

Abstract
Protein C, a secretory vitamin K-dependent anticoagulant serine protease, inactivates factors Va/VIIIa. It is exclusively synthesized in liver hepatocytes as an inactive zymogen (proprotein C). In humans, thrombin cleavage of the propeptide at PR221↓ results in activated protein C (APC; residues 222–461). However, the propeptide is also cleaved by a furin-like proprotein convertase(s) (PCs) at KKRSHLKR199↓ (underlined basic residues critical for the recognition by PCs), but the order of cleavage is unknown. Herein, we present evidence that at the surface of COS-1 cells, mouse proprotein C is first cleaved by the convertases furin, PC5/6A, and PACE4. In mice, this cleavage occurs at the equivalent site, KKRKILKR198↓, and requires the presence of Arg198 at P1 and a combination of two other basic residues at either P2 (Lys197), P6 (Arg193), or P8 (Lys191) positions. Notably, the thrombin-resistant R221A mutant is still cleaved by these PCs, revealing that convertase cleavage can precede thrombin activation. This conclusion was supported by the fact that the APC-specific activity in the medium of COS-1 cells is exclusively dependent on prior cleavage by the convertases, because both R198A and R221A lack protein C activity. Primary cultures of hepatocytes derived from wild-type or hepatocyte-specific furin, PC5/6, or complete PACE4 knock-out mice suggested that the cleavage of overexpressed proprotein C is predominantly performed by furin intracellularly and by all three proprotein convertases at the cell surface. Indeed, plasma analyses of single-proprotein convertase-knock-out mice showed that loss of the convertase furin or PC5/6 in hepatocytes results in a ∼30% decrease in APC levels, with no significant contribution from PACE4. We conclude that prior convertase cleavage of protein C in hepatocytes is critical for its thrombin activation.
Keywords: furin, hepatocyte, liver, serine protease, thrombin, vitamin K, anticoagulant serine pro, proprotein convertase, protein C, thrombin activation
Introduction
A large number of secretory proteins are produced as precursors that are cleaved at specific sites to generate mature bioactive products. Most of these specific cleavages occur after basic residues and are achieved by one or more of the seven basic amino acid-specific members of the proprotein convertase (PC)3 family, which share identities with bacterial subtilisin and yeast kexin (genes PCSK1 to PCSK7) (1). Four of them (furin, PC5/6, PACE4, and PC7) are widely or ubiquitously expressed and are responsible for most of the processing events at the consensus motif R/K(2X)nR/K↓ (where n = 0, 1, 2, or 3 spacer amino acids (aa); boldface and underlined basic residues critical for the recognition by proprotein convertases) occurring in the constitutive secretory pathway: trans-Golgi network (TGN), cell surface, and/or endosomes (1). This leads to the activation, and less frequently to the inactivation, of receptors, ligands, enzymes, viral glycoproteins, or growth factors (1, 2). Although these PCs exhibit a functional redundancy ex vivo (3), their inactivation in mice leads to specific phenotypes revealing that, in vivo, each PC fulfills unique processing events (1). Furin knock-out (KO) in mice resulted in numerous embryonic malformations, including the absence of axial rotation and heart looping, leading to death around embryonic days 10.5–11 (4). PC5/6 KO leads to death at birth with an altered antero-posterior pattern, including extra vertebrae and lack of tail, kidney agenesis, hemorrhages, collapsed alveoli, and retarded ossification (5, 6). PACE4 KO resulted in an altered left-right patterning, including cyclopism and craniofacial and cardiac malformations in some embryos (7). Finally, PC7 KO mice exhibit anxiolytic and novelty-seeking phenotypes (8).
Human protein C, a natural anticoagulant protein (9), is a circulating vitamin K-dependent serine protease that is mostly expressed in liver hepatocytes and undergoes extensive post-translational modifications, including γ-carboxylation, β-hydroxylation, and N-/O-glycosylation (10–13). It circulates as a two-chain polypeptide with a light N-terminal chain (aa 43–197) and a heavy C-terminal catalytic chain (aa 200–461) connected by a disulfide bond. This zymogen is activated upon cleavage by thrombin, a blood coagulation factor, at Arg221↓, resulting in the removal of the highly acidic activation peptide (aa 200–221) (Fig. 1A). This cleavage activation is greatly enhanced by thrombomodulin and the endothelial protein C receptor (14). Activated protein C and protein S then assemble on the platelet membrane, where the activated form of protein C (APC) proteolytically inactivates the coagulation factors Va and VIIIa. This process regulates blood clotting, inflammation, and cell death and maintains the permeability of blood vessel walls (15, 16). Indeed, APC provides physiologic homeostasis via its antithrombotic and anti-inflammatory actions through its ability to process protease-activated receptor 1 (PAR1) at Arg46↓ to regulate vascular integrity and inflammatory response (17). Interestingly, Arg46 in PAR1 is critical for its ability to inhibit furin in the TGN (18).
Figure 1.

Processing of mouse protein C in COS-1 cells. A, schematic representation of C-terminal V5-tagged mouse protein C is shown, as well as its canonical PCs (Arg198↓) and thrombin (Arg211↓) cleavage sites. Upon its cleavage by PCs, protein C circulates as a disulfide-linked inactive heterodimer of a light N-terminal chain (Pro-domain) and a heavy catalytic chain. We also show the alignment of the mouse and human proprotein C primary sequences around the two cleavage sites. B, COS-1 cells were transiently transfected with an empty vector (V) or vectors encoding the indicated proteins: V5-tagged mouse protein C (mProtein C-V5) and either PC5/6A, furin, PC7, or PACE4. C, COS-1 cells were transiently transfected with V5-tagged mouse protein C and either PC5/6A or furin. At 24 h post-transfection, cells were incubated for 16 h either with fresh medium (control; CTL) or with one containing the cell-permeable RVKR (50 μm) or the cell surface PC inhibitor D6R (20 μm). In both B and C, conditioned media were subjected to Western blotting using a V5-mAb. The migration positions of the full-length protein C (mProtein C) and its CT are shown. These data are representative of at least three independent experiments.
Several studies have indicated that the conversion of the zymogen form of human protein C into the active APC form involves an endoproteolytic cleavage at PR221↓ (Fig. 1A) located at the junction of the light and heavy chains (19, 20). Inspection of the sequences of circulating protein C and of its prodomain in humans and mice (lacking Lys41 in the prodomain compared to human; supplemental Fig. S1) revealed the presence of two putative PC-processing sites, RVRR41↓ and KKRKILKR198↓, upstream from the thrombin PR211↓ cleavage site (Fig. 1A), suggesting that PCs may cleave this prodomain twice. Indeed, in mammalian cell lines, co-expression of human protein C and Kex2p, the yeast homolog of PCs, resulted in cleavage at KKRSHLKR199↓ (21). In addition, engineering of mouse mammary gland to express furin and human protein C led to an efficient conversion of the precursor into the mature active form APC in milk (22). These data indicated that activation of protein C requires cleavages by both a furin-like proprotein convertase(s) and thrombin.
Herein, we used COS-1 cells and mice lacking specifically furin or PC5/6 in hepatocytes (hepatocyte-specific knock-out mice; hKO) (23, 24) to unravel the specificity of these convertases to process mouse protein C. Our data demonstrated that in COS-1 cells, mouse protein C is processed extracellularly mainly by furin, PC5/6A, the soluble isoform of PC5/6, and PACE4. Using an in vitro assay for the protein C activity, we further demonstrated that conversion of protein C to its active APC form by thrombin requires a prior cleavage by convertases at KKRKILKR198↓. Site-directed mutagenesis showed that the P1 Arg198 is critical, as well the presence of two other basic residues at P2, P6, or P8. Finally, mice lacking furin or PC5/6 in hepatocytes exhibit a ∼30% decrease in APC levels in plasma, whereas those completely lacking PACE4 do not show significant changes in circulating APC levels.
Results
Processing of mouse protein C in COS-1 cells
It has been shown previously that upon overexpression of human protein C with furin in mouse mammary gland, it undergoes cleavage at Arg199↓ (22). Such cleavage occurs at the C terminus of a typical basic amino acid PC-like recognition sequence KKRSHLKR199↓ located in a linker region between the light and heavy chain domains of the zymogen proprotein C. This PC-like site is highly conserved between humans and mice (Fig. 1A and supplemental Fig. S1). Because we aimed to analyze the activation of protein C in vivo in mice, we next tested the ability of PCs to process mouse proprotein C.
