMed] [Google Scholar]
- 97. Cheng C., Yaffe M. B., and Sharp P. A. (2006) A positive feedback loop couples Ras activation and CD44 alternative splicing. Genes Dev. 20, 1715–1720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Orian-Rousseau V., Chen L., Sleeman J. P., Herrlich P., and Ponta H. (2002) CD44 is required for two consecutive steps in HGF/c-Met signaling. Genes Dev. 16, 3074–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Tremmel M., Matzke A., Albrecht I., Laib A. M., Olaku V., Ballmer-Hofer K., Christofori G., Héroult M., Augustin H. G., Ponta H., and Orian-Rousseau V. (2009) A CD44v6 peptide reveals a role of CD44 in VEGFR-2 signaling and angiogenesis. Blood 114, 5236–5244 [DOI] [PubMed] [Google Scholar]
- 100. Hinz B., Phan S. H., Thannickal V. J., Galli A., Bochaton-Piallat M. L., and Gabbiani G. (2007) The myofibroblast: one function, multiple origins. Am. J. Pathol. 170, 1807–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Rao G. N., Katki K. A., Madamanchi N. R., Wu Y., and Birrer M. J. (1999) JunB forms the majority of the AP-1 complex and is a target for redox regulation by receptor tyrosine kinase and G protein-coupled receptor agonists in smooth muscle cells. J. Biol. Chem. 274, 6003–6010 [DOI] [PubMed] [Google Scholar]
- 102. Vaz E. R., Fujimura P. T., Araujo G. R., da Silva C. A., Silva R. L., Cunha T. M., Lopes-Ferreira M., Lima C., Ferreira M. J., Cunha-Junior J. P., Taketomi E. A., Goulart L. R., and Ueira-Vieira C. (2015) A short peptide that mimics the binding domain of TGF-β1 presents potent anti-inflammatory activity. PLoS One 10, e0136116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Pierer M., Wagner U., Rossol M., and Ibrahim S. (2011) Toll-like receptor 4 is involved in inflammatory and joint destructive pathways in collagen-induced arthritis in DBA1J mice. PLoS One 6, e23539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Southcott A. M., Jones K. P., Li D., Majumdar S., Cambrey A. D., Pantelidis P., Black C. M., Laurent G. J., Davies B. H., and Jeffery P. K. (1995) Interleukin-8: differential expression in lone fibrosing alveolitis and systemic sclerosis. Am. J. Respir. Crit. Care Med. 151, 1604–1612 [DOI] [PubMed] [Google Scholar]
- 105. Luster A. D. (1998) Chemokines: chemotactic cytokines that mediate inflammation. N. Engl. J. Med. 338, 436–445 [DOI] [PubMed] [Google Scholar]
- 106. Beck-Schimmer B., Oertli B., Pasch T., and Wüthrich R. P. (1998) Hyaluronan induces monocyte chemoattractant protein-1 expression in renal tubular epithelial cells. J. Am. Soc. Nephrol. 9, 2283–2290 [DOI] [PubMed] [Google Scholar]
- 107. White B. (1996) Immunopathogenesis of systemic sclerosis. Rheum. Dis. Clin. North Am. 22, 695–708 [DOI] [PubMed] [Google Scholar]
- 108. Izbicki G., Segel M. J., Christensen T. G., Conner M. W., and Breuer R. (2002) Time course of bleomycin-induced lung fibrosis. Int. J. Exp. Pathol. 83, 111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ghatak S., Hascall V. C., Berger F. G., Penas M. M., Davis C., Jabari E., He X., Norris J. S., Dang Y., Markwald R. R., and Misra S. (2008) Tissue-specific shRNA delivery: a novel approach for gene therapy in cancer. Connect. Tissue Res. 49, 265–269 [DOI] [PubMed] [Google Scholar]
- 110. Ley B., and Collard H. R. (2012) Risk prediction in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 185, 6–7 [DOI] [PubMed] [Google Scholar]