C-terminally V5-tagged mouse protein C was co-expressed in COS-1 cells with either furin, PC5/6A, PC7, or PACE4 (1). Western blot analysis of the media with a V5-monoclonal antibody (V5-mAb) revealed two forms corresponding to the ∼65-kDa full-length protein and the ∼48-kDa C-terminal (CT) catalytic domain of mouse protein C (Fig. 1B). In COS-1 cells, mouse protein C was processed mainly by PC5/6A and furin, to a lesser extent by PACE4, and not at all by PC7. In addition, processing of mouse protein C by PC5/6A and furin was completely abrogated by two general PC inhibitors: the cell-permeable decanoyl-RVKR-chloromethylketone (RVKR) irreversible inhibitor and the poorly cell-permeable (25) and hence primarily cell surface-reversible inhibitor hexa-d-arginine (D6R) (26) (Fig. 1C). The latter is a very weak inhibitor of PCs within the cells (e.g. Golgi and endosomes (see supplemental Fig. S3 in Ref. 27)) but inhibits very well cell surface PCs (27, 28). These data indicate that, in COS-1 cells, convertase cleavage of mouse protein C occurs almost exclusively at the cell surface, as was the case for the growth factor BMP10 (27).
To support this finding, we performed a similar experiment where we compared the inhibition of the furin processing of proprotein C by RVKR and D6R with that of a potent cell-impermeable protein inhibitor, α1-antitrypsin Portland variant (α1-PDX) (29) (Fig. 2). The data show that whereas incubation of cells with RVKR completely (100%) abrogated the furin processing, incubations with D6R or α1-PDX incompletely inhibited such processing by 80 and 88%, respectively. These data support the notion that in COS-1 cells furin processing occurs mostly at the cell surface.
Figure 2.
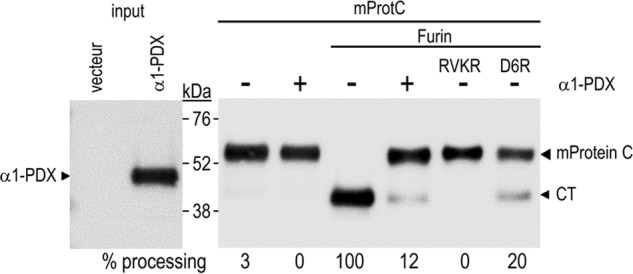
Cellular protein C processing is abrogated by incubation of cells with α1-PDX. The left panel represents the production levels of V5-tagged α1-PDX in COS-1 cells. In the right panel, full-length protein C (mProtein C) (tagged with hexa-His at the C terminus) was co-expressed in COS-1 cells with either empty vector or that encoding human furin, and the cells were then incubated for 16 h with media containing α1-PDX, RVKR (50 μm), or D6R (20 μm). The media were then analyzed for protein C by Western blotting using anti-His tag antibody. Quantifications were done using image analysis and are representative of two independent experiments.
We next wished to define the residues within the PC-like motif that are critical for mouse protein C cleavage. Accordingly, we generated Ala mutants at the P1 (Arg198), P2 (Lys197), P6 (Arg193), and P8 (Arg191) positions (Fig. 3A). The most critical residue occurs at the P1 site, because R198A is completely resistant to processing by PC5/6A (Fig. 3A). Analysis of the other single mutants, K191A, R193A, and K197A, revealed that mutation of the Lys197 at the P2 position into Ala reduced by ∼20% the extent of cleavage. In contrast, the P6 or P8 basic residues are not critical in the presence of the native P1 and P2 residues (Fig. 3A). However, the double mutant P8+P6 (K191A/R193A) or P6+P2 (R193A/K197A) completely prevented PC5/6A from processing mouse protein C (Fig. 3B). We conclude that although the P1 position is the most critical one, an additional combination of two basic residues at P2, P6, or P8 is also needed for maximal cleavage.
Figure 3.

Convertase cleavage of mouse protein C (mProtein C) is thrombin-independent. COS-1 cells were transiently transfected with an empty vector (V) or vectors encoding PC5/6A and WT mouse protein C or its PC cleavage site mutants at positions P1 (R198A), P2 (K197A), P6 (R193A), or P8 (K191A) or a mutant at the thrombin P1 site (R221A). Proteins in 24-h post-transfection conditioned media were subjected to Western blotting using a V5-mAb. A, Western blots indicating the analysis of the single mutants for PC sites and that of thrombin (thr). B, Western blot showing the impact of double Ala mutants (P8+P6 or P6+P2) on the processing of protein C by PC5/6A. Quantitation of the percentage of processing is shown below each lane. These data are representative of two independent experiments.
Processing of mouse protein C by convertases is thrombin-independent
In plasma, human protein C circulates predominantly as a disulfide-linked inactive heterodimer, suggesting an earlier cleavage in its prodomain, probably at the N terminus of the hinge region between the prodomain and the catalytic domain (30). The very acidic (50% Glu/Asp residues) hinge region is composed of 12 or 13 residues in humans or mice, respectively (Fig. 1A). Activation of the circulating form of protein C into APC at the surface of endothelial cells is achieved by thrombin cleavage at Arg221↓. To determine whether thrombin cleavage is required for PCs to process mouse protein C, a thrombin site mutant (R221A) was expressed in COS-1 cells with a control empty vector or with PC5/6A. The data in Fig. 3A show that the R221A mutation does not prevent PC5/6A from processing mouse protein C, indicating that PC cleavage is thrombin-independent.
Protein C activation by thrombin requires prior cleavage by PCs
Because it is difficult to discriminate between PCs and thrombin cleavage products by SDS-PAGE, as they only differ by ∼1.2 kDa, we used an activity test to evaluate the role of PCs in mouse protein C activation into APC. Protein C activity was measured in 24-h conditioned media from COS-1 cells expressing PC5/6A with either WT mouse protein C, its PC-resistant mutant R198A at P1, or its likely thrombin-resistant mutant R221A (Fig. 4A). Thus, we incubated these media with a chromogenic substrate of APC and a protein C activator (ProtAc), which is a snake venom-derived serine proteinase that rapidly converts inactive protein C into APC (31). Based on this assay, cleavage of WT mouse protein C with PC5/6A is associated with a significant increase (∼6.3-fold) of the activity of protein C in the presence of the protein C activator added to the media (Fig. 4B). This activity is not detectable in the R198A mutant, indicating that processing of mouse protein C at Arg198↓ is a prerequisite for protein C activation via thrombin cleavage at Arg221↓. Whereas the thrombin R221A mutant can still be processed by PC5/6A (Figs. 3 and 4), it cannot be activated (Fig. 4B), suggesting that ProtAc has a cleavage preference similar to that of thrombin for Arg221.
Figure 4.
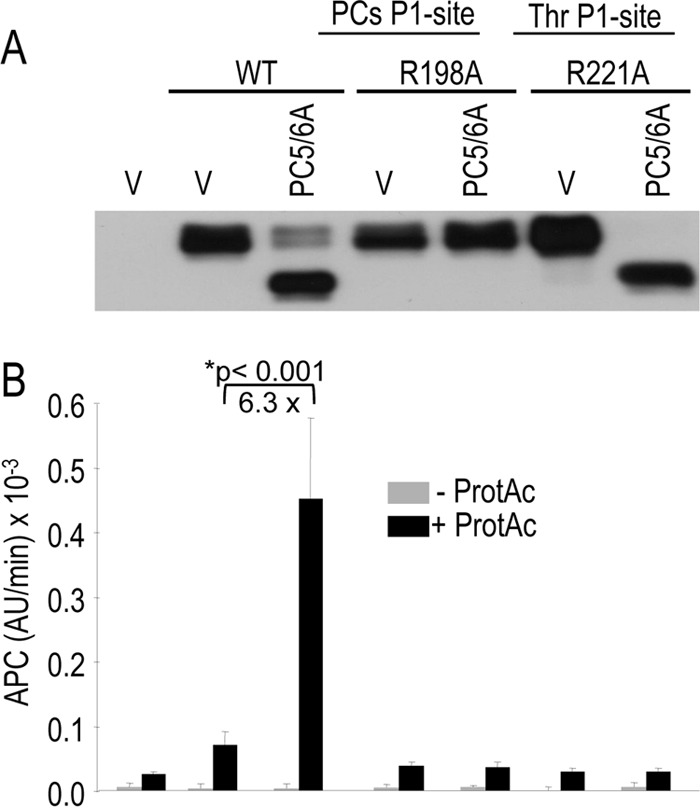
Activation of protein C requires cleavage by a proprotein convertase before that of thrombin. WT protein C and its processing site mutants (PCs site R198A, thrombin site: R221A) were co-expressed in COS-1 cells with either an empty vector or one encoding PC5/6A. A, Western blot showing the production and processing of protein C in 24-h conditioned media. B, protein C activity was assessed using a chromogenic substrate and a ProtAc, and the data are expressed as arbitrary units (AU)/min × 10−3. These data are representative of four independent experiments.
Protein C processing by furin is inhibited by PAR-1
The seven-transmembrane domain PAR-1 is processed by thrombin at Arg41↓ and by APC, PC5/6, and PACE4 at Arg46↓ (Fig. 5A), and the latter residue is critical for its ability to selectively inhibit furin activity, but not that of PC5/6A or PACE4 (18, 32). Thus, we tested the role of PAR-1 in inhibiting the processing of mouse protein C by furin in COS-1 cells. The data in Fig. 5B show that PAR-1 inhibits ∼97% of the endogenous processing of mouse protein C in these cells, suggesting that furin is the major endogenous mouse protein C processing enzyme in COS-1 cells, because PC5/6 and PACE4 cleave PAR-1 and are not inhibited by it (18). Overexpression of furin and mouse protein C in these cells in the presence or absence of overexpressed PAR1 also shows that under these conditions, the latter inhibits by > 50% the furin processing of mouse protein C (Fig. 5B).
Figure 5.
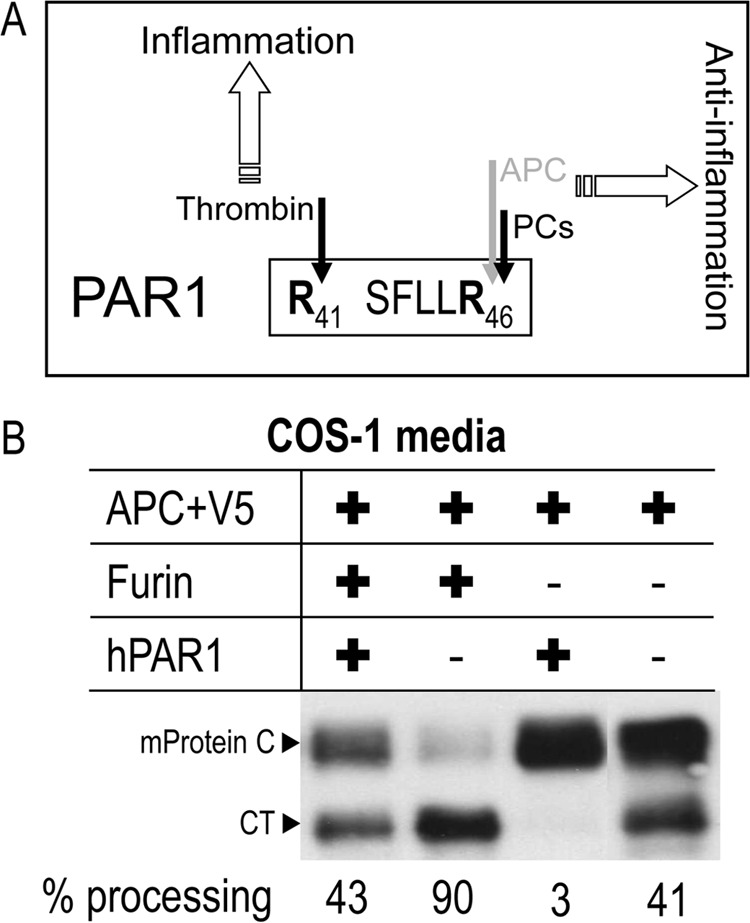
PAR1 inhibits the ability of furin to process protein C. A, schematic representation of PAR1 cleavage sites. B, WT protein C was co-expressed in COS-1 cells with either an empty vector or one encoding furin and/or PAR1, and the 24-h conditioned media were then analyzed by Western blotting using a V5 tag mAb. These data are representative of two independent experiments.
Protein C processing in primary hepatocytes from hepatocyte-specific hKO mice
To evaluate the role of PCs in protein C activation in cultured hepatocytes, primary hepatocytes from WT, PC5/6-hKO, or Fur-hKO mice were transiently transfected with an empty vector or one that codes for V5-tagged mouse protein C, and the cells were then treated with PCs inhibitors. The 48-h conditioned media were analyzed by Western blotting using an anti-V5 antibody (Fig. 6). The CT generated was not eliminated in any of the two hKO hepatocytes analyzed, suggesting a redundancy in the processing of protein C. However, treatment of the cells with the cell-permeable PC inhibitor decanoyl-RVKR-cmk (RVKR) completely eliminated the CT in WT and hKO hepatocytes of the two genotypes, implicating one or more PCs in the generation of the CT fragment. Furthermore, incubation of WT cells with the poorly cell-permeable (25) inhibitor D6R (27, 28), revealed that most of the overexpressed mouse protein C cleavage happens at the cell surface, with only ∼19% occurring intracellularly. Such intracellular processing is primarily due to furin, because it is no longer detectable in hKO cells lacking furin and treated with D6R (Fig. 6). We tried to examine the impact of endogenous convertases on the endogenous processing of mouse protein C using an anti-mouse protein C antibody (Abcam; ab201089). Unfortunately, this antibody does not detect endogenous mouse protein C in the conditioned media of primary hepatocytes, even when the protein is overexpressed, whereas the V5 antibody works very well (not shown). Thus, under conditions of overexpressed mouse protein C, we conclude that in primary hepatocytes and in COS-1 cells (Fig. 1B), furin plays a major extracellular role in mouse protein C cleavage at Arg198↓.
Figure 6.

Processing of mouse protein C (mProtein C) in hepatocytes lacking PC5/6 or furin. Mouse primary hepatocytes isolated from either WT mice or from those lacking PC5/6 (PC5/6-hKO) or furin (Fur-hKO) were transiently transfected with a vector expressing V5-tagged mouse protein C (mProtein C-V5). Cells were treated for 6 h with no inhibitor (−) or with the PC inhibitor RVKR (50 μm) or D6R (20 μm). Media were then replaced with fresh media not containing or containing the inhibitors for an additional 24 h and finally analyzed by Western blotting using a V5-mAb. The percentage of processing is shown below each lane. These data are representative of at least two independent experiments.
In the absence of PC5/6, D6R had a similar inhibitory effect on mouse protein C processing compared with WT cells (Fig. 6; 16% left versus 19%), emphasizing its minor role, if any, in the presence of furin, as previously observed in many furin-like substrates (27, 33). Finally, it should be noted that the absence of PC5/6 in hepatocytes does not affect the mRNA levels of furin, as demonstrated by qPCR analyses of mouse primary hepatocytes isolated from WT and PC5/6 hKO mice (Fig. 7). Altogether, our data demonstrate that, in primary hepatocytes and COS-1 cells, furin is the major endogenous convertase that processes overexpressed mouse protein C at Arg198↓. Under these conditions, such processing primarily occurs at the cell surface, with some activity occurring intracellularly.
Figure 7.