- 111. Raghu G., Collard H. R., Egan J. J., Martinez F. J., Behr J., Brown K. K., Colby T. V., Cordier J. F., Flaherty K. R., Lasky J. A., Lynch D. A., Ryu J. H., Swigris J. J., Wells A. U., Ancochea J., et al. (2011) An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 183, 788–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Rangarajan S., Locy M. L., Luckhardt T. R., and Thannickal V. J. (2016) Targeted therapy for idiopathic pulmonary fibrosis: where to now? Drugs 76, 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Fernandez I. E., and Eickelberg O. (2012) The impact of TGF-β on lung fibrosis: from targeting to biomarkers. Proc. Am. Thorac. Soc. 9, 111–116 [DOI] [PubMed] [Google Scholar]
- 114. Asano Y., Ihn H., Jinnin M., Mimura Y., and Tamaki K. (2006) Involvement of αvβ5 integrin in the establishment of autocrine TGF-β signaling in dermal fibroblasts derived from localized scleroderma. J. Invest. Dermatol. 126, 1761–1769 [DOI] [PubMed] [Google Scholar]
- 115. Ismail S., Sturrock A., Wu P., Cahill B., Norman K., Huecksteadt T., Sanders K., Kennedy T., and Hoidal J. (2009) NOX4 mediates hypoxia-induced proliferation of human pulmonary artery smooth muscle cells: the role of autocrine production of transforming growth factor-β1 and insulin-like growth factor binding protein-3. Am. J. Physiol. Lung Cell Mol. Physiol. 296, L489–L499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Hocevar B. A., Brown T. L., and Howe P. H. (1999) TGF-β induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. EMBO J. 18, 1345–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Gabbiani G. (2003) The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500–503 [DOI] [PubMed] [Google Scholar]
- 118. Zha L., Chen J., Sun S., Mao L., Chu X., Deng H., Cai J., Li X., Liu Z., and Cao W. (2014) Soyasaponins can blunt inflammation by inhibiting the reactive oxygen species-mediated activation of PI3K/Akt/NF-kB pathway. PLoS One 9, e107655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Galuppo M., Esposito E., Mazzon E., Di Paola R., Paterniti I., Impellizzeri D., and Cuzzocrea S. (2011) MEK inhibition suppresses the development of lung fibrosis in the bleomycin model. Naunyn Schmiedebergs Arch. Pharmacol. 384, 21–37 [DOI] [PubMed] [Google Scholar]
- 120. Amara N., Goven D., Prost F., Muloway R., Crestani B., and Boczkowski J. (2010) NOX4/NADPH oxidase expression is increased in pulmonary fibroblasts from patients with idiopathic pulmonary fibrosis and mediates TGFβ1-induced fibroblast differentiation into myofibroblasts. Thorax 65, 733–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Sturrock A., Cahill B., Norman K., Huecksteadt T. P., Hill K., Sanders K., Karwande S. V., Stringham J. C., Bull D. A., Gleich M., Kennedy T. P., and Hoidal J. R. (2006) Transforming growth factor-β1 induces Nox4 NAD(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L661–L673 [DOI] [PubMed] [Google Scholar]
- 122. Jarman E. R., Khambata V. S., Cope C., Jones P., Roger J., Ye L. Y., Duggan N., Head D., Pearce A., Press N. J., Bellenie B., Sohal B., and Jarai G. (2014) An inhibitor of NADPH oxidase-4 attenuates established pulmonary fibrosis in a rodent disease model. Am. J. Respir. Cell Mol. Biol. 50, 158–169 [DOI] [PubMed] [Google Scholar]
- 123. Atmuri V., Martin D. C., Hemming R., Gutsol A., Byers S., Sahebjam S., Thliveris J. A., Mort J. S., Carmona E., Anderson J. E., Dakshinamurti S., and Triggs-Raine B. (2008) Hyaluronidase 3 (HYAL3) knockout mice do not display evidence of hyaluronan accumulation. Matrix Biol. 27, 653–660 [DOI] [PubMed] [Google Scholar]
- 124. Hemming R., Martin D. C., Slominski E., Nagy J. I., Halayko A. J., Pind S., and Triggs-Raine B. (2008) Mouse Hyal3 encodes a 45- to 56-kDa glycoprotein whose overexpression increases hyaluronidase 1 activity in cultured cells. Glycobiology 18, 280–289 [DOI] [PubMed] [Google Scholar]
- 125. Flannery C. R., Little C. B., Hughes C. E., and Caterson B. (1998) Expression and activity of articular cartilage hyaluronidases. Biochem. Biophys. Res. Commun. 251, 824–829 [DOI] [PubMed] [Google Scholar]