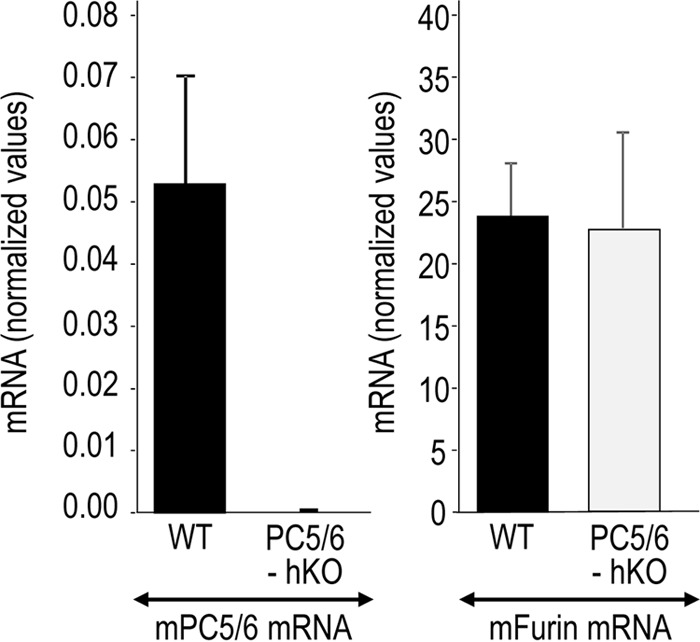
Furin mRNA levels in primary hepatocytes are not affected by the lack of PC5/6. Furin and PC5/6 transcript levels were determined by qPCR in WT and hKO in primary hepatocytes isolated from three different mice for each genotype (WT and PC5/6 hKO mice) and shown as mean ± S.E. (error bars).
Protein C-processing HepG2 cells and mouse primary hepatocytes lacking PACE4
We then assessed the implication of endogenous PACE4 in the processing of APC in the human hepatocyte cell line HepG2. Knockdown of PACE4 in these cells using lentiviral shRNA expression resulted in at least 80% reduction in PACE4 mRNA (Fig. 8A) (34). Accordingly, mouse protein C cDNA transfection of these cells versus control ones expressing unrelated shRNA revealed that reduction of PACE4 decreases the endogenous processing of mouse protein C by ∼24% (Fig. 8B). As in COS-1 cells, the processing of mouse protein C in naive HepG2 cells or those lacking PACE4 also occurs primarily at the cell surface, because treatment with either PC inhibitor RVKR or D6R inhibited >90% of the endogenous processing (Fig. 8B). We conclude that in cell lines, mouse protein C processing occurs primarily at the cell surface of HepG2 and COS-1 cells and that endogenous PACE4 plays a minor role.
Figure 8.
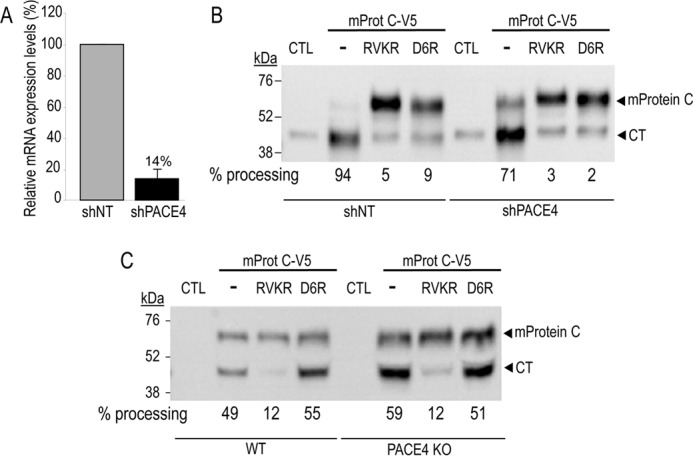
Role of PACE4 in the processing of mouse protein C (mProtein C). Stable knockdown of PACE4 in HepG2 cells was obtained by infection of the cells with a lentivirus encoding for nonspecific shRNA control (non-target; shNT) or an shRNA targeting PACE4 (shPACE4). A, the levels of PACE4 mRNA expression were determined by qPCR analysis and expressed relative to a β-actin control. Note that the PACE4 mRNA levels left after knockdown were 14% of control. ShPACE4 HepG2 cells (B) and primary hepatocytes lacking PACE4 (C) and their control cells were transiently transfected with a control empty vector (CTL) or one encoding V5-tagged mouse protein C (mProtC-V5). The cells were then treated with no inhibitor (−) or with the PC inhibitor RVKR (50 μm) or D6R (20 μm). Media were analyzed by Western blotting using a V5-mAb. The percentage of processing is shown below each lane. The data are representative of two independent experiments.
We next tested the processing of mouse protein C in primary hepatocytes isolated from complete PACE4 KO mice (Fig. 8C). These data supported those obtained in cells lines, in that PACE4 has a minor role in the processing of mouse protein C, which, if anything, is slightly better-processed in the absence of PACE4 (Fig. 8C). We further demonstrated that in livers from mice lacking PACE4, the expression of furin, PC5/6, and PC7 mRNAs does not significantly differ from their WT littermates (supplemental Fig. S2). Overall, these results revealed that PACE4 has a minor, if any, contribution in the processing of mouse protein C in hepatocytes.
In vivo activation of protein C by proprotein convertases
Because the complete furin (4) or PC5/6 (5) KO mice, but not PACE4 KO (7), resulted in lethal phenotypes, we investigated the APC activity in the blood of mice lacking each enzyme from hepatocytes (23), where mouse protein C originates, or from complete PACE4 KO mice. The data demonstrated that the absence of either furin or PC5/6 in hepatocytes results in a ∼30% reduction in circulating APC levels (Fig. 9A), suggesting that in the absence of furin, PC5/6 could partially process mouse protein C. We next analyzed the possibility that PACE4 would also participate in this process in hepatocytes. Because the complete PACE4 KO mice are viable, we assessed the APC activity in the plasma of WT and PACE4 KO mice (Fig. 9B). The data indicated that the absence of PACE4 does not significantly affect the APC activity in vivo. We thus conclude that in vivo furin and PC5/6 are implicated in mouse protein C processing that ultimately leads to its thrombin activation into APC and that PACE4 has a negligible role.
Figure 9.
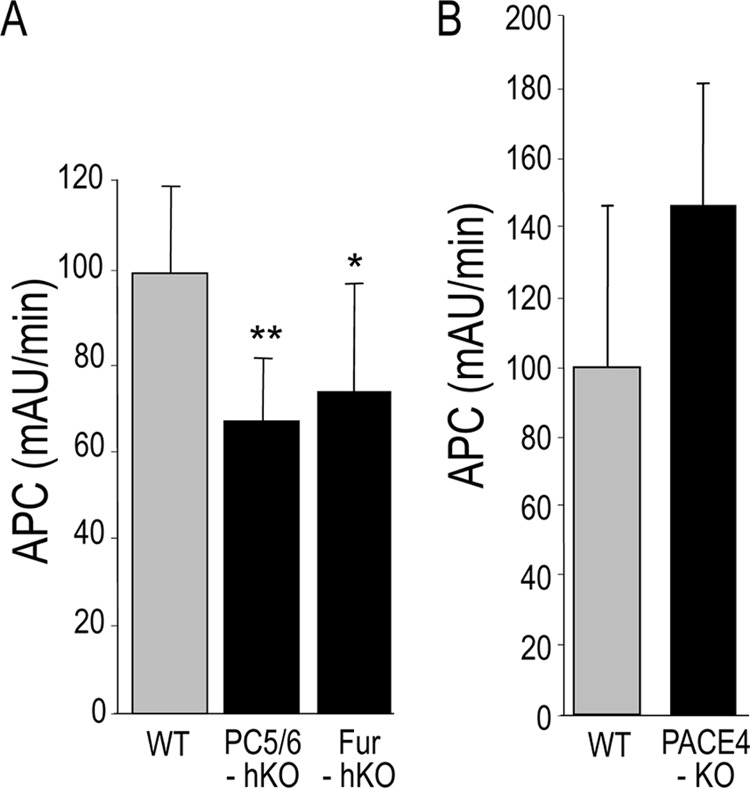
Measurement of protein C activity in the plasma of hepatocyte-specific KO mice (PC5/6-hKO and Fur-hKO) (A) or from complete PACE4-KO mice (B). The sodium citrate treated plasma was obtained from WT and KO mice. The APC activity was measured using a chromogenic substrate and a ProtAc. We used n = 4–8 mice for each experiment, and the values represent the mean ± S.D. (error bars) The data are representative of two independent experiments. AU, arbitrary units.
The coagulation rate in mouse lacking furin or PC5/6 in hepatocytes
To further define the in vivo role of the protein convertases furin and PC5/6 in blood coagulation, the activated partial thromboplastin time (aPTT) and prothrombin time (PT) were measured in blood from mice lacking either furin or PC5/6 in hepatocytes. The aPTT and PT reflect the combined activities of many coagulation factors, including those of APC. Our data show that the aPTT and PT were not significantly affected in Fur-hKO or PC5/6-hKO (supplemental Fig. S3). These data point out the lack of defects in blood clotting in the absence of either furin or PC5/6 in hepatocytes and that the partial reduction of APC processing in the furin hKO (Fig. 6) and the ∼30% reduction in the activity of circulating APC in either KO mice (Fig. 9A) are not enough to affect the blood coagulation rate.
Discussion
Of the ∼20,000 genes that comprise the mammalian genome, it is estimated that ∼20% encode secretory proteins that transit through the lumen of the endoplasmic reticulum to reach their final destination (35). Post-translational modifications of these secretory proteins can be reversible (e.g. phosphorylation, Cys-palmitoylation) or irreversible (e.g. N- and O-glycosylation, sulfation, Glu-γ-carboxylation, and proteolytic cleavage). Such modifications fashion the final form of the protein and often result in an enhanced biological activity and/or stability (36).
Protein C is an extensively modified secretory serine protease originating from liver of which the final active form, APC, is post-translationally modified by N- and O-glycosylations, disulfide bond formation, Glu-γ-carboxylation, Asp-β-hydroxylation, and proteolytic cleavage. The final activation of its circulating zymogen form is greatly enhanced by thrombomodulin and the protein C receptor at the surface of endothelial cells. This subject has been extensively studied (14). However, proteolytic cleavages of its prodomain are relatively less well defined, and the cognate proteases were not unambiguously identified.
Circulating human protein C is mostly inactive and composed of a light and heavy chain linked together by a disulfide bridge (Fig. 1A and supplemental Fig. S1). Sequence analysis of each chain revealed that the light chain starts at Ala43 and the heavy chain starts at Asp200 (30). This suggests that following signal peptidase cleavage of the nascent chain at Gly18↓, the immature human protease is further cleaved within the prodomain at Arg42↓ and Arg199↓ (Fig. 1A and supplemental Fig. S1), which are putative PC cleavage sites occurring at the recognition motif R/K(2X)nR/K↓ (1).
Cleavage at RIRKR42↓ANS generates a light chain with an N-terminal sequence starting with ANS LEE49 close to two critical γ-carboxylated residues (Glu48-Glu49). Interestingly, it was reported that a natural mutation R42S (known as Osaka 10) results in an extra Ser at the N terminus of the light chain that now starts with SANSFLEE49 (37), probably following cleavage at RIRK41↓SANS. This results in much reduced anti-coagulant activity of APC, possibly by affecting either the conformation of the γ-carboxylation-containing prodomain or the efficacy of recognition of the prodomain by the vitamin K-dependent γ-carboxylase (38). Thus, the cleavage of the prodomain by a PC-like enzyme at RIRKR42↓ANS (RVRR41↓ANS in mice; supplemental Fig. S1) seems to be important to ensure optimal conformation of the prodomain implicated in the inactivation of the coagulation factors Va and VIIIa (39). To address this point, we generated two Ala mutants of mouse protein C, namely R41A and R40A/R41A. These should abrogate the PC processing at RVRR41↓ANS. Upon co-expression of furin with either WT protein C or its R41A and R40A/R41A mutants, we did not observe any difference in the processing of these recombinant proteins by furin to generate the CT fragment (Fig. 10). This suggests that processing at KKRKILKR198↓ is independent from that occurring at RVRR41↓. Finally, transgenic mice overexpressing human protein C in mammary glands did not result in APC until furin was co-expressed, revealing that furin (and possibly other PCs) is needed for the activation of protein C in vivo (22).
Figure 10.
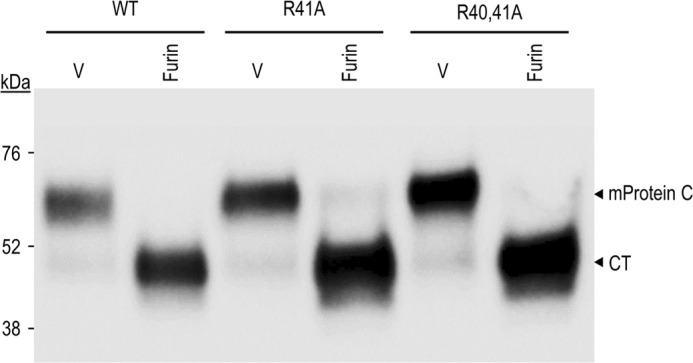
Ala mutation of the putative Arg40-Arg41 prodomain cleavage site does not affect processing of protein C by furin. COS-1 cells were transiently transfected with an empty vector (V) or vectors encoding furin and WT mouse protein C (mProtein C) or its prodomain PC cleavage site mutants R41A and R40A/R41A. Proteins in 24-h post-transfection conditioned media were analyzed by Western blotting using a V5-mAb. The data are representative of at least two independent experiments.
In the present study, we focused on the cleavage occurring at the equivalent site, KKRKILKR198↓, in mouse protein C (Fig. 1A and supplemental Fig. S1). Our data in COS-1 cells revealed that this motif is well-recognized by PC5/6 and furin, less so by PACE4, and not at all by PC7 (Fig. 1B). Cleavage mostly occurs at the cell surface, because it is almost completely inhibited by D6R and α1-PDX (Figs. 1C and 2). Site-directed mutagenesis then showed that whereas the P1 Arg198 is absolutely necessary for cleavage, a combination of two other basic residues at the P2, P6, or P8 positions is also needed for efficacious processing (Fig. 3). Thus, optimal processing of mouse protein C at Arg198↓ requires three basic residues. This property, while rare, is not unique to protein C, because a similar conclusion was also reached in studies of prorenin mutants, whereby in addition to the P1 Arg, at least two of the three basic residues at positions P2, P4, and P6 are required for efficient cleavage, and the presence of all three basic residues results in the most effective processing (40). We also demonstrated that the thrombin site mutant R221A is still well processed by PC5/6 (Fig. 3A). We conclude that processing of mouse protein C at Arg198↓ is independent from and precedes that at Arg221↓.
Finally, the GPCR protein PAR1 is an inhibitor of the membrane-bound furin but not of the soluble PC5/6A and PACE4 (18). The furin-PAR1 complex remains in the TGN and does not exit this compartment (18). In the present work, we also demonstrated that overexpression of PAR1 in COS-1 cells completely abrogated the processing of mouse protein C by endogenous furin and inhibited by ∼50% that generated by overexpressed furin (Fig. 5).
We next investigated the importance of a prior PC cleavage for the subsequent thrombin cleavage/activation of mouse protein C at Arg221↓ into APC (Fig. 1A). The data in COS-1 cells revealed that cleavage at Arg198↓ is a prerequisite for the subsequent thrombin activation of mouse protein C into APC, because the P1 mutant R198A is not activated by the thrombin-like protein C activator ProtAc (Fig. 4). Convertase cleavage of mouse protein C results in the exposure of a 12–13-mer activation peptide at the N terminus of human and mouse protein C, respectively (Fig. 1A). Thus, as long as the activation peptide is not free from the N-terminal prodomain, it prevents thrombin cleavage at PR221↓. So far, no crystal structure is available for proprotein C, but only for convertase-cleaved protein C. It is a matter of speculation whether in proprotein C, the highly basic C terminus of the prodomain (e.g. RWIEKKRKILKR198 in mice) interacts with the highly acidic activation peptide and possibly hinders the thrombin cleavage site at its C terminus (e.g. DTDLEDELEPDPR221 in mice), thereby preventing its processing by thrombin.