- 126. Stern R. (2003) Devising a pathway for hyaluronan catabolism: are we there yet? Glycobiology 13, 105R–115R [DOI] [PubMed] [Google Scholar]
- 127. Deguine V., Menasche M., Ferrari P., Fraisse L., Pouliquen Y., and Robert L. (1998) Free radical depolymerization of hyaluronan by Maillard reaction products: role in liquefaction of aging vitreous. Int. J. Biol. Macromol. 22, 17–22 [DOI] [PubMed] [Google Scholar]
- 128. Saari H. (1991) Oxygen derived free radicals and synovial fluid hyaluronate. Ann. Rheum Dis. 50, 389–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Casalino-Matsuda S. M., Monzon M. E., Conner G. E., Salathe M., and Forteza R. M. (2004) Role of hyaluronan and reactive oxygen species in tissue kallikrein-mediated epidermal growth factor receptor activation in human airways. J. Biol. Chem. 279, 21606–21616 [DOI] [PubMed] [Google Scholar]
- 130. Casalino-Matsuda S. M., Monzón M. E., and Forteza R. M. (2006) Epidermal growth factor receptor activation by epidermal growth factor mediates oxidant-induced goblet cell metaplasia in human airway epithelium. Am. J. Respir. Cell Mol. Biol. 34, 581–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Pascal M., Abdallahi O. M., Elwali N. E., Mergani A., Qurashi M. A., Magzoub M., de Reggi M., and Gharib B. (2000) Hyaluronate levels and markers of oxidative stress in the serum of Sudanese subjects at risk of infection with Schistosoma mansoni. Trans. R. Soc. Trop. Med. Hyg. 94, 66–70 [DOI] [PubMed] [Google Scholar]
- 132. Uchiyama H., Dobashi Y., Ohkouchi K., and Nagasawa K. (1990) Chemical change involved in the oxidative reductive depolymerization of hyaluronic acid. J. Biol. Chem. 265, 7753–7759 [PubMed] [Google Scholar]
- 133. Moeller A., Ask K., Warburton D., Gauldie J., and Kolb M. (2008) The bleomycin animal model: a useful tool to investigate treatment options for idiopathic pulmonary fibrosis? Int. J. Biochem. Cell Biol. 40, 362–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Kessel A., Ammuri H., Peri R., Pavlotzky E. R., Blank M., Shoenfeld Y., and Toubi E. (2007) Intravenous immunoglobulin therapy affects T regulatory cells by increasing their suppressive function. J. Immunol. 179, 5571–5575 [DOI] [PubMed] [Google Scholar]
- 135. McAnulty R. J., and Laurent G. J. (1995) Pathogenesis of lung fibrosis and potential new therapeutic strategies. Exp. Nephrol. 3, 96–107 [PubMed] [Google Scholar]
- 136. Liu S., Tobias R., McClure S., Styba G., Shi Q., and Jackowski G. (1997) Removal of endotoxin from recombinant protein preparations. Clin. Biochem. 30, 455–463 [DOI] [PubMed] [Google Scholar]
- 137. Bogatkevich G. S., Ludwicka-Bradley A., Highland K. B., Hant F., Nietert P. J., Singleton C. B., and Silver R. M. (2007) Down-regulation of collagen and connective tissue growth factor expression with hepatocyte growth factor in lung fibroblasts from white scleroderma patients via two signaling pathways. Arthritis Rheum. 56, 3468–3477 [DOI] [PubMed] [Google Scholar]
- 138. Misra S., Ghatak S., Patil N., Dandawate P., Ambike V., Adsule S., Unni D., Venkateswara Swamy K., and Padhye S. (2013) Novel dual cyclooxygenase and lipoxygenase inhibitors targeting hyaluronan-CD44v6 pathway and inducing cytotoxicity in colon cancer cells. Bioorg. Med. Chem. 21, 2551–2559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Elbashir S. M., Harborth J., Lendeckel W., Yalcin A., Weber K., and Tuschl T. (2001) Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 411, 494–498 [DOI] [PubMed] [Google Scholar]
- 140. Song E., Lee S. K., Wang J., Ince N., Ouyang N., Min J., Chen J., Shankar P., and Lieberman J. (2003) RNA interference targeting Fas protects mice from fulminant hepatitis. Nat. Med. 9, 347–351 [DOI] [PubMed] [Google Scholar]