We then extended the above studies to mouse primary hepatocytes isolated from either Fur-hKO or PC5/6-hKO. As in COS-1 cells, processing of mouse protein C is afforded by PC-like convertases, because the pan-PC inhibitor RVKR completely blocks its processing by endogenous convertases (Fig. 6). In these cells, mouse protein C is cleaved mostly at the cell surface and less so intracellularly. The extent of intracellular processing is achieved only by endogenous furin, whereas PC5/6 seems to exclusively process mouse protein C at the cell surface in the absence of furin (Fig. 6). These data are in agreement with the observation that the zymogen activation of furin occurs in the TGN from which the active enzyme cycles to the cell surface and back (41), whereas PC5/6A is mostly active at the cell surface (1, 33). The contribution of the intracellular furin cleavage is probably related to the different cell types, because we reported that pro-BMP10 is processed exclusively by furin at the cell surface of COS-1 cells, like mouse protein C, but at the cell surface and intracellularly in HEK293 cells (27). Whether the difference is due to the recycling rate of furin in these cells from the cell surface to the TGN that is regulated by PACS1 (41) or via some other mechanism is unknown.
We also tested the contribution of PACE4 to mouse protein C processing in control HepG2 cells and in cells where the expression of PACE4 mRNA was effectively knocked down (Fig. 8A). The data showed that whereas processing of overexpressed mouse protein C occurred mostly at the cell surface, as in COS-1 cells, the contribution of PACE4 was not important (Fig. 8B). We corroborated this conclusion in primary hepatocytes isolated from wild-type and complete PACE4 KO mice (Fig. 8C).
We next assessed the effect of the loss of either furin, PC5/6, or PACE4 on the activity of circulating mouse APC. The data showed that furin-hKO and PC5/6-hKO mice each exhibited a ∼30% reduction in the levels of circulating APC activity (Fig. 9A). In contrast, analysis of PACE4 total KO mice revealed no significant difference in circulating APC activity (Fig. 9B). Thus, for mouse protein C activation, these data suggest the presence of a partial redundancy between furin and PC5/6 in hepatocytes, but not PACE4, as was also previously suggested from studies of furin-hKO mice with other liver substrates (24, 42). However, the absence of furin or PC5/6 from hepatocytes may not preclude processing of immature circulating proprotein C at the cell surface of other tissues (e.g. endothelial cells) that express both enzymes (43, 44).
In furin-hKO and PC5/6-hKO mice, we observed that the aPTT blood coagulation time was not significantly altered (supplemental Fig. S3). This may be due to the fact that APC activity was only reduced by ∼30% in each hKO genotype and that processing of mouse protein C by other tissues is also possible. Indeed, it has been reported that heterozygote mouse protein C KO mice expressing 50% mouse protein C levels do not exhibit a coagulation defect, whereas mice expressing 18% of normal mouse protein C levels showed ∼50% increased aPTT (45). This means that APC levels significantly lower than 50% are needed to observe an increased coagulation time.
In conclusion, we clearly demonstrated that the convertases furin and PC5/6 contribute to the processing and activation of mouse protein C, through cleavage at KR198↓ in mice and probably KR199↓ in humans. Interestingly, very rare SNPs K198Q (rs765517843) and R199Q (rs773327173) have been identified in the human genome SNP database (https://www.ncbi.nlm.nih.gov/projects/SNP/, gene ID 5624), where all known protein C (gene 5624) SNPs are described. In view of the critical importance of the human P1 Arg199 and the redundancy of the P2 Lys198, P6 Arg194, and P8 Lys192, our data suggest that the subject exhibiting the R199Q mutant may have more severe blood coagulation or inflammation defects as compared with the K198Q mutant. Finally, high scale production of the Protac-activated APC may be achieved either by co-expression of proprotein C with membrane-bound furin or better still by its co-expression with the soluble PC5/6A, which, like APC, activates the cytoprotective/anti-inflammatory activity of PAR-1 by cleaving it at Arg46↓ (18).
Experimental procedures
Western blotting and antibodies
The V5-tagged protein C in culture media of transfected COS-1 cells (27) was analyzed by Western blotting using a V5-mAb (1:5000; Invitrogen). A commercial anti-mouse protein C antibody (Abcam, ab201089) was also tested. The antigen-antibody complexes were visualized using appropriate HRP-conjugated secondary antibodies and an enhanced chemiluminescence kit (ECL, Amersham Biosciences or Pierce), as described (23, 24).
Inhibitor treatment
Twenty-four hours post-transfection, hepatocytes were washed, and culture media were then replaced with a fresh serum-free medium containing or not the specified inhibitors for 5 h (pretreatment period). Subsequently, the media were replaced with fresh ones containing or not the specified inhibitors, and the cells were incubated for an additional 12–16 h. The media were then analyzed for processing by Western blotting. The inhibitors consisted of the pan-PC inhibitor RVKR (50 μm) or D6R (20 μm) (26), a cell-surface PC inhibitor (27), or α1-PDX-V5 produced in the media of COS-1 cells.
Protein C activity assay
Protein C activity was determined using a Coamatic® protein C activity assay kit, as recommended by the manufacturer (Chromogenix, catalog no. 82209863). Mouse plasma obtained from blood collected under 0.01 m sodium citrate or conditioned media from COS-1 cells expressing protein C were preincubated for 1 h at 37 °C with 50 μl of protein C activator (ProtAC, from the above kit) or water control. This was followed by the addition of 50 μl of the APC substrate (pyro-Glu-Pro-Arg-p-nitroaniline; S-2366 from the above kit), and the rate of hydrolysis was measured over 4 h at 405 nm on a spectrophotometer (SpectraMax i3, Molecular Devices). Comparison with a standard curve allowed the estimation of the amount of APC.
PACE4 stable shRNA knockdown in HepG2 cells
HepG2 cells were plated, 1 day before infection, at 200,000 cells/well in a 6-well plate in complete (10% FBS) Eagle's minimum essential medium. Lentiviral infection was performed as described previously (46) using a multiplicity of infection of 3 in the presence of 4 μg/μl Polybrene over a period of 24 h. Puromycin (2 μg/ml) was used for stable selection 48 h post-infection. RNA isolation was performed using RNeasy kit (Qiagen), and qPCR was carried out with RNA samples (1 μg) using SYBR Green and qPCR system Mx3005p (Stratagene). The primer sequences were 5′-CACCTGCTAGTGAAGACATCC-3′ and 5′-AACGAGAGCTTCTGCGTCCAC-3′ for hPACE4 forward and reverse oligonucleotides, respectively.
Quantitative PCR analysis of mouse primary hepatocytes
RNA from primary hepatocytes isolated from three different mice of each genotype (WT and hPC5/6 KO mice) was extracted as recommended by the manufacturer (Invitrogen), and cDNA synthesis and qPCR were performed as described previously (47). Specific primers in different exons were used to amplify mRNAs: PC5/6 sense and antisense primers, 5′-TGACCACTCTTCAGAGAATGGATAC-3′ and 5′-GAGATACCCACTAGGGCAGC-3′; furin sense and antisense primers, 5′-CATGACTACTCTGCTGATGG-3′ and 5′-GAACGAGAGTGAACTTGGTC-3′; S16 sense and antisense primers, 5′-GCTACCAGGGCCTTTGAGATG-3′ and 5′-AGGAGCGATTTGCTGGTGTGG-3′.
Mice and genotyping
The Furinflox/flox (42) or Pcsk5flox/flox (5) mice carrying or not a copy of Tg(Alb-cre) (23) were described previously. The complete PACE4 knock-out mice used were those reported previously (24). Mice were housed in a 12-h light/dark cycle and fed a standard diet (2018 Teklad global 18% protein rodent diet, Harlan Laboratories). All procedures were approved by the Institut de Recherches Cliniques de Montréal bioethics committee for animal care.
Preparation of primary hepatocytes, culture, and transfection
Hepatocytes were prepared from 8–12-week-old male livers using a two-step collagenase perfusion method (23). After anesthesia of mice using 2% isoflurane inhalation, the peritoneal cavity was opened, and the liver was perfused in situ via the inferior vena cava for 6 min at 37 °C with calcium-free HEPES buffer-I (142 mm NaCl, 6.7 mm KCl, 10 mm Hepes, pH 7.6), and for 8 min with calcium-supplemented HEPES buffer-II (4.7 mm CaCl2, 66.7 mm NaCl, 6.7 mm KCl, 100 mm Hepes, pH 7.4) containing 0.5 mg/ml collagenase type V (Sigma-Aldrich). The perfusion rates were set to 8 and 6 ml/min, respectively. In 3.5-cm Petri dishes coated with fibronectin (0.5 mg/ml; Sigma-Aldrich), 5 × 105 cells were seeded in Williams' medium E supplemented with 10% fetal bovine serum (Gibco BRL). After 2 h, the medium was replaced with hepatozyme medium (Gibco BRL) for 12 h before treatment. All primary hepatocytes transfections were performed using 6 μg of DNA and Effectene transfection reagent, as recommended by the manufacturer (Qiagen). All results are representative of at least two independent experiments.
Mouse bleeding and coagulation assay
Mice were fasted for 4 h before final bleeding by heart puncture. Both the PT and PTT measurements were carried out with a BCS coagulation analyzer (Dade Behring, Marburg, Germany). The samples were collected in sodium citrate BD tubes (BD Biosciences). The plasma was then analyzed at 37 °C. An excess calcium was added to reverse the effects of citrate in the case of PT, enabling the blood to clot again. To activate the intrinsic pathway of coagulation to assess PTT, phospholipid, silica activator, and calcium were mixed into the plasma samples, as routinely done in hematology clinics (48). The time required for clot formation was measured photo-optically. The PT and PTT results were reported as the time required for clot formation after the addition of calcium.
Author contributions
R. E. conducted most of the experiments and analyzed the results. D. S.-R. determined the enzymatic activity of protein C. J. G. participated in hepatocyte primary culture and transfection. M.-C. A. was responsible for all cell cultures. Western blots on the PAR1 and protein were performed by K. W. J. and V. S., whereas A. C. did mouse genotyping and qPCR. Z. A. participated in the coagulation test. K. L., R. Desjardins, and R. Day provided the HepG2 cells PACE4 and PACE4 KO mice. A. P. and N. G. S. analyzed the data and wrote the manuscript with R. E.
Supplementary Material
Acknowledgments
We are grateful to all of the members of the Seidah laboratory for helpful discussions and to Mathieu Ferron (Institut de Recherches Cliniques de Montréal) for insights into γ-carboxylation as well as to Brigitte Mary for efficacious editorial assistance.
This work was supported by CIHR Foundation Scheme Grant 148363 and Canada Research Chair 216684. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S3.
- PC
- proprotein convertase
- APC
- active protein C
- TGN
- trans-Golgi network
- hKO
- hepatocyte-specific knockout
- qPCR
- quantitative RT-PCR
- CT
- C-terminal fragment
- RVKR
- decanoyl-RVKR-chloromethylketone
- D6R
- hexa-d-arginine
- ProtAc
- protein C activator
- PT
- prothrombin time
- PTT
- partial thromboplastin time
- aPTT
- activated partial thromboplastin time
- α1-PDX
- α1-antitrypsin Portland variant.
References
- 1. Seidah N. G., and Prat A. (2012) The biology and therapeutic targeting of the proprotein convertases. Nat. Rev. Drug Discov. 11, 367–383 [DOI] [PubMed] [Google Scholar]
- 2. Seidah N. G., Sadr M. S., Chrétien M., and Mbikay M. (2013) The multifaceted proprotein convertases: their unique, redundant, complementary, and opposite functions. J. Biol. Chem. 288, 21473–21481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Seidah N. G., Mayer G., Zaid A., Rousselet E., Nassoury N., Poirier S., Essalmani R., and Prat A. (2008) The activation and physiological functions of the proprotein convertases. Int. J. Biochem. Cell Biol. 40, 1111–1125 [DOI] [PubMed] [Google Scholar]
- 4. Roebroek A. J., Umans L., Pauli I. G., Robertson E. J., van Leuven F., Van de Ven W. J., and Constam D. B. (1998) Failure of ventral closure and axial rotation in embryos lacking the proprotein convertase furin. Development 125, 4863–4876 [DOI] [PubMed] [Google Scholar]
- 5. Essalmani R., Zaid A., Marcinkiewicz J., Chamberland A., Pasquato A., Seidah N. G., and Prat A. (2008) In vivo functions of the proprotein convertase PC5/6 during mouse development: Gdf11 is a likely substrate. Proc. Natl. Acad. Sci. U.S.A. 105, 5750–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Szumska D., Pieles G., Essalmani R., Bilski M., Mesnard D., Kaur K., Franklyn A., El Omari K., Jefferis J., Bentham J., Taylor J. M., Schneider J. E., Arnold S. J., Johnson P., Tymowska-Lalanne Z., et al. (2008) VACTERL/caudal regression/Currarino syndrome-like malformations in mice with mutation in the proprotein convertase Pcsk5. Genes Dev. 22, 1465–1477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Constam D. B., and Robertson E. J. (2000) SPC4/PACE4 regulates a TGFβ signaling network during axis formation. Genes Dev. 14, 1146–1155 [PMC free article] [PubMed] [Google Scholar]
- 8. Wetsel W. C., Rodriguiz R. M., Guillemot J., Rousselet E., Essalmani R., Kim I. H., Bryant J. C., Marcinkiewicz J., Desjardins R., Day R., Constam D. B., Prat A., and Seidah N. G. (2013) Disruption of the expression of the proprotein convertase PC7 reduces BDNF production and affects learning and memory in mice. Proc. Natl. Acad. Sci. U.S.A. 110, 17362–17367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Esmon C. T. (1987) The regulation of natural anticoagulant pathways. Science 235, 1348–1352 [DOI] [PubMed] [Google Scholar]
- 10. DiScipio R. G., and Davie E. W. (1979) Characterization of protein S, a γ-carboxyglutamic acid containing protein from bovine and human plasma. Biochemistry 18, 899–904 [DOI] [PubMed] [Google Scholar]
- 11. Fernlund P., and Stenflo J. (1982) Amino acid sequence of the light chain of bovine protein C. J. Biol. Chem. 257, 12170–12179 [PubMed] [Google Scholar]
- 12. McMullen B. A., Fujikawa K., and Kisiel W. (1983) The occurrence of β-hydroxyaspartic acid in the vitamin K-dependent blood coagulation zymogens. Biochem. Biophys. Res. Commun. 115, 8–14 [DOI] [PubMed] [Google Scholar]
- 13. Foster D. C., Rudinski M. S., Schach B. G., Berkner K. L., Kumar A. A., Hagen F. S., Sprecher C. A., Insley M. Y., and Davie E. W. (1987) Propeptide of human protein C is necessary for γ-carboxylation. Biochemistry 26, 7003–7011 [DOI] [PubMed] [Google Scholar]
- 14. Navarro S., Bonet E., Estellés A., Montes R., Hermida J., Martos L., España F., and Medina P. (2011) The endothelial cell protein C receptor: its role in thrombosis. Thromb. Res. 128, 410–416 [DOI] [PubMed] [Google Scholar]
- 15. Griffin J. H., Fernández J. A., Gale A. J., and Mosnier L. O. (2007) Activated protein C. J. Thromb. Haemost. 5, 73–80 [DOI] [PubMed] [Google Scholar]
- 16. Jackson C. J., and Xue M. (2008) Activated protein C: an anticoagulant that does more than stop clots. Int. J. Biochem. Cell Biol. 40, 2692–2697 [DOI] [PubMed] [Google Scholar]
- 17. Riewald M., Petrovan R. J., Donner A., Mueller B. M., and Ruf W. (2002) Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 296, 1880–1882 [DOI] [PubMed] [Google Scholar]
- 18. Kim W., Zekas E., Lodge R., Susan-Resiga D., Marcinkiewicz E., Essalmani R., Mihara K., Ramachandran R., Asahchop E., Gelman B., Cohen É. A., Power C., Hollenberg M. D., and Seidah N. G. (2015) Neuroinflammation-induced interactions between protease-activated receptor 1 and proprotein convertases in HIV-associated neurocognitive disorder. Mol. Cell Biol. 35, 3684–3700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grinnell B. W., Gerlitz B., and Berg D. T. (1994) Identification of a region in protein C involved in thrombomodulin-stimulated activation by thrombin: potential repulsion at anion-binding site I in thrombin. Biochem. J. 303, 929–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Foster D. C., Sprecher C. A., Holly R. D., Gambee J. E., Walker K. M., and Kumar A. A. (1990) Endoproteolytic processing of the dibasic cleavage site in the human protein C precursor in transfected mammalian cells: effects of sequence alterations on efficiency of cleavage. Biochemistry 29, 347–354 [DOI] [PubMed] [Google Scholar]