- 141. Sledz C. A., Holko M., de Veer M. J., Silverman R. H., and Williams B. R. (2003) Activation of the interferon system by short-interfering RNAs. Nat. Cell Biol. 5, 834–839 [DOI] [PubMed] [Google Scholar]
- 142. Bridge A. J., Pebernard S., Ducraux A., Nicoulaz A. L., and Iggo R. (2003) Induction of an interferon response by RNAi vectors in mammalian cells. Nat. Genet. 34, 263–264 [DOI] [PubMed] [Google Scholar]
- 143. Jackson A. L., Bartz S. R., Schelter J., Kobayashi S. V., Burchard J., Mao M., Li B., Cavet G., and Linsley P. S. (2003) Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 21, 635–637 [DOI] [PubMed] [Google Scholar]
- 144. Manche L., Green S. R., Schmedt C., and Mathews M. B. (1992) Interactions between double-stranded RNA regulators and the protein kinase DAI. Mol. Cell Biol. 12, 5238–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Yew N. S., and Scheule R. K. (2005) Toxicity of cationic lipid-DNA complexes. Adv. Genet. 53, 189–214 [PubMed] [Google Scholar]
- 146. Rao D. D., Senzer N., Cleary M. A., and Nemunaitis J. (2009) Comparative assessment of siRNA and shRNA off target effects: what is slowing clinical development. Cancer Gene Ther. 16, 807–809 [DOI] [PubMed] [Google Scholar]
- 147. Rao D. D., Vorhies J. S., Senzer N., and Nemunaitis J. (2009) siRNA vs. shRNA: similarities and differences. Adv. Drug Deliv. Rev. 61, 746–759 [DOI] [PubMed] [Google Scholar]
- 148. Zender L., Hutker S., Liedtke C., Tillmann H. L., Zender S., Mundt B., Waltemathe M., Gosling T., Flemming P., Malek N. P., Trautwein C., Manns M. P., Kuhnel F., and Kubicka S. (2003) Caspase 8 small interfering RNA prevents acute liver failure in mice. Proc. Natl. Acad. Sci. U.S.A. 100, 7797–7802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Roth S. J., Carr M. W., and Springer T. A. (1995) C-C chemokines, but not the C-X-C chemokines interleukin-8 and interferon-γ inducible protein-10, stimulate transendothelial chemotaxis of T lymphocytes. Eur. J. Immunol. 25, 3482–3488 [DOI] [PubMed] [Google Scholar]
- 150. Zhang C., Zhang J., Hao J., Shi Z., Wang Y., Han L., Yu S., You Y., Jiang T., Wang J., Liu M., Pu P., and Kang C. (2012) High level of miR-221/222 confers increased cell invasion and poor prognosis in glioma. J. Transl. Med. 10, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Lawson W. E., Polosukhin V. V., Zoia O., Stathopoulos G. T., Han W., Plieth D., Loyd J. E., Neilson E. G., and Blackwell T. S. (2005) Characterization of fibroblast-specific protein 1 in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 171, 899–907 [DOI] [PubMed] [Google Scholar]
- 152. Tanjore H., Xu X. C., Polosukhin V. V., Degryse A. L., Li B., Han W., Sherrill T. P., Plieth D., Neilson E. G., Blackwell T. S., and Lawson W. E. (2009) Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 180, 657–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Misra S., Hascall V. C., Karamanos N. K., Markwald R. R., and Ghatak S. (2012) Delivery Systems Targeting Cancer at the Level of ECM, pp. 855–872, DeGruyter, Berlin, Germany [Google Scholar]
- 154. Vittal R., Horowitz J. C., Moore B. B., Zhang H., Martinez F. J., Toews G. B., Standiford T. J., and Thannickal V. J. (2005) Modulation of prosurvival signaling in fibroblasts by a protein kinase inhibitor protects against fibrotic tissue injury. Am. J. Pathol. 166, 367–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Hattori N., Degen J. L., Sisson T. H., Liu H., Moore B. B., Pandrangi R. G., Simon R. H., and Drew A. F. (2000) Bleomycin-induced pulmonary fibrosis in fibrinogen-null mice. J. Clin. Invest. 106, 1341–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]