- 21. Foster D. C., Holly R. D., Sprecher C. A., Walker K. M., and Kumar A. A. (1991) Endoproteolytic processing of the human protein C precursor by the yeast Kex2 endopeptidase coexpressed in mammalian cells. Biochemistry 30, 367–372 [DOI] [PubMed] [Google Scholar]
- 22. Drews R., Paleyanda R. K., Lee T. K., Chang R. R., Rehemtulla A., Kaufman R. J., Drohan W. N., and Luboń H. (1995) Proteolytic maturation of protein C upon engineering the mouse mammary gland to express furin. Proc. Natl. Acad. Sci. U.S.A. 92, 10462–10466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Essalmani R., Susan-Resiga D., Chamberland A., Abifadel M., Creemers J. W., Boileau C., Seidah N. G., and Prat A. (2011) In vivo evidence that furin from hepatocytes inactivates PCSK9. J. Biol. Chem. 286, 4257–4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Essalmani R., Susan-Resiga D., Chamberland A., Asselin M.-C., Canuel M., Constam D., Creemers J. W., Day R., Gauthier D., Prat A., and Seidah N. G. (2013) Furin is the primary in vivo convertase of angiopoietin-like 3 and endothelial lipase in hepatocytes. J. Biol. Chem. 288, 26410–26418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wender P. A., Mitchell D. J., Pattabiraman K., Pelkey E. T., Steinman L., and Rothbard J. B. (2000) The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc. Natl. Acad. Sci. U.S.A. 97, 13003–13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peinado J. R., Kacprzak M. M., Leppla S. H., and Lindberg I. (2004) Cross-inhibition between furin and lethal factor inhibitors. Biochem. Biophys. Res. Commun. 321, 601–605 [DOI] [PubMed] [Google Scholar]
- 27. Susan-Resiga D., Essalmani R., Hamelin J., Asselin M. C., Benjannet S., Chamberland A., Day R., Szumska D., Constam D., Bhattacharya S., Prat A., and Seidah N. G. (2011) Furin is the major processing enzyme of the cardiac-specific growth factor bone morphogenetic protein 10. J. Biol. Chem. 286, 22785–22794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Guillemot J., Canuel M., Essalmani R., Prat A., and Seidah N. G. (2013) Implication of the proprotein convertases in iron homeostasis: proprotein convertase 7 sheds human transferrin receptor 1 and furin activates hepcidin. Hepatology 57, 2514–2524 [DOI] [PubMed] [Google Scholar]
- 29. Anderson E. D., Thomas L., Hayflick J. S., and Thomas G. (1993) Inhibition of HIV-1 gp160-dependent membrane fusion by a furin-directed α 1-antitrypsin variant. J. Biol. Chem. 268, 24887–24891 [PubMed] [Google Scholar]
- 30. Kisiel W. (1979) Human plasma protein C: isolation, characterization, and mechanism of activation by α-thrombin. J. Clin. Invest. 64, 761–769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gempeler-Messina P. M., Volz K., Bühler B., and Müller C. (2001) protein C activators from snake venoms and their diagnostic use. Haemostasis 31, 266–272 [DOI] [PubMed] [Google Scholar]
- 32. Schuepbach R. A., Madon J., Ender M., Galli P., and Riewald M. (2012) Protease-activated receptor-1 cleaved at R46 mediates cytoprotective effects. J. Thromb. Haemost. 10, 1675–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mayer G., Hamelin J., Asselin M. C., Pasquato A., Marcinkiewicz E., Tang M., Tabibzadeh S., and Seidah N. G. (2008) The regulated cell surface zymogen activation of the proprotein convertase PC5A directs the processing of its secretory substrates. J. Biol. Chem. 283, 2373–2384 [DOI] [PubMed] [Google Scholar]
- 34. Couture F., Ly K., Levesque C., Kwiatkowska A., Ait-Mohand S., Desjardins R., Guérin B., and Day R. (2015) Multi-Leu PACE4 inhibitor retention within cells is PACE4 dependent and a prerequisite for antiproliferative activity. Biomed. Res. Int. 2015, 824014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ladunga I. (2000) Large-scale predictions of secretory proteins from mammalian genomic and EST sequences. Curr. Opin. Biotechnol. 11, 13–18 [DOI] [PubMed] [Google Scholar]
- 36. Seidah NG and Guillemot J. (2016) Posttranslational Processing of Secretory Proteins. In Molecular Neuroendocrinology: From Genome to Physiology (Murphy D., and Gainer H., eds) pp. 171–193, Wiley-Blackwell, New York [Google Scholar]
- 37. Miyata T., Zheng Y. Z., Sakata T., and Kato H. (1995) Protein C Osaka 10 with aberrant propeptide processing: loss of anticoagulant activity due to an amino acid substitution in the protein C precursor. Thromb. Haemost. 74, 1003–1008 [PubMed] [Google Scholar]
- 38. Gandrille S., Alhenc-Gelas M., Gaussem P., Aillaud M. F., Dupuy E., Juhan-Vague I., and Aiach M. (1993) Five novel mutations located in exons III and IX of the protein C gene in patients presenting with defective protein C anticoagulant activity. Blood 82, 159–168 [PubMed] [Google Scholar]
- 39. Preston R. J., Ajzner E., Razzari C., Karageorgi S., Dua S., Dahlbäck B., and Lane D. A. (2006) Multifunctional specificity of the protein C/activated protein C Gla domain. J. Biol. Chem. 281, 28850–28857 [DOI] [PubMed] [Google Scholar]
- 40. Watanabe T., Murakami K., and Nakayama K. (1993) Positional and additive effects of basic amino acids on processing of precursor proteins within the constitutive secretory pathway. FEBS Lett. 320, 215–218 [DOI] [PubMed] [Google Scholar]
- 41. Thomas G. (2002) Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 3, 753–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roebroek A. J., Taylor N. A., Louagie E., Pauli I., Smeijers L., Snellinx A., Lauwers A., Van de Ven W. J., Hartmann D., and Creemers J. W. (2004) Limited redundancy of the proprotein convertase furin in mouse liver. J. Biol. Chem. 279, 53442–53450 [DOI] [PubMed] [Google Scholar]
- 43. Marchesi C., Essalmani R., Lemarié C. A., Leibovitz E., Ebrahimian T., Paradis P., Seidah N. G., Schiffrin E. L., and Prat A. (2011) Inactivation of endothelial proprotein convertase 5/6 decreases collagen deposition in the cardiovascular system: role of fibroblast autophagy. J. Mol. Med. 89, 1103–1111 [DOI] [PubMed] [Google Scholar]
- 44. Kim W., Essalmani R., Szumska D., Creemers J. W., Roebroek A. J., D'Orleans-Juste P., Bhattacharya S., Seidah N. G., and Prat A. (2012) Loss of endothelial furin leads to cardiac malformation and early postnatal death. Mol. Cell Biol. 32, 3382–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lay A. J., Liang Z., Rosen E. D., and Castellino F. J. (2005) Mice with a severe deficiency in protein C display prothrombotic and proinflammatory phenotypes and compromised maternal reproductive capabilities. J. Clin. Invest. 115, 1552–1561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ly K., Saavedra Y. G., Canuel M., Routhier S., Desjardins R., Hamelin J., Mayne J., Lazure C., Seidah N. G., and Day R. (2014) Annexin A2 reduces PCSK9 protein levels via a translational mechanism and interacts with the M1 and M2 domains of PCSK9. J. Biol. Chem. 289, 17732–17746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Essalmani R., Hamelin J., Marcinkiewicz J., Chamberland A., Mbikay M., Chrétien M., Seidah N. G., and Prat A. (2006) Deletion of the gene encoding proprotein convertase 5/6 causes early embryonic lethality in the mouse. Mol. Cell Biol. 26, 354–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Junker R., Käse M., Schulte H., Bäumer R., Langer C., and Nowak-Göttl U. (2005) Interferences in coagulation tests: evaluation of the 570-nm method on the Dade Behring BCS analyser. Clin. Chem. Lab. Med. 43, 244–252 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.